Program
ENZYMOLOGIA
Biotechnologia, III rok
W ramach zajęć odbędą się:
Wyznaczanie parametrów kinetycznych (KM, kcat, kcat/KM) hydrolizy syntetycznego substratu (BApNA) katalizowanej przez trypsynę.
Wyznaczanie stężenia aktywnego inhibitora trypsyny (BPTI) metodą miareczkowania mianowanej trypsyny za pomocą inhibitora.
Pomiar stałej asocjacji (Ka) oddziaływania chymotrypsyna - BPTI metodą oznaczania resztkowej aktywności enzymatycznej chymotrypsyny.
Specyficzna, chemiczna modyfikacja łańcuchów bocznych reszt Ser w trypsynie i chymotrypsynie za pomocą PMSF (fenylometylosulfofluorku) i TLCK oraz jej wpływ na aktywność enzymu.
Wyznaczanie parametrów kinetycznych (KM, kcat, kcat/KM) hydrolizy syntetycznego substratu (BApNA) katalizowanego przez trypsynę
WSTĘP:
Parametry kinetyczne reakcji katalizowanych enzymatycznie są charakterystyczne dla danej pary enzym - substrat i warunków prowadzenia reakcji. Wyznacza się je w celu porównania zdolności katalitycznych różnych enzymów i/lub podatności różnych substratów na katalizę.
Celem ćwiczenia jest wyznaczenie parametrów:
stałej Michaelisa (KM), stałej szybkości reakcji (kcat) i stałej specyficzności reakcji (kcat/KM), katalizowanego przez β-trypsynę hydrolitycznego rozkładu syntetycznego, drobnocząsteczkowego substratu (Nα-Benzoilo-DL-Arginylo-p-Nitroanilidu - BApNA). Trypsyna w obojętnym lub lekko zasadowym środowisku efektywnie przyspiesza hydrolizę BApNA do Nα-benzoilo-DL-argininy i p-nitroaniliny (pNA), której ilość oznacza się spektrofotometrycznie przez pomiar absorpcji światła o długości fali równej - λ = 405 nm (εpNA405nm = 9620 M-1 cm-1, Mr = 434,89 g/mol).
W tym celu dokonujemy pomiaru prędkości początkowej reakcji enzymatycznej w funkcji stężenia substratu (v = f([BApNA]), w warunkach dużego nadmiaru substratu w stosunku do enzymu ([BApNA] >> [E0]) i przy stałym stężeniu enzymu ([E0] = constant). Początkowa prędkość reakcji enzymatycznej jest wyznaczana jako przyrost ilości jednego z produktów reakcji - p-nitroaniliny (pNA) w czasie - (v = δ{pNA}/δt). Do otrzymanych doświadczalnie punktów dopasowuje się krzywą zgodnie w modelem (równaniem) Michaelisa-Menten i wyznacza parametry równania, jakimi są stała Michaelisa (KM) i prędkość maksymalna reakcji (vmax). kcat wyznacza się jako iloraz prędkości maksymalnej i stężenia enzymu - kcat = vmax/[E0].
Wykonanie ćwiczenia:
W ramach ćwiczenia należy: przygotować niezbędne odczynniki, wyznaczyć dokładnie stężenia roztworów enzymu (β-trypsyny - [E0]) i substratu ([BApNA]), przeprowadzić właściwe doświadczenie i oszacować żądane parametry.
Przygotowanie odczynników:
100 ml buforu weronalowego: 0.1 M weronal, 20 mM CaCl2, pH = 8.3 - na całą grupę ćwiczeniową
200 ml buforu reakcyjnego: 0.1 M TRIS, 20 mM CaCl2, 5% DMSO, pH = 8.3 - na całą grupę ćwiczeniową
20 ml 0.001M HCl, 20 mM CaCl2 - na całą grupę ćwiczeniową
roztwór trypsyny - naważkę około 50 mg trypsyny rozpuścić w 1ml 1 mM HCl, 20 mM CaCl2; podczas ćwiczeń przechowywać na lodzie lub w lodówce, po wykonaniu ćwiczenia rozpipetować po 100 μl do ependorfek dokładnie opisać (stężenie) i zamrozić. Roztwór będzie wykorzystywany na kolejnych ćwiczeniach.- przygotowuje każdy zespół.
Roztwór BApNA - naważkę około 60 mg rozpuścić w około 600 μl DMSO, opisać, po zakończonych ćwiczeniach przechowywać w lodówce - roztwór będzie wykorzystywany na kolejnych ćwiczeniach - przygotowuje każdy zespół.
1 ml 10mM (3,4 mg/ml) r-ru NPGB w DMSO
Wyznaczanie stężenia roztworu trypsyny:
przez pomiar absorpcji światła o λ = 280 nm:
Do kuwety kwarcowej dodać 2 ml wody i zarejestrować widmo absorpcyjne w zakresie 240 nm do 400nm, odczytać i zapisać absorpcję przy 280 nm i 300 nm. Następnie do kuwety dodać 50 μl roztworu trypsyny (1d) (10 mg/ml), dokładnie wymieszać i podobnie jak wyżej zarejestrować widmo i absorpcje. Odpowiednio odjąć od siebie zmierzone absorpcje i przyjmując ktrp280nm = 1,5 oraz Mtrp = 24 000 g/mol obliczyć stężenie wyjściowego r-ru enzymu w mg/ml i M.
przez miareczkowanie centrów aktywnych trypsyny za pomocą wybuchowego, syntetycznego, drobnocząsteczkowego substratu - nitrofenylo-4-guanidyno-benzoesanu (NPGB).
TEORIA: Jednym z etapów mechanizmu hydrolizy wiązań przez trypsynę jest tworzenie przejściowego, kowalencyjnego kompleksu enzymu z substratem - acyloenzymu. W przypadku zastosowania NPGB jako substratu enzym szybko tworzy z nim acyloenzym i hydrolizuje wiązanie estrowe uwalniając do roztworu tylko barwny p-nitrofenol, podczas gdy druga część substratu (guanidyno-benzoesan) bardzo wolno zwalnia się z acyloenzymu blokując trwale kieszeń wiążącą trypsyny przed dostępem kolejnej cząsteczki substratu. Tym sposobem ilość wolnego p-nitrofenolu w roztworze wyznaczana przez pomiar absorpcji światła λ = 410 nm (εpNP410nm = 16595 M-1 cm-1) jest miarą ilości aktywnych cząsteczek enzymu w tym samym roztworze. Stężenia substratu i enzymu w mieszaninie reakcyjnej muszą być tak dobrane aby [NPGB] ≥ 0.9 [E].
Przebieg ćwiczenia:
Doświadczenie wykonać w kilku powtórzeniach, dlatego do czterech plastikowych kuwet dodać:
2 ml buforu weronalowego (1a),
10 μl r-ru NPGB (3.4 mg/ml, 10 mM) w DMSO
zakleić parafilmem, wymieszać, włożyć do spektrofotometru, wyzerować wskazanie aparatu i przez około 20 sekund rejestrować absorpcję światła A410, nie przerywając rejestracji wyjąć kuwetę z aparatu, dodać
50 μl r-ru trypsyny (1d)
wymieszać, włożyć do spektrofotometru i dalej rejestrować absorpcję przez około minutę. Odczytać absorpcję przed i po dodaniu enzymu do kuwety; z różnicy policzyć stężenie trypsyny w kuwecie i w roztworze wyjściowym. Policzyć średnią z czterech oznaczeń. Porównać stężenie oszacowane przez pomiar A280 z wyznaczonym przez miareczkowanie za pomocą NPGB licząc procentowy udział aktywnego enzymu w całkowitej ilości białka.
Wyznaczenie stężenia substratu - BApNA, przez całkowitą hydrolizę porcji r-ru substratu z wykorzystaniem trypsyny.
TEORIA: Gdy mamy do czynienia z układem enzym - substrat takim, że enzym sprawnie katalizuje przemianę substratu i gdy spełniony zostanie warunek [E] ≥ [S], to w krótkim czasie dochodzi do całkowitej hydrolizy porcji substratu w mieszaninie reakcyjnej. Miarą stężenia substratu (BApNA) jest zatem różnica A405 przed dodaniem odpowiedniej ilości trypsyny do kuwety z niewielką zawartością BApNA i po całkowitej jego hydrolizie. Stężenie substratu w mieszaninie reakcyjnej musi być tak dobrane aby po całkowitej hydrolizie uzyskać A405 w miarodajnym zakresie, tzn.
0.05 < A405 < 1.
Wykonanie ćwiczenia:
Przed wykonaniem ćwiczenia należy je zaplanować, licząc jakie porcje wyjściowych roztworów substratu (1e) i enzymu (1d) należy dodać do plastikowej kuwety z 2 ml buforu reakcyjnego, aby spełnić warunki określone wyżej (teoria). Skonsultować wyniki z prowadzącym.
Eksperyment przeprowadzić w kilku powtórzeniach, dlatego do trzech kuwet dodać:
2ml buforu reakcyjnego (1b)
odpowiednią porcję r-ru substratu (BApNA),
kuwetę zakleić parafilmem, wymieszać zawartość, umieścić w spektrofotometrze, wyzerować, rozpocząć rejestrację A405, po około 20 sekundach wyjąć z aparatu i dodać
odpowiednią porcję r-ru trypsyny (1d)
wymieszać, włożyć ponownie do aparatu i dalej rejestrować A405 dopóty, dopóki osiągnie stałą wartość wskazującą na fakt, że porcja substratu w kuwecie została całkowicie zhydrolizowana do pNA. Odczytać A405 przed dodaniem enzymu do kuwety i A405 końcową, z różnicy obliczyć stężenie substratu w kuwecie i r-rze wyjściowym.
Zależność początkowej prędkości reakcji enzymatycznej od stężenia substratu
Wykonanie ćwiczenia:
przygotować 25 ml r-ru trypsyny: Do probówki typu falcon o pojemności 50 ml nalać buforu reakcyjnego(1b), dodać odpowiednią porcję (skonsultować z prowadzącym) wyjściowego r-ru trypsyny(1d), aby stężenie enzymu w r-rze wynosiło około 2.5 × 10-7 M i dopełnić do kreski buforem reakcyjnym. Dla podniesienia dokładności rozcieńczenia warto dodawać porcję wyjściowego r-ru enzymu na wadze.
ćwiczenie zasadnicze: Do 10 plastikowych kuwet dodać po 2 ml buforu reakcyjnego z trypsyną, zalepić parafilmem i bezpośrednio przed pomiarem dodać do każdej kuwety z osobna odpowiednią porcję wyjściowego r-ru BApNA(1e), tak aby jego stężenie w poszczególnych kuwetach rozłożone było równomiernie między 2,5 × 10-4M < [BApNA] < 5 × 10-3M (skonsultować z prowadzącym). Po dodaniu porcji r-ru substratu do każdej kuwety należy energicznie wymieszać jej zawartość, włożyć do spektrofotometru, wyzerować, mierzyć A405 przez około 60 do 90 sekund i odczytać wartość początkowej prędkości reakcji (δA405/δt).
UWAGA: Przy wyższych stężeniach substrat może wypadać z r-ru; aby tego uniknąć bufor reakcyjny nie może zawierać detergentów (np. Tritonu), powinien zawierać do kilku procent DMSO, należy bardzo energicznie wymieszać zawartość kuwety po dodaniu roztworu substratu.
Opracowanie wyników:
obliczyć stężenie substratu w każdej z kuwet,
odczytaną ze spektrofotometru wartość początkowej prędkości enzymatycznej (δA405/δt) wyrazić w - δ{pNA} [mol] / δt [s],
obliczyć żądane parametry przeprowadzając nieliniową analizę danych według modelu Michaelisa-Menten.
Wyznaczanie stężenia kompetycyjnego inhibitora trypsyny - BPTI przez miareczkowanie mianowanego roztworu enzymu roztworem inhibitora
WSTĘP:
Eksperyment polega na wykonaniu krzywej hamowania enzymu przez inhibitor. Przy założeniu, że inhibitor wiąże się z enzym w stechiometrycznym stosunku 1 : 1 prosta, liniowa zależność aktywności enzymu o dokładnie znanym stężeniu od objętości dodanego r-ru inhibitora (Atrp = f{Vr-ruBPTI}) pozwala wyznaczyć dokładne stężenie inhibitora w dodawanym r-rze. Ekstrapolacja zależności Atrp = f{Vr-ruBPTI} do punktu o zerowej aktywności enzymu pozwala obliczyć objętość r-ru inhibitora potrzebną do całkowitego zahamowania aktywności znanej porcji enzymu w roztworze.
UWAGA: Stężenia enzymu i inhibitora do eksperymentu muszą być tak dobrane aby były wyższe (co najmniej 10-kronie) od stężenia równego stałej inhibitorowej (dysocjacji) (KI) dla konkretnej pary enzym-inhibitor (dla pary BPTI - β-trypsyna, w pH = 8.0, 25 °C, KI = 6.0 × 10-14 M).
Wykonanie ćwiczenia:
Na ćwiczenie składa się przygotowanie odczynników, w tym roztworu trypsyny o znanym stężeniu i r-ru BPTI, wykonanie zasadniczego doświadczenia i opracowanie wyników.
Przygotowanie odczynników:
bufor reakcyjny: 0.1 M TRIS, 20 mM CaCl2, pH = 8.3 - 200 ml - dla całej grupy ćwiczeniowej
r-r trypsyny - przygotowuje każdy zespół jak w punkcie I.1.d), wyznaczyć jego stężenie jak w punkcie I.2.) i dalej przygotować 50 ml r-ru trypsyny o stężeniu około 2.5 × 10-7 M w buforze reakcyjnym (1a) do zasadniczego doświadczenia podobnie jak w punkcie I.4.a).
r-r BPTI o stężeniu 5mg/ml: naważkę około 2,5 mg inhibitora rozpuścić w 0,5 ml wody - przygotowuje każdy zespół. Po zakończonych ćwiczeniach r-r rozpipetować po 100 μl do ependorfek, dokładnie opisać i zamrozić. Roztwór będzie wykorzystywany na kolejnych ćwiczeniach
d) 0.1 M r-r BApNA w DMSO - 150 μl dla każdego zespołu ćwiczeniowego
Wyznaczenie stężenia BPTI przez pomiar A280: do kuwety kwarcowej o pojemności 1 ml dodać 1 ml wody i zarejestrować widmo absorpcyjne w zakresie 240nm do 400nm, odczytać i zapisać absorpcje przy 280nm i 360nm. Następnie do kuwety dodać 50 μl roztworu BPTI (1c) o stężeniu 5mg/ml, dokładnie wymieszać i podobnie jak wyżej zarejestrować widmo i absorpcje. Odpowiednio odjąć od siebie zmierzone absorpcje i przyjmując εBPTI280nm = 5400 M-1 cm-1 oraz MBPTI = 6500 g/mol obliczyć stężenie wyjściowego r-ru inhibitora w mg/ml i M.
Rozcieńczony r-r inhibitora opisać i pozostawić do kolejnych ćwiczeń.
Miareczkowanie mianowanego r-ru trypsyny r-rem BPTI:
do 10 plastikowych kuwet dodać po 2 ml r-ru trypsyny o stężeniu około [E0] = 2.5 × 10-7M w buforze reakcyjnym (1b),
do pierwszej kuwety nie dodawać r-ru inhibitora
do kolejnych kuwet dodać odpowiednie porcje r-ru inhibitora, tak aby jego stężenie w poszczególnych kuwetach rozkładało się równomiernie w granicach 0.1 [E0] ≤ [BPTI] ≤ 0.9 [E0]. W tym celu należy odpowiednio rozcieńczyć na wadze wyjściowy r-r inhibitora (1c) tak aby dodawanie do kuwet planowanych jego porcji było obciążone możliwie małym błędem. Wszystkich obliczeń należy dokonać opierając się na stężeniu BPTI w wyjściowym roztworze wyznaczonym przez pomiar absorbancji A280. Skonsultować z prowadzącym
kuwety zakleić parafilmem i wymieszać ich zawartość
do każdej kuwety bezpośrednio przed pomiarem dodać 10 μl 0,1M r-ru BApNA (1d), wymieszać, włożyć do spektrofotometru, wyzerować aparat i mierzyć przyrost A405 przez około 60 sekund, zapisać wynik w postaci początkowej prędkości reakcji (δA405/δt)
Opracowanie wyników:
Wykreślić zależność początkowej prędkości reakcji od objętości dodawanego r-ru inhibitora, wykreślić (policzyć) prostą przechodzącą przez punkty doświadczalne i na podstawie równania prostej obliczyć objętość r-ru inhibitora, jaka powinna być dodana do r-ru enzymu aby go całkowicie zahamować. Znając dokładne stężenie enzymu w kuwecie i zakładając, że trypsyna oddziaływuje z BPTI w stosunku 1:1 policzyć stężenie inhibitora w r-rze dodawanym do kuwet i w r-rze wyjściowym. Porównać wyznaczone stężenie z oszacowanym na podstawie A280 licząc procentowy udział aktywnego inhibitora w ilości białka wyznaczonej przez A280.
III. Pomiar równowagowej stałej asocjacji (Ka) oddziaływania
α-chymotrypsyna - BPTI metodą oznaczania resztkowej aktywności enzymu
WSTĘP: Stała równowagi reakcji tworzenia kompleksu enzym - inhibitor kompetycyjny:
E + I ⇔ EI
dana jest wzorem: K = [EI] / [E] × [I]] (1) i nazywana stałą asocjacji (Ka). Odwrotność stałej asocjacji nazywana stałą inhibicji (KI) : 1/Ka = KI = [E] × [I] / [EI] (2), jest stałą równowagi reakcji rozpadu kompleksu enzym - inhibitor ( EI ⇔ E +I ). Przekształcając wzór (1) w kierunku wyprowadzenia wyrażenia na stężenie wolnego enzymu [E] otrzymujemy równanie:
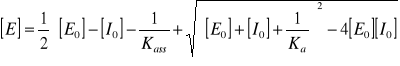
(3)
gdzie [E0] to całkowite stężenie enzymu równe - [E0] = [E] + [EI], a [I0] to całkowite stężenie inhibitora - [I0] = [I] + [EI]
Stałą asocjacji można zatem eksperymentalnie wyznaczyć jako parametr rónania (3) mierząc stężenie wolnego enzymu [E] w kolejnych próbach, w funkcji całkowitego stężenia inhibitora [I0] przy danym, stałym całkowitym stężeniu mianowanego enzymu [E0] - [E] = f([I0]){[E0]}. Stężenie wolnego enzymu pozostającego w równowadze z kompleksem enzym-inhibitor można wyznaczyć mierząc jego aktywność i porównując ją z aktywnością mianowego r-ru samego enzymu, bez dodatku inhibitora.
Eksperyment polega na tym, że do każdej z serii kuwet z r-rem enzymu o stałym, znanym stężeniu [E0] w buforze reakcyjnym dodać należy odpowiednich wzrastających porcji r-ru inhibitora. Następnie w celu wykazania aktywności wolnego enzymu do każdej z kuwet dodać należy tej samej porcji substratu i zarejestrować początkową prędkość reakcji enzymatycznej. Planując taki eksperyment spełnić należy kilka dodatkowych warunków:
eksperyment należy prowadzić w dolnym regionie krzywej hamowania enzymu przez inhibitor, przy takim stałym stężeniu całkowitym enzymu [E0], aby był spełniony warunek
2 < Ka × [E0] < 40stężenie całkowite inhibitora [I0] w kolejnych próbach powinno być równomiernie rozłożone w granicach - 0 ≤ [I0] ≤ 2 × [E0]. W przypadku bardzo słabego oddziaływania enzym-inhibitor (Ka ≤ 103 M-1) można zapomnieć o tym warunku i wymuszać tworzenie kompleksu wysokim stężeniem inhibitora
przed dodaniem substratu do mieszaniny reakcyjnej należy być pewnym, że układ jest w stanie równowagi chemicznej (rónanie1), tzn. należy inkubować enzym z inhibitorem w buforze reakcyjnym przez czas (tink) około dziesięciokrotnie dłuższy od czasu połówkowego (t1/2) tworzenia kompleksu EI - tink = 10 × t1/2 (4), przy czym t1/2 = 1/kon × [I0], gdzie kon jest stałą szybkości tworzenia kompleksu (dla oddziaływania chymotrypsyna - BPTI w warunkach prowadzonej reakcji kon = 3.7 × 105 M-1 sek-1)
substrat i jego stężenie muszą być dobrane adekwatnie do całkowitego stężenia enzymu [E0] przy jakim prowadzimy reakcje. Gdy [E0] jest niskie (wysoka Ka) stosować należy substrat o bardzo dobrych parametrach kinetycznych (niskie KM, wysokie kcat), gdy [E0] jest wysokie (niska Ka) stosować należy substraty o słabych parametrach kinetycznych(wysokie KM, niskie kcat). Stężenie substratu w mieszaninie reakcyjnej [S] powinno być od kilkuset do tysiąca razy wyższe od stężenia [E0].
Wykonanie ćwiczenia:
W ramach ćwiczenia należy: przygotować niezbędne odczynniki, wyznaczyć dokładnie stężenia wyjściowych roztworów enzymu (α-chymotrypsyny) i inhibitora (BPTI), przeprowadzić właściwe doświadczenie i opracować wyniki w celu wyznaczenia stałej asocjacji (Ka)
Przygotowanie odczynników:
0.5 litra buforu reakcyjnego: 0.05 M HEPES, 20 mM CaCl2, pH 7.5 - na całą grupę ćwiczeniową
roztwór chymotrypsyny - naważkę około 10 mg chymotrypsyny rozpuścić w 1ml 0.001M HCl, 20 mM CaCl2, po wykonaniu ćwiczenia rozpipetować po 50 μl do ependorfek dokładnie opisać i zamrozić. Roztwór będzie wykorzystywany na kolejnych ćwiczeniach.- przygotowuje każdy zespół
Roztwór BPTI - wykorzystać r-r przygotowany i zmiareczkowany na poprzednich ćwiczeniach. Jeśli trzeba przygotować nowy r-r inhibitora to postępować zgodnie z punktem (II.1c) - przygotowuje każdy zespół
0.8 ml roztworu substratu (Suc-Phe-pNA) w DMSO o stężeniu z naważki równym 250mM - przygotować na całą grupę;
2. Wyznaczanie stężenia roztworu chymotrypsyny:
przez pomiar absorpcji światła o λ = 280 nm:
Przebieg ćwiczenia:
Do kuwety kwarcowej dodać 2 ml wody i zarejestrować widmo absorpcyjne w zakresie 240 nm do 400nm, odczytać i zapisać absorpcję przy 280 nm i 300 nm. Następnie do kuwety dodać 50 μl roztworu chymotrypsyny (1b) (10 mg/ml), dokładnie wymieszać i podobnie jak wyżej zarejestrować widmo i absorpcje. Odpowiednio odjąć od siebie zmierzone absorpcje i przyjmując ε280 = 51000 M-1 cm-1 oraz Mchtrp = 25 000 g/mol obliczyć stężenie wyjściowego r-ru enzymu w mg/ml i M.
przez miareczkowanie centrów aktywnych chymotrypsyny za pomocą wybuchowego, syntetycznego, drobnocząsteczkowego substratu - nitrofenylo-4-guanidyno-benzoesanu (NPGB). Postępować analogicznie podobnie jak w przypadku wyznaczania stężenia trypsyny w punkcie (I.2b) pierwszych ćwiczeń, z pewną modyfikacją wynikającą z nietrwałego charakteru acyloenzymu. Acyloenzym rozkłada się dużo szybciej niż w przypadku miareczkowania trypsyny i dlatego po dodaniu chymotrypsyny do kuwety obserwujemy wzrost A410 (patrz niżej).
Przebieg ćwiczenia:
Doświadczenie wykonać w kilku powtórzeniach, dlatego do trzech plastikowych kuwet dodać:
2ml buforu weronalowego
10 μl r-ru NPGB (st.3.4 mg/ml, 10 mM) w DMSO
zakleić parafilmem, wymieszać, włożyć do spektrofotometru, wyzerować wskazanie aparatu i przez około 20 sekund rejestrować absorpcję światła A410, nie przerywając rejestracji wyjąć kuwetę z aparatu, dodać
50 μl r-ru chymotrypsyny
wymieszać, włożyć do spektrofotometru i dalej rejestrować absorpcję przez około minutę. Odczytać absorpcję przed dodaniem substratu do kuwety. Absorpcję po dodaniu enzymu do kuwety wyznaczyć ekstrapolując krzywą opisującą wzrost A410 w czasie do momentu dodania enzymu do kuwety. Z różnicy A410 policzyć stężenie chymotrypsyny w kuwecie i w roztworze wyjściowym. Policzyć średnią z trzech oznaczeń. Porównać stężenie oszacowane przez pomiar A280 z wyznaczonym przez miareczkowanie za pomocą NPGB licząc procentowy udział aktywnego enzymu w całkowitej ilości białka.
3. Wyznaczenie stężenia BPTI
Jeśli używamy r-ru przygotowanego na poprzednich zajęciach (II.) to należy posługiwać się stężeniem inhibitora ustalonym na poprzednich zajęciach.
Jeśli przygotowujemy nowy r-r BPTI to w celu wyznaczenia jego stężenia postępować jak w ćwiczeniu II.
4. Zależność stężenia wolnego enzymu [E] pozostającego w równowadze z inhibitorem od całkowitego stężenia inhibitora [I0]
Wykonanie ćwiczenia:
przygotować 30 ml roboczego r-ru chymotrypsyny: Do falkona o pojemności 50 ml nalać buforu reakcyjnego(1a), dodać odpowiednią porcję (skonsultować z prowadzącym) wyjściowego r-ru chymotrypsyny(1b), aby stężenie enzymu w r-rze wynosiło około [E0] ≅ 5 × 10-6 M i dopełnić na wadze buforem reakcyjnym. Przelać zawartość kolby do probówki typu falkon.
ćwiczenie zasadnicze:
Do 10 plastikowych kuwet dodać po 2 ml roztworu roboczego chymotrypsyny w buforze reakcyjnym (1a), zamknąć parafilmem. Do trzech pierwszych kuwet nie dodawać niczego ([E] = [E0]), a do kolejnych dodać takich porcji r-ru BPTI (1c) aby stężenie inhibitora w kolejnych kuwetach wzrastało równomiernie w granicach 0,2 [E0] ≤ [BPTI] ≤ 2 [E0] - skonsultować z prowadzącym ćwiczenia. Kuwety wymieszać i bezpośrednio przed pomiarem dodać do każdej 20 μl r-ru substratu (1d)(250mM Suc-Phe-pNA). Po dodaniu porcji r-ru substratu do każdej kuwety należy energicznie wymieszać jej zawartość, włożyć do spektrofotometru, wyzerować, mierzyć A405 przez około 60 do 90 sekund i odczytać wartość początkowej prędkości reakcji (δA405/δt).
UWAGA: Bardzo starannie dodawać do kuwet zarówno r-ru inhibitora jak i substratu. Od dokładności tego pipetowania zależy wynik ćwiczenia.
Opracowanie wyników:
obliczyć całkowite stężenie inhibitora [I0] w każdej z kuwet
obliczyć stężenie wolnego enzymu [E] w każdej kuwecie porównując zmierzoną początkową prędkość reakcji enzymatycznej (aktywność) w każdej kuwecie ze średnią z początkowych prędkości reakcji uzyskanych dla kuwet bez inhibitora, gdzie [E] = [E0].
obliczyć stałą asocjacji (Kass) oddziaływania α-chymotrypsyny z BPTI przeprowadzając nieliniową analizę danych [E] = f([I0]) według równania (3)
O poprawności wyznaczonej tą metodą stałej asocjacji świadczy zgodność (dopuszczalny błąd rzędu 2-3%) wyznaczonych eksperymentalnie (wystandaryzowanych) całkowitych stężeń enzymu ([E0]) i inhibitora ([I0]) z obliczonymi na podstawie równania (3).
V. Specyficzna, chemiczna modyfikacja enzymów (trypsyny, chymotrypsyny) za pomocą nieodwracalnych drobnocząsteczkowych inhibitorów: PMSF (fenylometylosulfofluorku), TLCK (tozylolizynochlorometyloketonu)
WSTĘP:
Istnieje wiele syntetycznych związków chemicznych, które reagują z reaktywnymi grupami (np.: -OH, -NH2, -SH) łańcuchów bocznych reszt aminokwasowych tworząc nieodwracalne, kowalencyjne kompleksy (bardziej lub mniej trwałe) modyfikują właściwości białek w tej liczbie enzymów. W zależności od budowy chemicznej związki te mogą być mniej lub bardziej selektywne w stosunku do reaktywnych grup białek i w stosunku do kieszeni wiążących substraty w enzymach. Tego typu modyfikacje mogą służy identyfikacji reszt zaangażowanych w mechanizm katalizy enzymatycznej i klasyfikacji enzymów.
Wykonanie ćwiczenia: W ramach ćwiczenia należy: przygotować niezbędne odczynniki, dokonać modyfikacji enzymów drobnocząsteczkowymi inhibitorami i sprawdzić aktywność enzymów niemodyfikowanych i modyfikowanych
Przygotowanie odczynników:
50 ml buforu do modyfikacji PMSF-em: 0.1M TRIS, 0,1M NaCl, 0,12M CaCl2, 10%(V/V) metanol, pH = 8,0 - na całą grupę ćwiczeniową.
200 ml buforu reakcyjnego: 0.1 M TRIS, 20mM CaCl2, 5% (V/V) DMSO, pH = 8,3 - na całą grupę ćwiczeniową.
20 ml buforu do modyfikacji TLCK- na całą grupę ćwiczeniową: do buforu reakcyjnego (1b) dodać DMSO do końcowego stężenia 10% (V/V).
1 ml 50mM PMSF w acetonie - na całą grupę ćwiczeniową.
R-ry TLCK- przygotowuje każdy zespół ćwiczeniowy - zważyć około 1mg każdego z inhibitorów w osobnych ependorfkach i rozpuścić w 100μl DMSO.
Roztwór trypsyny do modyfikacji PMSF-em - przygotowuje każdy mały zespół ćwiczeniowy - odpowiednio rozcieńczyć roztwór enzymu przygotowany do ćwiczenia I (1d), tak aby uzyskać 0,5 ml roztworu enzymu o stężeniu 5mg/ml (około 4,2 × 10-5M) w buforze do modyfikacji PMSF-em (1a). W przypadku braku r-ru enzymu z poprzednich ćwiczeń, naważyć 5mg trypsyny i rozpuścić w 1ml buforu do modyfikacji (1a).
roztwór trypsyny do modyfikacji TLCK- przygotowuje każdy mały zespół ćwiczeniowy - odpowiednio rozcieńczyć roztwór enzymu przygotowany do ćwiczenia I (1d), tak aby uzyskać 0,5 ml roztworu enzymu o stężeniu 5 mg/ml (około 4,2 × 10-5 M) w buforze do modyfikacji TLCK (1c). R-r włożyć do łaźni lodowej (2°C). W przypadku braku r-ru enzymu z poprzednich ćwiczeń, naważyć 2.5 mg trypsyny i rozpuścić w 0.5 ml buforu (1c).
roztwór chymotrypsyny do modyfikacji PMSF-em - przygotowuje każdy mały zespół ćwiczeniowy - naważkę 10 mg enzymu rozpuścić w 1 ml buforu do modyfikacji PMSF-em(1a).
roztwór chymotrypsyny do modyfikacji TLCK- przygotowuje każdy mały zespół ćwiczeniowy - 10 mg enzymu rozpuścić w 1 ml buforu do modyfikacji TLCK (1c). R-r włożyć do łaźni lodowej (2°C).
0,6 ml 0.1 M roztworu substratu do wykrywania aktywności trypsyny (BApNA) w DMSO - przygotowuje każdy zespół ćwiczeniowy - odpowiednio rozcieńczyć roztwór BApNA przygotowany do ćwiczenia I (1e) lub wykonać świeży r-r z odpowiedniej naważki BApNA
0,1 ml 250 mM roztworu substratu do wykrywania aktywności chymotrypsyny (Suc-Phe -pNA) w DMSO - przygotowuje każdy zespół ćwiczeniowy.
Modyfikacja enzymów PMSF-em:
modyfikacja trypsyny: r-r trypsyny do modyfikacji (1f) rozdzielić do ependorfek na dwie porcje po około 0,25 ml, do jednej dodać sześć 5 μl porcji 50 mM PMSF-u (1d) w odstępach około 10 minutowych i mieszać od czasu do czasu. Do drugiej porcji stanowiącej kontrolę niemodyfikowanego enzymu dodać 30 μl acetonu
modyfikacja chymotrypsyny: r-r chymotrypsyny do modyfikacji (1h) rozdzielić do probówek na dwie porcje po około 0,5 ml, do jednej dodać sześć 10 μl porcji 50 mM PMSF-u (1d) w odstępach około 10 minutowych i mieszać od czasu do czasu. Do drugiej porcji stanowiącej kontrolę niemodyfikowanego enzymu dodać 60 μl acetonu
Modyfikacja enzymów TLCK:
modyfikacja trypsyny: r-r trypsyny do modyfikacji (1g) rozdzielić do ependorfek na dwie porcje i włożyć do łaźni lodowej (2°C). Do jednej porcji dodać 50 μl r-ru TLCK (1e), do drugiej porcji stanowiącej kontrolę niemodyfikowanego enzymu dodać 50 μl DMSO. Inkubować 60 minut w łaźni lodowej mieszając od czasu do czasu.
modyfikacja chymotrypsyny: r-r chymotrypsyny do modyfikacji (1i) rozdzielić do ependorfek na dwie porcje i włożyć do łaźni lodowej (2°C). Do jednej porcji dodać 50 μl r-ru TLCK (1e) i wymieszać, do drugiej porcji stanowiącej kontrolę niemodyfikowanego enzymu dodać 50 μl DMSO. Inkubować 60 minut w łaźni lodowej mieszając od czasu do czasu.
Aktywność enzymów po modyfikacji.
aktywność trypsyny:
Przygotować cztery plastikowe kuwety z 2 ml buforu reakcyjnego (1b).
do pierwszej dodać 50 μl r-ru trypsyny niemodyfikowanej PMSF-em (2a),
do drugiej 50 μl r-ru trypsyny modyfikowanej PMSF-em (2a),
do trzeciej 50 μl r-ru trypsyny niemodyfikowanej TLCK (3a),
do czwartej 50 μl r-ru trypsyny modyfikowanej TLCK (3a)
Kuwety zakleić parafilmem i wymieszać ich zawartość. Przy spektrofotometrze do każdej kuwety dodać 10 μl 0,1 M r-ru BApNA (1k), wymieszać, włożyć do aparatu, wyzerować i przez jedną minutę śledzić przyrost absorbcji A410, zapisać początkową prędkość reakcji enzymatycznej, która jest miarą aktywności enzymu. Porównać wyniki dla prób z modyfikowaną i niemodyfikowaną trypsyną licząc procent zahamowanego enzymu.
aktywność chymotrypsyny:
Postępować analogicznie jak w przypadku wykazywania aktywności trypsyny z tą różnicą, że do kuwet należy dodawać odpowiednio niemodyfikowanych i modyfikowanych r-rów chymotrypsyny, a jako substratu - 20 μl 250 mM r-ru Suc-Phe -pNA (1l).
BApNA
NPGB
TLCK
PMSF
.
1
Wyszukiwarka
Podobne podstrony:
Enzymologia materiały do ćwiczeń
Instrukcja do cwiczenia 1
Instrukcje do ćwiczeń 2013
Ćw.1 Wybrane reakcje chemiczne przebiegające w roztworach wodnych ćwiczenie 1, Chemia ogólna i żywno
INSTRUKCJA do ćwiczenia pomiar temperatury obrabiarek v3 ver robocza
instrukcja 06, sem 3, Podstawy elektrotechniki i elektroniki, Laboratoria, instrukcje do cwiczen 201
Instrukcja do cwiczenia 2
Instrukcja do ćwiczenia laboratoryjnego PDH
instrukcja 09, sem 3, Podstawy elektrotechniki i elektroniki, Laboratoria, instrukcje do cwiczen 201
Instrukcja do ćwiczenia8
Instrukcja do ćwiczenia(8)
Ćwiczenia, Instrukcja do ćwiczenia 7, Instrukcja do ćwiczenia 11:
Instrukcja do ćwiczenia(12), ZESPÓŁ SZKÓŁ Nr 9 im
Chromatografia TLC Instrukcja do cwiczenia
instrukcja do cwiczenia t1 dla Nieznany
Instrukcja do ćwiczenia nr 6
Instrukcja do ćwiczenia(16), Badanie stopni mocy wzmacniaczy m
Instrukcja do ćwiczenia(14), ZESPÓŁ SZKÓŁ NR 9 im
więcej podobnych podstron