Autorzy przedstawiaj¹ dalszy ci¹g
badañ zwi¹zanych z opracowaniem
metodyki profilowania konopi na pod-
stawie sk³adu pierwiastkowego. Pod-
stawy metody oraz wyniki wstêpne zo-
sta³y ju¿ opisane na ³amach „Proble-
mów Kryminalistyki”
1,2,3
. Ci¹g dalszy
prac pokaza³, jak du¿ym wyzwaniem
jest ten temat badawczy. Ze wzglêdów
oczywistych elementy badañ ju¿ opi-
sane przez autorów w poprzednich pu-
blikacjach bêd¹ w niniejszym artykule
traktowane marginalnie, przy czym
czêsto bêd¹ stanowiæ punkt wyjœcia do
rozwi¹zywanych i opisywanych zagad-
nieñ. W tej czêœci tematu badawczego
autorzy opisali takie zagadnienia jak:
stabilnoœæ roztworów analitów w prób-
kach przygotowanych do analizy,
stabilnoϾ aparatu, efekty matrycowe
oraz sposoby ich kompensacji.
Warto przypomnieæ, ¿e w bada-
niach wykorzystywany jest spektro-
metr ICP-OES Optima 3100XL firmy
Perkin Elmer, podczas pracy którego
stosowano dwa zestawy warunków
operacyjnych: warunki standardowe
oraz odporne. Charakterystykê tych
warunków zamieszczono w tab. 1.
Próbki ziela konopi przygotowy-
wane s¹ do badañ przez poddanie
ich rozk³adowi na mokro z udzia³em
energii mikrofa-lowej w uk³adzie
zamkniêtym za pomoc¹ systemu
Multiwave firmy Anton Paar (Perkin
Elmer) z u¿yciem mieszaniny kwasu
azotowego i wody utlenionej.
Stabilnoœæ analitów w próbkach
konopi po mineralizacji
Przed przyst¹pieniem do badañ
sprawdzono stabilnoœæ analitów
w materiale po mineralizacji, tzn. jak
d³ugo przygotowane roztwory mog¹
byæ wykorzystywane do oznaczeñ
obecnych w nich pierwiastków. Ze
wzglêdu na fakt, ¿e czas przygotowa-
nia szeœciu próbek wynosi ok. 2 godz.
(mineralizacji mo¿na jednoczeœnie
poddaæ szeœæ próbek), a ich analiza
za pomoc¹ ICP-OES trwa oko³o 20
minut, mo¿na przyj¹æ, ¿e minimalna
stabilnoœæ pierwiastków w próbce po-
winna wynosiæ co najmniej tyle samo
czasu. Z praktycznego i ekonomiczne-
go punktu widzenia po¿¹dane by³oby
analizowanie nie mniej ni¿ kilkunastu
próbek w jednym rzucie. Wymaga³oby
to rozdzielenia w czasie etapów przy-
gotowania próbek oraz ich analizy.
Wykonanie tego jest mozliwe pod wa-
runkiem, ¿e pierwiastki obecne w uzy-
skanych roztworach próbek konopi s¹
stabilne przez kilka dni.
W celu zbadania stabilnoœci pier-
wiastków na wstêpie przygotowano
próbki zgodnie z procedur¹. Otrzyma-
ny roztwór poddawano analizie
w pierwszym, drugim, trzecim, czwar-
tym i dziewi¹tym dniu po mineraliza-
cji. Nale¿y zaznaczyæ, ¿e pos³ugiwa-
no siê intensywnoœciami analitów (nie
przeprowadzano ka¿dorazowo kali-
bracji). W celu korekcji ewentualnej
niestabilnoœci metody analitycznej
ka¿dego dnia oznaczano dodatkowo
pierwiastki w roztworze tego samego
wzorca wielopierwiastkowego o sta-
bilnoœci d³u¿szej ni¿ dziewiêæ dni,
a wyniki uzyskane dla próbki konopi
odpowiednio korygowano. Otrzymane
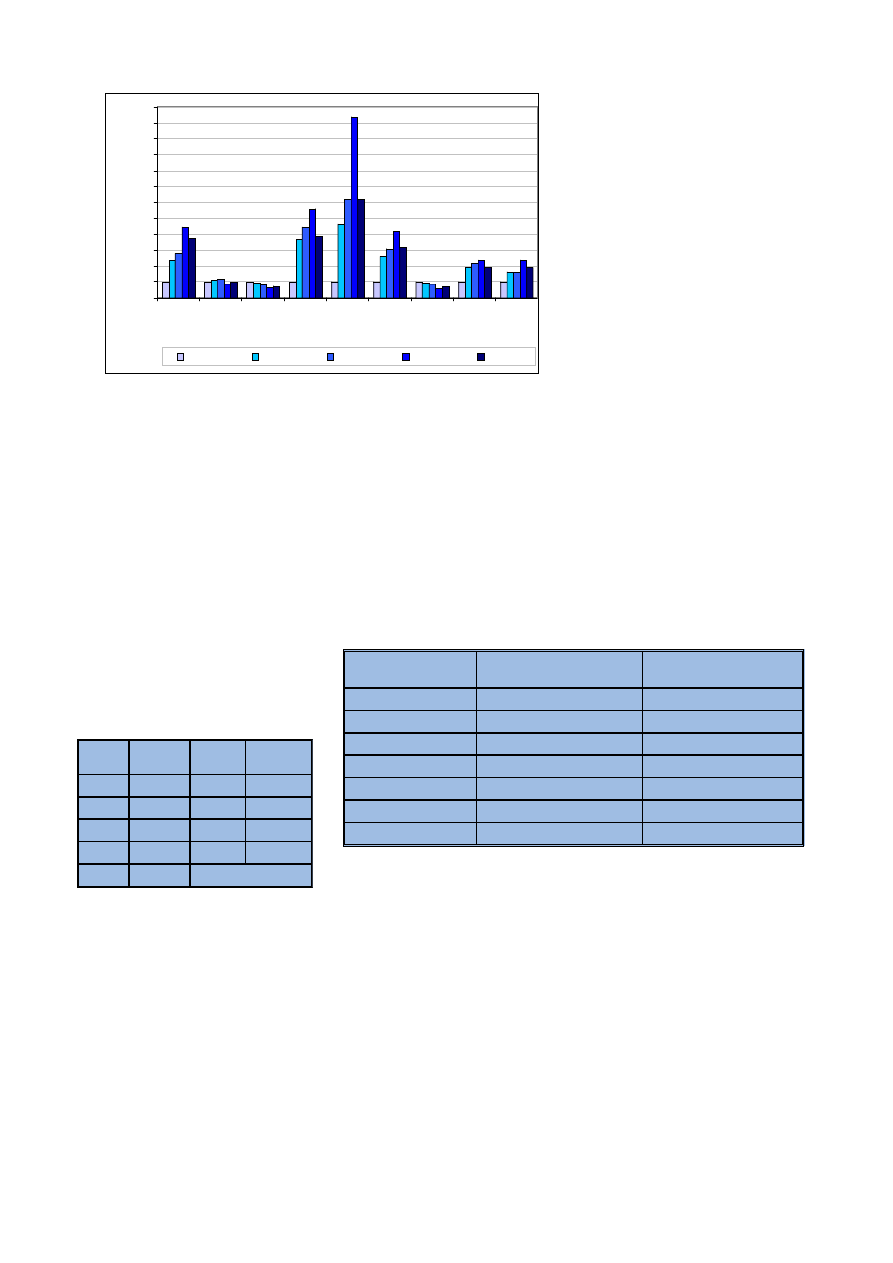
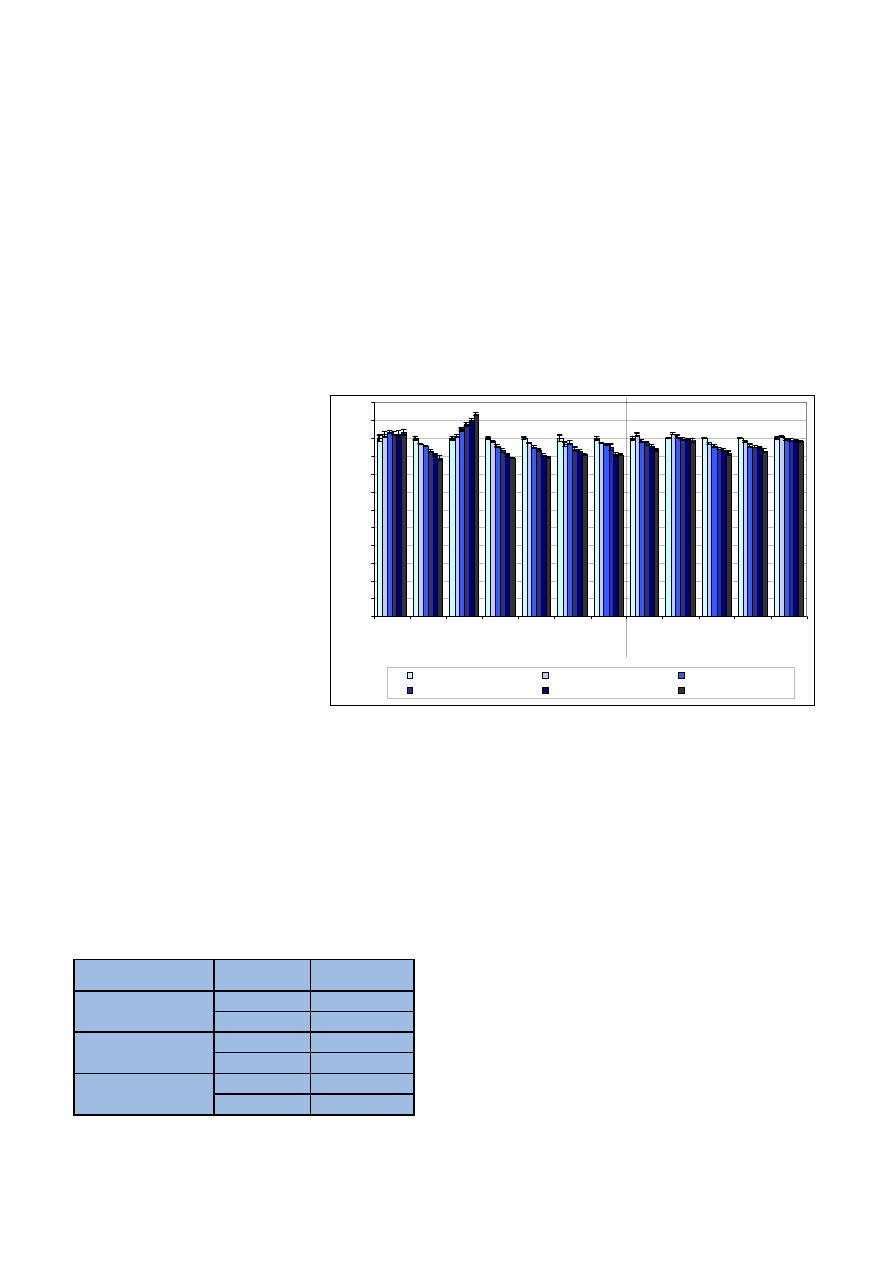
wyniki przedstawiono na rycinie 1.
Jak widaæ na wykresie, niewiele
oznaczanych pierwiastków jest stabil-
nych w próbce konopi po mineraliza-
cji. Nale¿¹ do nich tylko wapñ i bar,
które s¹ stabilne praktycznie w ca³ym
badanym czasie. Mangan mo¿na
uznaæ za stabilny do trzech dni po mi-
neralizacji. Reszta pierwiastków jest
bardzo niestabilna – oznaczone ich
zawartoœci dochodz¹ do 308% warto-
œci rzeczywistej. Badania wskazuj¹
na koniecznoϾ natychmiastowej
analizy próbek konopi po procesie mi-
neralizacji.
21
PROBLEMY KRYMINALISTYKI 252/06
Marzena Kuras
Marek Wachowicz
Profilowanie konopi
na podstawie sk³adu
pierwiastkowego – cz. I
(efekty matrycowe)
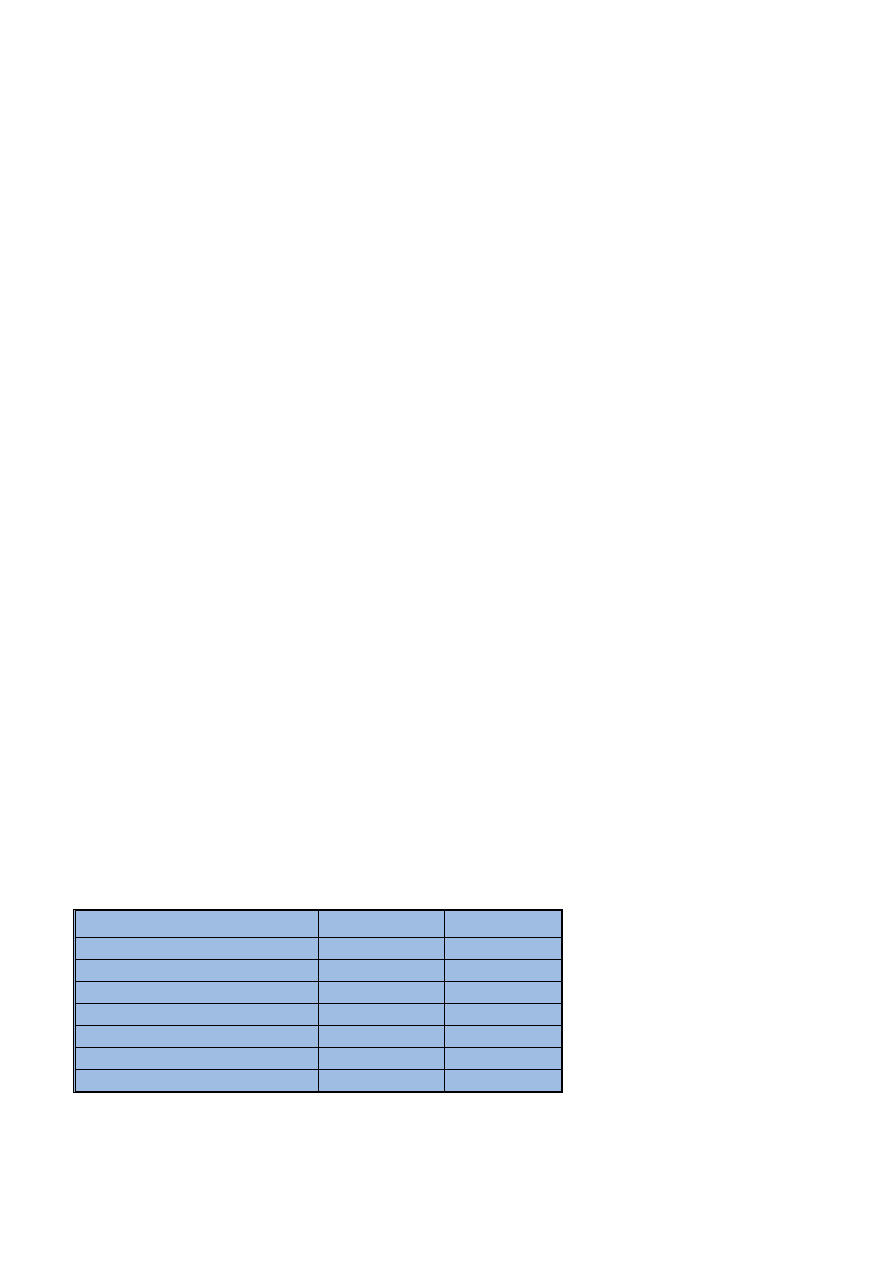
Tabela 1
Warunki operacyjne pracy spektrometru
Spectrometer operating conditions
P
Paarraam
me
ettrr
S
Sttaan
nd
daarrd
do
ow
we
e
O
Od
dp
po
orrn
ne
e
P
Prrzze
ep
p³³y
yw
w g
gaazzu
u p
pllaazzm
mo
ow
we
eg
go
o [[ll//m
miin
n]]
15
15
P
Prrzze
ep
p³³y
yw
w g
gaazzu
u p
po
om
mo
occn
niicczze
eg
go
o [[ll//m
miin
n]]
0,5
0,5
P
Prrzze
ep
p³³y
yw
w g
gaazzu
u p
prrzze
ezz rro
ozzp
py
yllaacczz [[ll //m
miin
n]]
0,8
0,5
M
Mo
occ p
pllaazzm
my
y [[W
W]]
1300
1450
W
Wy
ysso
ok
ko
oœœææ o
ob
bsse
errw
waaccjjii p
pllaazzm
my
y [[m
mm
m]]
15
15
P
Prrzze
ep
p³³y
yw
w p
prró
ób
bk
kii [[m
mll//m
miin
n]]*
*
1,5
1,5
C
Czzaass o
op
pó
óŸŸn
niie
en
niiaa [[ss]]
60
60
* W metodzie wzorca wewnêtrznego przep³yw próbki wynosi 0,65 ml/min, a czas opóŸnienia 90 s
StabilnoϾ aparatu
w trakcie trwania analizy
StabilnoϾ aparatu podczas trwa-
nia analizy wyznaczono, analizuj¹c
próbkê wzorca wielopierwiastkowego.
Próbkê kontroln¹ analizowano na po-
cz¹tku cyklu i po ka¿dych 5 próbkach,
czyli w pierwszej, osiemnastej, trzy-
dziestej siódmej i szeœædziesi¹tej trze-
ciej minucie od rozpoczêcia analizy
próbek. Na podstawie uzyskanych wy-
ników obliczono wspó³czynnik zmien-
noœci (CV), który podano w tab. 2.
Jak mo¿na by³o przypuszczaæ,
stabilnoœæ warunków panuj¹cych
w spektrometrze podczas analizy
próbek w jednym cyklu analitycznym
jest bardzo dobra. Œwiadczy o tym ni-
ska wartoœæ CV – dla wiêkszoœci
pierwiastków poni¿ej 2%. Tylko dla
cynku CV wynios³o 3,5%.
Identyfikacja efektów
matrycowych
Efekty matrycowe wystêpuj¹, gdy
matryca badanych próbek, œlepych
próbek oraz wzorców do kalibracji nie
jest taka sama. Powoduje to, ¿e
w plazmie mog¹ istnieæ inne warunki
podczas wprowadzania wzorców
i próbek. Mo¿e skutkowaæ to znacz¹-
cymi b³êdami analitycznymi. Efekty
matrycowe, skutkuj¹ce os³abieniem
lub wzmocnieniem sygna³u pocho-
dz¹cego od analitu, mog¹ byæ wywo-
³ywane przez wiele czynników, a w
szczególnoœci przez obecnoœæ kwa-
sów i pierwiastków ³atwo jonizuj¹-
cych siê. W badanych próbkach ro-
œlin obecne s¹ te dwa czynniki, za-
tem zostan¹ one opisane szczegó³o-
wo.
W ramach pracy magisterskiej
4
napisanej na Wydziale Chemii Uni-
wersytetu Warszawskiego przy
wspó³pracy z CLK KGP przeprowa-
dzono wstêpne badania konopi.
Analizie poddano 10 próbek, w któ-
rych oznaczono 12 pierwiastków.
Okaza³o siê, ¿e stê¿enia wapnia
i magnezu s¹ o kilka rzêdów wielko-
œci wy¿sze od stê¿eñ pozosta³ych
oznaczanych pierwiastków. Co wiê-
cej, ich zawartoœci ró¿ni¹ siê w sze-
rokim zakresie stê¿eñ (Ca 0,5
÷5%,
Mg 0,3
÷0,8%).
Poza wapniem i
magnezem
w próbkach konopi nale¿y spodzie-
waæ siê du¿ej zawartoœci potasu,
poniewa¿ jest on pierwiastkiem po-
wszechnie obecnym w próbkach ro-
œlinnych. W tabeli 3 zamieszczono
przyk³adowe zawartoœci potasu
w kilku roœlinnych materia³ach od-
niesienia.
Wp³yw kwasu azotowego (V)
Próbki roœlinne, jako próbki sta³e,
musz¹ zostaæ przeprowadzone do
roztworu. Odbywa siê to za pomoc¹
rozk³adu na mokro, zatem analizo-
wana próbka, oprócz pierwiastków
bêd¹cych sk³adnikami konopi, za-
PROBLEMY KRYMINALISTYKI 252/06
22
80
100
120
140
160
180
200
220
240
260
280
300
320
B
Ba
Ca
Cu
Fe
Mg
Mn
Sr
Zn
pierwiastek
względna intensywność [%]
dzień 1
dzień 2
dzień 3
dzień 4
dzień 9
Ryc. 1. Stabilnoœæ pierwiastków w analitach
Fig. 1. Elemental stability in analytes
A
An
naalliitt
C
CV
V
A
An
naalliitt
C
CV
V
B
B
0,3
M
Mg
g
1,5
B
Baa
0,4
M
Mn
n
1,3
C
Caa
2,1
S
Srr
2,0
C
Cu
u
0,9
Z
Zn
n
3,5
F
Fe
e
0,4
Tabela 2
StabilnoϾ aparatu w czasie cyklu
analitycznego
Instrument stability during
analytical cycle
CV – wspó³czynnik zmiennoœci (inaczej %RSD) –
ang. coefficient of variation
Tabela 3
Zawartoœæ potasu w certyfikowanych materia³ach roœlinnych
Potassium content in certified plant material
S
Sy
ym
mb
bo
oll m
maatte
erriiaa³³u
u
R
Ro
od
dzzaajj m
maatte
erriiaa³³u
u
Z
Zaaw
waarrtto
oœœææ p
po
ottaassu
u
[[%
%]]
C
CT
TA
A--O
OT
TL
L--1
1
Orientalne liœcie tytoniu
1,56
C
CT
TA
A--V
VT
TL
L--2
2
Liœcie tytoniu z Wirginii
1,03
N
NIIS
ST
T--1
15
51
15
5
Liœcie jab³oni
1,61
IIN
NC
CT
T --T
TL
L--1
1
Liœcie herbaty
1,70
B
BC
CR
R--1
12
29
9
Siano
3,38
IIN
NC
CT
T --M
MP
PH
H --2
2
Mieszanka polskich zió³
1,91
N
NIIS
ST
T--1
15
57
70
0aa
Liœcie szpinaku
2,90
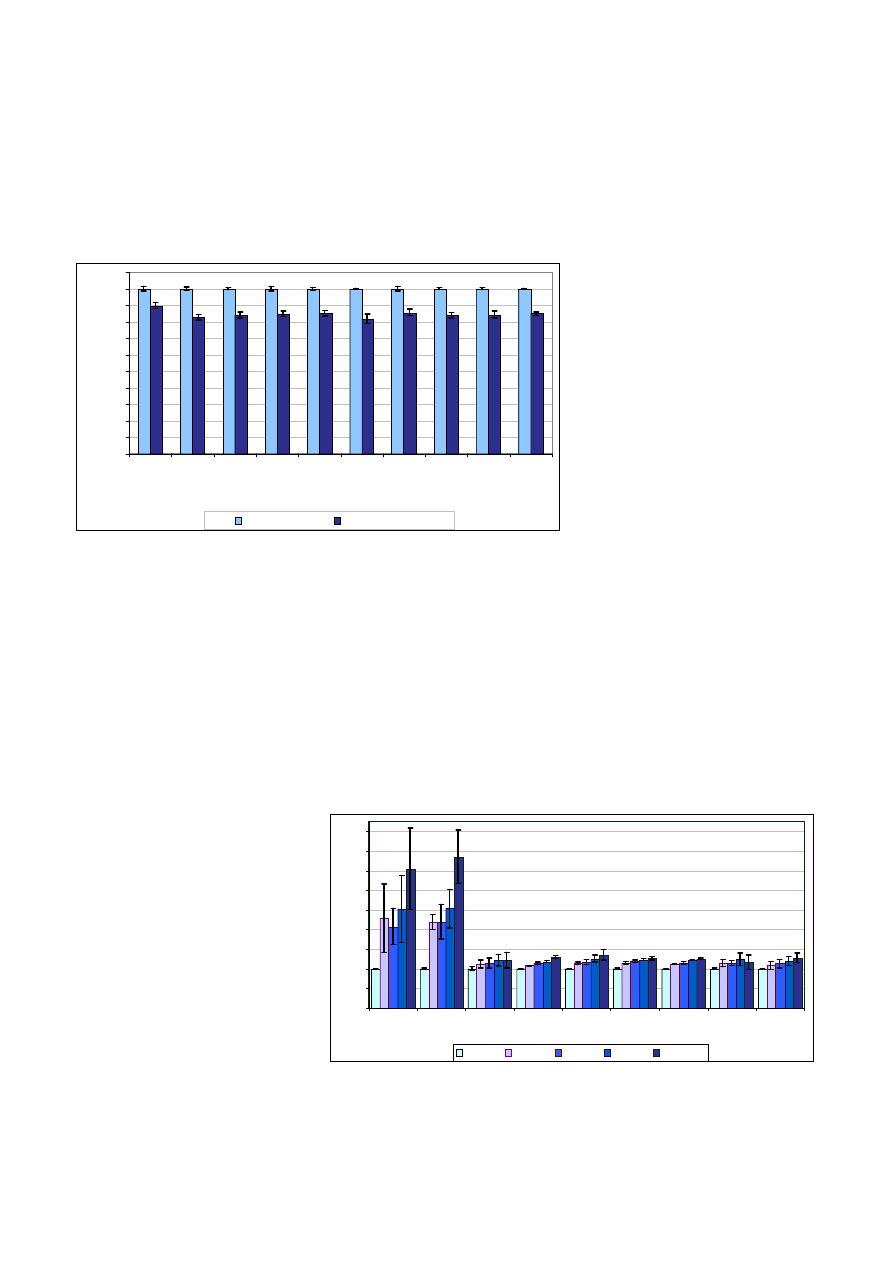
wiera znacz¹ce iloœci kwasu azoto-
wego (V), który stosowano w proce-
sie mineralizacji próbek. Wp³yw ma-
trycy tylko kwasu azotowego (V) na
intensywnoϾ emisji wybranych pier-
wiastków zobrazowano na rycinie 2.
Kwas azotowy (V) powoduje obni-
¿enie intensywnoœci sygna³u dla
wszystkich pierwiastków w porówna-
niu z roztworem wodnym. Os³abienie
sygna³u mieœci siê w granicach od
–10% dla baru do –18% dla strontu.
Jest to spowodowane ró¿nic¹ w lep-
koœci i gêstoœci roztworów wodnych
i kwaœnych. Wiêksza lepkoœæ i gê-
stoϾ roztworu kwasu azotowego (V)
ogranicza efektywnoϾ rozpylania
w porównaniu z roztworem wodnym,
co w konsekwencji prowadzi do os³a-
bienia sygna³u oznaczanego pier-
wiastka. Przedstawione wyniki badañ
s¹ zgodne z wczeœniejszymi publika-
cjami.
Wszystkie substancje bêd¹ce ma-
tryc¹ próbki mog¹ przeszkadzaæ
w precyzyjnym i dok³adnym oznacza-
niu pierwiastków na poziomie œlado-
wym. Rutynowym sposobem wykry-
wania wp³ywów matrycy jest analiza
próbek rozcieñczonych
5,6
. Po
uwzglêdnieniu wspó³czynnika roz-
cieñczenia wyniki porównywane s¹
z uzyskanymi dla próbek nierozcieñ-
czonych. O braku efektów matryco-
wych œwiadczy brak istotnej ró¿nicy
w otrzymywanych wynikach. Innym
sposobem interpretacji otrzymanych
wyników jest wykreœlenie zale¿noœci
wspó³czynnika rozcieñczenia od in-
tensywnoœci uzyskanego sygna³u.
Jeœli zale¿noœæ ta jest prostoliniowa,
matryca nie ma istotnego wp³ywu na
procesy zachodz¹ce w plazmie,
a w konsekwencji – na uzyskiwane
wyniki analityczne.
By to sprawdziæ, przeprowadzono
analizê trzech próbek. Dwie z nich sta-
nowi³y próbki konopi, a trzecia to ma-
teria³ certyfikowany INCT-MPH-2
(mieszanina polskich zió³). Dla ka¿dej
z próbek przygotowano seriê czterech
rozcieñczeñ. Analizowano próbki, dla
których wspó³czynniki rozcieñczenia
wynosi³y: 0 (bez rozcieñczenia), 3, 4,
6, 11. Otrzymane wyniki przemno¿one
przez wspó³czynnik rozcieñczenia
przedstawiono na rycinie 3.
Najwiêksze odchylenia intensyw-
noœci sygna³u dla próbki o wspó³-
czynniku rozcieñczenia równym 11
zaobserwowano dla baru i boru, dla
których intensywnoœæ sygna³u jest
odpowiednio o 285% i 255% wy¿sza
od prawid³owej (100%). Dla pozosta-
³ych pierwiastków zmiany mieszcz¹
siê w zakresie 17
÷36%. Nale¿y za-
znaczyæ, ¿e dla wszystkich oznacza-
nych pierwiastków wp³ywy matrycy
s¹ znacz¹ce. Zatem w celu zwiêk-
szenia precyzji i dok³adnoœci metody
niezbêdna jest minimalizacja efektów
matrycowych.
Wp³yw wapnia i magnezu
Jak to zosta³o ju¿ przedstawione,
g³ównymi sk³adnikami matrycy pró-
bek konopi, mog¹cymi powodowaæ
efekty uboczne, s¹ wapñ, magnez
i potas. Ze wzglêdu na fakt, ¿e potas
jako pierwiastek ³atwo ulegaj¹cy joni-
zacji zachowuje siê jak bufor, posta-
nowiono sprawdziæ, jak na dok³ad-
noœæ oznaczeñ pierwiastków na po-
ziomie œladowym wp³ywaj¹ wapñ
i magnez, a nastêpnie, w jakim stop-
niu dodatek pierwiastka ³atwo jonizu-
j¹cego siê minimalizuje te efekty.
Przede wszystkim sprawdzono,
w jaki sposób obecnoœæ wapnia i ma-
gnezu wp³ywa na intensywnoœæ sy-
gna³ów uzyskanych dla pozosta³ych
analitów. W tym celu przygotowano
PROBLEMY KRYMINALISTYKI 252/06
23
0
10
20
30
40
50
60
70
80
90
100
110
B Ba Cu
Fe
Mn
Sr
Zn
Be
Be
Y
pierwiastek
względna intensywność [%]
woda
kwas azotowy (V)
Ryc. 2. Wp³yw kwasu azotowego (V) o stê¿eniu 20% na intensywnoœæ emisji wybranych linii
Fig. 2. Effect of nitric acid (V) of 20% conc. on emission intensity of selected lines
0
50
100
150
200
250
300
350
400
450
B
Ba
Ca
Cu Fe Mg
Mn Sr
Zn
analit
% oryginalnego sygnału
rozc. 0x
rozc. 3x
rozc. 4x
rozc. 6x
rozc. 11x
Ryc. 3. Wzglêdna zmiana intensywnoœci sygna³ów uzyskanych dla kolejnych rozcieñczeñ próbek
Fig. 3. Relative change of signal intensity obtained for subsequent sample dilutions
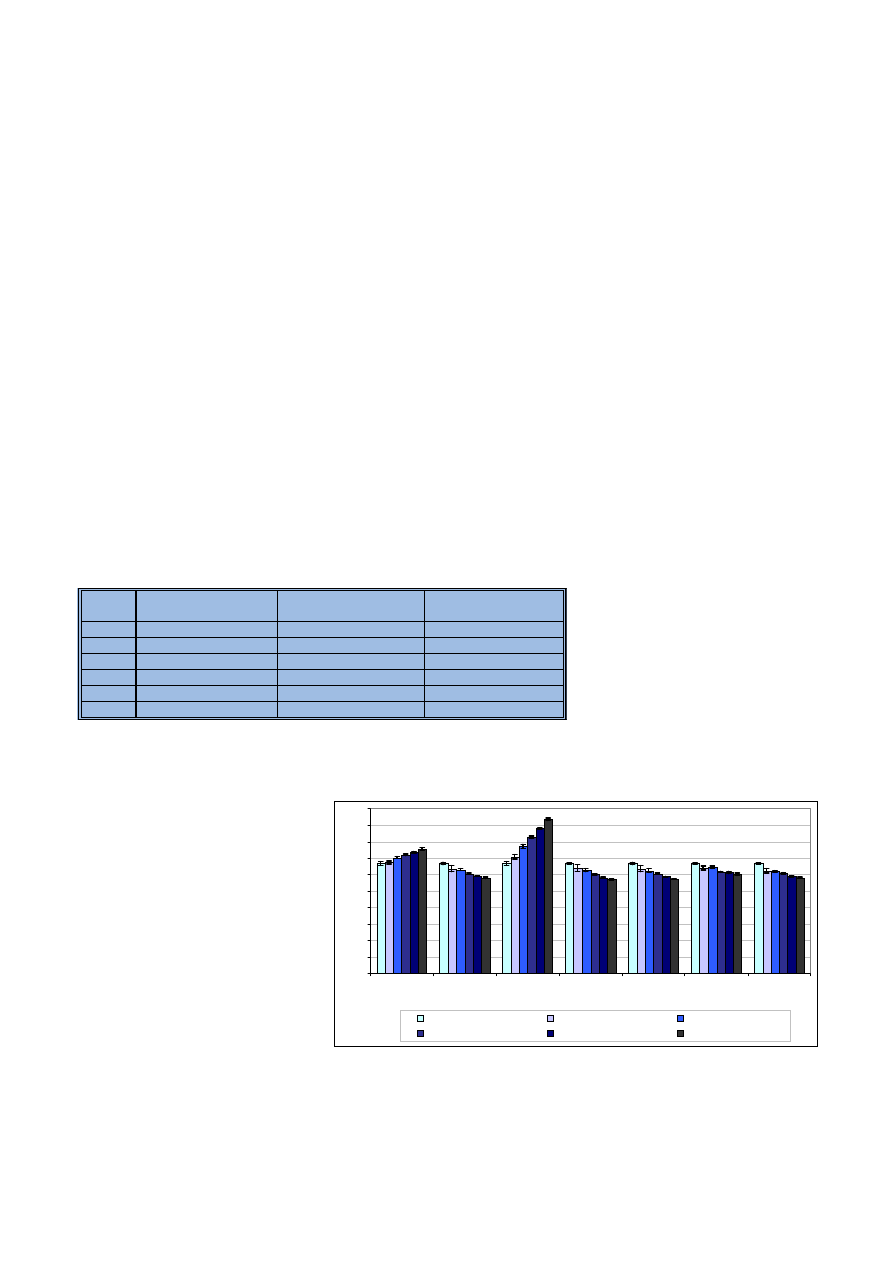
seriê roztworów wzorcowych, w któ-
rych stê¿enia oznaczanych pierwiast-
ków poza Ca i Mg wynosi³y po 10 mg/l.
Roztwory ró¿ni³y siê zawartoœci¹ wap-
nia i magnezu.
Badania wstêpne próbek konopi
wykaza³y, ¿e stê¿enie magnezu jest
oko³o dziesiêciu razy mniejsze od
stê¿enia wapnia. Znaj¹c – z wcze-
œniejszych badañ – maksymalne stê-
¿enie wapnia i magnezu obecne
w analizowanych próbkach konopi,
dobrano ich stê¿enia w próbkach
wzorcowych tak, aby pokrywa³y
w miarê równomiernie ca³y ich rze-
czywisty zakres. Matryc¹ wszystkich
próbek by³ 20% kwas azotowy (V).
Zawartoœci wapnia i magnezu w po-
szczególnych seriach roztworów po-
dano w tabeli 4.
Ka¿dy z roztworów przygotowano
trzykrotnie. Podczas analizy próbek
zastosowano warunki standardowe
pracy spektrometru (tab. 1). Uzyska-
ne dla analitów intensywnoœci sygna-
³ów przeliczono na wzglêdne procen-
towe intensywnoœci. Intensywnoœci
równe 100% przyjêto dla roztworów
niezawieraj¹cych sk³adników matry-
cy. Uzyskane wyniki zilustrowano na
rycinie 4.
Jak mo¿na zauwa¿yæ, dla wszyst-
kich oznaczanych pierwiastków
wzrost stê¿enia matrycy powoduje
zwiêkszenie niekorzystnych jej wp³y-
wów. W konsekwencji porównywanie
zawartoœci na przyk³ad miedzi
w próbkach ró¿ni¹cych siê znacz¹co
zawartoœci¹ wapnia i magnezu bê-
dzie prowadziæ do b³êdnych wnio-
sków. Dla wszystkich analitów oprócz
boru i miedzi zaobserwowano os³a-
bienie sygna³u wraz ze wzrostem stê-
¿enia matrycy. Jest to zwi¹zane z jo-
nowym/atomowym charakterem linii
analitycznych. Dodatek pierwiastka
³atwo jonizuj¹cego siê powoduje
wzrost liczby wolnych atomów, zatem
linie atomowe ulegaj¹ wzmocnieniu.
Dla boru wzmocnienie wynios³o do
13%, a dla miedzi 40%. Wraz ze
wzrostem iloœci elektronów zwi¹za-
nym z obecnoœci¹ pierwiastka ³atwo
ulegaj¹cego jonizacji nastêpuje os³a-
bienie linii jonowych od 10% dla
strontu do 15% dla ¿elaza.
Jak zosta³o pokazane, efekty ma-
trycowe maj¹ znacz¹cy wp³yw na
koñcowe wyniki iloœciowe. Aby mo¿li-
we by³o wiarygodne porównywanie
wyników analitycznych uzyskanych
dla ró¿nych próbek konopi, a w kon-
sekwencji wieloletnie ich gromadze-
nie i porównywanie w ramach progra-
mu profilowania konopi, niezbêdne
jest opracowanie metody ich minima-
lizacji. Ograniczenie ich wp³ywów po-
zwoli równie¿ na porównywanie wyni-
ków w ramach wspó³pracy miêdzyla-
boratoryjnej.
Metody korekcji
efektów matrycowych
Minimalizacja efektów matryco-
wych powodowanych obecnoœci¹
kwasów, pierwiastków pierwszej
i drugiej grupy uk³adu okresowego
jest niezbêdna dla uzyskania dok³ad-
nych wyników oraz dobrej powtarzal-
noœci i odtwarzalnoœci metody. Dlate-
go te¿ du¿y nacisk k³adziono na
opracowanie metody umo¿liwiaj¹cej
ograniczenie niekorzystnych wp³y-
wów matrycy. Najprostsz¹ metod¹
eliminacji wp³ywów matrycy jest do-
pasowanie matrycy wszystkich anali-
zowanych roztworów, tj. roztworów
wzorcowych, próbek oraz œlepych
prób. Zazwyczaj jednak ta metoda
nie jest mo¿liwa do zastosowania, bo
matryca jest zbyt skomplikowana lub
zbyt ró¿norodna w danej serii próbek.
Wtedy mo¿na zastosowaæ analizê
próbek rozcieñczonych. Wraz ze
wzrostem stopnia rozcieñczenia pró-
bek rozcieñczeniu ulega równie¿ ma-
tryca, a jej wp³yw na sygna³ oznacza-
nego pierwiastka nie jest ju¿ tak istot-
ny. Du¿¹ wad¹ tego sposobu minima-
lizacji efektów matrycowych jest
zmniejszenie czu³oœci metody,
a w konsekwencji brak mo¿liwoœci
oznaczania pierwiastków na niskim
poziomie stê¿eñ. Dlatego te¿ poszu-
PROBLEMY KRYMINALISTYKI 252/06
24
Tabela 4
Zawartoœæ wapnia i magnezu w poszczególnych seriach roztworów
Calcium and magnesium contents across diluent batches
N
Nrr sse
erriiii
O
Ozzn
naacczze
en
niie
e
Z
Zaaw
waarrtto
oœœææ w
waap
pn
niiaa
[[m
mg
g//ll]]
Z
Zaaw
waarrtto
oœœææ m
maag
gn
ne
ezzu
u
[[m
mg
g//ll]]
1
1
0 mg/l Ca+Mg
0
0
2
2
220 mg/l Ca+Mg
200
20
3
3
440 mg/l Ca+Mg
400
40
4
4
880 mg/l Ca+Mg
800
80
5
5
1320 mg/l Ca+Mg
1200
120
6
6
1760 mg/l Ca+Mg
1600
160
0
15
30
45
60
75
90
105
120
135
150
B Ba
Cu
Fe
Mn
Sr
Zn
pierwiastek
intensywność względna [%]
0 mg/l Ca+Mg
220 mg/l Ca+Mg
440 mg/l Ca+Mg
880 mg/l Ca+Mg
1320 mg/l Ca+Mg
1760 mg/l Ca+Mg
Ryc. 4. Wzglêdna zmiana intensywnoœci sygna³ów uzyskanych dla analitów w obecnoœci wzrastaj¹cego
stê¿enia matrycy wapnia i magnezu
Fig. 4. Relative change of analyte signal intensity in presence of increasing concentrations of calcium
and magnesium matrix

kuje siê innych metod kompensacji
interferencji fizycznych, a jest ich wie-
le. Dosyæ obszerna literatura na ten
temat opisuje minimalizacje efektów
matrycowych wywo³anych ró¿nymi
czynnikami, w tym obecnoœci¹ pier-
wiastków I i II grupy uk³adu okreso-
wego, a tak¿e obecnoœci¹ kwasów.
W niniejszym opracowaniu postano-
wiono przeanalizowaæ i porównaæ
trzy z nich:
a) zastosowanie zmodyfikowa-
nych warunków operacyjnych
spektrometru (tzw. warunków
odpornych),
b) zastosowanie odpornych wa-
runków plazmy w po³¹czeniu
z metod¹ dodatku buforu matry-
cy,
c) zastosowanie odpornych wa-
runków plazmy w po³¹czeniu
z metod¹ dodatku wzorca we-
wnêtrznego.
Ad a. Wystêpowanie efektów ma-
trycowych w du¿ym stopniu zale¿y
od warunków operacyjnych spektro-
metru (warunków pracy), a zw³asz-
cza od mocy oraz przep³ywu gazu
noœnego. Zmniejszenie przep³ywu
gazu noœnego prowadzi do d³u¿-
szych czasów przebywania analitu
w plazmie ze wzglêdu na redukcjê
prêdkoœci gazu przy wylocie wtryski-
wacza i spadku iloœci aerozolu, nie-
zbêdnego do odparowania, a w kon-
sekwencji do zmniejszenia wp³ywów
matrycy. Takie warunki operacyjne
pracy spektrometru, w których zmia-
na matrycy próbki nie powoduje
zmiany w intensywnoœci sygna³u
analitycznego, nazywa siê warunka-
mi odpornymi plazmy. By uzyskaæ
takie warunki, nale¿y zastosowaæ
wysok¹ moc, zwykle powy¿ej 1,4
kW, oraz d³ugi czas przebywania
analitu w plazmie, co uzyskuje siê
poprzez zmniejszenie przep³ywu ga-
zu przez nebulizer, zwykle poni¿ej
0,6 l/min. Nieodpornych warunków
plazmy powinno siê unikaæ w rutyno-
wych analizach
7, 8, 9
.
Jako kryterium odpornoœci plazmy
wybrano stosunek intensywnoœci linii
magnezu Mg II 280,270 nm do
Mg I 285,213 nm. Linia Mg II 280 to
linia jonowa, a linia Mg I 285 – atomo-
wa. Wykazano, ¿e w przeciwieñstwie
do linii jonowych wiêkszoœæ linii ato-
mowych jest stosunkowo nieczu³a na
zmiany warunków operacyjnych
10,
11
. Zatem stosunek linii jonowej do
atomowej pozwala na weryfikacjê
optymalizacji parametrów operacyj-
nych plazmy. Wzór na stosunek linii
jonowej do atomowej I
i
/I
a
ma nastê-
puj¹c¹ postaæ
12,13
.
gdzie:
Eexc – energia wzbudzenia,
Eion – energia jonizacji,
g – waga statystyczna,
A – prawdopodobieñstwo przejœcia,
λ
– d³ugoœæ fali,
k – sta³a Boltzmanna,
Te – temperatura elektronów,
Texc – temperatura wzbudzenia,
ne – iloœæ elektronów.
Stan lokalnej równowagi termicz-
nej jest osi¹gniêty, gdy T
e
= T
exc
= T.
Wartoœci g i A s¹ sta³e i wynosz¹
5,32 * 10
8
s
-1
i 14,85 * 10
8
s
-1
dla
Mg II 280,270 i Mg I 285,213
14
. Po
podstawieniu tych wartoœci do wzoru
uzyskana zostanie zale¿noœæ pomiê-
dzy T, iloœci¹ elektronów i stosunkiem
I
i
/I
a
. Wynika z niej, ¿e równowaga
w plazmie, a w konsekwencji jej od-
pornoœæ, jest osi¹gniêta przy wartoœci
tego stosunku co najmniej 10 przy
pionowej obserwacji plazmy i powy-
¿ej 8 dla obserwacji poziomej.
Efektywnoœæ korekcji efektów ma-
trycowych dziêki zastosowaniu od-
pornych warunków plazmy zosta³a
potwierdzona przez wielu badaczy.
Literatura naukowa podaje
15
, ¿e za-
stosowanie warunków odpornych
wraz z wiêksz¹ œrednic¹ iniektora
(wtryskiwacza) umo¿liwia minimaliza-
cjê wp³ywu sodu na intensywnoœæ in-
nych pierwiastków. W nieodpornych
warunkach plazmy matryca 10 g/l so-
du powodowa³a os³abienie lub
wzmocnienie sygna³u dla jonowych
linii analitycznych. Znaczne os³abie-
nie sygna³u (41
÷44%) zaobserwowa-
no np. dla linii Ni (II) 231 nm, Ni (II)
221 nm, Pb (II) 220 nm i Cu (II) 224
nm. Natomiast dodatek sodu spowo-
dowa³ wzrost intensywnoœci sygna³u
(do 209%) dla takich linii jak: Ba (II)
230 nm, Mg (II) 279,6 nm, Mg (II) 280
nm, Sr (II) 216 nm, Cd (II) 214 nm
i Zn (II) 202 nm. Dla linii atomowych
wp³yw sodu by³ równie nieregularny,
poniewa¿ nie jest mo¿liwe proste od-
niesienie zmiany sygna³u do energii
wzbudzenia. Zmiana intensywnoœci
wszystkich atomowych linii
analitycznych mieœci³a siê
w zakresie 76
÷273%.
Po zastosowaniu warun-
ków odpornych zmiana wy-
nosi³a od 62 do 103%. Jak mo¿na za-
uwa¿yæ, rozrzut zmiany intensywno-
œci w tych warunkach jest du¿o ni¿-
szy ni¿ w warunkach nieodpornych.
Nale¿y podkreœliæ, ¿e dla wielu linii
korekcja by³a na tyle efektywna, ¿e
mo¿na j¹ zastosowaæ w rutynowych
analizach. Przyk³adem mo¿e byæ li-
nia Sr (I) 460,773 nm. W nieodpor-
nych warunkach intensywnoϾ linii
wynosi³a 273%, a po zastosowaniu
warunków odpornych plazmy –
103%.
Zastosowanie odpornych warun-
ków plazmy umo¿liwi³o równie¿ mini-
malizacjê wp³ywów innych matryc,
np. matrycy Ca
16
oraz negatywnego
wp³ywu matrycy kwasów
17
.
Zastosowanie warunków odpor-
nych ogranicza wp³ywy matrycowe,
jednak czêsto nie do tego stopnia, by
mo¿na by³o zastosowaæ jedynie tê
metodê w rutynowych analizach.
Ad b. Jak wspomniano wczeœniej,
nawet w odpornych warunkach nie
jest mo¿liwa pe³na kompensacja
zmian w plazmie spowodowanych
zmianami w sk³adzie matrycy. Alter-
natyw¹ jest zastosowanie buforu, tj.
pierwiastka dodanego w du¿ym stê-
¿eniu, którego wp³yw bêdzie dra-
stycznie wiêkszy od wp³ywu innych
pierwiastków takich jak: Ca i Na
18
.
W konsekwencji zmiana stê¿enia Ca
lub Na nie bêdzie mia³a wp³ywu na
warunki plazmy. Jako bufor najbar-
dziej odpowiednie s¹ pierwiastki ³a-
two ulegaj¹ce jonizacji, a wœród nich
cez. Ma on najni¿sz¹ energiê joniza-
cji (3,894 eV), emituje tylko kilka linii
i zwykle nie jest oznaczany metod¹
ICP-OES ze wzglêdu na zbyt ma³¹
PROBLEMY KRYMINALISTYKI 252/06
25
(
)
exc
a
exc
i
exc
e
ion
kT
E
E
kT
E
e
i
a
a
a
i
i
e
a
i
e
e
T
A
g
A
g
n
I
I
−
−
−
×
×
×
=
2
3
21
10
83
.
4
λ
λ

czu³oœæ metody podczas oznaczania
tego pierwiastka. Nale¿y podkreœliæ,
¿e dodatek Cs obni¿a wartoœæ sto-
sunku Mg II/Mg I, a wiêc odpornoœæ
plazmy, lecz z drugiej strony utrzymu-
je j¹ na sta³ym poziomie, a¿ do stê¿e-
nia Na 1 g/l. Zatem dodatek 10 g/l Cs
umo¿liwia tolerancjê stê¿enia Na
o wartoœci 1 g/l bez znacz¹cych
zmian warunków plazmy, niezale¿nie
od warunków operacyjnych.
Ad c. Metoda wzorca wewnêtrz-
nego stosowana jest do korekcji in-
terferencji, gdy sk³ad wzorców i pró-
bek ró¿ni siê znacznie i nie mo¿e byæ
³atwo odtworzony. W tej metodzie do
roztworów kalibracyjnych i do próbek
rzeczywistych dodaje siê jednakow¹,
znan¹ iloœæ substancji chemicznej
wybranej w taki sposób, aby zapew-
niæ podobne zachowanie wzorca we-
wnêtrznego i analitu we wszystkich
etapach procedury. Wa¿ne jest rów-
nie¿, ¿eby sygna³y wzorca wewnêtrz-
nego i analitu w danej technice po-
miarowej by³y uzyskiwane w takich
samych warunkach pomiarowych,
mia³y podobn¹ wartoœæ i by³y dobrze
rozdzielone. Pierwiastek, który ma
pe³niæ funkcjê wzorca wewnêtrznego,
musi spe³niaæ nastepuj¹ce warunki:
– nie mo¿e byæ sk³adnikiem anali-
zowanych próbek,
– musi byæ stabilny w roztworze,
– stosowane linie analityczne po-
winny byæ wolne od interferencji
spektralnych,
– powinien zachowywaæ siê po-
dobnie do oznaczanych pier-
wiastków.
Wy¿ej podane wymagania odno-
sz¹ siê do wszystkich metod anali-
tycznych, w przypadku których mo¿li-
we jest stosowanie techniki wzorca
wewnêtrznego. W metodzie ICP-OES
dodatkowe wymagania to dopasowa-
nie atomowego/jonowego charakteru
wybranej linii analitycznej wzorca we-
wnêtrznego i oznaczanego pierwiast-
ka. Poza tym nale¿y wzi¹æ pod uwa-
gê energie wzbudzenia i jonizacji
ka¿dej pary pierwiastków, które po-
winny byæ zbli¿one.
Sygna³ analityczny, zdefiniowany
jako stosunek sygna³u uzyskanego
dla analitu do sygna³u wzorca we-
wnêtrznego, umo¿liwia wyeliminowa-
nie wp³ywów, które w taki sam spo-
sób oddzia³uj¹ na sygna³y obu sub-
stancji (np. efektywnoϾ rozpylania
i transportu próbek). Zasad¹ metody
wzorca wewnêtrznego jest to, ¿e do
wszystkich roztworów/próbek doda-
wana jest sta³a iloœæ wzorca we-
wnêtrznego, sygna³ analityczny po-
zostaje proporcjonalny do zawartoœci
analitu w próbce
19
. Metoda ta umo¿-
liwia kompensacjê b³êdów zwi¹za-
nych z interferencjami fizycznymi –
dryftem termicznym, zró¿nicowaniem
lepkoœci próbek i roztworów kalibra-
cyjnych oraz niestabilnoœci¹ rozpyla-
nia. Zauwa¿ono, ¿e efektywnoœæ we-
wnêtrznej standaryzacji w du¿ej mie-
rze zale¿y od zastosowanych warun-
ków operacyjnych pracy spektrome-
tru. W warunkach odpornych zasto-
sowanie wzorca wewnêtrznego daje
lepsze efekty
20
.
Metoda wzorca wewnêtrznego jest
szeroko stosowana w ICP-OES w ce-
lu udoskonalenia precyzji i dok³adno-
œci. Zastosowanie pojedynczego
wzorca wewnêtrznego jest efektyw-
ne, gdy celem jest kompensacja
efektów fizycznych zwi¹zanych
z systemem wprowadzania próbki.
Funkcjê wzorców wewnêtrznych
pe³ni³y ró¿ne pierwiastki. Brenner
i wsp. stosowali jonow¹ liniê skandu
jako wzorzec wewnêtrzny w ozna-
czeniach pierwiastków w próbkach
geologicznych
21
. Stwierdzono, ¿e
pojedynczy wzorzec wewnêtrzny ca³-
kowicie kompensuje zmiany inten-
sywnoœci sygna³u analitu zwi¹zane
ze zmianami w systemie wprowadza-
nia próbki, gdy energie wzbudzenia
dla analitu i wzorca wewnêtrznego s¹
zbli¿one, lecz nie wykonano badañ
potwierdzaj¹cych tê teoriê.
Ivaldi i Tyson zastosowali liniê
Y II 371,030 nm jako wzorzec we-
wnêtrzny
22
. Ich badania wykaza³y, ¿e
stosowanie tego pierwiastka w roli
wzorca wewnêtrznego powoduje
czterokrotne ulepszenie %RSD, któ-
re osi¹ga wartoœæ 0,1
÷0,2%.
Wiele publikacji porusza temat sto-
sowania pojedynczego wzorca we-
wnêtrznego do kompensacji interfe-
rencji zwi¹zanych z obecnoœci¹ pier-
wiastków ³atwo ulegaj¹cych joniza-
cji
23,24,25
. Zaobserwowano, ¿e wp³y-
wy te mo¿na istotnie zminimalizowaæ,
stosuj¹c jako wzorzec wewnêtrzny li-
niê Sc II. Pierwiastek ten zasto-
sowano jako wzorzec wewnêtrzny do
ograniczenia wp³ywów matrycy Ca
i Na. Okaza³o siê jednak, ¿e pojedyn-
czy wzorzec wewnêtrzny nie kompen-
suje w pe³ni tego rodzaju efektów ma-
trycowych. W innych publikacjach opi-
sana jest rola Ni II 231 nm, jako wzor-
ca wewnêtrznego przy oznaczaniu
pierwiastków w matrycy sodu
26
. Ba-
dania przeprowadzono, stosuj¹c od-
porne warunki spektrometru. Nie jest
jednak mo¿liwa pe³na kompensacja
efektów matrycowych wywo³anych
obecnoœci¹ ³atwo jonizuj¹cych siê
pierwiastków z zastosowaniem poje-
dynczego wzorca wewnêtrznego.
Do poprawy dok³adnoœci niezbêd-
ne jest zastosowanie kilku wzorców
wewnêtrznych. Brenner i wsp.
27
za-
stosowali Sb, Be, Y i Sc jako wzorce
wewnêtrzne w oznaczaniu analitów
w obecnoœci wapnia i sodu o stê¿e-
niu dochodz¹cym do 0,5% (v/v).
Oprócz znacz¹cej poprawy odtwa-
rzalnoœci metody wzorca wewnêtrz-
nego zaobserwowano znacz¹c¹ po-
prawê dok³adnoœci.
Bardzo dok³adn¹ metod¹ ozna-
czania pierwiastków g³ównych i œla-
dowych w ska³ach krzemianowych
okaza³a siê metoda PRISM (ang. Pa-
rameter Related Internal Standard
Method)
28
. Ta procedura umo¿liwia
kompensacjê b³êdów systematycz-
nych i losowych za pomoc¹ dwóch
wzorców wewnêtrznych: Cd i Rb.
Jednak¿e matryca próbek i wzorców
by³a podobna – zawiera³a zbli¿one
zawartoœci Li, który prawdopodobnie
zachowywa³ siê jak bufor
29
.
Inn¹ modyfikacj¹ metody wzorca
wewnêtrznego jest technika CAIS
(ang. Common Analyte Internal Stan-
darization)
30
. Opiera siê ona na jed-
noczesnych pomiarach dwóch linii
analitycznych tego samego pier-
wiastka. Jedna z tych linii s³u¿y do
pomiaru stê¿enia (linia analityczna),
a druga pe³ni funkcjê wzorca we-
wnêtrznego, za pomoc¹ którego
mo¿na minimalizowaæ efekty matry-
cowe. Podstawowym wymogiem tej
techniki jest to, by efekt matrycowy
wp³ywa³ na liniê analityczn¹ w inny
PROBLEMY KRYMINALISTYKI 252/06
26
sposób ni¿ na liniê odniesienia. Me-
todê zastosowano do korekcji wp³y-
wów trzech matryc: HNO
3
, H
2
SO
4
i NaCl. Badanymi pierwiastkami by³y
mangan, magnez, lantan i bar. Korek-
cja za pomoc¹ tej techniki da³a bar-
dzo dobre wyniki.
Odporne warunki plazmy
– wyznaczanie stosunku Mg II/Mg I
w ró¿nych matrycach
Na pocz¹tku sprawdzono odpor-
noϾ plazmy przy wprowadzaniu
próbek o ró¿nych matrycach. Jako
kryterium odpornoœci plazmy
wybrano stosunek intensywnoœci
dwóch linii magnezu Mg II 280,270
nm i Mg I 285,213 nm. Analizie pod-
dano dwie próbki wzorców magnezu
o stê¿eniu 1 mg/l o ró¿nych matry-
cach. Matryc¹ pierwszej by³a woda,
a drugiej kwas azotowy (V) o stê¿e-
niu 20%. Po analizie wzorców
sprawdzono, w jaki sposób zmienia
siê odpornoœæ plazmy przy wprowa-
dzaniu próbki roœlinnej – materia³u
certyfikowanego CTA-OTL-1 (orien-
talne liœcie konopi), rozcieñczonego
10-krotnie. Rozcieñczenie tej próbki
by³o niezbêdne ze wzglêdu na prze-
³adowanie linii Mg II 280,271 nm
podczas analizy próbki nierozcieñ-
czonej. Nale¿y zaznaczyæ, ¿e
wszystkie analizy wykonano w dwóch
seriach. W pierwszej serii próbki ana-
lizowano, stosuj¹c standardowe wa-
runki pracy spektrometru, a w drugiej
– warunki odporne. Uzyskane wyniki
zamieszczono w tabeli 5.
Jak mo¿na zauwa¿yæ, stosunki Mg
(II)/Mg (I) dla matrycy wodnej i kwaso-
wej s¹ porównywalne. W warunkach
standardowych wynios³y one odpo-
wiednio 5,2 i 5,1 dla matrycy wodnej
i kwaœnej. Zastosowanie warunków
odpornych znacznie poprawi³o odpor-
noœæ plazmy, gdy¿ wartoœæ stosunku
Mg (II)/Mg (I) wzros³a znacz¹co do
wartoœci oko³o 8,7. Natomiast odpor-
noϾ plazmy w warunkach standardo-
wych podczas analizy próbki roœlinnej
CTA-OTL-1 x 10 jest s³absza, o czym
œwiadczy ni¿szy stosunek Mg (II) /Mg
(I) równy 4,7. Po zastosowaniu warun-
ków odpornych wzrós³ on istotnie, lecz
matryca próbki by³a
na tyle z³o¿ona, ¿e
osi¹gniêcie wartoœci
takich jak dla wzor-
ców wodnych i kwa-
œnych by³o niemo¿li-
we. WartoϾ stosun-
ku Mg (II)/Mg (I) dla
tej próbki analizowa-
nej z zastosowaniem
warunków odpornych
wynios³a 8,2.
Badania praktycz-
ne potwierdzi³y tezê,
¿e zastosowanie wa-
runków odpornych
mo¿e zminimalizowaæ wp³yw matrycy.
Dlatego te¿ postanowiono zbadaæ,
czy ich zastosowanie minimalizuje
wp³ywy matrycy w próbce konopi
w zadowalaj¹cym stopniu. W tym celu
przygotowano serie roztworów, tak jak
podano w tabeli 4. W analizie tych roz-
tworów zastosowano warunki odporne
plazmy. Uzyskane wyniki zamieszczo-
no na rycinie 5.
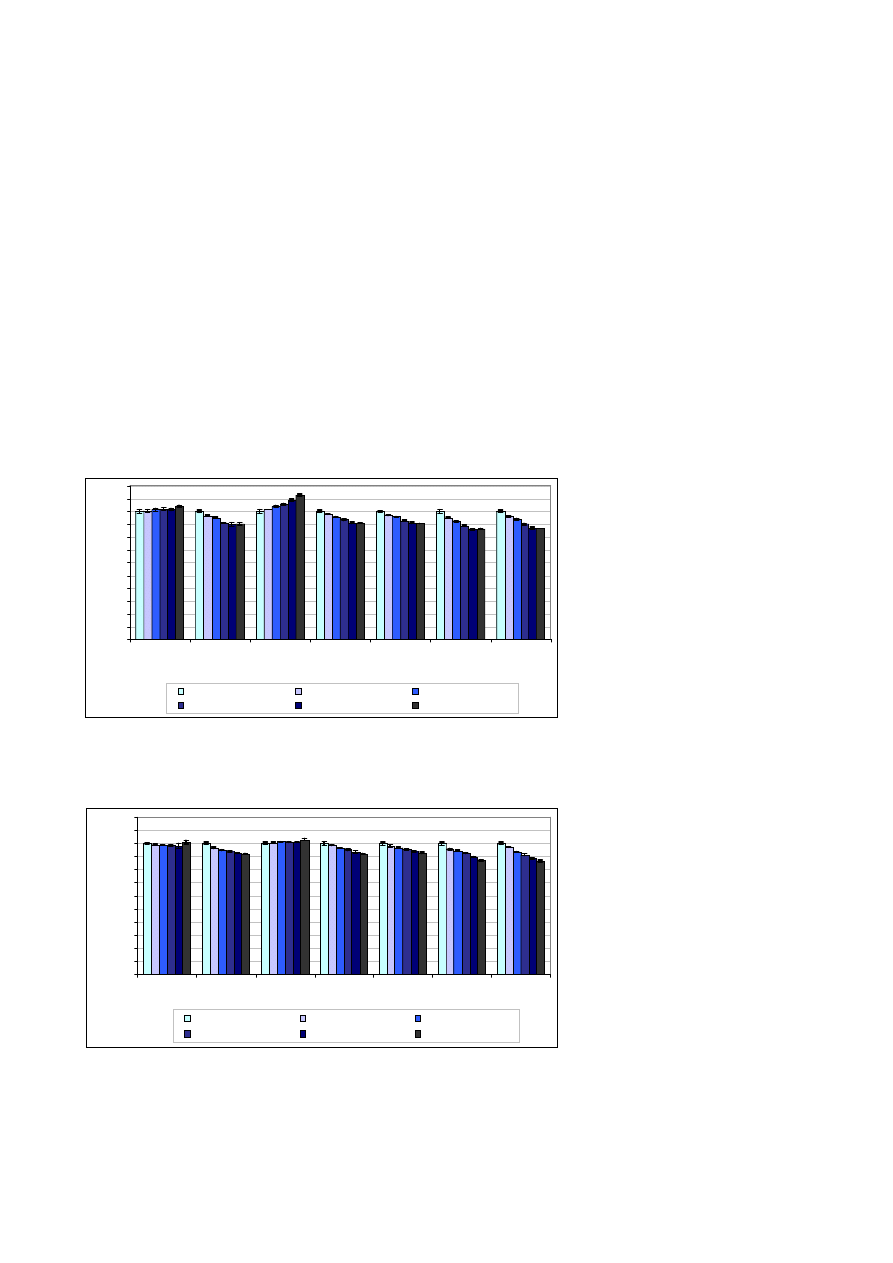
Tendencje zmian intensywnoœci
sygna³ów przy wzrastaj¹cym stê¿e-
niu matrycy z zastosowaniem odpor-
nych warunków plazmy s¹ podobne
jak przy stosowaniu warunków stan-
dardowych (ryc. 4).
W obecnoœci matrycy obserwowa-
ny jest wzrost intensywnoœci sygna³u
dla linii boru oraz miedzi. Dla pozo-
sta³ych pierwiastków wzrost stê¿enia
wapnia i magnezu w próbce powo-
duje os³abienie sygna³u. Nale¿y jed-
nak zaznaczyæ, ¿e zmiany intensyw-
noœci sygna³ów nie s¹ tak drastycz-
ne jak w przypadku warunków stan-
dardowych i wynosz¹ od -11% dla
¿elaza do +14% dla miedzi. Zastoso-
wanie odpornych warunków plazmy
umo¿liwia dok³adne oznaczanie je-
dynie boru, gdy¿ w tym przypadku
wp³yw matrycy na intensywnoœæ sy-
gna³u wynosi maksymalnie +3%. Dla
PROBLEMY KRYMINALISTYKI 252/06
27
0
10
20
30
40
50
60
70
80
90
100
110
120
B Ba
Cu
Fe
Mn
Sr Zn
Be
313.107
Be
234.861
Cr
267.716
Cr
205.560
Y
371.029
pierwiastek
intensywność względna [%]
0 mg/l Ca+Mg
220 mg/l Ca+Mg
440 mg/l Ca+Mg
880 mg/l Ca+Mg
1320 mg/l Ca+Mg
1760 mg/l Ca+Mg
Ryc. 5. Wzglêdna zmiana intensywnoœci sygna³ów uzyskanych dla analitów w obecnoœci wzrastaj¹cego
stê¿enia matrycy wapnia i magnezu oraz dla metody wzorca wewnêtrznego (wyniki uzyskano, stosuj¹c
odporne warunki plazmy)
Fig. 5. Relative change of analyte intensity in presence of increasing concentration of calcium and
magnesium matrix and for internal standardisation (results obtained by using resistant plasma
conditions)
Tabela 5
Zestawienie wartoœci Mg (II) /Mg (I) uzyskanych
dla ró¿nych próbek w ró¿nych warunkach
operacyjnych spektrometru
Mg (II)/MG (I) values obtained for various samples in
different operating conditions of spectrometer
M
Maattrry
yccaa
W
Waarru
un
nk
kii
M
Mg
g((IIII))//M
Mg
g((II))
Standardowe
5,2
H
H
2
2
O
O
Odporne
8,6
Standardowe
5,1
2
20
0%
% H
HN
NO
O
3
3
Odporne
8,7
Standardowe
4,7
C
CT
TA
A--O
OT
TL
L--1
1 x
x 1
10
0
Odporne
8,2
WZORCE WEWNĘTRZNE
pozosta³ych pierwiastków jest on
wiêkszy od 10%.
Zastosowanie
warunków odpornych
i dodatku buforu jonizuj¹cego
Zastosowanie warunków odpor-
nych plazmy doprowadzi³o do znacz-
nej redukcji wp³ywów matrycy, nie
zminimalizowa³o ich jednak na tyle,
by mo¿na by³o je zaakceptowaæ. Po-
stanowiono wiêc zbadaæ, w jaki spo-
sób na zmianê intensywnoœci sygna-
³ów w obecnoœci wapnia i magnezu
wp³ywa obecnoœæ pierwiastka I grupy
uk³adu okresowego – sodu –
w dwóch ró¿nych stê¿eniach. Zasto-
sowano odporne warunki plazmy.
Nadmiar buforu matrycy, a w konse-
kwencji elektronów w plazmie, jest
tak du¿y, ¿e jakiekolwiek zmiany stê-
¿enia wapnia i magnezu nie powinny
powodowaæ istotnych zmian inten-
sywnoœci sygna³ów pierwiastków œla-
dowych. W próbkach roœlinnych,
w tym konopi, pierwiastkiem obec-
nym w próbce, mog¹cym pe³niæ funk-
cjê bufora, jest potas.
Nale¿y zaznaczyæ, ¿e intensyw-
noœæ dla pierwiastków, dla których
oznaczeñ wykorzystano linie atomo-
we, powinna wzrastaæ w obecnoœci
sodu, a dla analitów, dla których wy-
brano linie jonowe – maleæ. Stê¿enie
sodu powinno byæ tak dobrane, aby
sygna³y analitów w obecnoœci wzra-
staj¹cego stê¿enia matrycy nie ulega-
³y zmianie, co wi¹¿e siê z os³abieniem
czu³oœci dla linii jonowych, a poprawê
dla linii atomowych. Uzyskane wyniki
przedstawiono na rycinach 6 i 7.
Jak mo¿na zauwa¿yæ na wykre-
sach, sód dodany w stê¿eniach 800
i 1200 mg/l nie ustabilizowa³ stê¿enia
elektronów w plazmie. Nale¿a³oby
zastosowaæ jeszcze wy¿sze stê¿enie
sodu, co jednak jest niekorzystne ze
wzglêdu na os³abianie czu³oœci dla
wiêkszoœci oznaczanych pierwiast-
ków, z których np.: Sr i Zn oznaczane
s¹ na bardzo niskim (dla metody
ICP-OES) poziomie stê¿eñ. Dodatek
buforu matrycy w stê¿eniu zarówno
800 mg/l, jak i 1200 mg/l umo¿liwia
dok³adne i precyzyjne oznaczenia tyl-
ko boru.
Dodatek 1200 mg/l sodu oprócz
boru minimalizuje wp³ywy matrycy
równie¿ dla miedzi do 3%, dlatego
mo¿na go stosowaæ w oznaczeniach
rutynowych. Dla pozosta³ych pier-
wiastków dodatek sodu jako buforu
matrycy nie minimalizuje niestety
wp³ywów matrycy w takim stopniu, by
metodê mo¿na by³o zastosowaæ
w codziennej pracy analitycznej. In-
tensywnoœci sygna³ów dla takich pier-
wiastków jak stront i cynk s¹ ni¿sze
o oko³o 13% w obecnoœci najwy¿sze-
go stê¿enia wapnia, magnezu i sodu.
Z wykresów 5, 6 wynika równie¿,
¿e dla pierwiastków takich jak bor,
¿elazo, mangan, stront i cynk stabili-
zuj¹cy efekt dodatku sodu obserwo-
wany jest dla stê¿enia matrycy powy-
¿ej 880 mg/l. Oznacza to, ¿e wapñ
i magnez, jako pierwiastki zwiêksza-
j¹ce stê¿enie elektronów w plazmie,
wzmacniaj¹ efekt buforuj¹cy sodu,
ich stê¿enie musi jednak przekraczaæ
880 mg/l.
Zastosowanie
warunków odpornych
i metody wzorca wewnêtrznego
W celu wyboru wzorca wewnêtrz-
nego, najlepiej minimalizuj¹cego
efekty matrycowe, postanowiono
przeanalizowaæ zachowanie kilku
pierwiastków, które nie s¹ obecne
w matrycy roœlinnej, a zatem mog¹
pe³niæ funkcjê wzorców wewnêtrz-
nych. Zbadano zachowanie trzech
pierwiastków Be, Cr i Y w obecnoœci
matrycy wapnia i magnezu o wzra-
PROBLEMY KRYMINALISTYKI 252/06
28
0
10
20
30
40
50
60
70
80
90
100
110
120
B
Ba Cu
Fe
Mn Sr
Zn
pierwiastek
intensywność względna [%]
0 mg/l Ca+Mg
220 mg/l Ca+Mg
440 mg/l Ca+Mg
880 mg/l Ca+Mg
1320 mg/l Ca+Mg
1760 mg/l Ca+Mg
0
10
20
30
40
50
60
70
80
90
100
110
120
B
Ba Cu
Fe
Mn Sr
Zn
pierwiastek
intensywność względna [%]
0 mg/l Ca+Mg
220 mg/l Ca+Mg
440 mg/l Ca+Mg
880 mg/l Ca+Mg
1320 mg/l Ca+Mg
1760 mg/l Ca+Mg
Ryc. 6. Wzglêdna zmiana intensywnoœci sygna³ów uzyskanych dla analitów w obecnoœci wzrastaj¹cego
stê¿enia matrycy wapnia i magnezu oraz z dodatkiem sodu o stê¿eniu 800 mg/l
Fig. 6. Relative change of analyte intensity in presence of increasing concentration of calcium and
magnesium matrix and with addition of 800 mg/l conc. sodium
Ryc. 7. Wzglêdna zmiana intensywnoœci sygna³ów uzyskanych dla analitów w obecnoœci wzrastaj¹cego
stê¿enia matrycy wapnia i magnezu oraz z dodatkiem sodu o stê¿eniu 1200 mg/l
Fig. 7. Relative change of analyte intensity in presence of increasing concentration of calcium and
magnesium matrix and with addition of 1200 mg/l sodium
staj¹cym stê¿eniu. Wyniki analiz za-
mieszczono wczeœniej na rycinie 5 ob-
razuj¹cym zachowanie pierwiastków
w obecnoœci Ca i Mg z zastosowa-
niem warunków odpornych pracy
spektrometru.
Wybór tych trzech pierwiastków
podyktowany by³ przede wszystkim
faktem, ¿e nie s¹ one sk³adnikami
próbek konopi. Ponadto dane pi-
œmiennicze donosz¹, ¿e s¹ one efek-
tywne w metodzie wewnêtrznej stan-
daryzacji.
Na pocz¹tku sprawdzono efek-
tywnoœæ wewnêtrznej standaryzacji
dwóch linii analitycznych chromu:
Cr 205,560 i Cr 267,716. Obydwie
te linie maj¹ charakter jonowy. Z te-
go wzglêdu nie by³o mo¿liwe dobra-
nie ich jako wzorców wewnêtrznych
dla linii B 249,677 i Cu 327,393, któ-
re maj¹ charakter atomowy. Ponad-
to zbyt du¿a ró¿nica w energii joni-
zacji chromu i baru uniemo¿liwi³a
dobranie Cr jako wzorca wewnêtrz-
nego dla tego pierwiastka. W tabeli
6 zamieszczono oznaczane pier-
wiastki oraz dobrane dla nich linie
chromu jako wzorca wewnêtrznego.
By zbadaæ, jak efektywnie chrom
kompensuje efekty matrycowe wywo-
³ane ró¿nymi czynnikami, przygoto-
wano próbki oznaczanych pierwiast-
ków w ró¿nych matrycach. Pierwsza
seria próbek zawiera³a wzorce wod-
ne, matryc¹ drugiej by³ kwas azotowy
(V) o stê¿eniu 20%. Trzecia seria
próbek oprócz oznaczanych pier-
wiastków i kwasu azotowego (V)
o stê¿eniu 20% zawiera³a 1600 mg/l
wapnia i 160 mg/l magnezu – czyli
najwy¿sze stê¿enia tych pierwiast-
ków wystêpuj¹ce w próbkach konopi.
Uzyskane wyniki kompensacji efek-
tów matrycowych za pomoc¹ chromu
jako wzorca wewnêtrznego przed-
stawiono na ryc. 8.
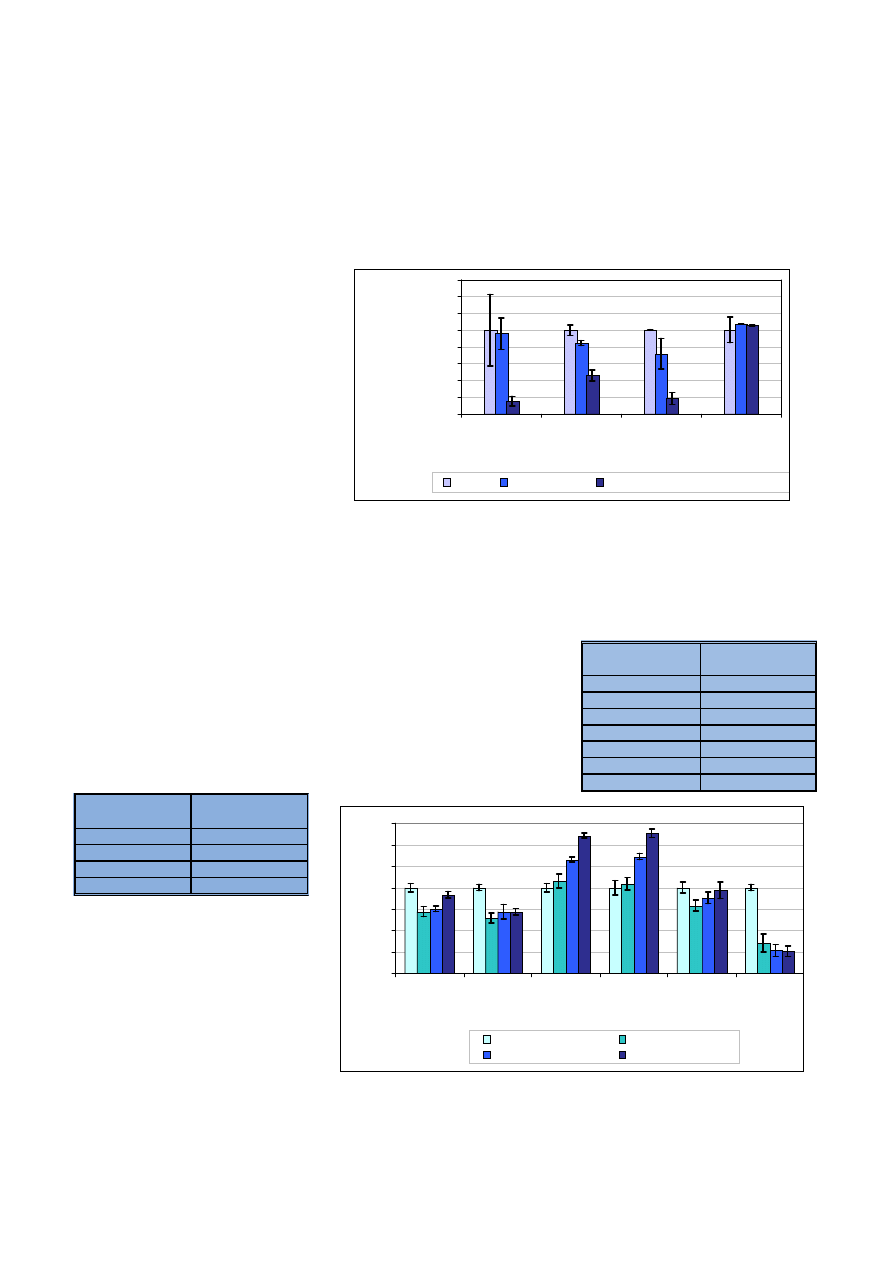
Wyniki, które mo¿na uznaæ za za-
dowalaj¹ce, uzyskano dla manganu,
dla którego maksymalny b³¹d powo-
dowany obecnoœci¹ matrycy wynosi³
+0,8%. Dla pozosta³ych pierwiastków
uzyskane wyniki obarczone s¹ zbyt
du¿ym b³êdem, dochodz¹cym do
wartoœci -8,5% dla cynku, -8,2% dla
¿elaza i -5,4% dla strontu. Uniemo¿li-
wia to zastosowanie chromu jako
wzorca wewnêtrznego przy rutyno-
wych oznaczeniach tych pierwiast-
ków w skomplikowanej matrycy.
Chrom jest dobrym wzorcem we-
wnêtrznym dla niewielu oznaczanych
pierwiastków w próbkach konopi.
Dlatego te¿ prowadzono dalsze po-
szukiwania pierwiastków, które spe³-
nia³yby funkcjê mo¿liwie wszech-
stronnych wzorców wewnêtrznych.
Ze wzglêdu na oszczêdnoœæ czasu
przygotowania próbek oraz odczynni-
ków po¿¹dane by³o zastosowanie jak
najmniejszej liczby wzorców we-
wnêtrznych do mo¿liwie du¿ej liczby
analitów bez uszczerbku dla dok³ad-
noœci oznaczeñ. Postanowiono zba-
daæ, jak zadanie to spe³ni¹ beryl i itr.
Po analizie wartoœci energii wzbu-
PROBLEMY KRYMINALISTYKI 252/06
29
Tabela 6
Cr jako wzorzec wewnêtrzny
Cr as internal standard
O
Ozzn
naacczzaan
ny
y
p
piie
errw
wii aasstte
ek
k
W
Wzzo
orrzze
ecc
w
we
ew
wn
nê
êttrrzzn
ny
y
Zn 206,200
Cr 205,560
Sr 232,235
Cr 205,560
Fe 238,204
Cr 205,560
Mn 257,610
Cr 267,716
Ryc. 8. Wzglêdna zmiana intensywnoœci sygna³ów uzyskanych dla analitów, dla których wzorcem we-
wnêtrznych by³ chrom
Fig. 8. Relative change of analyte intensity with Cr as internal standard
Tabela 7
Be i Y jako wzorce wewnêtrzne
Be and Y as internal standards
O
Ozzn
naacczzaan
ny
y
p
piie
errw
wiiaasstte
ek
k
W
Wzzo
orrzze
ecc
w
we
ew
wn
nê
êttrrzzn
ny
y
B 249,677
Be 234,861
Ba 233,527
Y 371,029
Cu 327,393
Be 234,861
Fe 238,204
Be 313,107
Mn 257,610
Y 371,029
Sr 232,235
Y 371,029
Zn 206,200
Be 313,107
92
94
96
98
100
102
104
106
B Ba Cu Fe
Mn
Sr
pierwiastek
względna intensywność [%]
0 mg/l Ca+Mg
440 mg/l Ca+Mg
1100 mg/l Ca+Mg
1760 mg/l Ca+Mg
Ryc. 9. Wzglêdna zmiana intensywnoœci sygna³ów uzyskanych dla analitów, dla których wzorcami we-
wnêtrznymi by³y beryl i itr
Fig. 9. Relative change of analyte intensity with Be and Y as internal standards
90
92
94
96
98
100
102
104
106
Zn Sr Fe Mn
pierwiastek
względna intensywność
[%]
H2O
20% HNO3
20% HNO3 1760 mg/l Ca+Mg

dzenia i jonizacji linii oraz ich charak-
teru atomowego/jonowego dobrano
wzorce wewnêtrzne do analitów, co
pokazuje tabela 7. By zbadaæ efek-
tywnoœæ wybranych wzorców we-
wnêtrznych, podobnie jak wczeœniej
przygotowano serie roztworów
o zmieniaj¹cej siê matrycy. Wyniki
przedstawiono na rycinie 9. Beryl i itr
to pierwiastki dobrze spe³niaj¹ce wy-
magania stawiane wzorcom we-
wnêtrznym. Dla wiêkszoœci oznacza-
nych pierwiastków kompensacja
efektów matrycowych za ich pomoc¹
jest zadowalaj¹ca, a b³¹d nie prze-
kracza 5%. Najwiêkszy b³¹d uzyska-
no dla strontu -5,9%. Uznano, ¿e taki
dobór wzorców wewnêtrznych mo¿e
byæ zastosowany w rutynowych ana-
lizach próbek konopi.
Podsumowanie
Wykonane badania s¹ kolejnym
etapem w opracowywaniu metody
profilowania konopi na podstawie
sk³adu pierwiastkowego. Pozwoli³y
one na okreœlenie wp³ywu efektów
matrycowych obecnych podczas
oznaczania pierwiastków w próbkach
konopi oraz ich minimalizacje. Po-
równano trzy techniki korekcji interfe-
rencji fizycznych: zastosowanie wa-
runków operacyjnych odpornych, do-
datek buforu jonizuj¹cego oraz meto-
dê wzorca wewnêtrznego. Stwierdzo-
no, ¿e metoda wzorca wewnêtrznego
najlepiej kompensuje wp³yw wapnia
i magnezu na dok³adnoœæ oznaczeñ.
By sprawdziæ dok³adnoœæ tej metody,
przeprowadzono analizê czterech
certyfikowanych materia³ów roœlin-
nych. Procentowy odzysk mieœci³ siê
w granicach 95
÷106%, co mo¿na
uznaæ za wartoœci zadowalaj¹ce. Wy-
niki oznaczania pierwiastków w prób-
kach materia³ów roœlinnych metod¹
wzorca wewnêtrznego charakteryzu-
j¹ siê du¿¹ dok³adnoœci¹. Opracowy-
wana metoda mo¿e zatem s³u¿yæ do
rutynowych analiz próbek konopi.
PRZYPISY
1 M. Kuras: praca magisterska „Anali-
za elementarna wybranych narkoty-
ków oraz pó³produktu i produktu syn-
tezy siarczanu 4-etoksyamfetaminy”,
Wydzia³ Chemii UW, W-wa 2002;
2 M. Wachowicz, M. Kuras: Wstêp do
profilowania konopi na podstawie
sk³adu pierwiastkowego, „Problemy
Kryminalistyki” 2003, nr 240, s. 10–19;
3 M. Wachowicz, M. Kuras: Minerali-
zacja mikrofalowa jako jedna z tech-
nik przygotowania próbek do badañ
porównawczych, „Problemy Krymi-
nalistyki” 2002, nr 238, s. 8–22;
4 M. Kuras, praca magisterska, op.cit.;
5 L. Pszonicki, W. Skwara: The stan-
dard addition and successive dilu-
tion method for evaluation and verifi-
cation of results in atomic – absorp-
tion analysis, „Talanta”, 12 (1989), s.
1265–1276;
6 L. Pszonicki: Significance of extra-
polation methods for the evaluation
of analytical signals, [w:] A. Kabata-
-Pendias, B. Szteke: Quality pro-
blems in trace analysis in environ-
mental studies, Wydawnictwo Edu-
kacyjne Zofii Dobkowskiej, Warsaw
1998, s. 79–111;
7 X. Romero, E. Poussel, J.M. Mer-
met: The effect of sodium on analy-
te line intensities in inductively co-
upled plasma atomic emission spec-
trometry: influence of the operating
conditions, „Spectrochim. Acta” Part
B 52 (1997), s. 495–502;
8 I. Novotny, J.C. Farińas, J.L. Wan,
E. Poussel, I.M. Mermet: Effect on
power and carrier gas flow rate on the
tolerance to water loading in inducti-
vely coupled plasma atomic emission
spectrometry, „Spectrochim. Acta”
Part B 51 (1996), s. 1517–1526;
9 I.M. Mermet: Ionic to atomic line in-
tensity ratio and residence time in in-
ductively coupled plasma – atomic
emission spectrometry, „Spectrochim.
Acta” Part B 44 (1989), s. 1109–1116;
10 P.W.J.M. Boumans, „ICP Inf.
Newsl.”, 4 (1978), s. 89;
11 T. Edmonds, G. Horlick: „Appl.
Spectrosc.”, 31 (1977) 536;
12 J.M. Mermet: Use of magnesium as
a test element for inductively coupled
plasma atomic emission spectrome-
try diagnostics, „Anal. Chim. Acta”
250 (1991), s. 85–94;
13 J. Dennaud, A. Howes, E. Poussel,
J.M. Mermet: Study of ionic-to-a-
tomic line intensity ratios for two
axial viewing-based inductively co-
upled plasma atomic emission spec-
trometers, „Spectrochim. Acta” Part
B 56 (2001), s. 101–112;
14 W.L. Wiese, M.W. Smith, B.M. Mi-
les: Atomic transition probabilities.
Sodium through calcium, National
Bureau Standards, Washington DC,
National Standards Reference Data
Series 2, 1969;
15 M. Stepan, P. Musil, E. Poussel,
J.M. Mermet: Matrix-induced shift ef-
fects in axially viewed inductively co-
upled plasma atomic emission spec-
trometry, „Spectrochim. Acta” Part B
56 (2001), s. 443–454;
16 Ibidem;
17 A.C. Fernandez, M. Murillo, N. Car-
rion, J.M. Mermet: „J. Anal. At.
Spectrom.” 9 (1994) 217;
18 J. Dennaud, A. Howes, E. Poussel,
J. M. Mermet: Study of ionic-to-a-
tomic line in-tensity ratios for two
axial viewing-based inductively co-
upled plasma atomic emission spec-
trometers, „Spectrochim. Acta” Part
B 56 (2001), s. 101–112;
19 K. Wróbel: Analityka, nr 1, 2001;
20 X. Romero, E. Poussel, J.M. Mer-
met: Influence of the operating con-
ditions on the efficiency of internal
standardization in inductively co-
upled plasma atomic emission spec-
trometry, „Spectrochim. Acta” Part B
52 (1997), s. 487–393;
21 I.B. Brenner, A. Zander, M. Cole, A.
Wiseman: „J. Anal. At. Spectrom”,
12 (1997), 897;
22 J.C. Ivaldi, J.F. Tyson: „Spectro-
chim. Acta”, 51B, 1443, 1996;
23 I.B. Brenner, H. Eldad, S. Erlich, N. Dal-
man: „Anal. Chim. Acta”, 1984, 66, 51;
24 I.B. Brenner, A. E Watson, G.M.
Roussel, M. Gonclaves, „Chem.
Geol.”, 1980, 28, 321;
25 I.B. Brenner, A. Zander, M. Cole, A.
Wiseman: „J. Anal. At. Spectrom.”,
12 (1997), 897;
26 M. Stepan, P. Musil, E. Poussel,
J.M. Mermet: op.cit.;
27 I.B. Brenner, A. Le Marchand, C.
Daraed, L. Chauvet: Compensation
of Ca and Na interference effects in
axially and radially viewed inductive-
ly coupled plasmas, Microchemical
Journal 63 (1999), s. 344–355;
28 M.H. Ramsey, M. Thompson: „J.
Anal. At. Spectr.”, 2 (1987), s. 497;
29 I.B. Brenner, A. Le Marchand, C.
Daraed, L. Chauvet: op.cit.,
s. 344–355;
30 A.S Al-Ammar, R.M Barnes: Cor-
rection for non-spectroscopic ma-
trix effects in inductively coupled
plasma atomic emission spectro-
scopy by internal standardization
using spectral lines of the same
analyte, „Spectrochim. Acta” Part B
(1998), s. 1583–1593.
PROBLEMY KRYMINALISTYKI 252/06
30
Wyszukiwarka
Podobne podstrony:
Profilowanie konopii na podstawie składu pierwiastkowego Część II walidacja metody
0krślanie struktury stali specjalnych na podstawie składu chemicznego
Efekty matrycowe w profilowaniu konopi (elementy walidacji metody)
I . ANALIZA POJĘĆ NA PODSTAWIE LITERATURY, szkoła, Rady Pedagogiczne, wychowanie, profilaktyka
9.Rysunki złożeniowe, Rysunek zlozeniowy, Rysunek wykonawczy-rysunek na podstawie ktorego wykonana b
ukladokres, Układ okresowy pierwiastków, układ periodyczny pierwiastków, tablica grupująca pierwiast
Efekty matrycowe w profilowaniu konopi (elementy walidacji metody)
5 Sałacka A Profilaktyka zakażeń pneumokokowych na podstaw
ING Lojalność wobec klientów na podstawie ING Banku Śląskiego S A
PDW na podstawie obserwacji pedagogicznej
Lęk i samoocena na podstawie Kościelak R Integracja społeczna umysłowo UG, Gdańsk 1995 ppt
Prognozowanie na podstawie modeli autoregresji
Uczucia Juliusza Słowackiego na podstawie utworów, Notatki, Filologia polska i specjalizacja nauczyc
Status producenta na podstawie przepisów prawa w oparciu o praktykę, BHP I PRAWO PRACY, PORADY PRAWN
więcej podobnych podstron