
PROCESY KATALITYCZNE I ADSORPCYJNE W OCHRONIE
RODOWISKA
Laboratorium
wiczenie Nr 3
Adsorpcyjne usuwanie zanieczyszcze na w glu aktywnym

2
ADSORPCJA NA W GLU AKTYWNYM
I. C z t e o r e ty c z n a
1. Wst p
Problemy zwi zane z zanieczyszczeniem rodowiska naturalnego oraz wdra aniem
w przemy le nowych, efektywnych technologii spowodowały wzrost zainteresowania
procesami adsorpcji i adsorbentami w glowymi (w glami aktywnymi).
W giel aktywny jest otrzymywany w procesie
karbonizacji i aktywacji materiałów
głównie pochodzenia ro linnego. Produktem procesu karbonizacji, tj. ogrzewania w gla
bez dost pu powietrza i bez udziału czynników chemicznych, jest praktycznie
nieaktywny adsorpcyjnie materiał o niewielkiej powierzchni wła ciwej. Materiał o du ej
porowato ci i silnie rozwini tej powierzchni - setek m
2
/g, a nawet powy ej kilku tysi cy
m
2
/g, jest otrzymywany z karbonizatu w procesie jego aktywacji. Taki adsorbent, maj cy
silnie rozbudowany system porów, nazywa si
w glem aktywnym.
Przez aktywacj rozumie si procesy, które prowadz do rozwini cia powierzchni
wewn trznej ciała stałego przez rozbudowanie istniej cej ju pierwotnie struktury
kapilarnej lub jej modyfikacj .
Ze wzgl du na budow układu kapilarnego, ciała stałe mo na podzieli na: porowate
i nieporowate.
Ciała nieporowate maj niewielk powierzchni wła ciw , zwi zan głównie
z powierzchni zewn trzn , któr mo na zwi kszy przez rozdrobnienie tych substancji.
Ciała porowate natomiast maj silnie rozbudowany system porów. Ich powierzchnia
wła ciwa jest sum powierzchni zewn trznej i wewn trznej, przy czym powierzchnia
wewn trzna jest wielokrotnie wi ksza od powierzchni zewn trznej. Oprócz tego, wskutek
wyst puj cych specyficznych krzywizn porów, ma ona zwi kszony potencjał adsorpcyjny,
dzi ki czemu ma zdolno , w wi kszym lub mniejszym stopniu, wi zania (adsorbowania)
z otoczenia cz steczek, atomów lub jonów. Proces adsorpcji znajduje szerokie zastosowanie
w wielu gał ziach przemysłu.
Ciało stałe, na którego powierzchni zachodzi adsorpcja, nazywa si
adsorbentem,
a substancja adsorbowana
adsorbatem.
W giel aktywny znany był ju wiele lat temu, a jego pierwsze praktyczne
zastosowanie zwi zane było z medycyn . Ju Hipokrates i jego uczniowie zalecali
zasypywanie ran w glem drzewnym w celu ich szybszego gojenia i usuwania przykrego
zapachu. W Egipcie ok. 1500 roku p.n.e. stosowano w giel drzewny w dolegliwo ciach
oł dkowych a w Indiach do oczyszczania wody pitnej.

3
wiadome jego wykorzystanie w skali przemysłowej wi e si z przemysłem
cukrowniczym (odbarwianie syropów cukrowych), a nast pnie z przemysłem zbrojeniowym
(maski przeciwgazowe, twórca maski prof. Zieli ski z Politechniki Petersburskiej; w 1915
roku Niemcy, pod koniec pierwszej wojny wiatowej, u yli po raz pierwszy gazów
bojowych we Francji i w Polsce, przeciwko wojskom francuskim, angielskim i rosyjskim-
st d gor czkowe poszukiwania ochrony dróg oddechowych). Po drugiej wojnie wiatowej
nast puje gwałtowny rozwój przemysłu i nauki zwi zanej z zjawiskiem adsorpcji na w glach
aktywnych.
W ci gu ostatnich lat obserwuje si gwałtowny rozwój zapotrzebowania na w giel
aktywny, zwi zany z rosn cym zanieczyszczeniem rodowiska naturalnego człowieka. Jest
on coraz cz ciej stosowany jako adsorbent w oczyszczaniu powietrza, wód i cieków,
a tak e jako materiał elektrodowy w chemicznych ródłach pr du, czy jako no nik
katalizatorów. Intensywnie s rozwijane prace technologiczne nad zastosowaniem w gli
aktywnych do usuwania z powietrza SO
2
, H
2
S, CS
2
i tlenków azotu.
Obecnie wiatowa produkcja w gli aktywnych wynosi ok.400 tys. Mg/rok, z czego
ponad 90 % stanowi w gle aktywne do adsorpcji z fazy gazowej i ciekłej. Ocenia si ,
e roczne zapotrzebowanie na w gle aktywne wzrasta o ok. 7 %.
2. Otrzymywanie w gli aktywnych
2.1. Surowce stosowane do produkcji w gli aktywnych
Dobór surowca do produkcji w gla aktywnego jest zagadnieniem bardzo istotnym.
Podstawowe własno ci otrzymywanych w gli aktywnych, takie jak budowa i wielko
układu kapilarnego, wytrzymało mechaniczna czy zdolno ci adsorpcyjne, w du ej mierze
zale od u ytych surowców.
Surowcem o podstawowym znaczeniu s w gle kopalne, poczynaj c od w gli
brunatnych, a ko cz c na antracytach. Ich udział w produkcji w gli aktywnych wynosi
ok. 60%. Wytwarzanie krajowych w gli aktywnych opiera si głównie na w glu
kamiennym. Wybór w gla kamiennego jako surowca do otrzymywania w gla aktywnego
powinien by podyktowany po danymi własno ciami produktu ko cowego. W gle
kamienne wysokouw glone stanowi dobry surowiec do otrzymania w gli aktywnych do
sorpcji par i gazów. W celu otrzymania w gli aktywnych o szerokim rozkładzie porów
w funkcji ich promienia zaleca si stosowanie w gli niskouw glonych o du ej zawarto ci
cz ci lotnych.
Spo ród surowców o małym stopniu uw glenia praktyczne znaczenie przemysłowe
maj : w giel brunatny, torf, materiały pochodzenia drzewnego, tworzywa sztuczne
i syntetyczne.
Materiał w glowy przeznaczony do produkcji w gli aktywnych powinien si
charakteryzowa :
• mał zawarto ci cz ci lotnych

4
• wysok zawarto ci w gla pierwiastkowego
• okre lon porowato ci
• odporno ci mechaniczn na rozkruszanie i cieranie.
W gle aktywne otrzymane z w gla brunatnego maj lepiej rozbudowany układ kapilarny
ni w gle aktywne uzyskane z w gli kamiennych, ale charakteryzuj si nisk
wytrzymało ci mechaniczn i s stosowane głównie jako w gle jednokrotnego u ycia,
np. do oczyszczania cieków.
W gle aktywne mog wyst powa w postaci pyłu, jako w gle ziarnowe, b d
granulowane. W gle aktywne pyłowe i ziarnowe s to najcz ciej adsorbenty
jednorazowego u ycia.
2.2. Granulowane w gle aktywne
Granulowane w gle aktywne wielokrotnego u ycia uzyskuje si za pomoc wst pnego
formowania pyłu surowca podstawowego z lepiszczem.
Najcz ciej stosowanymi, tradycyjnymi lepiszczami s : smoły drzewne, smoły w glowe
i paki. Obecnie s prowadzone prace zmierzaj ce do poszukiwania nowych rodków
wi
cych. Podj to próby zastosowania jako lepiszcza: ługów posulfitowych, smoły z w gla
brunatnego, smoły pogazowej, asfaltu czy soli sodowej karboksymetylocelulozy (nazwa
handlowa - glikocel).
W Polsce opracowano technologi otrzymywania w gli aktywnych bez u ycia lepiszcza,
przez tabletkowanie pyłu w gla kamiennego spiekaj cego. Prowadzi si tak e badania nad
otrzymaniem w gli aktywnych formowanych bez lepiszcza przez brykietowanie w gla
brunatnego pod wysokimi ci nieniami.
Podstawowe operacje technologiczne, stosowane w celu otrzymania granulowanych
w gli aktywnych, polegaj na:
• sporz dzeniu pasty smołowo-w glowej,
• tłoczeniu granul o kształcie cylindrycznym b d kulistym,
• suszeniu,
• karbonizacji,
• aktywacji.
2.3. Karbonizacja
Jednym z najwa niejszych etapów procesu otrzymywania w gli aktywnych jest
karbonizacja. Od parametrów tego procesu w du ej mierze zale y powstanie pierwotnej
struktury porowatej, która nast pnie jest jedynie
rozwijana lub modyfikowana
w aktywacji nast puj cej po procesie karbonizacji.
Materiał w glowy do wytwarzania w gli aktywnych musi spełnia okre lone warunki,
z których najistotniejsze to: du a zawarto C
a
, mała zawarto V
daf
, okre lona wst pna
porowato , a tak e wysoka odporno mechaniczna. Naturalne surowce w glowe nie
spełniaj jednocze nie wszystkich tych wymaga i dlatego musz by karbonizowane.

5
Proces karbonizacji prowadzi si najcz ciej w temperaturach 500-900 °C w atmosferze
beztlenowej (w obecno ci gazów pirolitycznych lub w przepływie gazów oboj tnych: argon,
azot). W tych warunkach, w wyniku wydzielania z w gli lotnych substancji, materiał
w glowy wzbogaca si w pierwiastek C i nabiera wytrzymało ci mechanicznej. Działanie
energii cieplnej prowadzi do destrukcji substancji organicznej w gla, jak te lepiszcza.
W rezultacie post puj cych procesów polimeryzacji i polikondensacji powstaje coraz
bardziej jednorodny materiał o okre lonej strukturze porowatej, która w du ej mierze
w trakcie obróbki termicznej ulega ró nym zmianom zwi zanym z rodzajem surowca
wyj ciowego i z warunkami prowadzenia procesu karbonizacji.
Karbonizaty o wymaganych własno ciach otrzymuje si przez odpowiedni dobór
warunków, w których odbywa si karbonizacja materiałów w glowych. Nale do nich:
1.
Ko cowa temperatura karbonizacji. Jest to najwa niejszy parametr procesu
karbonizacji. Wysoko tej temperatury wynika z konieczno ci doprowadzenia do
w glowych makrocz steczek takiej ilo ci energii, aby nast piło rozerwanie mniej trwałych
wi za chemicznych i oddestylowanie z otoczenia materiału w glowego lotnych
produktów jego termicznego rozkładu. Ponadto w karbonizacie musi nast pi pewne
uporz dkowanie zwartej substancji w glowej, znacznie wi ksze ni było w surowcu
wyj ciowym, a mo e ono nast pi tylko w wysokiej temperaturze.
2. Czas przetrzymywania w gla w ko cowej temperaturze karbonizacji. Jego
wydłu enie wpływa na wi ksze uporz dkowanie budowy zwartej substancji w glowej,
z ewentualnym tworzeniem krystalitów, ale powoduje te spadek reaktywno ci karbonizatu
i wielko ci obj to ci najdrobniejszych porów.
3.
Szybko osi gania temperatury ko cowej. Gdy wzrost temperatury jest szybki,
wówczas poszczególne fazy rozkładu termicznego w gla oraz wtórne reakcje
produktów pirolizy mi dzy sob nakładaj si , a przez to powstawanie struktury
porowatej w karbonizacie jest trudniejsze do kontrolowania. Du a szybko wzrostu
temperatury nie sprzyja wi c dalszemu uporz dkowaniu, w rezultacie otrzymujemy
karbonizat o wi kszej porowato ci i reaktywno ci, ale o mniejszym uporz dkowaniu zwartej
substancji w glowej i mniejszej wytrzymało ci mechanicznej.
4.
Atmosfera, w której odbywa si karbonizacja, wpływa na rozkład termiczny w gla
oraz na przebieg wtórnych reakcji produktów pirolizy mi dzy sob i mi dzy stałym
karbonizatem. Gdy wydzielaj ce si podczas pirolizy gazy i pary s szybko eliminowane
za pomoc przepływaj cego gazu oboj tnego, wówczas ilo uzyskanego karbonizatu
jest mniejsza, za to jego reaktywno jest wi ksza.
Karbonizacj materiału w glowego w przemy le prowadzi si najcz ciej w piecach
stacjonarnych ogrzewanych przeponowo, w piecach obrotowych z ogrzewaniem
wewn trznym lub zewn trznym, a tak e w reaktorach fluidalnych.

6
2.4. Aktywacja
Produkty karbonizacji maj słabo rozwini t struktur porowat i bez uzupełniaj cej
aktywacji s zazwyczaj słabymi sorbentami i nie mog by stosowane w procesach
sorpcyjnych.
W zale no ci od przebiegu procesu aktywacji i zastosowanego czynnika aktywuj cego
metody aktywacji dzieli si na:
- chemiczne
- fizykochemiczne (gazowo-parowe).
Aktywacja chemiczna. Najcz ciej stosowanymi czynnikami aktywuj cymi
w chemicznych metodach aktywacji s KOH i NaOH. Oprócz nich stosuje si substancje
o własno ciach utleniaj cych, takie jak: H
2
SO
4
, H
3
PO
4
, KCNS, KHSO
4
, K
2
SO
4
, K
2
S,
MgCl
2
, ZnCl
2
, A1C1
3
, CaCl
2
, NH
4
C1 i inne.
W procesie aktywacji chemicznej istotny wpływ na własno ci otrzymanych w gli
aktywnych maj warunki prowadzenia procesu, a przede wszystkim temperatura aktywacji
i stosunek masy suchego surowca w glowego do masy bezwodnego aktywatora. Materiały
uzyskane w wyniku aktywacji chemicznej odznaczaj si g bczast struktur porów
z w skimi przew eniami do ich wn trz. Przew enia te mo na usun przez dodatkowo
prowadzon aktywacj fizykochemiczn , w której nast puje przepalanie przew e
i polepszenie własno ci sorpcyjnych tych materiałów.
Aktywacja fizykochemiczna obejmuje cykl przemian fizykochemicznych substancji
w glowej, zachodz cych pod wpływem czynników utleniaj cych i temperatury. Surowcami
najcz ciej stosowanymi w aktywacji fizykochemicznej s produkty karbonizacji w gli
kopalnych, a czynnikami aktywuj cymi: para wodna, dwutlenek w gla, tlen, dwutlenek
siarki, siarkowodór, amoniak, chlorowodór i inne.
Proces aktywacji fizykochemicznej w zasadzie rozwija, b d modyfikuje, układ porów
istniej cych ju w surowcu po karbonizacji. Na własno ci w gli aktywnych otrzymanych
w procesie aktywacji fizykochemicznej maj wi c wpływ nie tylko parametry aktywacji
(temperatura, szybko podawania czynnika utleniaj cego), ale tak e rodzaj materiału
wyj ciowego (sposób przygotowania i warunki karbonizacji). Dla układu karbonizat-
czynnik aktywuj cy mo na dobra odpowiednie, optymalne warunki procesu aktywacji
w zale no ci od rodzaju materiału wyj ciowego. W celu uzyskania struktury porowatej
o po danych własno ciach aktywat mo na podda dodatkowej modyfikacji.
W przypadku aktywacji fizykochemicznej paliw stałych zakłada si , e jej przebieg
jest wieloetapowy. Najpierw czynnik utleniaj cy powoduje destrukcj struktury w glowej
najmniej odpornej na działanie utleniaczy (boczne ła cuchy spolimeryzowanej substancji
w glowej oraz boczne grupy mi dzy krystalitami), nast pnie reakcji ulegaj poszczególne,
coraz gł bsze warstwy krystalitów, w rezultacie powstaj pory o coraz wi kszych
wymiarach.
W reakcjach mi dzy w glem a czynnikiem utleniaj cym mo na wyró ni

7
nast puj ce stadia:
• stadium dyfuzyjne polegaj ce na transporcie reagentów z fazy gazowej do granicy
faz
• stadium adsorpcyjne, w którym cz steczki reagenta adsorbuj si na powierzchni
ciała stałego
• stadium reakcji chemicznej: nast puje reakcja mi dzy powierzchniowymi atomami
w gla a zaadsorbowanymi cz steczkami reagenta
• stadium desorpcji produktów reakcji z powierzchni w gla i ich transport do fazy
gazowej.
W procesach aktywacji fizykochemicznej najcz ciej stosowanymi czynnikami
aktywuj cymi s : para wodna i dwutlenek w gla, rzadziej tlen. Reakcje zachodz ce mi dzy
w glem a tymi czynnikami s reakcjami heterogenicznymi.
W zale no ci od temperatury reakcji zgazowania szybko ich mo e by
zdeterminowana przez szybko reakcji chemicznej lub przez dyfuzj cz steczek czynnika
utleniaj cego do powierzchni w gla, a nast pnie do wn trza jego porów. Dlatego dla tych
reakcji mo na wyró ni nast puj ce obszary heterogenicznego reagowania:
kinetyczne,
przej ciowe i dyfuzyjne.
Istotnym problemem jest dobranie takich warunków aktywacji w gla gazowymi
czynnikami utleniaj cymi, aby reakcja zachodziła w obszarze kinetycznym, co zapewnia
przebieg reakcji zgazowania na całej powierzchni ziaren. Niekorzystny jest natomiast proces
zgazowania biegn cy w obszarze dyfuzyjnym ze wzgl du na du szybko reakcji
reagentów z w glem, która przebiega cz sto w sposób niekontrolowany, głównie na
zewn trznej powierzchni ziaren, co prowadzi do pogorszenia parametrów
charakteryzuj cych struktur kapilarn w gli aktywnych.
Z wymienionych uprzednio utleniaczy najbardziej aktywny jest tlen, najmniej aktywny -
dwutlenek w gla.
W reakcji
tlenu z w glem powstaje równocze nie tlenek i dwutlenek w gla według
nast puj cych równa stechiometrycznych reakcji chemicznych:
C + O
2
→ CO
2
H = -387 kJ/mol
2C + O
2
→ 2 CO H = -226 kJ/mol
Obie reakcje s egzotermiczne, ich mechanizm nie jest jeszcze w pełni poznany. W gle
aktywowane tlenem zawieraj du o tlenowych powierzchniowych grup funkcyjnych.
Stosowanie tlenu jako czynnika aktywuj cego jest zwi zane z wieloma trudno ciami
i dlatego u ywa si go niezwykle rzadko.
Podstawow reakcj
w gla z par wodn jest proces endotermiczny, który mo e by
zapisany zgodnie z równaniem stechiometrycznym:
C + H
2
O
→ H
2
+ CO H= + 130 kJ/mol
Oprócz powy szej reakcji zachodz jednocze nie nast puj ce reakcje:
C + 2H
2
O
→ CO
2
+ 2H
2
H = +77 kJ/mol

8
CO + H
2
O
→ CO
2
+ H
2
H = -42 kJ/mol
Poniewa jednym z produktów badanych reakcji jest wodór, mo liwe jest równie tworzenie
metanu w reakcji:
C + 2H
2
→ CH
4
H = -86 kJ/mol
lub w reakcji:
2C + 2H
2
O
→ CH
4
+ CO
2
H = -9 kJ/mol
Proces ten jest badany wszechstronnie, poniewa jest on dominuj cy nie tylko
w reakcji aktywacji, ale tak e podczas produkcji gazu wodnego. Aktywacj par wodn
prowadzi si w temperaturze rz du 700-900 °C. Jest ona katalizowana tlenkami
i w glanami metali.
Reakcj
w gla z dwutlenkiem w gla mo na przedstawi równaniem:
C + CO
2
→ 2 CO H = + 172 kJ/mol
Reakcja dwutlenku w gla z powierzchni w glow jest wynikiem tworzenia si kompleksów
powierzchniowych, przy czym szybko reakcji w gla z dwutlenkiem w gla w danej
temperaturze jest ok. 30% wolniejsza ni z par wodn .
Aktywacji w gla czynnikami utleniaj cymi w przemy le dokonuje si w piecach
o ró nej konstrukcji i zasadzie działania. Mo na je podzieli na trzy zasadnicze typy: piece
komorowe, piece obrotowe i reaktory z warstw fluidaln .
3. Przemysłowa produkcja w gli aktywnych
Wytwarzaniem w gli aktywnych w Polsce zajmuj si w kraju dwie wytwórnie:
• GRYFSKAND sp. z o.o., Zakład produkcyjny w Hajnówce
• CARBON sp. z o.o., w Raciborzu
Roczna produkcja w gli aktywnych wynosi ł cznie
2400 – 2800 Mg.
GRYFSKAND – HAJNÓWKA
Jest przedsi biorstwem wielozakładowym o profilu produkcyjnym, na czele firmy stoi
jednoosobowy zarz d, którego organem wykonawczym jest Biuro Zarz du zajmuj ce si
zarz dzaniem, rozliczaniem finansowym, strategi gospodarcz i rozwojem.
Najstarszym zakładem spółki jest Zakład Produkcyjny w Gryfinie, wywodz cy si
z byłego Gryfi skiego Przedsi biorstwa Suchej Destylacji W gla. Zakład produkuje:
• w giel drzewny do celów gastronomicznych i przemysłowych w postaci naturalnej
i zbrykietowanej.
• popularne w kraju segmenty parkietowe

9
• posiada jedyn w Europie przemysłow instalacj do zw glania trocin działaj c
wg. my li patentowej własnych in ynierów.
• torebki do konfekcjonowania w gla i brykietów.
Kolejnym zakładem jest spółka handlowa z siedzib w Hamburgu, która spełnia
funkcj przedstawiciela handlowego na rynki Unii Europejskiej.
Zakład Produkcyjny w Hajnówce
Dawne Hajnowskie Przedsi biorstwo Suchej Destylacji Drewna, które w 1996r, zostało
wykupione od Skarbu Pa stwa. Zakład rozpocz ł produkcj w 1905r. Jako jedno
z najnowocze niejszych przedsi biorstw. Obecnie działaj cy w strukturze i pod szyldem
GRYFSKAND zatrudnia ok. 480 pracowników, w tym wielu in ynierów z bran y
chemicznej. Wła cicielami firmy s udziałowcy. Udziałowcami wi kszo ciowymi s firmy
skandynawskie i niemieckie, Skarb Pa stwa oraz osoby fizyczne, wywodz ce si z byłego
Gryfickiego Przedsi biorstwa Suchej Destylacji Drewna.
PRODUKCJA W GLI AKTYWNYCH W HAJNÓWCE
Produkcja w stosunku do lat ubiegłych została mocno ograniczona ze wzgl dów rynkowych.
Podstawowy profil produkcyjny to w gle aktywne produkowane na bazie w gla kamiennego,
w gla drzewnego oraz pewnych ilo ci w gla z łupin orzecha kokosowego. Stosownie do
zapotrzebowania rynku zakład oferuje ok. 30 rodzajów w gli aktywnych, wykorzystywanych
w najró niejszych procesach, np.
• usuwanie smaków, zapachów i detergentów.
• odsiarczanie spalin
• odbarwianie
• usuwanie chloru, fenolu, amin, pestycydów, substancji kancerogennych, rt ci,
arsenu etc.
• oczyszczanie powietrza
• uzdatnianie wody do celów konsumpcyjnych i przemysłowych
• doczyszczanie cieków.
W zwi zku z konieczno ci rozwoju nowoczesnych technologii ochrony rodowiska,
przewiduje si w najbli szych latach wzrost zapotrzebowania na w gle aktywne do
uzdatniania wody pitnej oraz na koksy aktywne do oczyszczania spalin.
Surowce do produkcji w gli aktywnych stosowane w Hajnówce:
• w giel kamienny: typ 33 lub 34, o wysokim współczynniku spiekalno ci, niskiej
zawarto ci cz ci niepalnych, głównie popiołu, niskiej zawarto ci siarki (<0,5%)
• w giel drzewny: produkowany na miejscu w retorcie ruchu ci głego, głównie
z brzozy, olchy i w niewielkich ilo ciach z d bu (21 pieców obrotowych, ka dy po
14,5 m długo ci i rednicy 1,5 m)
• skorupy orzecha kokosowego.

10
CARBON – RACIBÓRZ
W Raciborzu w 1909 roku otrzymano w tutejszych Zakładach Chemicznych pierwsz
parti w gla aktywnego na skal przemysłow . W giel ten nosił nazw EPONIT, a jego
produkcj prowadzono wg. patentu Ostrejki – na bazie w gla drzewnego. W ci gu kolejnych
dwóch lat wdro ono do produkcji dwa nast pne gatunki w gla aktywnego o nazwach NORIT
i PURIT- w gle, dla których podstawowym surowcem był torf. W Raciborzu powstała wi c
pierwsza fabryka w gli aktywnych w Europie Wschodniej. Od chwili rozpocz cia
działalno ci Zakłady Chemiczne w Raciborzu miały wielu wła cicieli, ale pomimo cz stych
zmian produkcj wci rozszerzano.
W 1951 roku wła cicielem wytwórni stały si Zakłady Elektrod W glowych, a w roku
1993 w miejsce Wydziału W gli Aktywnych Elektrod W glowych SA. powstała spółka
z o.o.„CARBON”, która kontynuuje profil produkcji swoich poprzedników, a wi kszo
wyrobów stanowi w gle aktywne. Ich jako jest zgodna z polskimi normami, ale
wytwarzane s równie w gle aktywne o parametrach uzgodnionych z klientem.
Obecnie głównym surowcem do produkcji w gli aktywnych jest w giel drzewny, na
bazie tego półproduktu otrzymywane s w gle ziarniste i pyłowe. Ponadto produkuje si
w giel aktywny z nisko uw glonego w gla kamiennego z Kopalni Wieczorek. Przez cały
okres istnienia, do chwili obecnej trwaj poszukiwania nowych surowców do produkcji w gli
aktywnych.
W zakładzie CARBON produkowane s głównie w gle pyliste i ziarnowe z w gla
drzewnego, stosowane przede wszystkim do adsorpcji z fazy ciekłej.
Ł czna produkcja w gli aktywnych obu tych zakładów (Gryfskand i Carbon) jest
niewielka i je liby powa nie potraktowa wykorzystanie w gla aktywnego w ochronie
rodowiska (oczyszczanie powietrza i wód ciekowych), to roczna produkcja tych zakładów
mogłaby zaspokoi jedynie zapotrzebowanie Górnego l ska.
W Europie Zachodniej, Stanach Zjednoczonych i Japonii produkuje si wiele
ró norodnych w gli aktywnych o specyficznych własno ciach, a ich produkcja systematycznie
wzrasta. Obecnie za jedn z najlepszych w Europie i na wiecie uwa a si wytwórni w gla
aktywnego w Holandii (fabryk Norit).
4. Ocena własno ci w gli aktywnych
Ocen własno ci w gli aktywnych dokonuje si zgodnie z Polskimi Normami PN-82/
C-97555.00 do 31. Ich ocena obejmuje oznaczenie ponad 30 ró nego rodzaju parametrów,
takich jak: analiza sitowa, oznaczenie g sto ci nasypowej, liczby metylenowej, jodowej,
miligramowej, fenolowej, detergentowej, oznaczenie zawarto ci popiołu, wody, pH
wyci gu wodnego, substancji rozpuszczalnych w wodzie i w HC1, zawarto ci jonów Cl
-
,
Ca
2+
, Fe
3+
, wykonanie prób na obecno jonów Cu
2+
, S
2-
, NO
3
-
, jonów metali ci kich
w przeliczeniu na Pb
4+
, próby na brak jonów CN
-
, oznaczenie aktywno ci katalitycznej
rozkładu H
2
O
2
, wytrzymało ci mechanicznej, cieralno ci, nasi kliwo ci wodnej, zjawiska
11
cieplnego, chłonno ci dynamicznej i statycznej w stosunku do par benzenu, aktywno ci
dynamicznej w stosunku do chlorku etylu, powierzchni wła ciwej, obj to ci porów
o promieniu poni ej 1,5 nm, oporu warstwy w gla aktywnego i inne.
Spo ród tak szerokiej gamy ró nego rodzaju oznacze istotne znaczenie maj takie
parametry, które s ci le zwi zane z wykorzystaniem w gla aktywnego do konkretnych
procesów.
5. Zastosowanie w gli aktywnych
• Oczyszczanie wody i cieków: usuwanie pestycydów, detergentów, substancji
odpowiedzialnych za smak, barw , i zapach, wody, w glowodorów alifatycznych
i aromatycznych, fenoli i ich pochodnych, metali ci kich, bakterii i wirusów,
niskocz steczkowych zwi zków organicznych, odchlorowania.
• Oczyszczanie przemysłowych gazów odlotowych: adsorpcja SO
2
, SO
3
, H
2
S, CS
2
, NH
3
,
NO
x
i innych gazów toksycznych. Uwzgl dniaj c obecno w gazach odlotowych tlenu
i pary wodnej to obok adsorpcji na w glu aktywnym zachodzi tak e m.in. katalityczne
utlenianie i inne reakcje wtórne.
• Odzyskiwanie rozpuszczalników organicznych: benzyny, benzenu, toluenu, ksylenów,
acetonu, ni szych alkoholi, eterów, estrów, w glowodorów parafinowych,
chlorowcopochodnych w glowodorów, (chloroform, tetrachlorek w gla, chlorek
metylenu, dichloroetan, chlorobenzen i inne).
• Oczyszczanie powietrza: ochrona dróg oddechowych przed substancjami
toksycznymi(we wszystkich dziedzinach przemysłu chemicznego i innych, technice
wojskowej; pochłanianie chloru, tlenków siarki, tlenków azotu, siarkowodoru, amoniaku
merkaptanów, rt ci, substancji promieniotwórczych i innych).
• Deodoryzacja powierza; usuwanie nieprzyjemnych zapachów z powietrza, np.
merkaptanów, amin, fenoli, pirydyny i innych.
• Rozdzielanie mieszanin gazowych: wydzielenie poszczególnych składników
z mieszaniny gazów, np. rozdział propanu i butanu z gazu ziemnego, rozdzielenie
powierza na tlen i azot i inne.
• Magazynowanie energii: Adsorpcyjne magazynowanie gazu ziemnego, metanu, wodoru
i innych no ników energii pod ekonomicznym i bezpiecznym ci nieniem.
• Oczyszczanie spalin samochodowych: na no niku w glowym z osadzonymi
katalizatorami nast puje dopalanie nie spalonego w silniku paliwa a tak e adsorpcja
tlenków siarki i azotu.
• Przemysł chemiczny: odbarwianie roztworów, odzyskiwanie cennych składników
z gazów produkcyjnych, eliminacja substancji szkodliwych itd.
• Przemysł petrochemiczny: wytwarzanie no ników w glowych, niezb dnych do
utrzymania fazy aktywnej katalizatora w stanie zdyspergowanym, w procesach

12
katalitycznych takich jak hydroodsiarczanie, hydroodazotowanie, izomeryzacja
i uwodornienie.
• Przemysł farmaceutyczny: synteza, oczyszczanie i rozdział niektórych preparatów.
• Przemysł spo ywczy: oczyszczanie i odbarwianie syropów cukrowych, odbarwianie
olejów i tłuszczów, odbarwianie oraz polepszanie własno ci i smaku napojów
alkoholowych (spirytus, koniak, piwo), dekofinacja kawy.
• Przemysł metalurgiczny: do odzyskiwania metali szlachetnych z ich rud.
• Przemysł elektrotechniczny: jako materiał elektrodowy.
• Medycyna: w zatruciach pokarmowych, do oczyszczania krwi, do dializy.
• Przemysł górniczy: utylizacja zasolonych wód kopalnianych.
• Rolnictwo: adsorpcja rodków ochrony ro lin, oczyszczanie cieków rolniczych.
6. Badania układu kapilarnego w gla aktywnego
6.1. Układ kapilarny w gla aktywnego
Wiele własno ci w gli aktywnych, istotnych z punktu widzenia ich ró norodnych
zastosowa , głównie jako adsorbentów, no ników katalizatorów i materiałów elektrodowych,
jest zdeterminowanych ich budow krystaliczn zale n zarówno od surowca, jak i sposobu
oraz warunków prowadzenia procesu karbonizacji i aktywacji. Dotyczy to zwłaszcza stopnia
rozwini cia i charakteru ich struktury kapilarnej. Przeci tne w gle aktywne maj na ogół
bardzo dobrze rozwini t powierzchni wła ciw , która cz sto przekracza 1000 m
2
/g
i zwykle odznaczaj si polidyspersyjn struktur kapilarn . Tworz j pory
o zró nicowanym kształcie i wielko ciach.
W układzie kapilarnym sorbentów wyró nia si , na podstawie szeroko ci porów
(podwójny efektywny promie porów) oraz mechanizmu adsorpcji par i gazów, nast puj ce
kategorie porów, których podział jest zgodny z zaleceniami IUPAC (Mi dzynarodowa
Unia Chemii Czystej i Stosowanej):
1.
Makropory - pory o efektywnych promieniach wi kszych ni 25 nm, które nie s
praktycznie zapełniane obj to ciowo adsorbatem zgodnie z mechanizmem kondensacji
kapilarnej. Ich obj to wynosi 0,2-0,8 cm
3
/g, a powierzchnia wła ciwa nie przekracza 0,5-
2,0 m
2
/g i jest bardzo mała w porównaniu z powierzchni pozostałych rodzajów porów.
Makropory nie odgrywaj wi c istotnej roli w procesie adsorpcji, spełniaj jedynie funkcj
arterii transportowych, dlatego makroporów w zasadzie nie charakteryzuje si metodami
sorpcyjnymi. Do badania ich własno ci stosuje si przede wszystkim porozymetri rt ciow ,
a tak e mikroskopi optyczn , elektronow i skaningow .
2.
Mezopory - pory o efektywnych promieniach w granicach od 1 nm do 25 nm (zwane

13
s tak e porami przej ciowymi). Zapełnianie ich obj to ci adsorbatem przebiega zgodnie
z mechanizmem kondensacji kapilarnej. Dla przeci tnych w gli aktywnych obj to ci
mezoporów s w granicach 0,1-0,5 cm
3
/g, a powierzchnie wła ciwe 20-400m
2
/g. Mezopory,
oprócz znacz cego udziału w adsorpcji, odgrywaj tak e rol głównych arterii
transportowych dla adsorbatu. Najcz ciej stosowanymi metodami badawczymi
mezoporów s : sorpcja par i gazów, porozymetria rt ciowa oraz mikroskopia elektronowa
i skaningowa.
3.
Mikropory - pory o efektywnych promieniach mniejszych ni 1 nm. S to pory
o wymiarach porównywalnych z wymiarami adsorbowanych cz steczek. Obj to ci
mikroporów dla przeci tnych w gli aktywnych wynosz 0,2-0,6 cm
3
/g i maj one
najwi kszy wkład w wielko powierzchni wła ciwej porów. Energia adsorpcji w mikroporach
jest znacznie wi ksza ni dla podobnych wielko ci adsorpcji w mezoporach czy na
powierzchni nieporowatej, poniewa w tak w skich porach nast puje nakładanie si
potencjałów adsorpcyjnych pochodz cych od przeciwległych cian porów, co prowadzi do
szczególnie du ego zwi kszenia zdolno ci adsorpcyjnych w obszarze małych ci nie
równowagowych adsorbatu. W mikroporach adsorpcja biegnie zgodnie z mechanizmem ich
obj to ciowego zapełniania (teoria Dubinina). Do charakterystyki struktury mikroporowatej
wykorzystuje si przede wszystkim adsorpcj par i gazów oraz metod rozpraszania
promieni rentgenowskich pod małymi k tami.
Dla kinetyki adsorpcji istotne znaczenie ma wzajemne poł czenie
trzech rozpatrywanych
rodzajów porów. Istnieje na ten temat wiele hipotez. Według jednej z nich mezopory s
odgał zieniem makroporów, a mikropory mezoporów (struktura drzewiasta), według innej -
ka dy z trzech rodzajów porów ma bezpo rednie poł czenie z powierzchni ziaren
w gla.
6.2. Techniki do wiadczalne badania struktury porowatej w gla
Wielko adsorpcji gazu lub pary zale y nie tylko od ich charakteru, ale równie od
warunków, w jakich przebiega proces sorpcji, a przede wszystkim od
temperatury, ci nienia
i struktury adsorbentu. Gdy inne warunki s zachowane, zazwyczaj ze wzrostem ci nienia
ro nie ilo zaadsorbowanego adsorbatu. Krzywe podaj ce zale no ilo ci zaadsorbowanego
adsorbatu od jego ci nienia równowagowego w stałej temperaturze nazywa si
izotermami
adsorpcji. Przebieg izoterm adsorpcji jest wi c odbiciem struktury kapilarnej ciała stałego, ich
interpretacja umo liwia okre lenie stanu rozwini cia i rodzajów porów, ustalenie typu
strukturalnego adsorbentu, wielko ci i charakteru jego powierzchni wła ciwej (metoda BET).
Wielko adsorpcji gazów i par na ciałach stałych i odpowiednie izotermy wyznacza
si metodami
dynamicznymi lub statycznymi.
Dynamiczne metody pomiaru adsorpcji polegaj na przepuszczaniu przez warstw
adsorbentu strumienia mieszaniny gazu oboj tnego (np. N
2
), zwanego gazem no nym,
z odpowiedni ilo ci adsorbowanego gazu lub pary. Nast pnie zaznacza si pojawienie

14
tego gazu za warstw adsorbentu, czyli tzw. przebicie. W dokładniejszych metodach
mierzy si stopniowy wzrost st enia gazu za warstw adsorbentu po przebiciu.
Metody statyczne polegaj na tym, e adsorbent umieszcza si w atmosferze gazu lub pary
i po ustaleniu si równowagi mierzy ci nienie oraz ilo pochłoni tego adsorbatu. Metody
statyczne stosuje si zwykle podczas badania adsorpcji indywidualnych gazów i par
w aparatach pró niowych, w których adsorbent został uprzednio poddany ogrzewaniu
w wysokiej pró ni w celu uwolnienia jego powierzchni z zaadsorbowanych substancji.
Najwi cej danych na temat struktury kapilarnej porowatych adsorbentów dostarcza
analiza izoterm adsorpcji i desorpcji par, wyznaczanych w warunkach statycznych. Do
najcz ciej stosowanych adsorbatów nale zarówno gazy, takie jak N
2
, Ar, Kr, CO
2
, jak
i pary w glowodorów niskowrz cych, alkoholi, benzenu, cykloheksanu, tetrachlorku w gla,
pary wodnej, itp. Nale y tu zauwa y , e na adsorpcj adsorbatów o charakterze polarnym
wpływa nie tylko struktura kapilarna adsorbentu, ale tak e chemiczna budowa jego
powierzchni {np. tlenowe powierzchniowe grupy funkcyjne w przypadku adsorpcji pary
wodnej). Dlatego te do charakteryzowania struktury kapilarnej najcz ciej wykorzystuje si
takie adsorbaty, jak N
2
, Ar, C
6
H
6
i CO
2
.
Do wyznaczania izoterm adsorpcji i desorpcji stosuje si obecnie wiele ró nych rodzajów
aparatury adsorpcyjnej, których działanie jest oparte na ró nych zasadach. Istota pomiaru
polega na okre leniu wielko ci adsorpcji w stanie równowagi i odpowiadaj cego jej
ci nienia. Wspomniana wielko mo e by wyznaczana albo na podstawie bezpo redniego
pomiaru zmian ilo ci adsorbatu zaadsorbowanego na adsorbencie, albo zmian jego ilo ci
w przestrzeni nad adsorbentem.
Spo ród wielu istniej cych obecnie rodzajów aparatury adsorpcyjnej, za pomoc której
mo na wyznaczy izotermy adsorpcji i desorpcji z fazy gazowej, najcz ciej s stosowane
wagi sorpcyjne McBaina i Bakra, mikrobiuretki cieczowe oraz manostaty sorpcyjne.
6.3. Izotermy adsorpcji i klasyfikacja adsorbentów
Izotermy sorpcji par (adsorpcji i desorpcji) pozwalaj na uzyskanie wa nych informacji
na temat wewn trznej struktury adsorbentów i ich interpretacja jest bardzo istotna.
Przebieg izotermy adsorpcji, b d cej odbiciem wewn trznej struktury adsorbentu,
umo liwia okre lenie stanu rozwini cia, rodzaju porów danego typu, ustalenie typu
strukturalnego adsorbentu, wielko ci i charakteru jego powierzchni wła ciwej.
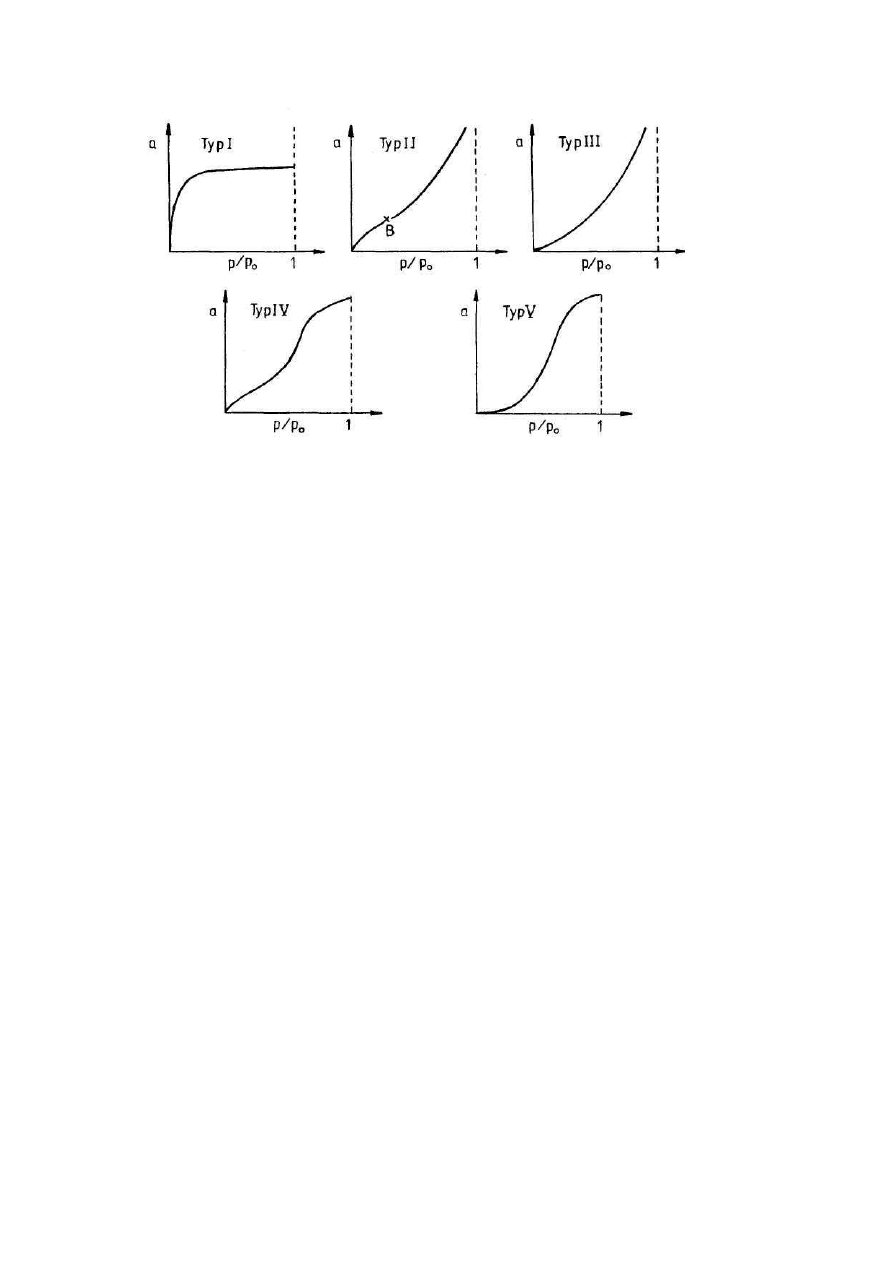
Według Brunauera istnieje pi zasadniczych kształtów izoterm adsorpcji par
i gazów. Przedstawiono je na poni szym rysunku:
15
Typy izoterm adsorpcji par i gazów
Typ I odpowiada izotermie Langmuira i odznacza si monotonicznym zbli aniem do
adsorpcji granicznej, która odpowiada kompletnej warstwie monomolekularnej.
Typ II jest bardzo rozpowszechniony w adsorpcji fizycznej i wi e si z powstawaniem
wielomolekularnej warstwy adsorpcyjnej.
Typ III izoterm adsorpcji jest stosunkowo rzadki W tym przypadku charakterystyczn
cech jest ciepło adsorpcji równe lub mniejsze, co do warto ci bezwzgl dnej, ciepłu
skraplania czystego adsorbatu.
Izotermy typów IV i V odpowiadaj krzywym typu II i III i ró ni si od nich jedynie
tym, e przy ni szych ci nieniach wzgl dnych biegn one równolegle do osi ci nie .
Przyczyn tego jest powstanie w porach adsorbentu tylko ograniczonej liczby warstw
adsorpcyjnych, ze wzgl du na szeroko porów.
W stałej temperaturze zazwyczaj ze wzrostem ci nienia ro nie ilo zaadsorbowanego
adsorbatu. Jednak dla poszczególnych odcinków izotermy wpływ ci nienia jest ró ny. Na
przykład dla drobnoporowatych adsorbentów wpływ ci nienia jest szczególnie widoczny
w obszarze niskich ci nie wzgl dnych, podczas gdy w przypadku szerokoporowatych
adsorbentów jego wpływ jest widoczny tylko dla ci nie zbli aj cych si do ci nienia pary
nasyconej. Gdy ci nienie dalej ro nie, ro nie tak e ilo zaadsorbowanej substancji na skutek
powstawania warstw polimolekulamych. Proces ten w przypadku ciał porowatych ko czy si
procesem kondensacji kapilarnej.
Cech charakterystyczn zapełniania porów zgodnie z mechanizmem kondensacji
kapilarnej jest mo liwo wyst powania p tli histerezy na izotermach sorpcji. Desorpcyjna
gał izotermy adsorpcji i szeroko p tli histerezy mog słu y do okre lania struktury
ciała porowatego i s w pewnym sensie odbiciem wielko ci i kształtu porów.
Na podstawie zarówno procesów adsorpcyjnych, jak i procesów kondensacji
kapilarnej, adsorbenty mo na podzieli na pi grup (tablica):

16
Klasyfikacja adsorbentów według typów strukturalnych
Grupa
Typ strukturalny
Podgrupa
Kształt izotermy
I
nieporowate
izoterma w kształcie litery S bez p tli histerezy
II
jednorodne
szerokoporowate
szkielet sztywny
szkielet
elastyczny
powolny wzrost izotermy w obszarze niskich
ci nie wzgl dnych i gwałtowny w obszarze
ci nie bliskich ci nieniu pary nasyconej
III
jednorodne
w skoporowate
ultra-porowate
drobno-
porowate
gwałtowny wzrost izotermy w obszarze niskich
warto ci p/p
0
, szybkie osi ganie równowagi, brak p tli
histerezy
nie wyst puje p tla histerezy dla substancji o wielkich
cz steczkach i pojawia si tylko podczas przej cia do
substancji o małych cz steczkach
IV
struktury
mieszane
maj ce dwa
rodzaje porów
lepiej rozwini ta
struktura
mikroporowata
lepiej rozwini te
mezopory
dwa gwałtowne wzrosty izotermy w obszarze
niskich i rednich warto ci p/p
0
gwałtowny wzrost izotermy w obszarze niskich
warto ci p/p
0
i mniej gwałtowny dla rednich
warto ci p/p
0
gwałtowny wzrost dla rednich warto ci p/p
0
i mniej gwałtowny dla niskich warto ci p/p
0
V
jednorodne
nieporowate
gwałtowny wzrost dla rednich warto ci p/p
0

17
ADSORPCJA NA W GLU AKTYWNYM
II. C z d o w i a d c z a l n a
Do wiadczenie 1.
Adsorpcja barwnych substancji z roztworu.
a) Adsorpcja bł kitu metylenowego
Do rozdzielacza wypełnionego w glem aktywnym wla roztwór wody destylowanej
zabarwionej bł kitem metylenowym (roztwór barwy niebiesko-granatowej). Otworzy kranik
w rozdzielaczu i obserwowa barw wypływaj cego do podstawionej zlewki roztworu.
b) Adsorpcja fuksyny
Probówk sto kow napełni do połowy jej obj to ci jasno ró owym roztworem
fuksyny. Wsypa do probówki kilka mikro- szpatułek formowanego w gla aktywnego.
Probówk zatka korkiem i wstrz sa energicznie w ci gu kilku minut. Oddzieli w giel od
roztworu przez odstanie lub s czenie na s czku z mi kkiej bibuły. Zwróci uwag na zmian
zabarwienia roztworu.
Do wiadczenie 2
. Adsorpcja substancji gazowych.
Probówk sto kow napełni gazowym bromem (pod wyci giem). W tym celu
zanurzy bagietk na gł boko ok. 1 cm w pojemniku z ciekłym bromem. Bagietk
zwil on bromem wprowadzi do probówki i dotkn jej dna, po czym szybko j wyci gn ,
a probówk zatka korkiem. Zwróci uwag na barw gazu. Do probówki wypełnionej
parami bromu, wsypa przygotowan porcj w gla aktywnego ziarnowego i szybko zatka j
korkiem. Probówk energicznie wstrz sa , obserwuj c odbarwianie si gazu.
Do wiadczenie 3.
Adsorpcja jonów z roztworu.
a) Adsorpcja jonów Pb
2+
z roztworu
Wykonanie próbki odniesienia: do probówki wprowadzi 3-4 krople 0,01 M roztworu
azotanu ołowiu (II) i doda 1 kropl 0,1M roztworu jodku potasu. Probówk z wytr conym
ółtym osadem PbI
2
zachowa .
Drug probówk napełni do połowy tym samym roztworem azotanu ołowiu (II)
i wsypa 3-4 mikro- szpatułki ziarnowego w gla aktywnego. Probówk zatka korkiem
i energicznie wytrz sa w ci gu 2-3 minut, a nast pnie pozostawi do odstania lub
przes czy . Pipetk pobra 3-4 krople klarownego roztworu z nad w gla i przenie do innej

18
probówki, po czym doda 1 kropl roztworu 0,1M KI. Porówna ilo otrzymanego osadu
jodku ołowiu (II) z próbk odniesienia.
b) Adsorpcja jonów Fe
3+
z roztworu
Do rozdzielacza wypełnionego w glem aktywnym wla roztwór wody
zanieczyszczonej zwi zkami elaza, zwróci uwag na barw tego roztworu. Otworzy kranik
w rozdzielaczu , obserwuj c barw wypływaj cego roztworu i zebra do zlewki kilka
mililitrów przes czu. Wykona reakcj charakterystyczn na obecno jonów Fe
3+
, w wodzie
przed adsorpcj i po adsorpcji. W tym celu pobra do dwóch probówek po ok.1 cm
3
wody
zanieczyszczonej zwi zkami elaza i wody oczyszczonej na w glu aktywnym , doda po 1-2
kropli 0,1 M tiocyjanianu amonu (NH
4
SCN). Jony SCN
-
w obecno ci Fe
3+
powoduj
zabarwienie krwistoczerwone (czuło reakcji jest bardzo du a). Opisa obserwacje.
III. L i t e r a t u r a
1. JANKOWSKA H., WI TKOWSKI A., C
HOMA
J., W giel aktywny, Warszawa, WNT,
1985.
2. D
UBININ
M. M., Structure and properties of activated carbons, Chemistry and Physics of
Carbon, London, P. L. Walker Jr., 1966.
3. TOMKÓW K., Zestaw automatycznej, rejestruj cej aparatury termograwimetrycznej do
bada procesów karbochemicznych, Pr. Nauk. Inst. Chem. Technol. Nafty PWr., nr 25,
Wrocław, 1975.
4. BRUNAUER S., EMMETT P. H., TELLER E., Adsorption of gasses in
monomolecularlayers, Journal of the American Chemical Society 60, 309-319,
1938.
5. SlNG K. S. W., EVERETT D. H., HAUL R. A. W., MOSCOU L., PlEROTI R. A.,
ROUQUEROL J., SlEMIENIEWSKA T., Reporting physisorption data for gas solid
systems with special reference to the determination of surface area and porosity,
International Union of Pure and Applied Chemistry vol. 57, No. 4, 1985.
Wyszukiwarka
Podobne podstrony:
24. Adsorpcja kwasu octowego na węglu aktywnym, chemia w nauce i gospodarce Uł, semestr V, sprawozda
24 Adsorpcja kwasu octowego na węglu aktywnym
Usuwanie?rwy ścieku metodą?sorpcji na węglu aktywnym sprawko
Sprawko+ +Adsorpcja+na+granicy+faz+ciało+stałe ciecz +Wyznaczanie+adsorpcji+barwnika+na+węglu+aktywn
ADSORPCJA PARACETAMOLU NA WEGLU AKTYWNYM
Adsorpcja na granicy faz ciało stałe ciecz Wyznaczanie izotermy adsorpcji na węglu aktywnym
ADSORPCJA W UKŁADZIE ROZTWÓR – CIAŁO STAŁE NA PRZYKŁADZIE?SORPCJI KWASU OCTOWEGO NA WĘGLU AKTYWNYMx
Wyznaczanie izoterm?sorpcji jednokarboksylowych kwasów organicznych na węglu aktywnym
19 ADSORBCJA NA GRANICY FAZ CIAŁO STAŁE CIECZ WYZNACZANIE ADSORBCJI BARWNIKA NA WĘGLU AKTYWNYM
Wyznaczanie izotermy?sorpcji ?osprcja kwasu octowego na węglu aktywnym
kubica,biofizyka L,?sorpcja błękitu metylenowego na węglu aktywnym w obecności?etonux
ćw.23.02.06, Samooczyszczanie się wód zanieczyszczonych ściekami bytowo gospodarczymi polega na biol
adsorpcja na węglu akt
więcej podobnych podstron