
1
INSTRUKCJA
do ćwiczeń laboratoryjnych
Przedmiot:
Chemia
Temat ćwiczenia:
Badania korozji metali
Miejsce ćwiczeń: Katedra Chemii i Technologii Nieorganicznej
Zakład Technologii Nieorganicznej i Elektrochemii
Gliwice, ul. B. Krzywoustego 6, III piętro, p. 328
Instrukcję opracował: dr inż. Ginter Nawrat
Gliwice, 2007 r.

2
KOROZJA METALI
1. CEL I ZAKRES ĆWICZENIA
Celem ćwiczenia jest zapoznanie się ze zjawiskiem korozji metali i
metodami jej badania oraz wyznaczenie wskaźników szybkości korozji stali
metodą grawimetryczną, zgodnie z zaleceniami polskich norm: PN-70/H-04600
− „Badanie odporności korozyjnej metali i stopów” oraz PN-78/H-04608
− „Skala odporności metali na korozję”.
Ć
wiczenie polega na wyznaczeniu ubytku masy V
C
[g/m
2
·doba], liniowej
szybkości korozji V
P
[mm/rok] oraz stopnia odporności korozyjnej [
°
k] dla stali
w środowisku kwasu siarkowego i solnego o stężeniu 1 ÷ 2 mol/dm
3
na
podstawie różnicy wagi ∆m [g] próbek stalowych w czasie τ [min] od ok.
20 ÷ 60 minut. Na podstawie wyników ubytku wagi uzyskanych dla różnych
czasów przebywania próbek w środowisku korozyjnym sporządza się wykresy:
∆
m = f(τ), V
C
= f(τ), V
P
= f(τ). Ponadto, prowadzi się test kontrolny zachowania
się stali w środowisku zbliżonym do środowiska panującego w betonie, to jest w
nasyconym roztworze wodorotlenku wapnia w czasie około 60 minut i
wyznacza takie same wskaźniki szybkości korozji.
2. WPROWADZENIE
Pojęcie: korozja, odnosi się do procesów niszczenia metali pod wpływem
chemicznego
lub
elektrochemicznego
oddziaływania
z
otaczającym
ś
rodowiskiem. Procesy niszczenia różnych materiałów pod wpływem zjawisk
fizycznych określa się pojęciem: erozja. W niektórych przypadkach zjawiskom
oddziaływania chemicznego towarzyszy oddziaływanie fizyczne i wówczas używa
się pojęć: korozja naprężeniowa, korozja cierna.
Termin: rdzewienie, stosuje się jedynie w przypadku korozji żelaza i stali,
ponieważ produkty korozji żelaza, czyli rdzę stanowią związki żelaza, głównie
uwodnione tlenki żelaza. Metale nieżelazne nie mogą więc rdzewieć, lecz
oczywiście mogą korodować. Proces korozji zawsze rozpoczyna się na
powierzchni metalu stykającego się z fazą ciekłą lub gazową i stopniowo
postępuje w głąb fazy stałej, przy czym zależnie od rodzaju tworzywa i
ś
rodowiska korozyjnego zniszczenia mogą mieć różny zasięg i charakter.
Zjawiska korozji metali można sklasyfikować ze względu na:

3
- mechanizm procesu korozji,
- charakter zniszczeń,
- rodzaj środowiska i warunki procesów korozyjnych.
2.1. Podział zjawisk korozji ze względu na mechanizm procesu
Ze względu na mechanizm przebiegu procesu, rozróżnia się dwa zasadnicze
typy korozji, tj. korozję elektrochemiczną i chemiczną oraz procesy korozji,
których przebieg jest przyśpieszany przez oddziaływania fizyczne.
2.1.1. Korozja elektrochemiczna
Korozja elektrochemiczna zachodzi na powierzchni metalu przebywającego
w środowisku wodnego roztworu elektrolitu, np. w wilgotnej ziemi - tzw. „korozja
ziemna” a także w środowisku wilgotnego gazu, np. powietrza atmosferycznego –
„korozja atmosferyczna”. Jest to najczęściej spotykany w praktyce rodzaj korozji.
Korozja ta wywoływana jest działaniem krótkozwartych ogniw korozyjnych
o zasięgu lokalnym, tzw. miejscowych mikro- i makroogniw korozyjnych, które
tworzą się po zetknięciu metalu z roztworem elektrolitu. Zachodzą w nich procesy
utleniania i redukcji podobnie jak w ogniwach galwanicznych.
Warunkiem koniecznym przebiegu korozji elektrochemicznej jest:
1 - styczność ze środowiskiem wodnego roztworu elektrolitu,
2 - przepływ ładunku elektrycznego w obszarze występowania korozji.
Wyidealizowanym
przykładem
działania
ogniwa
korozyjnego
jest
zachowanie układu dwóch metali: miedzi i cynku zanurzonych w roztworze
elektrolitu i połączonych przewodem elektrycznym przez amperomierz.
Otrzymany układ dwóch różnych metali zanurzonych w roztworze elektrolitu
stanowi ogniwo galwaniczne i w powstałym obwodzie elektrycznym popłynie
prąd elektryczny, przy czym metal mniej szlachetny, tj. cynk będzie ulegał
roztwarzaniu i będzie stanowił anodę tego ogniwa.
W ten sposób zbudowane są ogniwa: Daniella (Rys. 1) i Volty (Rys. 2).
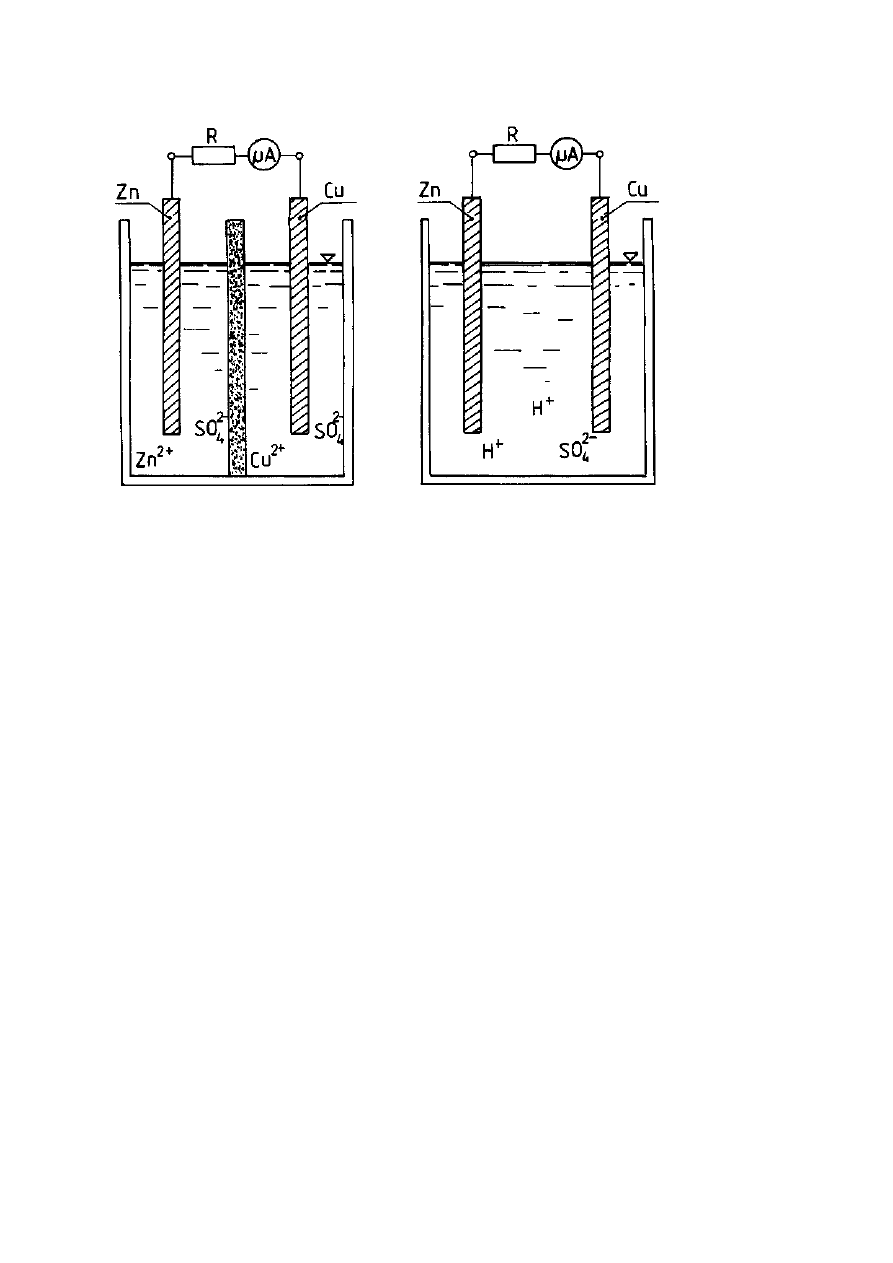
4
Rys. 1 Ogniwo Daniella
Rys. 2 Ogniwo Volty
Opis rysunków: R - opornik elektryczny,
µ
A - mikroamperomierz, Zn - elektroda
cynkowa, D – diafragma, Cu - elektroda miedziana, Zn
2+
, SO
4
2-
- roztwór
siarczanu(VI) cynku, Cu
2+
, SO
4
2-
- roztwór siarczanu(VI) miedzi, H
+
, SO
4
2-
-
roztwór kwasu siarkowego(VI).
W ogniwie Daniella zachodzą reakcje elektrodowe:
Anoda:
Zn - 2e
-
→
Zn
2+
(reakcja utleniania, czyli reakcja anodowa)
Katoda:
Cu
2+
+ 2e
-
→
Cu
(reakcja redukcji, czyli reakcja katodowa)
Ogniwo Daniella jest ogniwem odwracalnym - podłączenie przeciwnie
skierowanego, zewnętrznego źródła prądu elektrycznego powoduje przepływ
prądu w odwrotnym kierunku i powrót do stanu pierwotnego (akumulator):
Zn
2+
+ 2e
-
→
Zn
(katoda, czyli reakcja redukcji)
Cu – 2e
-
→
Cu
2+
(anoda, czyli reakcja utleniania)
W ogniwie Volty zachodzą reakcje elektrodowe:
Anoda:
Zn - 2e
-
→
Zn
2+
(reakcja utleniania, czyli reakcja anodowa)
Katoda:
2H
+
+ 2e
-
→
H
2
↑
(reakcja redukcji, czyli reakcja katodowa)
Ogniwo Volty jest ogniwem nieodwracalnym, wydzielony wodór opuszcza
ś
rodowisko reakcji. Ponadto metal katody, którym jest miedź (tak zbudowane było
pierwsze ogniwo Volty), może być zastąpiony dowolnym innym, odpornym
chemicznie przewodnikiem elektrycznym; może to być: platyna, złoto, srebro,
grafit, węgiel itp. Proces katodowy w tym ogniwie nie zależy od tworzywa katody
- ważne jest tylko to aby był to przewodnik elektryczny.
5
W realnych technicznie warunkach, jeżeli powierzchnia miedzi będzie
zanieczyszczona wtrąceniami cynku lub odwrotnie, powierzchnia cynku -
wtrąceniami miedzi, to procesy elektrochemiczne przebiegające w mikroskali na
takiej powierzchni będą analogiczne do opisanych wcześniej. Pomiędzy
obydwoma metalami stanowiącymi jeden wspólny element prąd może przepływać
swobodnie, praktycznie bez oporu, a wytworzone ogniwo będzie pracować w
warunkach zwarcia elektrycznego. Podobne procesy będą przebiegały w
przypadku każdych dowolnych układów dwóch różnych metali, np. stal - miedź,
stal - nikiel, cynk – stal i innych, a także układów metal – niemetal, który
przewodzi prąd, np. stal – grafit, stal – węglik żelaza, stal – tlenek żelaza i inne.
Najczęstsze przyczyny powstawania ogniw korozyjnych to:
- zanieczyszczenie powierzchni obcymi metalami lub tlenkami metali,
- obecność różnych składników stopowych lub różnych faz w strukturze metalu,
- niejednorodność materiałowa spowodowana nierównomierną obróbką cieplną
lub miejscowym przegrzaniem metali i stopów, np. w czasie szlifowania,
- różnice stężenia roztworu elektrolitu, w tym stopnia napowietrzenia roztworu,
- różnice temperatury roztworu stykającego się z metalem,
- różnice szybkości przepływu roztworu stykającego się z powierzchnią metalu.
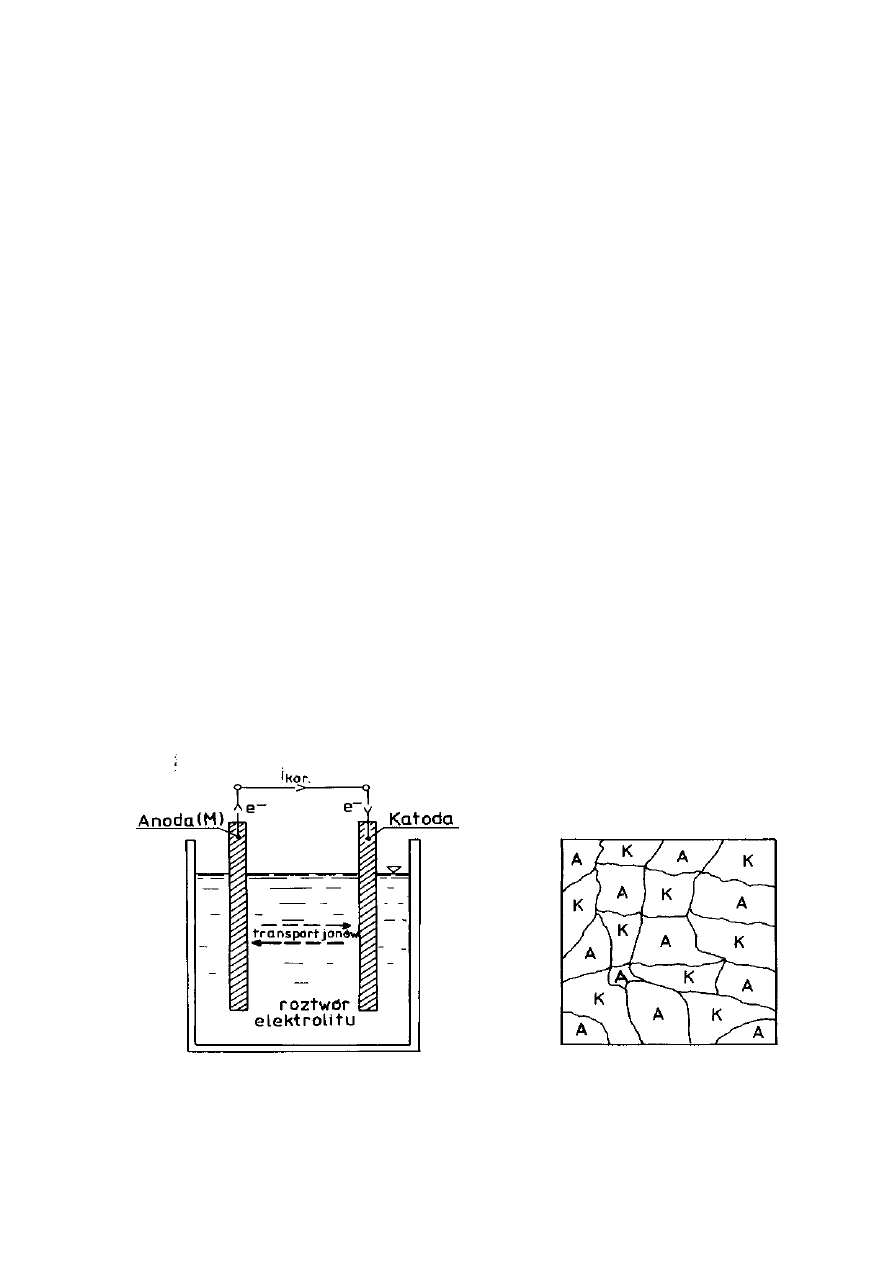
Korozją ogólną przebiegającą według mechanizmu elektrochemicznego
schematycznie przedstawiają rysunki 3 i 4. Na powierzchni ulegającej korozji
pojawiają się obszary anodowe (A) i katodowe (K), które ulegają ciągłemu
przemieszczaniu się; obszar anodowy może stać się po chwili katodowym i
odwrotnie. Jest to dynamiczny układ obszarów anodowych i katodowych.
Rysunek 3. Schemat ogólny ogniwa
Rysunek 4. Korodująca powierzchnia

6
2.1.2. Procesy elektrochemiczne przebiegające na powierzchni metali
Większość procesów elektrochemicznych, z którymi mamy do czynienia w
praktyce, przebiega w warunkach stykania się metali z różnymi środowiskami
wodnymi, a więc ich przebieg związany jest z jednoczesnym przepływem
ładunków elektrycznych (elektronów i jonów) przez granicę faz: metal-otaczające
ś
rodowisko korozyjne (roztwór elektrolitu).
Powierzchnia metalu nie jest zazwyczaj jednorodna, lecz ma miejsca o różnej
aktywności, czyli różnej skłonności do ulegania korozji. W zetknięciu z
roztworem elektrolitu, miejsca o większej aktywności ulegają roztwarzaniu i stają
się lokalnymi anodami, podczas gdy miejsca mniej aktywne elektrochemicznie
przybierają rolę katod w lokalnych ogniwach galwanicznych. Lokalne anody i
katody są pomiędzy sobą krótko zwarte elektrycznie poprzez masę metalu. Na
powierzchni anody przebiegają elektrochemiczne reakcje utleniania, tj. oddawania
elektronów przez atomy, cząsteczki lub jony do materiału anody, a na powierzchni
katody zachodzą elektrochemiczne reakcje redukcji, tj. pobierania elektronów od
materiału elektrody przez atomy, cząsteczki lub jony zawarte w roztworze.
Różnica między potencjałem wypadkowym ustalającym się na metalu, który
ulega działaniu roztworu elektrolitu, a elektrodą odniesienia (np. nasyconą
elektrodą kalomelową - NEK) nosi nazwę potencjału korozyjnego (względem
NEK), natomiast sumaryczne natężenie prądu, który przepływa między
anodowymi i katodowymi miejscami powierzchni metalu, nazywa się prądem
korozyjnym. Zarówno potencjał, jak i prąd korozyjny zależą od rodzaju metalu lub
stopu, składu chemicznego, struktury i stanu powierzchni próbki, jak też od składu
roztworu oraz panujących warunków fizycznych. Ponadto, czynniki te ulegają
zmianom w czasie, głównie w miarę postępowania procesów korozyjnych.
Można wyróżnić następujące trzy odrębne grupy czynników wywołujących
powstawanie i działanie ogniw lokalnych na granicy stykania się metalu z
roztworem elektrolitu:
1) niejednorodności występujące w metalu i na jego powierzchni,
2) niejednorodności składu roztworu,
3) różnice warunków fizycznych w różnych miejscach układu.
W pierwszym przypadku mamy do czynienia z typem ogniwa analogicznym
do ogniwa złożonego np. z dwóch metali zanurzonych w tym samym roztworze.
W drugim przypadku do ogniwa stężeniowego, natomiast z trzecim przypadkiem
mamy do czynienia wtedy, gdy poszczególne części zanurzonego w roztworze
metalu mają różną temperaturę bądź naprężenia mechaniczne.

7
Przyczyną występowania elektrochemicznej niejednorodności powierzchni
metalu są makro- i mikroskopowe różnice jego składu chemicznego, wywołane
segregacją składników i zanieczyszczeń, wydzieleniami obcych faz itp., a także
czynniki związane z obecnością defektów krystalicznej struktury metalu
(dyslokacje i inne defekty struktury, granice ziaren, naprężenia mechaniczne) i z
niejednorodnością składu i struktury warstw fazowych lub adsorpcyjnych na
powierzchni metalu. W przypadku działania lokalnych mikroogniw, miejsca
anodowe i katodowe ulegają w miarę przebiegu korozji częstemu przemieszczaniu
się, co może prowadzić do równomiernego atakowania metalu na całej jego
powierzchni kontaktu z roztworem, czyli do tzw. korozji ogólnej (równomiernej).
Jeżeli atakowaniu ulegają głównie granice ziaren, które tworzą w obrębie metalu
ciągłą siatkę, to wtedy mamy do czynienia z korozją międzykrystaliczną. Lokalne
makro- i mikroskopowe wydzielenia zanieczyszczeń i obcych faz stają się zwykle
zarodkami wżerów i zapoczątkowują w pewnych warunkach korozję wżerową.
Zestawienie układów połączeń stali z wtrąceniami niemetalicznymi obecnymi
w stali oraz stali z innymi metalami (np. miedzi ze szczotek mosiężnych
stosowanych do czyszczenia powierzchni stali) stanowiącymi lokalne ogniwa
korozyjne, w których lokalne anody ulegają korozyjnemu roztwarzaniu, stanowi
praktyczne rozszerzenie szeregu napięciowego metali (Tab. 1).
Do najważniejszych typów ogniw występujących w układach korozyjnych o
niejednorodnym składzie roztworu należą:
- ogniwa stężeniowe obejmujące roztwór o różnym stężeniu tych samych jonów,
- ogniwa stężeniowe o różnym stopniu napowietrzenia roztworu, czyli o różnym
stężeniu tlenu rozpuszczonego w różnych miejscach roztworu,
- ogniwa zawierające roztwór o różnych wartościach pH w różnych miejscach.
W przypadku ogniwa zawierającego roztwór o różnym stężeniu tych samych
jonów, elektroda stykająca się z bardziej rozcieńczonym roztworem własnych
kationów jest anodą ogniwa korozyjnego, a elektroda zanurzona w roztworze
bardziej stężonym stanowi jego katodę. Jednakże obecność agresywnych anionów
w roztworze, np. jonów chlorkowych, zmienia podany wyżej układ i anodą jest
elektroda zanurzona w roztworze bardziej stężonym.
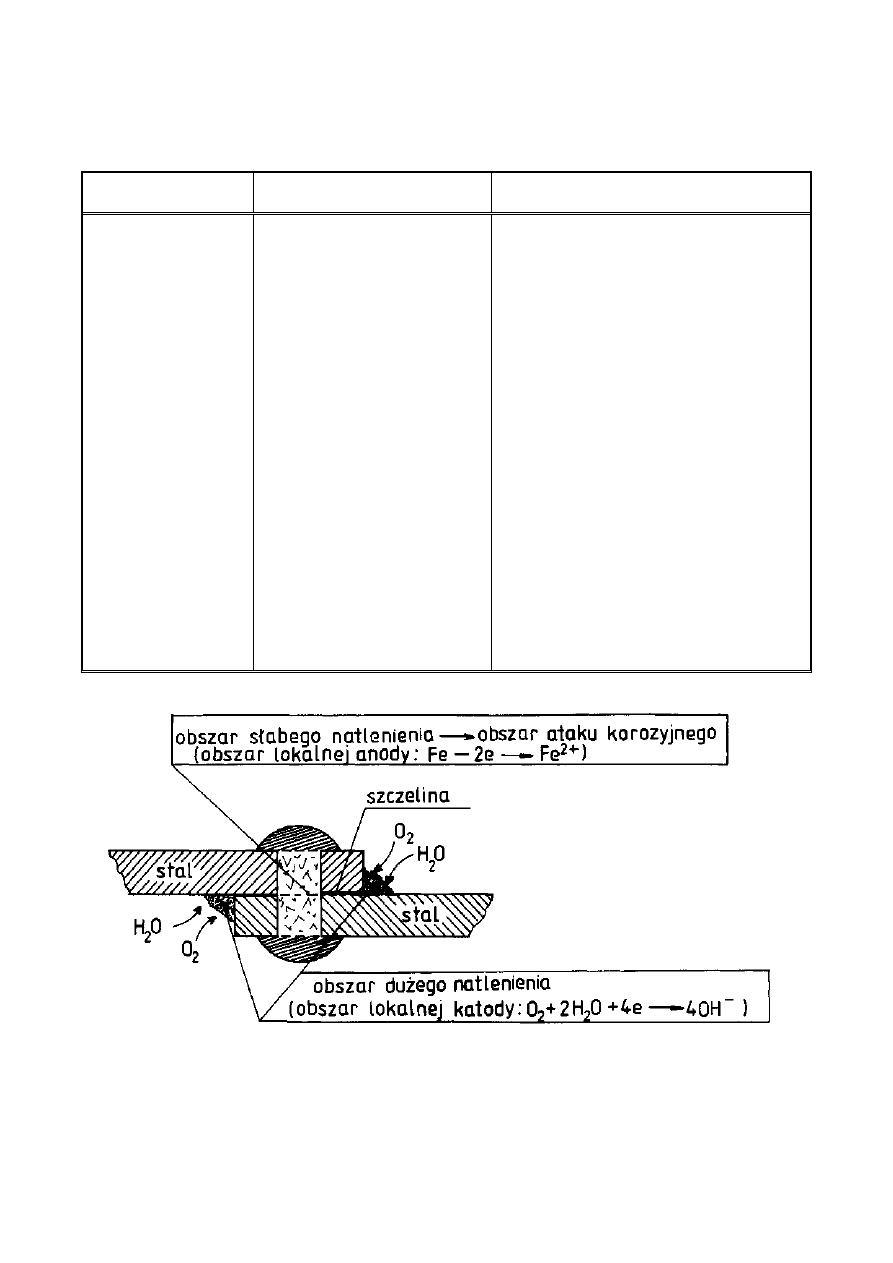
Ogniwa elektrochemiczne o różnym stopniu napowietrzenia są układami
często spotykanymi w praktyce. Dwie elektrody z tego samego metalu zanurzone
w roztworze o różnych stężeniach rozpuszczonego tlenu zachowują się z reguły
tak, że elektroda stykająca się z roztworem bogatszym w tlen działa jak katoda
(Rys. 5) a zniszczenia korozyjne występują w obszarze do których dostęp tlenu
jest utrudniony. Tlenowe ogniwo korozyjne nosi nazwę ogniwa Evansa.
8
Tabela 1. Zestawienie wybranych układów katodowo-anodowych w ogniwach
korozyjnych z udziałem stali
Lp.
Anoda
Katoda
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
ż
elazo
-"-
-"-
-"-
stal naprężona
stal
-"-
-"-
-"-
-"-
-"-
-"-
-"-
magnez
cynk
aluminium
węgliki, np. cementyt
wydzielone siarczki
zgorzelina
grafit
stal odpuszczona
miedź
mosiądz
ołów
nikiel
cyna
kobalt
kadm
stal
-"-
-"-
-"-
Rysunek 5. Przykład działania korozyjnego ogniwa tlenowego Evansa w
przypadku nieszczelnego połączenia nitowanego

9
Dla ogniwa z roztworem o różnej kwasowości, ważna jest wielkość obszaru
metali stykających się z roztworem o wyższej wartościowości pH, który działa
zwykle jako katoda. Przepływ prądu korozyjnego, związany z działaniem ogniw
lokalnych powoduje zmiany składu przyległych warstw roztworu, a tym samym
przyczynia się do podtrzymania, a nawet wzmożenia procesów korozyjnych, jeżeli
podczas korozji nie utworzą się nierozpuszczalne osady. Z tego względu raz
zapoczątkowany proces elektrochemiczny szczególnie w przypadku (korozji
punktowej) ma tendencję do dalszego coraz intensywniejszego przebiegu.
Czynnik ten odgrywa ważną rolę w przypadku korozji wżerowej.
W przypadku różnicy temperatur, to miejsca o podwyższonej temperaturze są
zwykle obszarami anodowymi lokalnych ogniw korozyjnych.
2.1.3. Korozja chemiczna
Korozja chemiczna występuje w przypadku oddziaływania suchych gazów
lub cieczy nieprzewodzących prądu na metal i obejmuje przypadki, którym nie
towarzyszy powstawanie i przepływ ładunku elektrycznego.
Najbardziej rozpowszechnionym rodzajem korozji chemicznej jest utlenianie
się powierzchni metali w warunkach uniemożliwiających skraplanie się wilgoci, w
suchych gazach zawierających tlen lub innych gazach utleniających, zwykle w
podwyższonych temperaturach. Produkt reakcji może być lotny, ciekły lub stały.
W pewnych przypadkach, w wyniku chemicznego oddziaływania tlenu na
powierzchnię metalu tworzy się szczelna warstewka tlenku metalu, która może
zabezpieczać przed dalszym utlenianiem i postępem korozji, co powoduje, że
metal przechodzi w stan pasywny, a warstewka ta nosi nazwę warstwy pasywnej.
Metal, który znajduje się w cieczy nie będącej elektrolitem może również
doznać zniszczeń korozyjnych w sposób chemiczny. Zwykle wytwarza się
warstewka produktów korozji hamująca częściowo postęp korozji w głąb metalu.
Jako przykład tego rodzaju środowiska korozyjnego można wymienić ropę
naftową zanieczyszczoną związkami siarki. Nie wykazuje ona przewodnictwa
elektrycznego, a pomimo tego rurociągi stalowe z ropą ulegają korozji.
Warunkiem koniecznym przebiegu korozji chemicznej jest:
1 - styczność z suchym i utleniającym środowiskiem,
2 - brak przepływu ładunku elektrycznego w obszarze korozji.
Warstewki tlenkowe na metalach mają najczęściej strukturę krystaliczną
jonową i wykazują z reguły przewodnictwo jonowe lub elektronowe. W procesie
tworzenia się i narastania zgorzeliny w podwyższonych temperaturach odbywa się
ruch jonów metalu i wolnych elektronów w jednym kierunku i jonów tlenu w
10
przeciwnym kierunku, przy czym jonizacji atomów tlenu dokonują na zewnętrznej
powierzchni warstewki tlenkowej elektrony, które się przez nią przedostały.
Przyjęty mechanizm nie wyklucza możliwości równoległego odbywania się
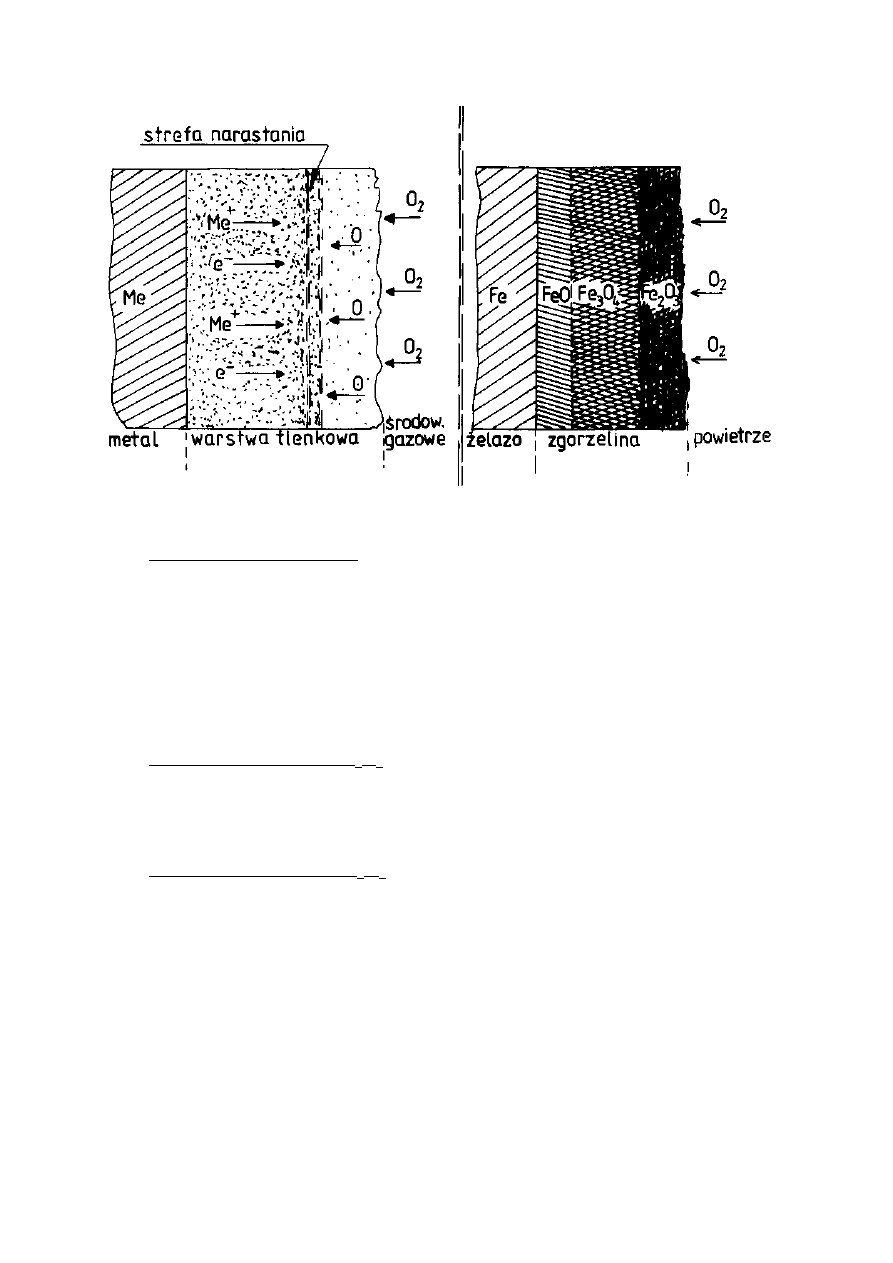
dyfuzji atomów (Rys. 6). Promienie jonów metali są znacznie mniejsze od
promieni jonów tlenu i tym tłumaczy się większą ruchliwość jonów metali w
procesie dyfuzji niż jonów tlenu oraz fakt narastania warstewki głównie w pobliżu
powierzchni zewnętrznej (Tab. 2). Jon metalu jest mniejszy od odpowiadającego
mu atomu, natomiast odwrotnie jest z utleniaczem; anion utleniacza jest większy
niż odpowiadający mu atom. Można zatem przypuszczać, że dla większości
przypadków od powierzchni metalu, poprzez warstewkę tlenkową wędrują
głównie jony metalu i elektrony (a nie atomy), natomiast od powierzchni
warstewki w głąb - atomy utleniacza (a nie jony). W przypadku narastania
zgorzeliny na powierzchni stali obserwuje się dyfuzję jonów żelaza z powierzchni
metalu przez warstwę tlenków na zewnątrz oraz dyfuzję jonów lub atomów tlenu
w kierunku przeciwnym. Narastanie grubości warstewki tlenkowej odbywa się
więc wewnątrz zgorzeliny.
Często się zdarza, że zgorzelina wykazuje wyraźną strukturę warstwową.
Skład chemiczny poszczególnych warstw zgorzeliny jest zbliżony do składu
tlenków żelaza: FeO, Fe
3
O
4
i Fe
2
O
3
. Warstwy te układają się w takiej kolejności,
ż
e bezpośrednio do metalu przylega tlenek o najniższej zawartości tlenu FeO, dalej
znajdzie się tlenek o składzie pośrednim Fe
3
O
4
i wreszcie powierzchnię
zewnętrzną tworzy tlenek o najwyższej zawartości tlenu Fe
2
O
3
. Stosunki ilościowe
grubości warstw zależne są przede wszystkim od warunków utleniania, głównie
temperatury (Rys. 7).
Tabela 2. Wielkość atomów i jonów wybranych pierwiastków
Atom
Jon
Pierwiastek
symbol
promień [Å]
oznaczenie
promień [Å]
1. Żelazo
2. Żelazo
3. Chrom
4. Nikiel
5. Kobalt
6. Glin
7. Cynk
Fe
Fe
Cr
Ni
Co
Al
Zn
1,26
1,26
1,25
1,25
1,25
1,43
1,31
Fe
2+
Fe
3+
Cr
3+
Ni
2+
Co
2+
Al
3+
Zn
2+
0,75
0,67
0,65
0,78
0,82
0,50
0,74
8. Tlen
9. Siarka
O
S
0,60
1,04
O
2-
S
2-
1,40
1,84
11
Rysunek 6. Narastanie zgorzeliny
Rysunek 7. Struktura zgorzeliny
Tlenek żelaza(II), FeO, (nazywany wustytem) jest trwały tylko w
temperaturach powyżej 575
o
C. W temperaturach niższych tlenek ten się nie
tworzy, a w czasie powolnego chłodzenia zgorzeliny powstałej w wyższych
temperaturach, rozkłada się według równania:
4FeO
→
Fe + Fe
3
O
4
Znajdująca się zwykle w warstwie zgorzeliny faza FeO występuje w stanie
pozornie trwałym (przechłodzonym).
Tetratlenek triżelaza, Fe
3
O
4
, (nazywany magnetytem) jest trwały w całym
zakresie temperatur, od temperatury pokojowej aż do temperatury topnienia żelaza
(t
t.Fe
=1538
o
C). Jest jednorodną fazą pod względem krystolagraficznym i
bynajmniej nie stanowi cząsteczkowej mieszaniny tlenków FeO + Fe
2
O
3
.
Tlenek żelaza(III), Fe
2
O
3
, (nazywany hematytem) jest trwały w
temperaturach poniżej 1100
o
C.
Podczas utleniania żelaza w powietrzu proces przebiega kolejno w stadiach:
1. W czasie ogrzewania w temperaturach do 200
o
C narasta warstewka tlenkowa
Fe
3
O
4
lecz po osiągnięciu niewielkiej grubości warstwy proces utleniania
praktycznie ustaje.
2. W czasie dalszego ogrzewania do temperatury 575
o
C następuje niewielki
wzrost grubości tej warstwy, a w warstwie zewnętrznej zachodzi przemiana
Fe
3
O
4
w Fe
2
O
3
. Tworzą się więc dwie warstwy: Fe
3
O
4
− Fe
2
O
3
.
3. W temperaturach powyżej 575
o
C tworzą się już trzy warstwy (Rys. 7):
FeO − Fe
3
O
4
− Fe
2
O
3
. Proces utleniania zachodzi przy tym znacznie szybciej.

12
Chcąc uzyskać warstewkę pasywną na żelazie składającą się wyłącznie z
magnetytu Fe
3
O
4
proces utleniania prowadzi się w temperaturach poniżej 575
o
C w
parze wodnej z niewielką zawartością tlenu - tak prowadzi się proces oksydowania
wyrobów stalowych, np. lufy karabinów.
Szczególne zainteresowanie budzą tlenki złożone powstające w czasie
utleniania stopów, szczególnie stali stopowych. Największe znaczenie praktyczne
mają roztwory stałe tlenków tworzące się na stalach żaroodpornych w czasie ich
ogrzewania. Przykładem mogą być FeO-NiO, FeO-CoO, FeO-MnO, Fe
2
O
3
-Cr
2
O
3
.
2.1.4. Odporność stali i metali nieżelaznych w różnych środowiskach
W praktyce przemysłowej najczęściej mamy do czynienia z przedmiotami
stalowymi, aluminiowymi i cynkowymi lub ze stali ocynkowanej, niekiedy z
miedzią. W środowisku silnie kwaśnym metale te nie są odporne, nawet miedź. W
ś
rodowisku obojętnym odporne jest aluminium i cynk oraz w niektórych
przypadkach stal. W środowisku alkalicznym odporna jest stal, natomiast
aluminium i cynk reagują gwałtownie z jonami wodorotlenkowymi OH
-
z
wydzieleniem wodoru. Z tego względu przedmioty aluminiowe, cynkowe i grubo
ocynkowane nie mogą być umieszczane w betonie, który jak wiadomo ma odczyn
alkaliczny (wskaźnik stężenia jonów wodorowych pH powyżej 12). Najbardziej
odpornym metalem w wymienionych środowiskach jest miedź, która nie jest
odporna w środowisku silnie kwaśnym i utleniającym jednocześnie, np. kwas
azotowy i w środowisku roztworów amoniaku, cyjanku (Tab. 3).
Reakcje stali, aluminium, cynku i miedzi w różnych środowiskach:
1. Kwaśne środowisko
Fe + 2HCl
→
FeCl
2
+ H
2
↑
Al + 3HCl
→
AlCl
3
+ 3/2H
2
↑
Zn + 2HCl
→
ZnCl
2
+ H
2
↑
Cu + HCl
→
nie reaguje
2. Alkaliczne środowisko
Fe + NaOH
→
pasywacja powierzchni
Al + NaOH + 3H
2
O
→
Na[Al(OH)
4
] + 3/2H
2
↑
Zn + 2NaOH + 2H
2
O
→
Na
2
[Zn(OH)
4
] + H
2
↑
2Cu + O
2
→
2CuO (warstwa pasywna)

13
3. Obojętne środowisko
3Fe + 2O
2
→
Fe
3
O
4
(środowisko suche)
4Fe + 3O
2
+ 2xH
2
O
→
2Fe
2
O
3
×xH
2
O – rdza (środowisko wilgotne)
4Al + 3O
2
→
2Al
2
O
3
(warstwa pasywna)
2Zn + O
2
→
2ZnO (warstwa powierzchniowa)
2Cu + O
2
→
2CuO (warstwa pasywna)
Tabela 3: Odporność metali w różnych środowiskach
Metal
Ś
rodowisko korozyjne
kwaśne
alkaliczne
obojętne
1. Stal i żelazo
2. Aluminium
3. Cynk
4. Miedź
−
−
−
+
+
−
−
+
±
+
+
+
gdzie: + - odporny,
−
- nieodporny, ± - inne warunki decydują o odporności
2.1.5. Korozja stali w różnych środowiskach
Stal zajmuje naczelne miejsce w grupie metali stosowanych w budownictwie,
głównie jako zbrojenie różnego typu budowli. Korozja stali i innych metali,
objawia się, oprócz zmiany wyglądu, spadkiem wytrzymałości mechanicznej. We
współczesnych konstrukcjach żelbetowych korozja zbrojenia coraz częściej jest
procesem, który decyduje o ich trwałości. W przypadku niektórych rodzajów
konstrukcji zniszczenia i awarie obserwowano jedynie w wyniku korozji
zbrojenia. Dotyczy to głównie konstrukcji cienkościennych i elementów z
betonów porowatych. Mechanizm korozji zależy jednak od środowiska (Tab. 4).
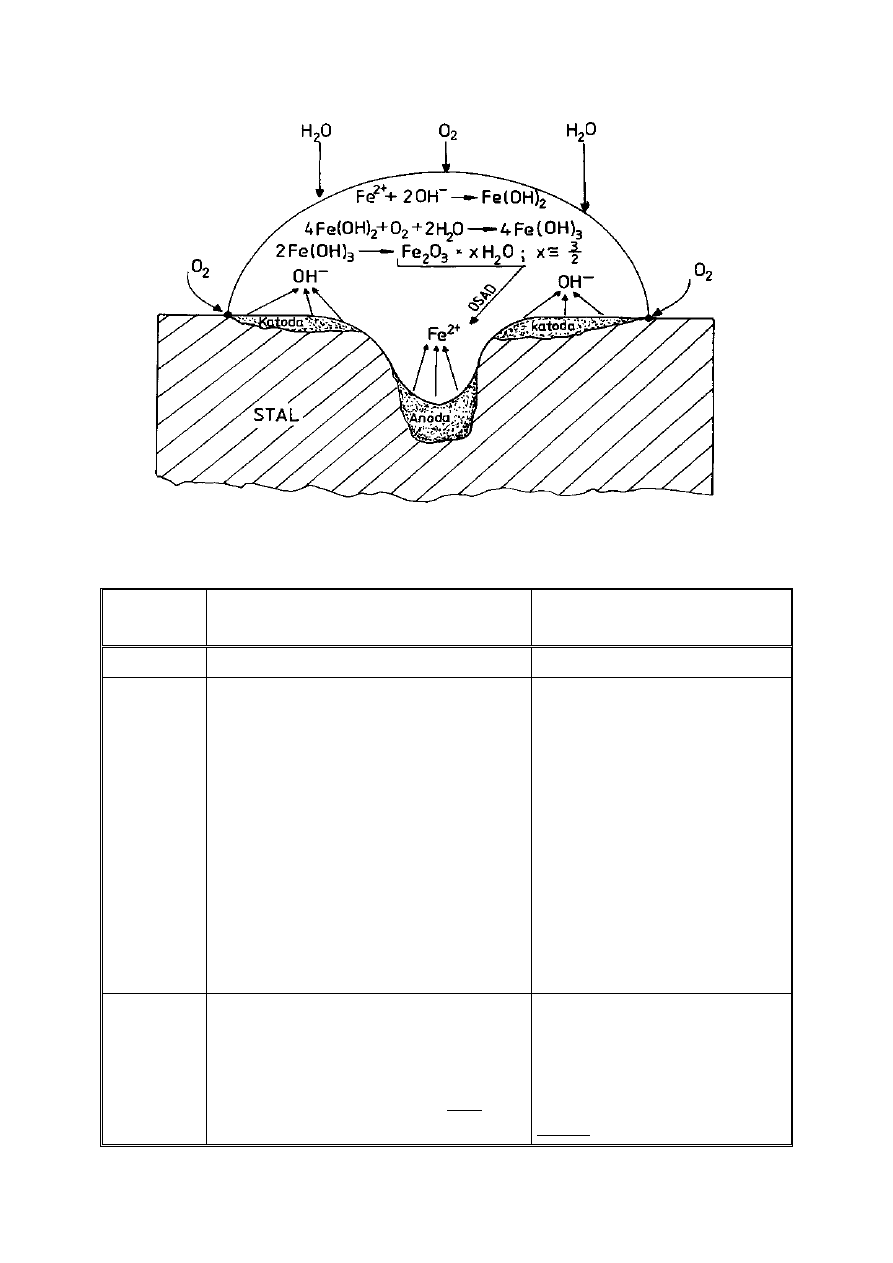
W przypadku korozji atmosferycznej żelaza (przy swobodnym dostępie
tlenu) na jego powierzchni zachodzą następujące reakcje (Rys. 8):
1 – proces anodowy: Fe – 2e
-
→ Fe
2+
2 – proces katodowy: O
2
+ 2H
2
O +4e
-
→ 4OH
-
3 – procesy chemiczne:Fe
2+
+ 2OH
-
→Fe(OH)
2
4Fe(OH)
2
+ O
2
+ 2H
2
O →4Fe(OH)
3
2Fe(OH)
3
→Fe
2
O
3
· xH
2
O,
gdzie: x=ok.3/2
14
Rysunek 8. Korozja stali w warunkach atmosferycznych (jak w ogniwie Evansa)
Tabela 4. Wpływ środowiska na korozję żelaza
Strefa
Reakcje
chemiczne,
przemiany
fazowe
Ś
rodowisko korozyjne
Anoda
Fe - 2e
-
→
Fe
2+
pH < 11
Katoda
1. O
2
+ 2H
2
O + 4e
-
→
4OH
-
2. O
2
+ 4H
+
+ 4e
-
→
2H
2
O
3. 2H
+
+ 2e
-
→
H
2
↑
4. 2H
2
O + 2e
-
→
2H + 2OH
-
→
→
H
2
+ 2OH
-
1. Środowisko obojętne ze
swobodnym dostępem tlenu
(pH =ok. 7)
2. Środowisko lekko kwaśne
(pH = 4 ÷ 7) ze swobodnym
dostępem tlenu
3. Środowisko silnie kwaśne
(pH = 0 ÷ 4) z dostępem tlenu
lub bez dostępu
4. Środowisko obojętne bez
dostępu tlenu
Otaczające
ś
rodowisko
Fe
2+
+ 2OH
-
→
Fe(OH)
2
4Fe(OH)
2
+ O
2
+ 2H
2
O
→
4Fe(OH)
3
4Fe
2+
+ O
2
+ 2H
2
O
→
4Fe
3+
+ 4OH
-
2Fe(OH)
3
→
Fe
2
O
3
×xH
2
O (rdza)
(FeO, Fe
2
O
3
, Fe
3
O
4
)×xH
2
O, x=3/2
Środowisko obojętne ze
swobodnym dostępem tlenu
(pH =ok. 7)
uwaga: V
rdzy
=3×V
metalu

15
2.2. Podział zjawisk korozji ze względu na charakter zniszczeń korozyjnych
W zależności od rozległości zniszczeń korozyjnych rozróżnia się korozję:
- równomierną, tzw. ogólną, obejmującą całą powierzchnię metalu,
- nierównomierną, tzw. miejscową, kiedy zniszczenia korozyjne zlokalizowane
są głównie w niektórych miejscach powierzchni.
Korozja nierównomierna może przejawiać się w różnych postaciach, wskutek
czego można ją podzielić na następujące rodzaje:
1) Korozja plamista, która odznacza się tym, że zniszczenia metalu występują w
poszczególnych miejscach w postaci plam o niewielkiej głębokości.
2) Korozja punktowa występuje w postaci głębokich miejscowych uszkodzeń.
3) Korozja wżerowa (tzw. pitting) występuje w postaci głębokich zniszczeń
zlokalizowanych na niewielkiej powierzchni, przy czym rozgraniczenie
pomiędzy tymi odmianami jest umowne i nie zawsze dostatecznie wyraźne.
4) Korozja podpowierzchniowa rozpoczyna się od powierzchni ale rozprzestrzenia
się w głąb metalu na niewielkiej głębokości pod powierzchnią.
5) Korozja międzykrystaliczna, której cechą charakterystyczną jest niszczenie
metalu wzdłuż granic ziaren. Produkty korozji pozostają wewnątrz metalu a
ś
rodowisko korozyjne przenika przez nie do coraz głębszych warstw metalu. W
wyniku tego na powierzchni metalu są minimalne zmiany, a wytrzymałość
metalu lub stopu znacznie maleje. Korozji międzykrystalicznej ulega, zależnie
od rodzaju środowiska, wiele metali. Szczególnie narażone są spawy i wąskie
przestrzenie przy nich, gdzie znajdują się węgliki chromu i żelaza.
2.3. Podział korozji ze względu na warunki przebiegu procesów korozyjnych
Ze względu na rodzaj środowiska i warunki przebiegu procesów korozji
rozróżnić można korozję: - w roztworach elektrolitów kwaśnych i alkalicznych,
atmosferyczną, wodną, ziemną, biologiczną i gazową.
Korozja w roztworach elektrolitów kwaśnych charakteryzuje się na ogół
bardziej intensywnym przebiegiem niż w roztworach alkalicznych. Ze względu
jednak na dużą różnorodność przypadków każdy układ: metal-roztwór elektrolitu
musi być rozpatrywany indywidualnie.
Korozja atmosferyczna, zwłaszcza konstrukcji metalowych eksploatowanych
pod gołym niebem, charakteryzuje się bardzo silnym wpływem składu
chemicznego atmosfery oraz jej wilgotności i temperatury. Najaktywniejszą pod
względem korozyjnym jest silnie zanieczyszczona agresywnymi składnikami
gazowymi atmosfera przemysłowa oraz tropikalna nadmorska.

16
O agresywności atmosfery decyduje:
- obecność pary wodnej i możliwość jej wykraplania na powierzchni,
częstotliwość opadów,
- stężenie zanieczyszczeń gazowych takich jak: SO
2
, SO
3
, CO, CO
2
, NO
x
,
- liczba cząstek stałych.
Znajdujące się w powietrzu cząstki węgla i popiołu (kurz przemysłowy)
osiadając na powierzchni metalu stają się ośrodkami kapilarnej kondensacji
wilgoci. Zawierają one rozpuszczalne aktywne składniki; w związku z tym należy
uważać je za czynniki podwyższające korozyjną agresywność atmosfery.
Korozja atmosferyczna jest wynikiem przebiegu reakcji elektrochemicznych
na powierzchni metalu. Zachodzi przy pełnym dostępie tlenu, a transport jonów
odbywa się w cienkich warstwach wilgoci lub kroplach wody na powierzchni.
Nie bez znaczenia jest również stan powierzchni metalu. Występujące na
powierzchni metalu szczeliny, rysy, stwarzają dogodne warunki do kondensacji
wilgoci i zatrzymywania zanieczyszczeń na powierzchni metalu.
Korozja wodna jest wynikiem niszczącego działania wody na urządzenia
metalowe, np. zbiorniki, rury, pompy itp. Zimna, chemicznie czysta woda nie
powoduje intensywnej korozji metalu. Wzrost temperatury i zawartości
rozpuszczonego w niej tlenu powoduje przyśpieszenie korozji. Korozja w
naturalnych wodach kwaśnych postępuje szybciej niż w obojętnych czy
alkalicznych. Korozja wodna ma także charakter elektrochemiczny, podobnie jak
atmosferyczna i ziemna.
Szczególny przypadek korozji wodnej stanowi korozja w wodzie morskiej.
Woda morska jest bardzo agresywnym środowiskiem. Największa intensywność
korozji występuje na granicy faz. Duża zawartość w wodzie morskiej dobrze
zdysocjowanych soli czyni z niej środowisko sprzyjające intensywnej korozji.
Szczególnie dużą aktywność korozyjną wykazuje jon chlorkowy, obecny w
stosunkowo dużym stężeniu.
Korozja ziemna metali występuje tylko w środowisku wilgotnym - w suchej
glebie korozja nie zachodzi. Każda gleba zawiera pewną ilość soli, kwasów i
gazów. które łącznie z wilgocią stanowią o agresywności środowiska. Szybkość
korozji w glebie zależy od: - struktury gleby, która wpływa na przepuszczalność
wody i powietrza, napowietrzenia gleby, ilości zawartej wody i obecności bakterii.
Grunty gliniaste najbardziej sprzyjają korozji, ponieważ są mało
przepuszczalne i zatrzymują wodę, a ponadto zawierają skupiska soli. Ponadto, w
przypadku korozji ziemnej, korozję przyspieszają tzw. prądy błądzące.

17
2.4. Szereg elektrochemiczny
Intensywność procesów korozyjnych przebiegających według mechanizmu
elektrochemicznego zależy od szybkości reakcji elektrodowych w krótkozwartych
ogniwach korozyjnych oraz od oporności elektrycznej wytworzonego układu
galwanicznego. Istotnym zagadnieniem jest zatem zaznajomienie się z procesami
elektrochemicznymi zachodzącymi na granicy faz: metal-roztwór elektrolitu.
2.4.1. Pojęcia podstawowe
Elektrodę, zwaną także półogniwem, stanowi układ stykających się ze sobą
przewodników: elektronowego (np. metalu Me) i jonowego (np. wodnego
roztworu soli zawierającego jony tego metalu Me
z+
), przy czym na granicy faz
występuje różnica potencjałów, zwana potencjałem elektrody. Ustala się przy tym
pewien stan równowagi dynamicznej pomiędzy procesem przechodzenia atomów
z metalu do roztworu (w postaci jonów) oraz procesem przeciwnym, wydzielania
jonów z roztworu na powierzchni metalu (w postaci atomów).
Ogniwem galwanicznym nazywamy układ złożony z dwóch elektrod, w
którym możliwe jest przenoszenie ładunków elektrycznych pomiędzy elektrodami
przez obecne w roztworach jony. Przenoszenie tych ładunków związane jest z
procesami utleniania i redukcji, zachodzącymi na powierzchni elektrod.
Anodę stanowi elektroda, na której zachodzi proces utleniania tj. wzrost
stopnia utleniania substancji biorącej udział w reakcji elektrodowej, np.
przechodzenie metalu w stan jonowy (anoda oddaje elektrony z ogniwa):
Me
→
Me
z+
+ ze
-
Katodą jest elektroda, na której zachodzi proces redukcji czyli obniżenie
stopnia utlenienia substancji czynnej w reakcji elektrodowej, np. wydzielanie
jonów metalu na jej powierzchni (katoda pobiera elektrony do ogniwa):
Me
z+
+ ze
→
Me
Napięcie ogniwa równa się iloczynowi natężenia prądu oddawanego przez
ogniwo i sumie oporów elektrycznych: zewnętrznego, które zwiera ogniwo i
wewnętrznego, wynikającego z budowy ogniwa. Gdy ogniwo znajduje się w
stanie równowagi, to jest wtedy gdy nie przepływa przez opór zewnętrzny żaden
prąd wypadkowy, różnica potencjałów pomiędzy elektrodami nosi nazwę siły
elektromotorycznej (SEM).
E
anody
- E
katody
= SEM

18
Elektrodę odniesienia (E
odn.
) stanowi elektroda o ściśle zdefiniowanej
wartości potencjału, która umożliwia wyznaczanie potencjału elektrody badanej
(E
bad.
) poprzez pomiar siły elektromotorycznej ogniwa złożonego z tych elektrod.
E
bad.
- E
odn.
= SEM
Jako elektrodę odniesienia w pomiarach najczęściej stosuje się nasyconą
elektrodę kalomelową (NEK) a niezwykle rzadko normalną elektrodę wodorową
(NEW). Jednak przy interpretacji wyników jest odwrotnie – wartość potencjału
elektrod podaje się zwykle względem normalnej elektrody wodorowej (NEW).
Według przyjętej konwencji, potencjał NEW wynosi zero gdy ciśnienie
parcjalne wodoru gazowego wynosi
hPa
1013
=
p
H
2
i aktywność jonów
wodorowych w roztworze wynosi
1
=
a
O
H
+
3
(tj.
dm
mol/
1
c
3
O
H
+
3
≈
).
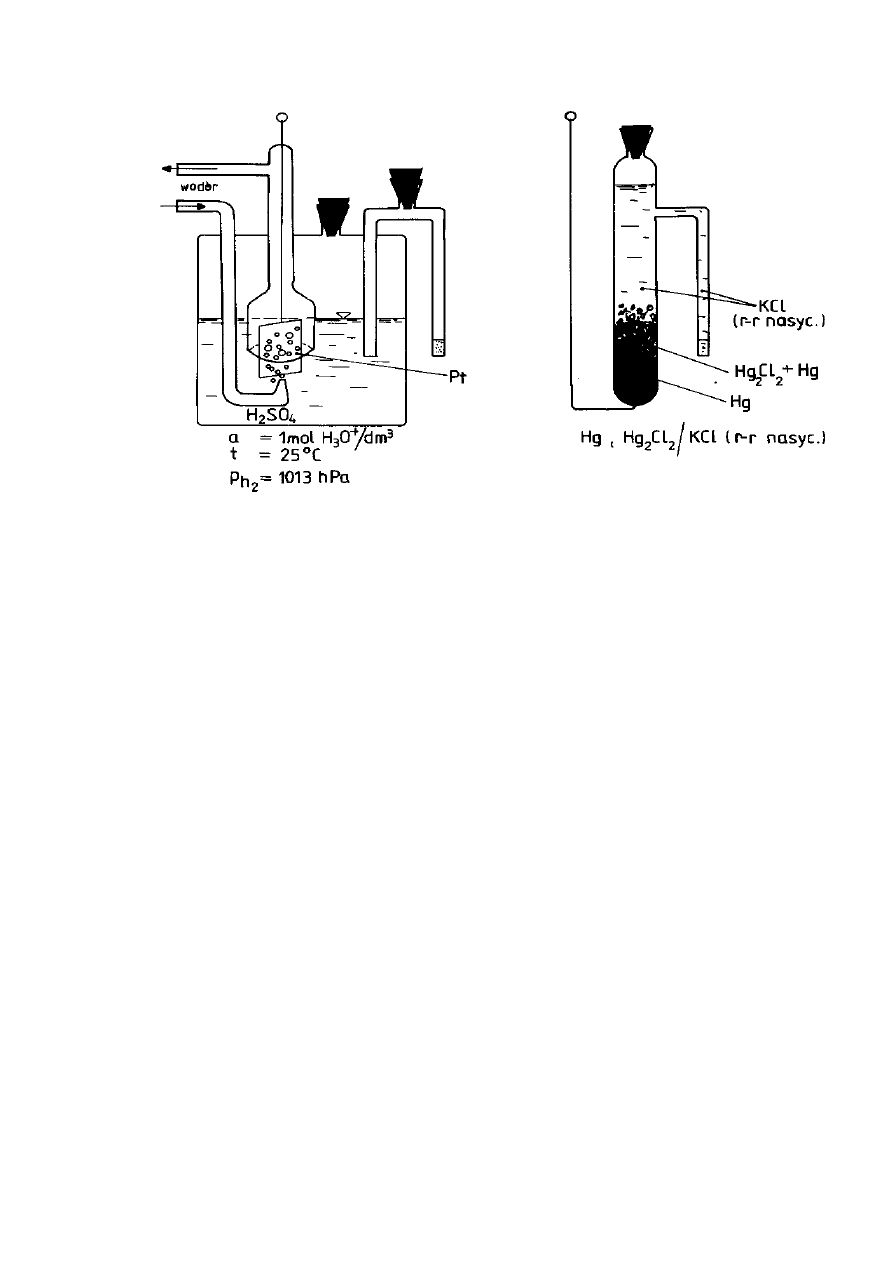
Równowagę elektrody wodorowej (Rys. 9) określa równanie:
2H
+
+ 2e
-
⇆
H
2 (g)
Potencjał elektrody wodorowej wynika z równowagi pomiędzy stężeniami
wodoru w fazie gazowej i w cieczy. Normalną elektrodę wodorową sporządza się
poprzez ciągłe przedmuchiwanie gazowego wodoru pod ciśnieniem 1013 hPa
przez roztwór kwasu siarkowego o aktywności jonów wodorowych równej
1 mol/dm
3
. Niezbędna przy tym elektroda metaliczna, pełniąca rolę przewodnika
ładunku elektrycznego, wykonana jest z platyny pokrytej czernią platynową.
Stosowanie takiej elektrody jest uciążliwe, dlatego w praktyce stosuje się
elektrody odniesienia innej konstrukcji, najczęściej nasyconą elektrodę
kalomelową (NEK). Potencjał NEK zależy od temperatury i wynosi od 0,257 V w
temperaturze O °C do 0,223 V w temperaturze 50
o
C (E
NEK
o
= 0,242 V dla 25 °C).
Równowagę elektrody określa równanie:
Hg
2
Cl
2
(s) + 2e
-
⇆
2Hg
(m)
+ 2Cl
-
Substancją elektrodową jest trudno rozpuszczalny chlorek rtęci(I) (Hg
2
Cl
2
)
zwany kalomelem i od niej pochodzi nazwa elektrody (Rys. 10).
19
Rysunek 9. Elektroda wodorowa
Rysunek 10. Elektroda kalomelowa
2.4.2. Szereg elektrochemiczny metali
W praktyce przemysłowej oraz technologicznej, często pojawia się problem
aktywności i odporności korozyjnej metali w różnych środowiskach. W takich
przypadkach można posłużyć się szeregiem elektrochemicznym metali, w którym
wielkością charakteryzującą metal jest jego potencjał elektrochemiczny. Potencjał
ten wyznacza się dla układu złożonego z elektrody metalowej (Me) zanurzonej w
roztworze soli tego metalu (Me
z+
). Na granicy faz: metal-roztwór elektrolitu
wytwarza się stan równowagi, który można opisać ogólnym równaniem reakcji
elektrodowej (wartościowość atomów metalu wynosi zero):
redukcja
Me
⇆
Me
z+
+ ze
-
utlenianie
gdzie: z - ilość elektronów (e
-
) biorących udział w reakcji elektrodowej.
Stan opisany równaniem reakcji elektrodowej jest odwracalny, lecz procesy
utleniania metali i redukcji ich jonów mogą przebiegać z różną intensywnością w
zależności od rodzaju metalu. Wielkość ładunku elektrycznego (ze
-
) będzie różna
dla poszczególnych metali i każdy układ metal-roztwór jego soli, zwany
półogniwem, posiada w stanie równowagi określoną wartość potencjału, zwaną
potencjałem elektrochemicznym tego metalu. Jak wiadomo, nie ma możliwości
pomiaru bezwzględnej wartości potencjału. W praktyce laboratoryjnej potencjał

20
elektrochemiczny metali wyznacza się na podstawie wyników pomiaru
względnego potencjału badanego w stosunku do tzw. elektrod odniesienia, których
wartości potencjału są ściśle określone i stałe w danej temperaturze - normalna
elektroda wodorowa (NEW) oraz nasycona elektroda kalomelowa (NEK).
W celu wyznaczenia wartości potencjału elektrochemicznego elektrody
metalowej, zestawia się ogniwo składające się z elektrody z badanego metalu i
NEK. Wartość potencjału mierzy się bezprądowo metodą mostkową lub
woltomierzem o odpowiednio dużej wartości oporu wewnętrznego (R
W
rzędu
10
12
Ω). Zmierzone wartości potencjałów elektrochemicznych metali, po
uwzględnieniu potencjału elektrody kalomelowej, przelicza się na wartości
względem elektrody wodorowej.
Jeżeli elektroda z badanego metalu zanurzona jest w roztworze jonów tego
metalu o
aktywności
)
dm
mol/
z
1
c
(
1
=
a
3
Me
M
z+
z+
≈
to
zmierzony
potencjał
elektrochemiczny metalu względem NEW w temperaturze 25 °C nosi nazwę
potencjału standardowego. Uszeregowanie metali według rosnących wartości
standardowych
potencjałów
elektrochemicznych
tworzy
tzw.
szereg
elektrochemiczny metali, wyrażający kolejność, w której metale wykazują
zdolność do wypierania się nawzajem z roztworu.
Potencjał układu metal-roztwór jego soli, opisuje równanie Nernsta:
a
a
ln
zF
RT
+
E
=
E
.
red
.
utl
o
,
gdzie: E - potencjał elektrody w warunkach pomiaru, V,
E
o
- potencjał standardowy elektrody, V,
R - stała gazowa (R=8,315 J/K×mol),
T - temperatura, K,
z - liczba elektronów wymienianych w procesie jednostkowym redoks,
F - stała Faradaya (F = 96500 C/mol = 26,8Ah/mol),
a
utl.
,
a
red.
- aktywność
utlenionej
i
zredukowanej
formy
jonów
uczestniczących w reakcji.
Wartość aktywności jonów, uwzględniającą wpływ stężenia i obecności
innych składników roztworu, można w pewnych przypadkach prostych roztworów
zastąpić wartością ich stężenia. Wstawiając do wzoru znane wartości stałych i
wprowadzając logarytmy dziesiętne w miejsce naturalnych oraz uwzględniając
fakt, że dla metali w stanie stałym i dla gazów pod ciśnieniem 1013 hPa
aktywność a=1, można podać uproszczoną postać wzoru dla temperatury 25
o
C:
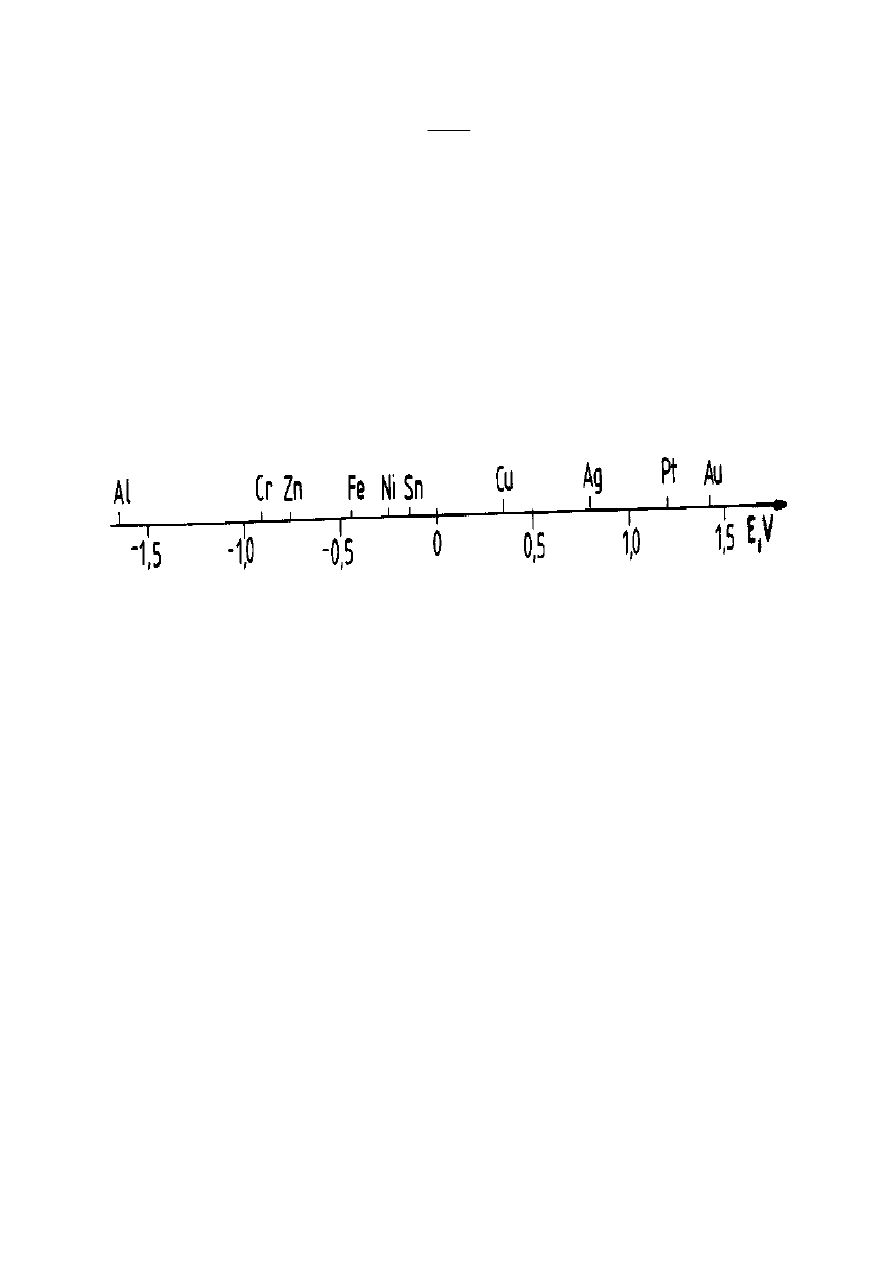
21
c
lg
z
0,059
+
E
=
E
2
Me
o
+
Wzór ten określa także warunki standardowe pomiaru (jeżeli aktywność
metalu a=1 mol/dm
3
i temperatura wynosi 25 °C, to potencjał E jest równy
potencjałowi standardowemu E
o
) i jednocześnie umożliwia wyliczenie wartości
potencjału metalu w zależności od zmian stężenia jego soli, w którym jest
zanurzony. W oparciu o zależność wartości potencjału metalu od stężenia soli
możliwe jest zbudowane tzw. ogniwa stężeniowego. W ogniwie tym, elektrody
wykonane są z tego samego metalu i zanurzone są w roztworach tej samej soli lecz
o różnych stężeniach.
Rysunek 11. Szereg elektrochemiczny metali na osi liczbowej
Szereg elektrochemiczny metali wykorzystuje się przy określaniu przebiegu
reakcji metali z roztworami soli innych metali i z kwasami nieutleniającymi oraz
zachowanie się układu dwóch metali tworzących ogniwo korozyjne.
2.4.3. Reakcje metali z roztworami soli
Reakcje takie określa się również jako wypieranie metali z roztworów.
Generalnie można stwierdzić, że metal aktywniejszy (o niższym potencjale
elektrochemicznym) będzie wypierał z roztworu soli metal mniej aktywny (o
wyższym potencjale). Można więc przewidywać, że będzie zachodziła reakcja:
Zn + CuSO
4
→
ZnSO
4
+ Cu
Nie będzie natomiast przebiegała reakcja, jeżeli przykładowo do roztworu
soli cynku zostanie wprowadzona metaliczna miedź względnie do roztworu soli
wapnia zostanie wprowadzone metaliczne żelazo, ponieważ cynk charakteryzuje
się niższym (bardziej ujemnym) potencjałem niż miedź. Wypieranie metali z
roztworów za pomocą metali aktywniejszych wykorzystuje się w technice, jako
tzw. metodę cementacyjnego otrzymywania metali.
22
2.4.4. Reakcje metali z kwasami
Występowanie wodoru w szeregu elektrochemicznym metali, względem
którego wyznacza się ich potencjał, umożliwia przewidywanie przebiegu reakcji
metali z kwasami nieutleniającymi zgodnie z ogólną zasadą, że czynnik
aktywniejszy wypiera mniej aktywny. Tak więc wszystkie metale o ujemnych
wartościach potencjałów mogą reagować z kwasami z wypieraniem wodoru, np.:
Mg + 2HCl
→
MgCl
2
+ H
2
↑
Metale o potencjałach dodatnich nie reagują z kwasami nieutleniającymi,
reagują jedynie z kwasami utleniającymi. Wówczas nie mamy do czynienia z
wypieraniem wodoru, lecz z utworzeniem tlenku metalu, właściwego dla użytego
kwasu i dalsza reakcja utworzonego tlenku z nadmiarem kwasu z wydzieleniem
wody. Przykładem mogą być reakcje miedzi w stężonych i rozcieńczonych
roztworach kwasu azotowego, które opisują równania:
Cu + 4HNO
3
→
Cu(NO
3
)
2
+ 2NO
2
↑
+ 2H
2
O (HNO
3
stężony)
3Cu + 8HNO
3
→
3Cu(NO
3
)
2
+ 2NO
↑
+ 4H
2
O (HNO
3
rozcieńczony)
2.4.5. Praktyczne wykorzystanie szeregu elektrochemicznego metali
Na podstawie kolejności usytuowania metali w szeregu napięciowym można
zrozumieć zachowanie się układu dwóch metali w warunkach korozyjnych, np.
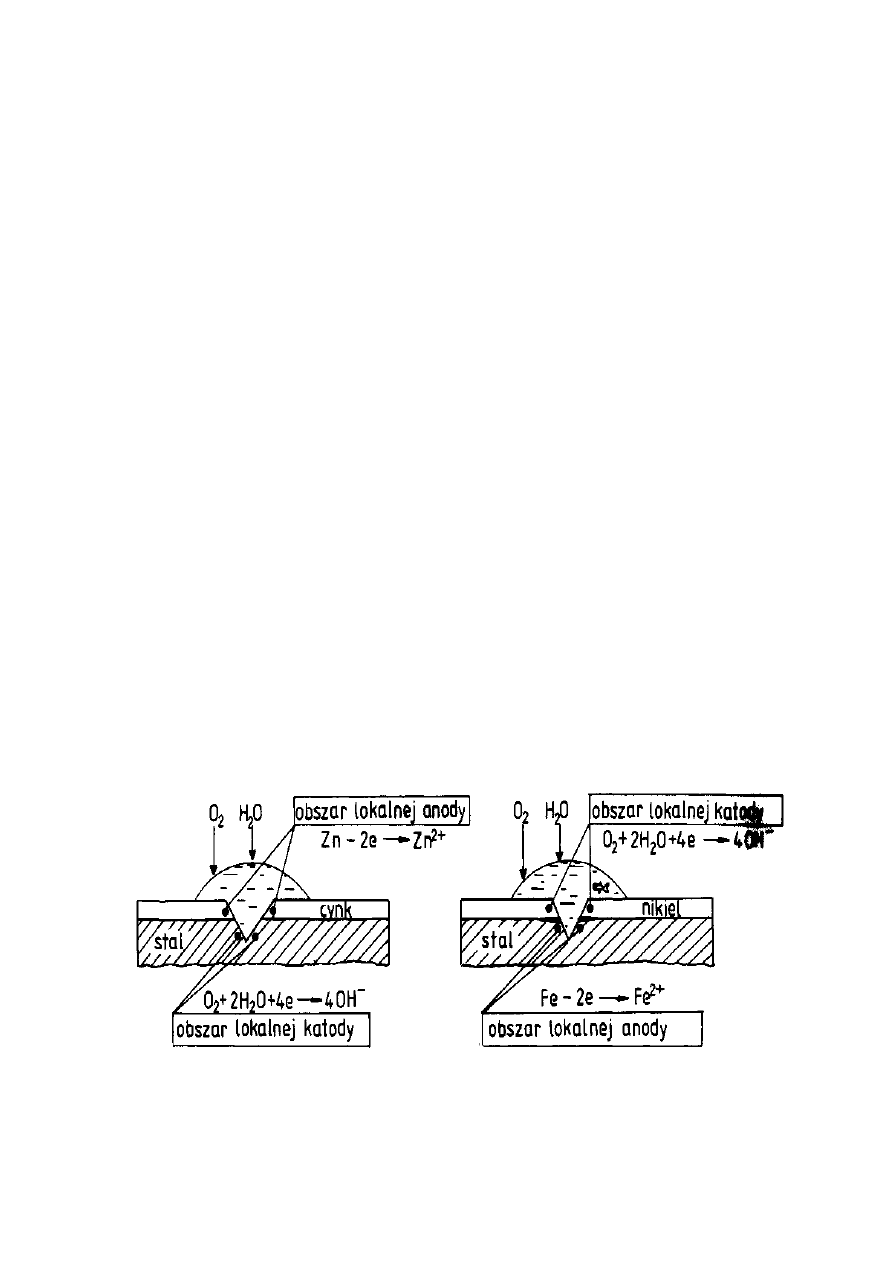
zachowanie się stali pokrytej powłokami: cynkową (Rys. 12) i niklową (Rys. 13).
Rysunek 12. Powłoka cynkowa na stali Rysunek 13. Powłoka niklowa na stali

23
Praktycznym zaleceniem wynikającym z szeregu elektrochemicznego metali
jest konieczność stosowania tych samych metali na elementy konstrukcyjne i
łączące, np. gwoździe miedziane w przypadku dachu z blachy miedzianej.
Zastosowanie
gwoździ
stalowych
do
mocowania
blachy
miedzianej
spowodowałoby bardzo szybką korozję gwoździ ze wszystkimi konsekwencjami
tego faktu. Odwrotnie, dopuszczalne jest stosowanie gwoździ stalowych do
mocowania blach cynkowych lub stalowych ocynkowanych gdyż są one
chronione protektorowo przez cynk.
2.5. Metody badania korozji
Badania laboratoryjne ze względu na potrzebny sprzęt i sposób postępowania
można podzielić na badania elektrochemiczne i nieelektrochemiczne.
Zaawansowanie procesu korozji metodami nieelektrochemicznymi można
oceniać na podstawie zmian fizycznych powierzchni korodującego metalu, zmian
składu środowiska lub badań warstwy produktów korozji tworzących się na
powierzchni metalu.
Metody elektrochemiczny polegają głównie na pomiarze potencjału
korodującego metalu przy zastosowaniu różnych procedur zmiany potencjału bądź
zmiany prądu i rejestracji wartości zmieniającego się potencjału.
2.5.1. Badania korodującego metalu
Najprostsze badania polegają na obserwacji wizualnej, najlepiej przy użyciu
mikroskopu optycznego, z powiększeniem od 10- do 500-krotnym, lub
elektronowego mikroskopu skaningowego, z powiększeniem od 100- do 20 000-
krotnym. Badania takie mają charakter jakościowy i dostarczają danych ogólnych.
Ocenę stopnia skorodowania powierzchni próbek metalowych na podstawie
produktów korozji oraz głębokości wżerów ujmuje norma: PN-78/H-046610.
Metoda grawimetryczna jest powszechnie stosowana do ilościowej oceny
procesów korozyjnych i jest zalecana przez Polską Normę: PN-70/H-04600.
Metoda ta polega na określaniu ubytku lub przyrostu masy próbki. Przy określaniu
ubytku masy próbki, produkty korozji powinny być usunięte w sposób zgodny z
normą. Oznaczanie przyrostu masy dokonuje się natomiast najczęściej do badania
korozji chemicznej w procesie utleniania metalu w wysokich temperaturach.
Przyrost masy próbki rejestruje się wtedy najczęściej w sposób automatyczny.
Wyniki oznaczeń badań ubytków korozyjnych metali i stopów metodą
grawimetryczną przedstawia się jako ubytek masy materiału przypadający na
jednostkę powierzchni i w jednostkowym czasie:

24
t
A
G
=
V
c
•
∆
gdzie: V
c
- ubytek masy, g/m
2
×doba,
∆
G - zmiana masy próbki, g,
A – wielkość powierzchni próbki przed korozją, m
2
,
t - czas trwania próby (korozji), doba.
Ubytki te można również przedstawić jako ubytek wymiaru poprzecznego w
czasie, jako tzw. liniową szybkość korozji:
V
p
= V
c
×
α
d
1000
365
=
•
α
gdzie: V
p
- liniowa szybkość korozji, mm/rok,
α
- współczynnik wiążący ubytek masy próbki z liniową szybkością
korozji,
d - gęstość tworzywa, g/cm
3
.
Jest to jedna ze starszych metod badawczych. Przydatna jest do oceny korozji
ogólnej lub powierzchniowej, a zawodzi w przypadku korozji lokalnej, np.
wżerowej, międzykrystalicznej lub w przypadku odcynkowania stali. Metoda
grawimetryczna jest niezastąpiona gdy chodzi o uzyskanie dostatecznie pewnych
danych dotyczących odporności metali w określonych warunkach, lecz powinna
być uzupełniona także o inne metody badania korozji. Metoda ta odznacza się
długim czasem trwania doświadczenia, które prowadzi się w stosunkowo dużej,
termostatowanej aparaturze, jest żmudna, wymaga zużywania znacznych ilości
materiału, a w dodatku nie daje wglądu w mechanizm procesów korozyjnych.
Metody prób mechanicznych są istotne w przypadku detali metalowych
stanowiących elementy konstrukcyjne, przy czym stosowane są głównie badania
wytrzymałości na rozciąganie i skręcanie.
Metoda pomiaru własności elektrycznych pozwala na ocenę stopnia
skorodowania próbek bez wyjmowania ich ze środowiska korozyjnego. Polega
ona na określeniu zmian oporu elektrycznego próbki, wynikających ze
zmniejszenia się jej przekroju poprzecznego.
Metody oparte na pomiarach grubości ścianek aparatów ulegających
korozji, np. metody ultradźwiękowe, rentgenowskie lub izotopowe.

25
2.5.2. Badania zmian składu środowiska
Badania te polegają na określeniu zmian składu fazy gazowej lub ciekłej,
które można określać na drodze analitycznej lub konduktometrycznej. W
przypadku gdy produkty korozji zabarwiają roztwór, stosować można metodę
kolorymetryczną, natomiast gdy zmieniają przewodnictwo - konduktometryczną.
2.5.3. Badania warstw produktów korozji
Badania warstwy powierzchniowej można przeprowadzić po selektywnym
roztworzeniu podłoża metalowego, a wyizolowane warstewki poddać badaniom
rentgenostrukturalnym lub spektrograficznym. Warstewki korozyjne można także
analizować bezpośrednio na metalu, lub po ich mechanicznym usunięciu z
metalicznego podłoża, przy czym istotnymi informacjami są:
- masa w odniesieniu do jednostki powierzchni i jednostki czasu procesu,
- skład chemiczny z uwzględnieniem stopnia utleniania metalu podłoża,
- skład fazowy.
Dobór właściwej metody określenia odporności korozyjnej zależy w każdym
przypadku od szeregu czynników i wymaga szczegółowego ich rozważenia.
Nieelektrochemiczne metody badania korozji w większości przypadków w
znikomym stopniu informują o mechaniźmie procesów korozji i stanowią
dokumentację dokonanych już zniszczeń. Bardziej wartościowe wydają się być
metody pozwalające na ocenę skłonności metalu lub stopu do korozji w danych
warunkach i przebiegającej w sposób nierównomierny, np. do korozji wżerowej,
lub międzykrystalicznej. Do grupy tej należą badania elektrochemiczne.
2.6. Elektrochemiczne metody badania korozji
Badania elektrochemiczne służą do oceny odporności materiału w
określonych warunkach korozyjnych (który będzie w tych warunkach przebywał).
Pozwalają one na zebranie w krótkim czasie danych o przydatności materiału do
danego (narzuconego) środowiska oraz pozwalają na ocenę zjawisk korozyjnych
zachodzących na powierzchni metali, stopów i w powłokach ochronnych w
określonym środowisku. Podstawowe badania elektrochemiczne obejmują:
- sporządzanie wykresów funkcji równowag elektrochemicznych metali i stopów
w funkcji pH roztworu i potencjału elektrodowego badanego metalu,
- pomiar potencjału korozyjnego metalu w określonych warunkach i rejestracja
tego potencjału w funkcji czasu,

26
- wyznaczanie krzywych polaryzacji anodowej i katodowej metodą: oporu
polaryzacyjnego, ekstrapolacji linii Tafela, przegięcia krzywej polaryzacji;
służą do określania gęstości prądu korozyjnego i szybkości korozji ogólnej,
- rejestrowanie krzywych polaryzacji anodowej pasywujących się metali,
szczególnie przydatne do badań stopów i stali nierdzewnych celem określenia
podatności na korozję wżerową i szybkości korozji w stanie pasywnym,
zakresu pasywnego, wartości potencjałów pasywacji (Fladego), potencjału
tworzenia się wżerów i potencjału ich repasywacji,
- utrzymywanie stałego potencjału anodowego dla przyśpieszonych badań
korozji międzykrystalicznej i naprężeniowej, a także do badań wpływu
inhibitorów korozji i zachowania się powłok metalowych.
Badania elektrochemiczne należą do badań porównawczych, a ich wyniki
powinny być sprawdzone w próbach eksploatacyjnych.
2.6.1. Wykresy równowagowe potencjału w funkcji pH
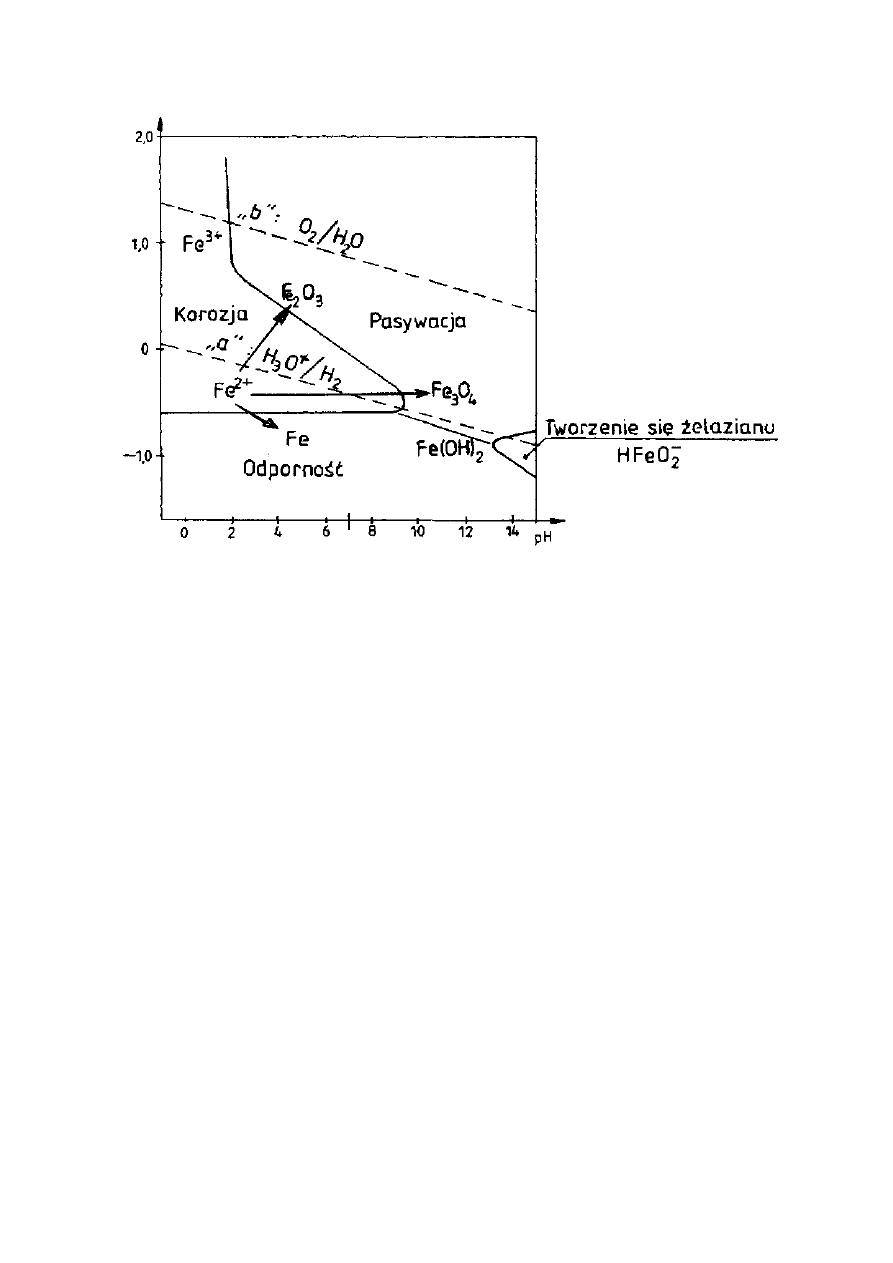
Z wykresu równowag elektrochemicznych metalu w funkcji pH roztworu i
potencjału elektrodowego w przypadku stali, tzw. wykresów Pourbaix, można
wyznaczyć obszary (w zależności od pH i potencjału elektrody), w których
utlenianie żelaza może prowadzić do powstawania produktów łatwo
rozpuszczalnych, takich jak: Fe
2+
, Fe
3+
i HFeO
2
-
oraz obszary w których tworzą się
produkty trudnorozpuszczalne, takie jak tlenki: Fe
3
O
4
i Fe
2
O
3
. Na tej podstawie
wyznaczyć można trzy podstawowe sposoby zachowania się żelaza:
- obszar korozji (tworzą się jony: Fe
2+
, Fe
3+
i HFeO
2
-
),
- obszar odporności (proces utleniania nie może zachodzić),
- obszar pasywacji (tworzą się tlenki: Fe
2
O
3
i Fe
3
O
4
).
W obszarze pasywacji tworzą się trudnorozpuszczalne i dostatecznie trwałe
produkty korozji - tlenki Fe
2
O
3
i Fe
3
O
4
, które tworzą na powierzchni żelaza zwartą
warstwę o dobrej szczelności i przyczepności, wystarczającej do całkowitego
zahamowania korozji, tzw. warstwę pasywną.
Ogólnie, przy niskich wartościach pH utlenianie żelaza przebiega z
roztwarzaniem się metalu (korozja), natomiast w miarę wzrostu wartości pH
wzrasta także tendencja do wytwarzania się warstewki tlenkowej na powierzchni
(możliwość pasywacji).
Powyżej wartości pH około 11,5 (wartość ta zależy od składu chemicznego i
gatunku stali) obszar korozji zanika i stal może być tylko w stanie odporności na
korozję bądź w stanie pasywnym. Dzięki temu zbrojenie stalowe w betonie
(pH = ok. 12) nie ulega korozji lecz przechodzi w stan pasywny.
27
Rysunek 14. Wykres Pourbaix dla żelaza
2.7. Pytania kontrolne
1.
Przebieg korozji chemicznej.
2.
Przebieg korozji elektrochemicznej.
3.
Rola tlenu w przebiegu procesów korozji przebiegającej w atmosferze
powietrza (chemicznej i elektrochemicznej) .
4.
Korozja atmosferyczna – jak powstaje rdza?
5.
Korozja atmosferyczna – korozja w kropli wody.
6.
Tworzenie się i pogłębianie wżerów – korozyjne ogniwo tlenowe Evansa.
7.
Korozja żelaza w środowisku kwaśnym i w środowisku obojętnym przy
dostępie tlenu.
8.
Wyjaśnić, co jest przyczyną, że stal nie koroduje w betonie – wykres
Pourbaix dla żelaza.
9.
Szereg elektrochemiczny metali – dlaczego nie można stosować gwoździ
stalowych do mocowania blachy miedzianej?
10.
Szereg elektrochemiczny metali – na czym polega ochronne działanie
powłok cynkowych i powłok niklowych na stali?
11.
Wskaźniki szybkości korozji i grawimetryczna metoda wyznaczania tych
wskaźników.

28
3. WYKONANIE ĆWICZENIA
3.1. Sprzęt i odczynniki
Do wykonania ćwiczenia potrzebne są:
1.
waga analityczna
2.
zestaw zlewek o pojemności od 400 do 800 cm
3
3.
tryskawka
4.
pinceta
5.
papier ścierny o uziarnieniu 320
6.
bibuła filtracyjna
7.
10 próbek stali o wymiarach 20
×
100
×
0,3 mm
8.
roztwory: - 1 M HCl
- 2 M HCl
- 1 M H
2
SO
4
- nasycony Ca(OH)
2
3.2. Praktyczne wykonanie ćwiczenia
Próbki stali w formie blaszek należy oczyścić papierem ściernym tak aby nie
było śladów rdzy i plam po trawieniu w kwasach, odtłuścić w acetonie i dobrze
wysuszyć zwracając uwagę aby powierzchnia próbek pozostała cały czas czysta
i odtłuszczona. Próbki należy zważyć z dokładnością do 0,1 mg i umieścić w
dwóch zlewkach o pojemności 600 cm
3
każda, przy czym w jednaj zlewce
umieścić należy osiem próbek a w drugiej dwie próbki. Próbki należy umieścić
tak aby nie było wzajemnego ekranowania ich powierzchni i aby był swobodny
dostęp roztworu do korodującej powierzchni. Do zlewki z ośmioma próbkami
należy ostrożnie wlać roztwór kwasu tak aby próbki pozostały na swoich
miejscach i zanotować godzinę według zegarka. Do drugiej zlewki (z dwiema
próbkami) ostrożnie wlać nasycony roztwór wodorotlenku wapnia i również
zanotować godzinę. Po upływie 15, 30, 45 i 60 minut należy wyjmować po dwie
próbki z kwasu, każdą z nich opłukać wodą bieżącą i destylowaną, a następnie
dobrze wysuszyć i zważyć. Próbki umieszczone w roztworze wodorotlenku
wapnia należy wyjąć po 60 minutach, także opłukać, wysuszyć i zważyć.
Następnie należy wyliczyć wartości średnie dla każdej pary próbek, przy czym
rozrzut wyników ubytku masy w poszczególnych parach próbek nie powinien
być większy niż 20 %. Z otrzymanych wyników obliczyć ubytek masy
V
C
[g/m
2
·godz] oraz liniową szybkość korozji V
P
[mm/rok]. Na podstawie
tabelki ze skalą odporności metali na korozję wyznaczyć stopień odporności na

29
korozję na podstawie wartości średnich wszystkich wyników dla określonego
ś
rodowiska. Sporządzić wykresy zależności: ∆m = f(τ), V
C
= f(τ), V
P
= f(τ).
Uwaga: Chcąc otrzymać powtarzalne wyniki należy przestrzegać zaleceń:
1. Próbki stalowe należy kłaść na czystą kartkę papieru i przekładać przy
użyciu pincety a jeżeli zachodzi taka potrzeba to próbki można przytrzymywać
palcami za brzegi, tak aby powierzchnia próbek nie uległa ponownemu
zabrudzeniu lub zatłuszczeniu.
2. Ubytki wagi wynoszą 10 ÷ 15 mg w czasie godzinnego przebywania w
roztworze trawiącym, wobec tego pierwsze próbki, które wyjmowane są po
upływie 15
÷
20 minut, będą miały ubytki wagi ok. 4 mg. Biorąc pod uwagę, że
ważyć można z dokładnością ±0,2 mg, to dokładność otrzymanych wyników na
ogół nie przekracza ±5 %. Samo ważenie wprowadza wobec tego możliwości
popełnienia największych błędów pomiarowych. W miarę wydłużania czasu
trwania próby dokładność wyników ulega polepszeniu.
3. Celem wykorzystania pełnych możliwości związanych z dokładnością
ważenia, wagę należy „zerować” przed każdą serią próbek i używać tylko
zakresu skali wagi od 0 do +10 mg. Nie używać zakresu od 0 do –10 mg.
4. Zwracać uwagę na dokładne wysuszenie ważonych próbek stalowych
przed zanurzeniem w roztworach oraz po wyjęciu i opłukaniu z roztworów.
4. ZALECENIA BHP
W czasie trwania ćwiczeń obowiązuje ścisłe przestrzeganie przepisów
zawartych w instrukcjach znajdujących się w każdym pomieszczeniu
laboratoryjnym w widocznym miejscu.
Obowiązuje przestrzeganie:
1. Instrukcji BHP dla studentów wraz ze znajomością listy trucizn.
2. Regulaminu pracy obowiązującego studentów Uczelni w pracowniach
dydaktycznych Wydziału Chemicznego Politechniki Śląskiej.
3. Instrukcji przeciwpożarowej dla pracowników i studentów w katedrach,
zakładach, pracowniach i administracji Politechniki Śląskiej.
Ponadto obowiązuje zarządzenie Dziekana Wydziału Chemicznego
zobowiązujące studentów do noszenia okularów ochronnych i fartuchów
ochronnych w pomieszczeniach laboratoryjnych.

30
5. PODSTAWA ZALICZENIA ĆWICZENIA
Zaliczyć należy część praktyczną i część literaturową.
Podstawę zaliczenia praktycznej części ćwiczenia jest aktywne uczestnictwo
w zajęciach laboratoryjnych, poprawne opracowanie wyników i oddanie
sprawozdania według zamieszczonego w punkcie 7 wzoru.
Ocena z ćwiczenia wpisywana do katalogu wystawiana jest na podstawie
sprawdzianu przygotowania się do zajęć w formie rozmowy ustnej lub pisemnie
w formie kartkówki z zakresu znajomości materiału dotyczącego korozji metali,
który obejmują pytania pomocnicze zamieszczone na zakończenie punktu 2.
6. LITERATURA UZUPEŁNIAJĄCA
1. N.O.Tomaszow: Teoria korozji i ochrony metali, PWN Warszawa, 1962
2. L.L.Shreira: Korozja metali i stopów - t.I, WNT Warszawa, 1966
3. L.L.Shreira: Korozja metali i stopów - t.II, WNT Warszawa, 1966
4. S.Mrowiec: Zarys teorii utleniania metali, Wyd. Śląsk Katowice, 1971
5. Z.Szklarska-Śmiałowska: Inhibitory korozji metali, WNT Warszawa, 1971
6. R.Juchniewicz: Technika przeciwkorozyjna, P.W.S.Z. Warszawa, 1973
8. H.H.Uhlig: Korozja i jej zapobieganie, WNT Warszawa, 1976
9. G.Wranglen: Podstawy korozji i ochrony metali, WNT Warszawa, 1985
10. Praca zbiorowa: Ochrona przed korozją - Poradnik WK i Ł Warszawa, 1986
11. Praca zbiorowa: Ochrona elektrochemiczna przed korozją, WNT Warszawa,
1991
12. J.Marciniak, G.Nawrat, Z.Paszenda: Ćwiczenia laboratoryjne z biomateriałów,
skrypt Pol.Śl. nr 1729, Wyd. Pol.Śl. Gliwice 1993
31
7. WZÓR SPRAWOZDANIA Z WYKONANIA ĆWICZENIA
Ć
WICZENIE 5 - KOROZJA METALI
Nazwisko i Imię
Grupa
Semestr
Data
1.
2.
3.
Badany metal:....................;gęstość:.....................;powierzchnia:......................
Ś
rodowisko korozyjne:
Roztwór A
Roztwór B
Roztwór C
Wyniki pomiarów:
Charakterystyka próbek
Obliczenia szybkości korozji
waga próbki
Lp.
Nr Środowisko/
czas trwania
próby
τ
[min]
przed
próbą
m
p
[g]
po
próbie
m
k
[g]
ubytek
wagi
∆
m [g]
ubytek
masy,
V
C
[g/m
2
·doba]
szybkość
liniowa,
V
P
[mm/rok]
Stopień
odporn.
koroz.
[k]
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
Sporządzić wykresy: 1. ∆m = f(τ)
2. V
C
= f(τ)
3. V
P
= f(τ)
Wnioski z przeprowadzonych pomiarów (porównanie badanych środowisk):
Wyszukiwarka
Podobne podstrony:
więcej podobnych podstron