
1
INSTRUKCJA DO ĆWICZEŃ LABORATORYJNYCH
Z OCHRONY ŚRODOWISKA
Kierunek: Inżynieria Środowiska
Semestr 2

2
Oznaczanie azotu amonowego metodą miareczkowania alkacymetrycznego
po wstępnej destylacji
Zawartość tzw. azotu ogólnego w wodzie jest sumą zawartości wszystkich form występowania
azotu:
N
og
= N
NH
4
+ N
NO
2
+ N
NO
3
+ N
org
gdzie:
N
og
- azot ogólny,
N
NH
4
- azot amonowy (amoniak i sole amonowe),
N
NO
2
- azot azotynowy (azotany(III)),
N
NO
3
- azot azotanowy (azotany(V)),
N
org
- azot w związkach organicznych.
Najczęściej azot ogólny oznacza się korzystając z automatycznych analizatorów, w których próbka
wprowadzana jest do wypełnionego katalizatorem i ogrzanego do temperatury ok. 800°C pieca
rurowego gdzie cały azot, niezależnie od formy występowania, przechodzi w NO i w tej postaci jest
oznaczany przez odpowiedni detektor.
Można również, korzystając ze specyficznych metod, oznaczyć oddzielnie każdą z form
występowania azotu, a azot ogólny obliczyć jako ich sumę. Ten sposób postępowania jest bardziej
pracochłonny, ale umożliwia uzyskanie informacji o tym w jakiej postaci azot występuje w wodzie, co
jest konieczne do prawidłowej oceny jej jakości.
Amoniak (azot amonowy) występujący w wodach powierzchniowych pochodzi zwykle
z biochemicznego rozkładu organicznych związków azotowych roślinnych lub zwierzęcych, jak białko
i produkty jego rozpadu, mocznik itp. Źródłem amoniaku mogą być także zrzuty ścieków
przemysłowych (np. z koksowni) lub ścieków miejskich. Zawartość amoniaku w ściekach miejskich
może dochodzić do kilkudziesięciu mg/dm
3
N
NH
4
. W wodach silnie zanieczyszczonych amoniak może
pochodzić także z biochemicznego procesu redukcji azotanów.
W wodach podziemnych amoniak może występować na skutek redukcji azotanów(III)
i azotanów(V) przez siarkowodór, piryty i inne związki redukujące. Podwyższone ilości amoniaku
w wodach podziemnych związane są z zwykle ze znaczną zawartością związków żelaza lub związków
humusowych.
Duża zawartość amoniaku w wodach użytkowych jest niepożądana. W procesie uzdatniania wody
amoniak stwarza trudności przy chlorowaniu wody, a poza tym zwiększa korozję rur wodociągowych.
Z punktu widzenia sanitarnego istotne znaczenie ma nie tylko zawartość amoniaku w wodzie, ale
i jego pochodne. Często obecność amoniaku jest wywołana rozkładem odpadków zwierzęcych, co
stwarza znaczne zagrożenie, szczególnie w przypadku użytkowania wód do celów komunalnych.
Zawartość azotu amonowego N
NH
4
w wodzie powierzchniowej klasy I nie może przekraczać
1,0 mg/dm
3
, w wodzie klasy II - 3 mg/dm
3
, w odzie klasy III - 6 mg/dm
3
(Dz. U. nr 16, z 5.11.1991 r.,
poz. 503).
Zawartość azotu amonowego w wodzie do picia nie może przekraczać 0,5 mg/dm
3
, dla wód
podziemnych niechlorowanych - 1,5 mg/dm
3
(Dz. U. nr 82, z 4.10.2000r., poz. 937).
Oznaczenie amoniaku powinno być wykonane jak najszybciej po pobraniu próbki.
Azot amonowy można oznaczyć następującymi metodami:
−
kolorymetrycznie, wprost z próbki po reakcji z odczynnikiem Nesslera,
−
po destylacji, którą należy stosować w przypadku występowania dużych ilości substancji
przeszkadzających,
oznaczenie
można
przeprowadzić
bądź
metodą
kolorymetryczną
z odczynnikiem Nesslera, bądź przy większych zawartościach amoniaku metodą miareczkowania
alkacymetrycznego.
Metodą destylacyjną można oznaczyć również azot organiczny po uprzednim rozkładzie związków
organicznych metodą Kiejdahla w stężonym kwasie siarkowym, wobec katalizatora - siarczanu rtęci.
W tych warunkach azot zawarty w związkach organicznych przechodzi w siarczan amonu i jest dalej
oznaczany jak amoniak.

3
Zasada oznaczenia
W środowisku alkalicznym słabe zasady są wypierane ze swoich soli. Amoniak należy do słabych
zasad, więc po dodaniu NaOH do wody, w której znajdują się sole amonowe, będzie zachodziła
reakcja wymiany, przykładowo dla chlorku amonu:
NH
4
Cl + NaOH = NaCl + NH
3
+ H
2
O
ś
eby całkowicie wydzielić z próbki amoniak należy część próbki oddestylować. Destylat
zawierający amoniak zbiera się w odmierzonej ilości roztworu kwasu solnego o znanym stężeniu.
Zachodzi reakcja:
NH
3
+ HCl = NH
4
Cl
Ilość kwasu musi być tak dobrana, żeby z amoniakiem przereagowała tylko jego część. Pozostały
nadmiar kwasu oznacza się metodą miareczkowania mianowanym roztworem NaOH wobec czerwieni
metylowej jako wskaźnika.
Wiedząc ile moli kwasu nie przereagowało i ile wzięto do pochłaniania amoniaku można z różnicy
obliczyć ilość moli kwasu, który przereagował z amoniakiem, a tym samym ilość moli amoniaku.
Odczynniki
−
kwas solny 0,1 mola HCl/dm
3
(z biuretą),
−
wodorotlenek sodu 0,1 mola NaOH/dm
3
(z biuretą),
−
wodorotlenek sodu 2 mole NaOH/dm
3
,
−
wskaźnik - czerwień metylowa 0,2% alkoholowy roztwór,
−
odczynnik Nesslera.
Przyrządy i naczynia
Zestaw do destylacji amoniaku, kolba stożkowa o poj. 300 cm
3
, pipeta jednomiarowa o poj. 50 cm
3
,
cylinder miarowy o poj. 50 cm
3
, szkiełko zegarkowe.
Sposób wykonania
Do kolby stożkowej będącej odbieralnikiem odmierzyć z biurety 20 cm
3
kwasu solnego o stężeniu
0,1 mol HCl/dm
3
i dodać kilka kropli wskaźnika. Przedłużacz chłodnicy powinien być zanurzony
w kwasie.
Do kolby destylacyjnej odmierzyć 50 cm
3
badanej wody i dodać 50 cm
3
wody dejonizowanej.
Wrzucić kilka kawałków porcelany aby uniknąć przegrzewania się cieczy i związanego z tym
podrzucania jej w kolbie w trakcie wrzenia. Następnie wlać 50 cm
3
(odmierzone cylindrem miarowym)
roztworu NaOH o stężeniu 2 mole/dm
3
, szybko połączyć kolbę z nasadką i chłodnicą i ogrzać roztwór
do wrzenia. Następnie tak regulować ogrzewanie, aby wrzenie miało spokojny przebieg. Po
oddestylowaniu ok. 2/3 objętości roztworu usunąć odbieralnik, zebrać na szkiełko zegarkowe kilka
kropli destylatu i sprawdzić odczynnikiem Nesslera czy destylat nie zawiera amoniaku. W razie
obecności amoniaku kontynuować destylację aż do całkowitego oddestylowania amoniaku z próbki.
Po zakończeniu destylacji nadmiar kwasu w odbieralniku odmiareczkować mianowanym
roztworem wodorotlenku sodu o stężeniu 0,1 mol NaOH/dm
3
do zmiany barwy wskaźnika z czerwonej
na żółtą. Zawartość azotu amonowego obliczyć wg wzoru:
(
)
3
p
NaOH
NaOH
HCl
HCl
N
mg/dm
V
1000
14
c
V
c
V
C
3
NH
⋅
⋅
⋅
−
⋅
=
gdzie:
V
HCl
- objętość mianowanego roztworu HCl wlana do odbieralnika, cm
3
c
HCl
- stężenie mianowanego roztworu HCl, mol/dm
3
V
NaOH
- objętość mianowanego roztworu NaOH zużyta na zmiareczkowanie próbki, cm
3
c
NaOH
- stężenie mianowanego roztworu NaOH, mol/dm
3
V
p
- objętość próbki, cm
3
14 - masa molowa azotu
1000 - przelicznik z dm
3
na cm
3

4
Oznaczanie utlenialności
Utlenialność, nazywana również indeksem nadmanganianowym, jest pojęciem umownym. Określa
zdolność do utleniania się w reakcji z silnym czynnikiem utleniającym, manganianem(VII) potasu,
w roztworze kwaśnym lub alkalicznym w ściśle określonych warunkach eksperymentalnych. Wynik
oznaczenia podawany jest jako ilość mg O
2
równoważna ilości zużytego manganianu(VII). Wartość ta
stanowi przybliżoną miarę zawartości w badanym roztworze związków organicznych i niektórych,
łatwo utleniających się, związków nieorganicznych. Jest ona z reguły niższa od rzeczywistej, gdyż
szereg związków organicznych utlenia się tylko częściowo lub w ogóle nie ulega działaniu
manganianu(VII) potasu. Oznaczenie to ma duże znaczenie przy charakteryzowaniu obciążenia wody
i ścieków związkami organicznymi. Utlenialność wody do picia nie powinna przekraczać 3 mg
O
2
/dm
3
.
Najczęściej oznaczanie utlenialności przeprowadza się w środowisku kwaśnym. Do próbki
wprowadza się znaną ilość manganianu(VII) potasu i przeprowadza reakcję utlenienia w temperaturze
wrzenia. Następnie wprowadza się nadmiar szczawianu sodu, który reaguje z pozostałym po reakcji
manganianem(VII) potasu:
2 MnO
4
–
+ 5 Na
2
C
2
O
4
+ 16 H
+
= 2 Mn
2+
+ 10 Na
+
+ 10 CO
2
+ 8 H
2
O
Pozostały szczawian sodu odmiareczkowuje się roztworem KMnO
4
. Zachodząca reakcja jest
analogiczna do podanej powyżej. Znając ilość manganianu(VII) potasu zużytego na miareczkowanie
i wprowadzoną ilość szczawianu sodu można wyliczyć ile szczawianu sodu przereagowało
z pozostałym po reakcji utlenienia manganianem, co po uwzględnieniu wprowadzonej początkowo
ilości KMnO
4
pozwala obliczyć ilość manganianu, który przereagował ze związkami organicznymi.
W obecności łatwo utleniających się związków nieorganicznych (np. soli żelaza(II), azotanów(III),
siarczków) należy wykonać dwa oznaczenia. Pierwsze oznaczenie na zimno pozwala oznaczyć ilość
tlenu zużytego przez związki nieorganiczne, drugie na gorąco, w celu określenia sumarycznej ilości
tlenu zużytego przez związki organiczne i nieorganiczne. Różnica wyników obu oznaczeń daje
utlenialność związków organicznych.
Coraz częściej, obok utlenialności, jako wskaźnik obciążenia związkami organicznymi podawana
jest zawartość tzw. węgla organicznego, oznaczana przy pomocy automatycznych analizatorów.
W analizatorze próbka wprowadzana jest do wypełnionego katalizatorem i ogrzanego do temperatury
ok. 800°C pieca rurowego gdzie cały węgiel, niezależnie od formy występowania, przechodzi w CO
2
i w tej postaci jest oznaczany przez odpowiedni detektor. Niezależnie oznaczana jest zawartość węgla
pochodzącego z obecnych w próbce węglanów. Zawartość węgla organicznego jest różnicą zawartości
węgla całkowitego i węgla nieorganicznego.
Odczynniki
−
mianowany roztwór manganianu(VII) potasu o stężeniu 0,0025 moli KMnO
4
/dm
3
(z biuretą),
−
mianowany roztwór szczawianu sodu o stężeniu 0,00625 moli Na
2
C
2
O
4
/dm
3
,
−
roztwór kwasu siarkowego(VI) 1:3.
Przyrządy i naczynia
Łaźnia wodna, 2 kolby stożkowe o poj. 300 cm
3
, pipety jednomiarowe o poj. 10 cm
3
- 1 szt.,
25 cm
3
- 1 szt., cylinder miarowy o poj. 25 cm
3
.
Sposób wykonania
Należy przeprowadzić równolegle co najmniej dwa oznaczenia.
Do kolby stożkowej o pojemności 300 cm
3
odmierzyć 25 cm
3
badanego roztworu i dodać 75 cm
3
wody dejonizowanej. Wlać (przy pomocy cylindra miarowego) 10 cm
3
roztworu kwasu
siarkowego(VI) 1:3, i z biurety 10 cm
3
roztworu KMnO
4
o stężeniu 0,0025 moli KMnO
4
/dm
3
.
Wymieszać, natychmiast wstawić do wrzącej łaźni wodnej i ogrzewać przez 30 minut. Poziom wody
w łaźni wodnej powinien być nieco niższy niż poziom w kolbie. Po wyjęciu próbki z łaźni dodać
pipetą 10 cm
3
roztworu szczawianu sodu o stężeniu 0,00625 mol Na
2
C
2
O
4
/dm
3
i po odbarwieniu

5
miareczkować na gorąco roztworem manganianu(VI) potasu o stężeniu 0,0025 moli KMnO
4
/dm
3
do
wystąpienia słabego różowego zabarwienia utrzymującego się przez kilka minut.
Utlenialność wyliczyć z wyrażenia:
(
)
3
2
p
O
C
Na
O
C
Na
KMnO
KMnO
2
KMnO
1
/dm
O
mg
1000
40
V
c
V
5
2
c
V
V
X
4
2
2
4
2
2
4
4
4
⋅
⋅
⋅
−
⋅
+
=
gdzie:
V
1 KMnO
4
- objętość roztworu KMnO
4
wprowadzona na początku do próbki, cm
3
V
2 KMnO
4
- objętość roztworu KMnO
4
zużyta do miareczkowania, cm
3
c
KMnO
4
- stężenie roztworu KMnO
4
, mol/dm
3
V
Na
2
C
2
O
4
- objętość roztworu Na
2
C
2
O
4
wprowadzona do próbki, cm
3
c
Na
2
C
2
O
4
- stężenie roztworu Na
2
C
2
O
4
, mol/dm
3
V
p
- objętość próbki, cm
3
2/5 - ilość moli KMnO
4
reagująca z jednym molem Na
2
C
2
O
4
40 - utlenialność odpowiadająca reakcji próbki z 1 milimolem KMnO
4
1000 - przelicznik z dm
3
na cm
3

6
Oznaczanie węgla organicznego w glebie
Zawartość materii organicznej w glebie można oznaczać metodą bezpośrednią jako straty masy
w wyniku prażenia w temp. 550°C po uprzednim wysuszeniu próbki do stałej masy w temp. 105°C,
bądź też pośrednio wyznaczając zawartość węgla organicznego w glebie i przeliczając ją na ilość
substancji organicznych. Biorąc pod uwagę fakt, że zawartość węgla w różnych rodzajach gleb,
a nawet w tej samej glebie ale na różnych poziomach, nie jest jednakowa i waha się w granicach
45-70%, powszechnie jako wskaźnik zawartości substancji organicznej podawana jest ilość węgla
organicznego w glebie.
Zawartość węgla organicznego w wierzchnich warstwach gleb Polski waha się w szerokich
granicach od 0,35 do 3,50 % i w dużym stopniu zależy od rodzaju gleby. Najmniej zasobne w materię
organiczną są gleby bielicowe i piaskowe, najbardziej mady, czarne ziemie i rędziny. Gleby brunatne
charakteryzuje pośrednia zawartość substancji organicznych.
Oznaczenie zawartości węgla organicznego przeprowadza się na drodze suchej, bądź też na drodze
mokrej. Pierwsza z metod polega na spaleniu próbki w temp. 800-950°C i oznaczeniu ilości
wydzielonego CO
2
. Proces ten najczęściej przeprowadzany przy pomocy automatycznego analizatora.
W metodzie analizy mokrej utlenia się zawarte w glebie substancje organiczne mieszaniną
chromową (K
2
Cr
2
O
7
+ H
2
SO
4
) i wyznacza zawartość węgla organicznego poprzez kolorymetryczny
pomiar ilości powstałego Cr(III) lub oznaczenie nie przereagowanego dichromianu(VI) potasu metodą
miareczkową.
Utlenianie węgla przy pomocy K
2
Cr
2
O
7
przebiega zgodnie z reakcją:
3C + 2K
2
Cr
2
O
7
+ 8H
2
SO
4
= 2K
2
SO
4
+ 2Cr
2
(SO
4
)
3
+ 8H
2
O + 3CO
2
.
W celu zapewnienia ilościowego przebiegu reakcji dodawany jest siarczan(VI) srebra lub rtęci(II),
który spełnia rolę katalizatora.
Odczynniki
−
roztwór 0,067 mol/dm
3
K
2
Cr
2
O
7
w stężonym H
2
SO
4
(1+1),
−
ś
lepa próba: roztwór powyższy rozcieńczony wodą 2,5 raza
−
siarczan(VI) srebra(I) - Ag
2
SO
4
lub siarczan(VI) rtęci(II) - HgSO
4
.
Przyrządy i naczynia
Spektrofotometr Spekol firmy Zeiss Jena, sito o średnicy oczek 0,75-0,80 mm, moździerz,
2 naczyńka wagowe, 2 kolby stożkowe na 200 cm
3
, 2 kolby miarowa na 50 cm
3
, pipeta jednomiarowa
na 20 cm
3
, 2 lejki szklane, palnik, siatka.
Sposób wykonania
Otrzymaną próbkę gleby przesiać przez sito o średnicy oczek 0,75-0,80 mm. Pozostałą na sicie
grubszą frakcję rozetrzeć w moździerzu i ponownie przesiać. Czynność powtarzać aż cała próbka
zostanie przesiana. Z przesianej próbki odważyć na wadze analitycznej z dokładnością do 0,0001 g
dwie odważki o masie w granicach 0,950 - 1,30 g. Do 2 kolb stożkowych o poj. 200 cm
3
wprowadzić
przygotowane odważki, dodać 20 cm
3
mieszaniny 0,067 M K
2
Cr
2
O
7
w stężonym kwasie
siarkowym(VI) (1:1) oraz parę kryształków Ag
2
SO
4
lub HgSO
4
. Do kolbek wstawić mały lejek służący
jako chłodnica powietrzna zwrotna i ogrzewać na siatce do wrzenia, utrzymując łagodne wrzenie przez
5 minut. Następnie oziębić roztwór i dodać około 20 cm
3
wody, ponownie oziębić przenieść do kolbki
miarowej o poj. 50 cm
3
, używając przy tym niewielkiej ilości wody. Dopełnić wodą do kreski,
wymieszać i pozostawić do opadnięcia osadu (cząstek gleby). W przypadku próbek zawierających
drobne cząstki proces opadania (sedymentacji) może trwać nawet 24 godziny. Klarowny roztwór z nad
osadu przenieść ostrożnie pipetką do kuwety o grubości 1 cm i zmierzyć absorbancję przy długości fali
λ
=600 nm. Jako próbę ślepą użyć znajdujący się w zestawie odczynników roztwór zawierający
K
2
Cr
2
O
7
w kwasie siarkowym(VI), o stężeniach analogicznych do otrzymanych przy sporządzaniu
próbki glebowej.

7
Opracowanie wyników
Wykonując opisany tok oznaczenia dla 3 mg węgla uzyskuje się absorbancję 0,235. Ze względu na
liniową zależność pomiędzy absorbancją A i zawartością węgla S w mg C, po odpowiednim
przekształceniu, można otrzymać zależność
S
A
= ⋅
3
0 235
,
mg C
W związku z tym, uwzględniając odważkę m [g] oznaczanej próbki glebowej, procentową
zawartość węgla organicznego w glebie wyraża się wzorem:
m
A
W
⋅
=
277
,
1
gdzie:
W – procentowa zawartość węgla organicznego w glebie,
A – absorbancja roztworu,
m – odważka oznaczanej próbki glebowej w g.

8
Oznaczanie fosforu w glebie
Fosfor, obok azotu i potasu, należy do podstawowych pierwiastków, co do których wymagania
pokarmowe roślin są szczególnie duże. Fosfor w glebie występuje w formie związków
nieorganicznych o różnej rozpuszczalności (są to głównie fosforany wapnia, żelaza, glinu i magnezu)
oraz połączeń organicznych, których ilość i jakość uzależniona jest od materii organicznej zawartej
w glebie. Zawartość fosforu ogólnego w warstwie ornej gleb szacowana jest na 0,01–0,2%, w tym
fosfor w połączeniach organicznych stanowi kilkanaście %. Pierwotnym źródłem fosforu w glebie są
ulegające wietrzeniu minerały. Dodatkowym źródłem łatwo przyswajalnych (dobrze rozpuszczalnych)
form fosforu są nawozy sztuczne i ścieki. W przyrodzie zachodzi nieustanny obieg fosforu pomiędzy
glebą i organizmami roślinnymi i zwierzęcymi, w których pierwiastek ten pełni niezastąpioną rolę
w procesach metabolicznych. Przy rekultywacji terenów zniszczonych przez przemysł konieczna jest
znajomość zasobności gleb w fosfor, azot i potas.
W celu określenia ilości fosforu dostępnego dla roślin oznacza się zawartość tzw. fosforu
przyswajalnego (rozpuszczalnych fosforanów), używając różnych roztworów ekstrahujących. Jednym
z powszechniej używanych roztworów jest bufor składający się z mleczanu amonu w kwasie octowym,
który ekstrahuje równocześnie z gleby związki fosforu i potasu. Wyekstrahowane fosforany oznacza
się metodą błękitu molibdenowego, która polega na reakcji, w roztworze kwaśnym, jonów
fosforanowych z molibdenianem amonu, w wyniku czego powstaje kwas fosforomolibdenowy
o żółtym zabarwieniu. Kwas ten ulega redukcji pod wpływem chlorku cyny(II), tworząc związek
kompleksowy - błękit molibdenowy - o intensywnym niebieskim zabarwieniu. Intensywność
zabarwienia
jest
zależna
od
zawartości
fosforanów.
Oznacza
się
ją
wizualnie
lub
spektrofotometrycznie.
Odczynniki
−
bufor mleczanu amonu w kwasie octowym:
10 g kwasu mlekowego rozpuścić na gorąco 20 cm
3
wody, a po oziębieniu dodać 17,8 cm
3
kwasu
octowego 96% (18,7 g) 77 g octanu amonowego i uzupełnić wodą destylowaną do 1000 cm
3
.
Roztwór ten jest 0,1 M wobec kwasu mlekowego i 0,4 M wobec jonów octanowych. Jego pH
powinno wynosić 3,7.
−
roztwór molibdenianu amonowego:
25 g (NH
4
)
6
Mo
7
O
27
·4H
2
O cz.d.a. rozpuścić w 175 cm
3
wody destylowanej. Do 400 cm
3
wody
destylowanej dodać ostrożnie 280 cm
3
stężonego H
2
SO
4
i ostudzić. Do roztworu kwasu siarkowego
dodać roztwór molibdenianu i rozcieńczyć wodą do 1 dm
3
.
−
chlorek cyny(II):
rozpuścić 2,5 g SnCl
2
·2H
2
O w 100 cm
3
gliceryny. Ogrzewać na łaźni wodnej, mieszając szklaną
bagietką, by przyspieszyć rozpuszczanie. Odczynnik jest trwały. Chlorek cyny(II), występujący
w postaci proszku, nie nadaje się do przygotowania odczynnika.
−
podstawowy roztwór wzorcowy fosforu - 1 mg P
2
O
5
/cm
3
:
rozpuścić w wodzie 1,9169 g fosforanu jednopotasowego KH
2
PO
4
wysuszonego w temp. 110°C,
dodać 1 cm
3
chloroformu (zapobiega tworzeniu się pleśni) i rozcieńczyć do 1 dm
3
.
Przyrządy i naczynia
Spektrofotometr Spekol firmy Zeiss Jena, 2 mieszadła magnetyczne, sito o średnicy oczek
0,75 - 0,80 mm, moździerz, 2 naczyńka wagowe, 4 zlewki o poj. 250 cm
3
, 1 kolba miarowa o poj.
100 cm
3
, 7 kolb miarowych o poj. 50 cm
3
, pipeta jednomiarowa o poj. 100 cm
3
, pipeta wielomiarowa
o poj.: 5 cm
3
; 2 lejki ilościowe, sączki średnie.
Sposób wykonania
Ekstrakcja fosforu z gleby
Otrzymaną próbkę gleby przesiać przez sito o średnicy oczek 0,75 - 0,8 mm. Pozostałą na sicie
grubszą frakcję rozetrzeć w moździerzu i ponownie przesiać. Czynność powtarzać aż cała próbka
zostanie przesiana. Z przesianej próbki odważyć na wadze analitycznej z dokładnością do 0,0001 g

9
dwie odważki o masie w granicach 0,950 - 1,30 g. Do 2 zlewek o poj. 250 cm
3
wprowadzić
przygotowane odważki, dodać po 100 cm
3
buforu mleczanu amonu w kwasie octowym i mieszać na
mieszadłach magnetycznych przez 30 minut (czas ten wykorzystać do przygotowania krzywej
wzorcowej). Każdą z próbek przesączyć przez sączek średni, odrzucając pierwsze 10–20 cm
3
przesączu.
Sporządzenie krzywej wzorcowej
Przygotować roboczy roztwór wzorcowy fosforu. W tym celu do kolbki o poj. 100 cm
3
odpipetować
5 cm
3
podstawowego roztworu wzorcowego fosforu i dopełnić kolbkę wodą destylowaną do kreski.
Wymieszać. Tak przygotowany wzorzec zawiera 0,05 mg P
2
O
5
/cm
3
.
Do 5 kolbek miarowych o poj. 50 cm
3
odpipetować kolejno 0; 0,5; 1; 1,5; 2,0 cm
3
roboczego
roztworu wzorcowego fosforu. Do każdej z kolbek dodać po 40 cm
3
wody destylowanej, a następnie
1,5 cm
3
roztworu molibdenianu, wymieszać i dodać 0,5 cm
3
roztworu cyny(II). Uzupełnić wodą do
kreski, a po wymieszaniu pozostawić na 10 minut. Po tym czasie zmierzyć absorbancję roztworów
przy długości fali
λ
=650 nm. Próbkę, która nie zawiera fosforu (próbkę ślepą) potraktować jako
roztwór odniesienia przy pomiarach kolorymetrycznych. Wykreślić zależność absorbancji od
zawartości fosforu w roztworach wzorcowych.
Oznaczanie fosforu
Z każdego przesączu wykonać dwa równoległe oznaczenia wg poniższego przepisu:
Do kolbki miarowej o poj. 50 cm
3
wprowadzić 5 cm
3
przesączu, dodać 40 cm
3
wody destylowanej,
a następnie 1,5 cm
3
roztworu molibdenianu, wymieszać i dodać 0,5 cm
3
roztworu cyny(II). Uzupełnić
wodą do kreski, a po wymieszaniu pozostawić na 10 minut. Po tym czasie zmierzyć absorbancję
powstałego roztworu o niebieskim zabarwieniu przy długości fali
λ
=650 nm. Zawartość fosforu
odczytać z krzywej wzorcowej. Wynik podać w % wag. uwzględniając rozcieńczenie próbki i masę
odważki.

10
Oznaczanie potasu w glebie
Potas, obok fosforu i azotu, należy do podstawowych pierwiastków, niezbędnych do prawidłowego
rozwoju roślin. Przeważająca ilość potasu glebowego jest pochodzenia pierwotnego, tj. pochodzi ze
skały macierzystej danej gleby. Nieporównanie mniejsza część dostaje się do gleby przez nawożenie
i z wód głębinowych oraz (w nieznacznych ilościach) z atmosfery. Potas zawarty w pierwotnych
minerałach glebowych jest, praktycznie biorąc, niedostępny dla roślin. Pod wpływem procesów
wietrzeniowych i glebotwórczych (działanie wody przy współudziale kwasów nieorganicznych
i organicznych oraz temperatury, procesy życiowe mikro- i makroorganizmów) minerały zawierające
potas ulegają przekształceniom, w toku których pierwiastek ten zostaje uwolniony.
Zawartość potasu ogólnego w glebach polskich waha się w szerokich granicach, od 0,013% gleby
torfowej do 2,06% w przypadku ciężkiej mady. Zawartość potasu ogólnego nie jest jednak miernikiem
ilości potasu dostępnego dla roślin, ponieważ większość tego składnika występuje w formie związanej
w minerałach glebowych. Potas przyswajalny określa się w badaniach agrochemicznych, dobierając
odpowiednie roztwory ekstrakcyjne, za pomocą których można wyodrębnić z gleby potas dostępny dla
roślin.
Odczynniki
−
bufor mleczanu amonu w kwasie octowym:
10 g kwasu mlekowego rozpuścić na gorąco 20 cm
3
wody, a po oziębieniu dodać 17,8 cm
3
kwasu
octowego 96% (18,7 g) 77 g octanu amonowego i uzupełnić wodą destylowaną do 1000 cm
3
.
Roztwór ten jest 0,1 M wobec kwasu mlekowego i 0,4 M wobec jonów octanowych. Jego pH
powinno wynosić 3,7.
−
roztwór wzorcowy potasu - 1 mg K/cm
3
:
do kolby miarowej o pojemności 1000 cm
3
wprowadzić 1,907 g KCl cz.d.a. wysuszonego w 105°C,
rozpuścić i uzupełnić wodą do kreski.
Przyrządy i naczynia
Fotometr płomieniowy Flapo, 2 mieszadła magnetyczne, sito o średnicy oczek 0,75 - 0,80 mm,
moździerz, 2 naczyńka wagowe, statyw do sączenia, 4 zlewki o poj. 250 cm
3
, 5 kolb miarowych
o poj. 100 cm
3
, pipeta jednomiarowa o poj. 100 cm
3
, pipeta wielomiarowa o poj.: 5 cm
3
; 2 lejki
ilościowe, sączki średnie.
Sposób wykonania
Ekstrakcja potasu z gleby
Otrzymaną próbkę gleby przesiać przez sito o średnicy oczek 0,75 - 0,80 mm. Pozostałą na sicie
grubszą frakcję rozetrzeć w moździerzu i ponownie przesiać. Czynność powtarzać aż cała próbka
zostanie przesiana. Z przesianej próbki odważyć na wadze analitycznej z dokładnością do 0,0001 g
dwie odważki o masie w granicach 0,950 - 1,30 g. Do 2 zlewek o poj. 250 cm
3
wprowadzić
przygotowane odważki, dodać po 100 cm
3
buforu mleczanu amonu w kwasie octowym i mieszać na
mieszadłach magnetycznych przez 30 minut (czas ten wykorzystać do przygotowania krzywej
wzorcowej). Każdą z próbek przesączyć przez sączek średni, odrzucając pierwsze 10–20 cm
3
przesączu.
Przygotowanie roztworów do krzywej wzorcowej
Do kolb miarowych o pojemności 100 cm
3
wprowadzić kolejno za pomocą pipety 1,0; 2,0; 3,0; 4,0;
5,0 cm
3
roztworu wzorcowego o zawartości 1 mg K w 1 cm
3
, uzupełnić wodą do kreski i dokładnie
wymieszać. Tak przygotowane roztwory zawierają 10; 20; 30; 40; 50 mg K w 1 dm
3
.
Wykonanie oznaczenia
Uruchomić fotometr płomieniowy zgodnie z instrukcją obsługi przyrządu, w obecności prowadzącego
ćwiczenia,
−
ustawić filtr potasowy,
−
ustawić przepływ powietrza zgodnie ze specyfikacją przyrządu,

11
−
ustawić przepływ gazu palnego zgodnie ze specyfikacją przyrządu,
−
wyzerować przyrząd używając wody dejonizowanej nie zawierającej K (ślepej próby),
−
ustawić wartość 100 na skali podczas wzbudzania wzorca o największym stężeniu,
−
zmierzyć kolejno emisję pozostałych roztworów wzorcowych,
−
zmierzyć emisję badanych próbek przesączu glebowego. Dla każdej próbki wykonać trzykrotny
odczyt. W przypadku, gdy wielkość emisji mierzonej próbki nie mieści się w zakresie krzywej
wzorcowej próbkę należy rozcieńczyć.
Obliczanie wyników
Wykreślić na papierze milimetrowym albo obliczyć metodą regresji, korzystając z komputera lub
kalkulatora, krzywą wzorcową przedstawiającą zależność wielkości emisji wzorców od stężenia potasu
we wzorcach. Odczytać z krzywej wzorcowej stężenie potasu odpowiadające średniej arytmetycznej
z 3 pomiarów emisji dla każdej próbki. Wynik podać w procentach wagowych uwzględniając objętość
buforu ekstrahującego i masę odważki.

12
Oznaczanie sodu w wodzie
Sód jest pierwiastkiem powszechnie występującym w wodzie. Przeciętne stężenie sodu
w naturalnych wodach powierzchniowych wynosi od kilku do 20 -30 mg Na/dm
3
. Wody podziemne
mogą zawierać znacznie większe ilości sodu. W wodach kopalnianych stężenie sodu może dochodzić
nawet do 100 g Na/dm
3
.
Zrzut tak silnie zasolonych wód bezpośrednio do rzeki może powodować
znaczące zachwianie równowagi biologicznej.
Do oznaczania sodu w wodzie stosuje się powszechnie metody: spektrometrii absorpcji atomowej
(AAS), spektrometrii emisyjnej ze wzbudzeniem plazmowym (spektrometria ICP) i fotometrii
płomieniowej. W poniższym ćwiczeniu została zastosowana ostatnia z wymienionych technik
z wykorzystaniem najczulszej linii Na 589,0 nm.
Na wyniki oznaczania sodu metodą fotometrii płomieniowej mają wpływ inne jony występujące
w wodzie, jeżeli ich stężenie przekracza następujące wartości: K - 10 mg/dm
3
, Ca -100 mg/dm
3
,
Mg - 100 mg/dm
3
. Analizowana w ćwiczeniu woda nie zawiera jonów przeszkadzających
w oznaczeniu.
Odczynniki
−
roztwór wzorcowy sodu - 1 mg Na/cm
3
:
do kolby miarowej o pojemności 1000 cm
3
wprowadzić 2,542 g NaCl cz.d.a. wysuszonego
w 130°C, rozpuścić i uzupełnić wodą do kreski.
Przyrządy i naczynia
Fotometr płomieniowy Flapo, 5 kolb miarowych o poj. 100 cm
3
, pipeta wielomiarowa o poj.: 5 cm
3
.
Sposób wykonania
Przygotowanie próbki do analizy
Jeżeli próbka jest klarowna i nie zawiera substancji organicznych można przeprowadzić oznaczenie
bez operacji wstępnych. W przeciwnym przypadku 100 cm
3
próbki ostrożnie odparować do sucha
w parownicy kwarcowej unikając przegrzania. W celu usunięcia substancji organicznych pozostałość
wyprażyć w piecu, w temperaturze 550°C przez 2 godziny. Po ostudzeniu osad rozpuścić w 2 cm
3
roztworu HCl (1:1) i dodać 30 cm
3
wody redestylowanej. Roztwór przesączyć do kolby miarowej
o pojemności 100 cm
3
, osad na sączku przemyć około 30 cm
3
wody i roztwór w kolbie uzupełnić wodą
redestylowaną do kreski.
Przygotowanie roztworów do krzywej wzorcowej
Do kolb miarowych o pojemności 100 cm
3
wprowadzić kolejno za pomocą pipety 1,0; 2,0; 3,0; 4,0;
5,0 cm
3
roztworu wzorcowego o zawartości 1 mg Na w 1 cm
3
, uzupełnić wodą do kreski i dokładnie
wymieszać. Tak przygotowane roztwory zawierają 10; 20; 30; 40; 50 mg Na w 1 dm
3
.
Wykonanie oznaczenia
Uruchomić fotometr płomieniowy zgodnie z instrukcją obsługi przyrządu, w obecności prowadzącego
ćwiczenia.
−
ustawić filtr sodowy,
−
ustawić przepływ powietrza zgodnie ze specyfikacją przyrządu,
−
ustawić przepływ gazu palnego zgodnie ze specyfikacją przyrządu,
−
wyzerować przyrząd używając wody dejonizowanej nie zawierającej Na (ślepej próby),
−
ustawić wartość 100 na skali podczas wzbudzania wzorca o największym stężeniu,
−
zmierzyć kolejno emisję pozostałych roztworów wzorcowych,
−
zmierzyć emisję badanej próbki wody. Dla każdej próbki wykonać trzykrotny odczyt. W przypadku,
gdy wielkość emisji mierzonej próbki nie mieści się w zakresie krzywej wzorcowej próbkę należy
rozcieńczyć.

13
Obliczanie wyników
Wykreślić na papierze milimetrowym albo obliczyć metodą regresji, korzystając z komputera lub
kalkulatora, krzywą wzorcową przedstawiającą zależność wielkości emisji wzorców od stężenia sodu
we wzorcach. Odczytać z krzywej wzorcowej stężenie sodu odpowiadające średniej arytmetycznej
z 3 pomiarów emisji dla każdej próbki. Wynik podać w mg Na/dm
3
. Uwzględnić przy obliczeniu
ewentualne rozcieńczenie analizowanej próbki.
14
Pomiar zasolenia wody metodą konduktometryczną
Czysta woda jest bardzo złym przewodnikiem elektryczności, ze względu na mały stopień
dysocjacji na jony H
+
i OH
–
. Jej przewodność właściwa w temperaturze 25
°
C wynosi 5
⋅
10
–8
µ
S
⋅
cm
–1
.
Wzrost przewodności świadczy o wzroście zanieczyszczenia wody związkami mineralnymi, gdyż
związki organiczne występujące w wodzie dysocjują przeważnie nieznacznie lub nie dysocjują wcale.
Przewodność właściwa wód naturalnych waha się przeciętnie od 50 do 1000
µ
S·cm
–1
,
a przewodność ścieków może przekroczyć 10000
µ
S cm
–1
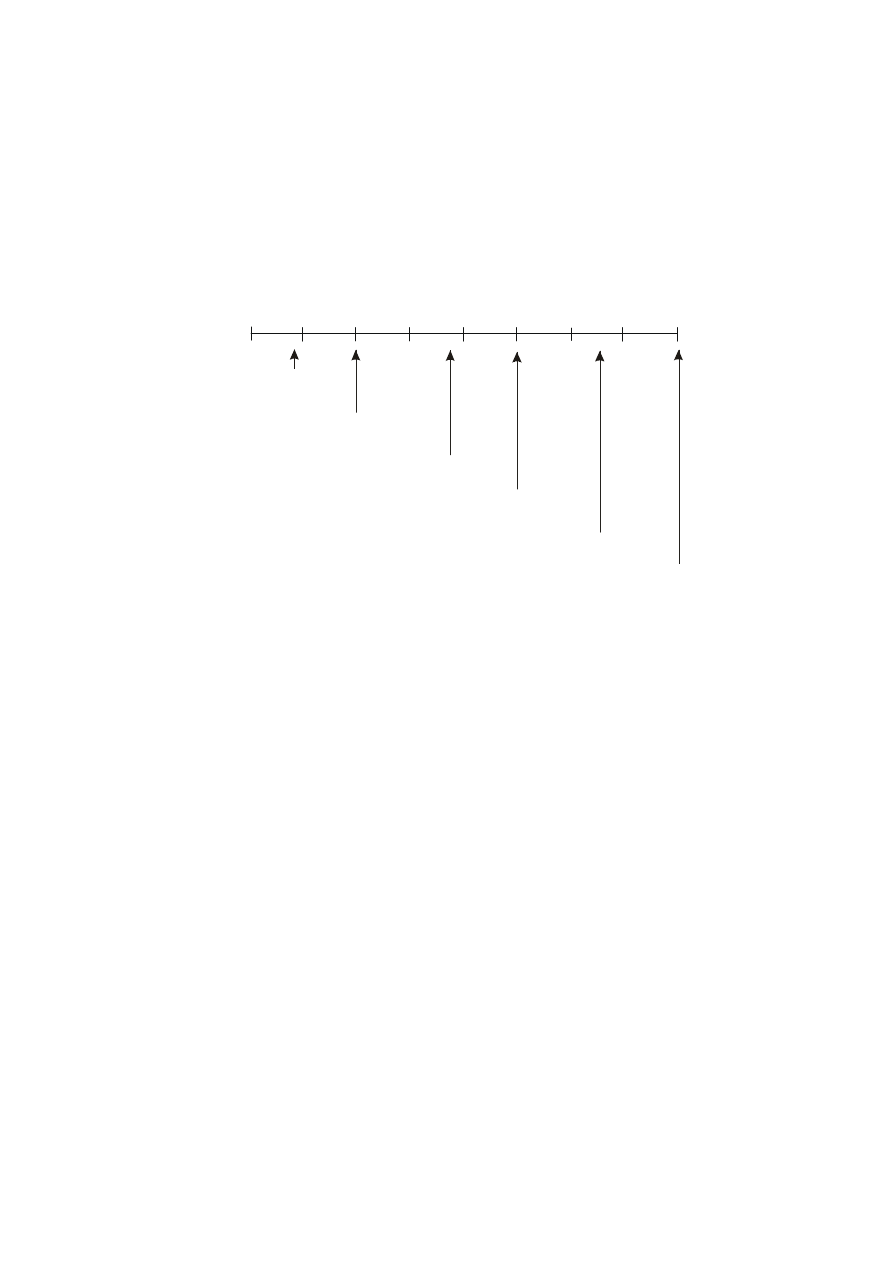
. Na poniższym rysunku przedstawiono
zakres przewodności właściwej niektórych roztworów wodnych.
10
-2
10
-1
1
10
1
10
2
10
3
10
4
10
5
10
6
Woda
superczysta
Woda
destylowana
Woda surowa
dobrej jako
ś
ci
0,05% NaCl
Woda morska
30% H SO
2
4
Przewodność właściwa niektórych roztworów wodnych (wg Beckman Instruments)
Na podstawie pomiaru przewodności można określić zawartość substancji mineralnych
rozpuszczonych w wodzie. Pomiar przewodności umożliwiają przyrządy zwane konduktometrami.
Próbki wody do pomiaru przewodności powinny być przechowywane w naczyniach szczelnie
zamkniętych, aby zapobiec wydzielaniu się lub dodatkowemu rozpuszczeniu CO
2
. Pomiar
przewodności powinno się wykonać w ciągu 24 godzin od chwili pobrania próbki. W przypadku wód
zawierających duże ilości zawiesin, próbki należy przesączyć lub pozostawić do opadnięcia zawiesin.
Jeżeli w próbkach występują oleje lub tłuszcze należy je przed pomiarem usunąć gdyż mogą
zanieczyścić elektrody i spowodować błędy pomiaru.
Prowadząc pomiary przewodności wód o bardzo dużym stężeniu elektrolitów (np. stężonych
solanek) należy pamiętać, że ze wzrostem stężenia maleje aktywność jonów i przewodnictwo przestaje
być liniową funkcją stężenia.
Istotny wpływ na wyniki pomiaru ma temperatura. Pomiary należy wykonywać w stałej
temperaturze np. 25
°
C lub stosować automatyczną kompensacją temperatury.
Pomiary przewodności właściwej (
κ
) mogą służyć do sprawdzenia wyników analizy w zakresie
jonowego składu wód. Dla większości wód naturalnych przewodność właściwa jest (w µS·cm
–1
w 25°C) pomnożona przez odpowiedni współczynnik daje zawartość substancji rozpuszczonych
w wodzie. Zależność ta przedstawiona jest równaniem:
κ
= b·c
gdzie:
κ
- przewodność właściwa, µS·cm
–1
,
b - współczynnik przeliczeniowy,
c - stężenie jonów, mmol/dm
3
.

15
Przewodność właściwa roztworu równa jest sumie przewodności właściwych jonów obecnych
w roztworze.
Metoda ta może być stosowana w przypadku próbek o pH w zakresie 6-9 i przewodności większej
niż 90 µS·cm
–1
.
W przypadku obecności w próbce głównie jednego rodzaju soli zależność ta może również posłużyć
do obliczenia stężenia tej soli.
Przykładowo dla Na
2
SO
4
wzór przyjmie postać:
c
2 b
b
mmol / dm
Na
SO
3
4
2
=
⋅
+
+
−
κ
Współczynniki do przeliczenia stężeń jonów występujących w wodzie na przewodność
właściwą (25°C)
Jon
b
Cl
–
75,9
NO
3
–
71,0
HCO
3
–
43,6
SO
4
2–
147,8
CO
3
2–
169,2
Mg
2+
93,2
Ca
2+
104,0
Na
+
48,9
K
+
72,0
Odczynniki
−
roztwór wzorcowy chlorku potasu o stężeniu: 0,0100 mol/dm
3
−
roztwory wzorcowe solanek o stężeniach 80 g NaCl/dm
3
i 100 g NaCl/dm
3
Przyrządy i naczynia
Konduktometr, elektroda zanurzeniowa, naczyńko pomiarowe, 5 kolbek miarowych o poj. 50 cm
3
.
Sposób wykonania
Kalibracja konuktometru za pomocą roztworu wzorcowego
Przeprowadzić kalibrację konduktometru zgodnie z instrukcją obsługi.
Pomiar przewodnictwa właściwego próbki wody
Po wypłukaniu naczyńka pomiarowego napełnić go badaną próbką wody i zanurzyć elektrodę. Po
ustaleniu się wskazań przyrządu odczytać wartość przewodnictwa właściwego. Pomiar przeprowadzić
na zakresie 1,999 mS. (1999 µS)
Przy założeniu, że w próbce znajduje się głównie jeden rodzaj soli (podany przez prowadzącego
ć
wiczenia) otrzymaną wartość przewodności właściwej przeliczyć na zawartość soli (mg/dm
3
).
Określenie zawartości NaCl w solankach
Krzywa wzorcowa
Przy dużym zasoleniu wody wartości przewodności właściwej nie są liniową funkcją stężenia i dla
szerokiego zakresu pomiarowego wymagane jest wykonanie odpowiedniej krzywej wzorcowej. Ze
względu na niemożliwość kalibracji konduktometru w całym zakresie pomiarowym wartości
wskazywane przez przyrząd nie odpowiadają dokładnie przewodności właściwej, a służą tylko do
wykonania krzywej wzorcowej. Przy pomiarach przełączyć zakres
Do pięciu kolbek o pojemności 50 cm
3
odmierzyć pipetą kolejno: 10, 20, 30, 40, 50 cm
3
wzorcowego roztworu NaCl, uzupełnić wodą dejonizowaną do objętości 50 cm
3
i wymieszać.
Tak przygotowanymi roztworami napełnić kolejno naczyńko pomiarowe zaczynając od roztworu
o największym stężeniu. Zanurzyć elektrodę pomiarową, ustalić właściwy zakres pomiarowy

16
(w przypadku migania cyfr na wyświetlaczu przełączyć zakres na wyższy) i notować wskazania
konduktometru. Pomiary realizować w stałej temperaturze. Aby wykreślić krzywą wzorcową należy
uzyskane wyniki pomiarowe nanieść na papier milimetrowy, odkładając na osi rzędnych wskazania
przyrządu, a na osi odciętych stężenia NaCl w odpowiednich roztworach wzorcowych solanki.
Pomiar właściwy
Naczyńko pomiarowe napełnić roztworem badanym w ilości niezbędnej do zanurzenia elektrody
pomiarowej. Pomiar realizować w tej samej temperaturze, w której przygotowano krzywą wzorcową.
Na podstawie wskazania konduktometru odczytać z krzywej wzorcowej stężenie soli w solance.

17
Oznaczanie zasadowości wód naturalnych metodą potencjometryczną
Zasadowość nadają wodom naturalnym wodorowęglany, węglany oraz rzadziej wodorotlenki,
borany, krzemiany oraz fosforany. W wodach naturalnych (o pH<8,3) występują przeważnie
wodorowęglany wapnia, magnezu i żelaza i niekiedy wodorowęglany sodu (zasadowość alkaliczna).
W wodach zanieczyszczonych ściekami przemysłowymi o odczynie zasadowym (pH > 8,3) oprócz
anionów słabych kwasów, jak np. HCO
3
–
, CO
3
2–
, H
2
PO
4
–
, HPO
4
2–
, SO
3
2–
, ulegających hydrolizie,
mogą występować również mocne zasady (NaOH, KOH). W związku z tym w zależności od wartości
pH wody rozróżnia się tzw. zasadowość ogólną (Z
og
) dla wód o pH > 4,5 i zasadowość mineralną (Z
m
)
- dla wód o pH > 8,3. Zasadowość ogólna (Z
og
) oznacza sumę wszystkich związków reagujących
zasadowo wobec oranżu metylowego jako wskaźnika. Zasadowość mineralna (Z
m
) oznacza sumę
wszystkich związków reagujących zasadowo wobec fenoloftaleiny. Różnica pomiędzy wartością
zasadowości a wielkością twardości ogólnej określana jest mianem zasadowości alkalicznej.
Zasadowość wody z punktu widzenia sanitarnego ma znaczenie drugorzędne, ma natomiast duże
znaczenie w ocenie wody do celów gospodarczych i technicznych (przemysłowych). Niepożądana jest
zasadowość ogólna w wodzie do zasilania kotłów energetycznych, a zasadowość alkaliczna powoduje
pienienie wody w kotle.
Oznaczanie zasadowości wody polega na określeniu zawartości w niej związków chemicznych,
które reagują zasadowo w obecności odpowiedniego wskaźnika. Zawartość tych związków określa się
za pomocą miareczkowania roztworem mocnego kwasu, początkowo do pH ok. 8,3, a następnie do pH
ok.4,5. Reakcje chemiczne podczas oznaczania zasadowości przebiegają następująco:
−
miareczkowanie do pH ok. 8,3 OH
–
+ H
+
= H
2
O
−
miareczkowanie do pH ok. 4,5 CO
3
2–
+ H
+
= HCO
3
–
HCO
3
–
+ H
+
= H
2
O + CO
2
W przypadku kiedy próbka jest barwna lub mętna nie jest możliwe wykonanie oznaczenia
z wykorzystaniem wskaźników alkacymetrycznych. Stosuje się wtedy miareczkowanie pH-metryczne
do osiągnięcia odpowiedniej wartości pH.
Odczynniki
−
roztwory buforowe o pH 4,0 i 7,0 do kalibracji pH-metru.
−
roztwór HCl (w biurecie) o stężeniu 0,05 mol/dm
3
.
Przyrządy i naczynia
Pehametr, elektroda kombinowana (szklana + chlorosrebrowa), naczyńko pomiarowe, zlewka o poj.
250 cm
3
, pipeta jednomiarowa o poj. 100 cm
3
.
Sposób wykonania
- Kalibracja pH-metru za pomocą roztworów buforowych
Przeprowadzić kalibrację pH-metru zgodnie z instrukcją obsługi.
Pomiar pH wody
Po wypłukaniu naczyńka pomiarowego napełnić go badaną próbką wody i zanurzyć elektrodę. Po
ustaleniu się wskazań przyrządu odczytać wartość pH.
Oznaczanie zasadowości wody
Oznaczanie zasadowości mineralnej (pH 8,3)
Do zlewki o pojemności 250 cm
3
odmierzyć pipetą 100 cm
3
badanej wody. Zanurzyć elektrodę
i mieszając dodawać kroplami z biurety HCl obserwując zmianę pH. W miarę zbliżania się do punktu
końcowego (pH 8,3) zmniejszyć szybkość dodawania kwasu. Zakończyć miareczkowanie
uzyskawszy pH=8,3±0,3. Zapisać objętość zużytego kwasu. Próbkę po zmiareczkowaniu pozostawić
do oznaczenia zasadowości ogólnej.
Oznaczanie zasadowości ogólnej (pH 4,5)
Próbkę, w której oznaczono zasadowość mineralną miareczkować dalej dodając kroplami z biurety
kolejne porcje HCl. W miarę zbliżania się do punktu końcowego (pH 4,5) zmniejszyć szybkość

18
dodawania kwasu. Zakończyć miareczkowanie uzyskawszy pH=4,5±0,3. Zapisać objętość zużytego
kwasu.
Obliczanie wyników
Kwasowość i zasadowość badanej wody obliczamy ze wzorów:
zasadowość mineralna:
Z
m
= (V
1
·C
M(HCl)
/V
p
)·1000 [mmol/dm
3
]
zasadowość ogólna:
Z
og
= (V
2
·C
M(HCl)
/V
p
)·1000 [mmol/dm
3
]
gdzie:
V
1
- ilość roztworu HCl zużyta do zmiareczkowania próbki wody badanej do pH 8,3; cm
3
V
2
- ilość roztworu HCl zużyta do zmiareczkowania próbki wody badanej do pH 4,5; cm
3
(w przypadku wykonania oznaczenia w próbce uprzednio wykorzystanej do określenia
zasadowości wobec fenoloftaleiny, ilość ta będzie równa sumie zużytego kwasu na oba
miareczkowania. tj. do pH = 8,3 i pH = 4,5),
V
p
- objętość próbki wody użytej do oznaczenia, cm
3
,
C
M(HCl)
- stężenie roztworu HCl, mol/dm
3
.
Uwaga: Często kwasowość i zasadowość wody (nawet w normach) podaje się w tzw. mval/dm
3
,
czyli miligramorównoważnikach chemicznych. Jednostek tych nie powinno się obecnie stosować,
ponieważ w układzie SI jednostką liczności materii jest mol lub jego podwielokrotności (najczęściej
mmol). W przypadku oznaczania kwasowości i zasadowości poprzez miareczkowanie roztworami
NaOH i HCl 1 mval = 1 mmol.
Wyszukiwarka
Podobne podstrony:
Instrukcje IŚ EK, 2 sem ochr srod
Grafik IŚ EK, 2 sem ochr srod
Sprawozdania z analizy instrumentalnej, ION (2), cwiczenie na ochr. srod.
9 ochr środ
ochr srod wyklad 3 biologia
Zagęszczanie osadów, PW IŚ, Inżynier, sem V, TOŚ
instrukcje IŚ - II rok, SPIS TREŚCI
prawo ochrony środowiska, Kary ochr srod
instrukcje IŚ - II rok, SPIS TREŚCI
prawo ochrony środowiska, Kary ochr srod
instrukcje budownictwo III sem
EGZAMIN-OCHR SROD, Ochrona Środowiska
Analiza fiz-chem sciekow, PW IŚ, Inżynier, sem V, TOŚ
scieki2, PW IŚ, Inżynier, sem V, TOŚ
więcej podobnych podstron