
Dr n. med. Emilian Snarski
Klinika Hematologii, Onkologii i Chorób
Wewnętrznych
Warszawa 27.09.2010
wersja 1.0
Hematologia - Przypadki kliniczne
Przypadek kliniczny 1
50 letni mężczyzna z przewlekłym
zmęczeniem i uczuciem wczesnej sytości
w wywiadach
Morfologia krwi
WBC 75 x 109/L (4-10 x 109/L)
Hgb 14 g% (14-16.5 g%)
Hct 42% (42%-50%)
MCV 88 fl (80-96 fl)
PLT 550 x 10
9
/L (150-400 x 10
9
/L)
Segmented neutrophils 33 x 109/L (1.8-7.0 x 109/L)
Pałki 15 x 109/L (0-0.7 x 109/L)
Metamielocyty 11 x 109/L (0)
Mielocyty 7.5 x 109/L (0)
Bazofile 3.75 x 109/L (0-0.2 x 109/L)
Limfocyty 3 x 109/L (1.0-4.8 x 109/L)
Monocyty 0.75 x 109/L (0-0.8 x 109/L
)
Jakie nieprawidłowości możemy zauważyć?
Leukocytoza z
przesunięciem w
lewo
Trombocytoza
Bazofilia
Historia – O co zapytamy?
Chudnięcie, nocne poty, gorączki
Alkohol
Kawa
Infekcje
Wywiad rodzinny
Swędzenie
Bolesne węzły chłonne
Bóle palców
Podróże
Bóle stawów
Krwawienia
Zakrzepica
Leki
Praca
Podsumowanie: Brak przewlekłych stanów
zapalnych mogących prowadzić do reaktywnej
granulocytozy lub trombocytozy
Badanie przedmiotowe: śledziona wyczuwalna –
4 cm
Jakie znaczenie ma powiększenie
śledziony u tego chorego?
Nie ma znaczenia, bo śledziona może być wyczuwalna?
Powiększenie śledziony oznacza małe szanse infekcyjnej
przyczyny nieprawidłowości?
Choroby mieloproliferacyjne często wiążą się z
powiększeniem śledziony?
Prawidłowa odpowiedź…
Mechanizmy powiększenia śledziony
Endothelial or immune system (lymphocyte and macrophage) hyperplasia from
infections, immune disorders or chronic hemolysis.
Infections associated with splenomegaly:
Bacterial endocarditis
Malaria
Schistosomiasis
Tuberculosis
Immune disorders associated with splenomegaly:
Systemic lupus erythematosis
Rheumatoid arthritis
Chronic hemolysis associated with splenomegaly:
Hereditary: thalassemias, spherocytosis, hemoglobin SC disease (NB: young
children with sickle cell disease will have splenomegaly, but recurrent infarctions
ultimately result in necrosis, fibrosis and functional asplenia)
Acquired: autoimmune hemolytic anemia
Altered splenic blood flow: cirrhosis; splenic, hepatic or portal vein thrombosis
Primary or metastatic malignancies: lymphoma, Hodgkin's disease, chronic
lymphocytic leukemia
Extramedullary hematopoiesis (myeloproliferative disorders)
Infiltration: amyloid, Gaucher's disease
Jakie badania wykonamy u chorego?
Kreatynina
Wapń
RTG kl piersiowa
Enzymy wątrobowe
Kwas moczowy
Krew utajona w kale
Badania gospodarki żelazem
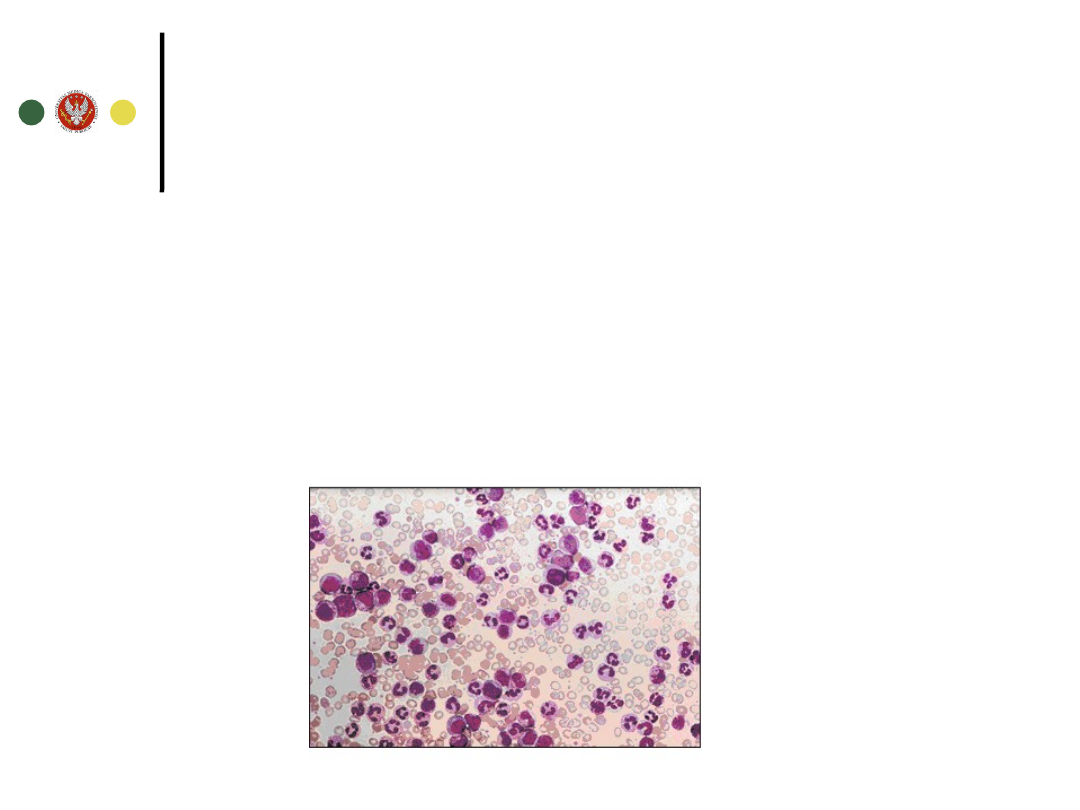
Rozmaz krwi
prawidłowy
prawidłowe
9 mg/dL
Wynik ujemny
Wynik prawidłowy
Zwiększona liczba
granulocytów
Przesunięcie w
lewo
Zwiększona liczba
płytek krwi
Jakie rozpoznanie jest najbardziej
prawdopodobne?
Ostra białaczka szpikowa
Czerwienica prawdziwa
Nadpłytkowość samoistna
Przewlekła białaczka szpikowa
Nadpłytkowość i granulocytowa w
przebiegu infekcji
Nadpłytkowość i granulocytowa w
przebiegu nowotworu
9
Jak odróżnić PBSz od odczynowej
hiperleukocytozy?
Splenomegalia
Bazofilia
Fosfataza alkaliczna granulocytów (30-90)
0 - 30 - PBSz
30 – 90 – i więcej – Odczyn białaczkowy
Jakie badania wykonamy u chorego?
Mielogram
Cytogenetyka
FISH (fluorescence in situ hybridization) genu
BCR-ABL
QPCR genu bcr-abl
Badanie cytofotometryczne krwi obowdowej
Badanie mutacji JAK-2
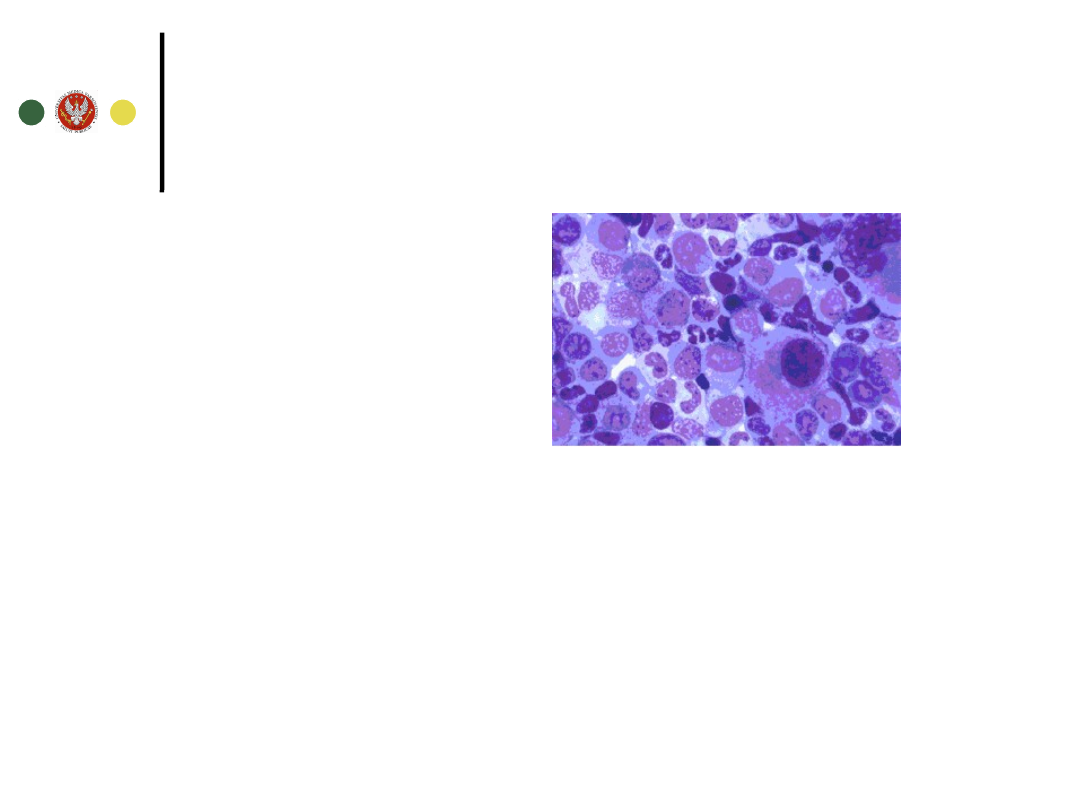
Wyniki badań szpiku
Mielogram
Szpik bogatokomórkowy
75-95% - zmniejszona ilość tkanki tłuszczowej
Dominuje granulopoeza (granulocytów do
erytrocytów 10:1 – 30:1 przy normie 2:1 – 4:1)

Wyniki badań szpiku
Cytogenetyka
13
Wyniki badań szpiku
FISH (fluorescence
in situ
hybridization) genu
BCR-ABL
QPCR – obecność
genu BCR - ABL
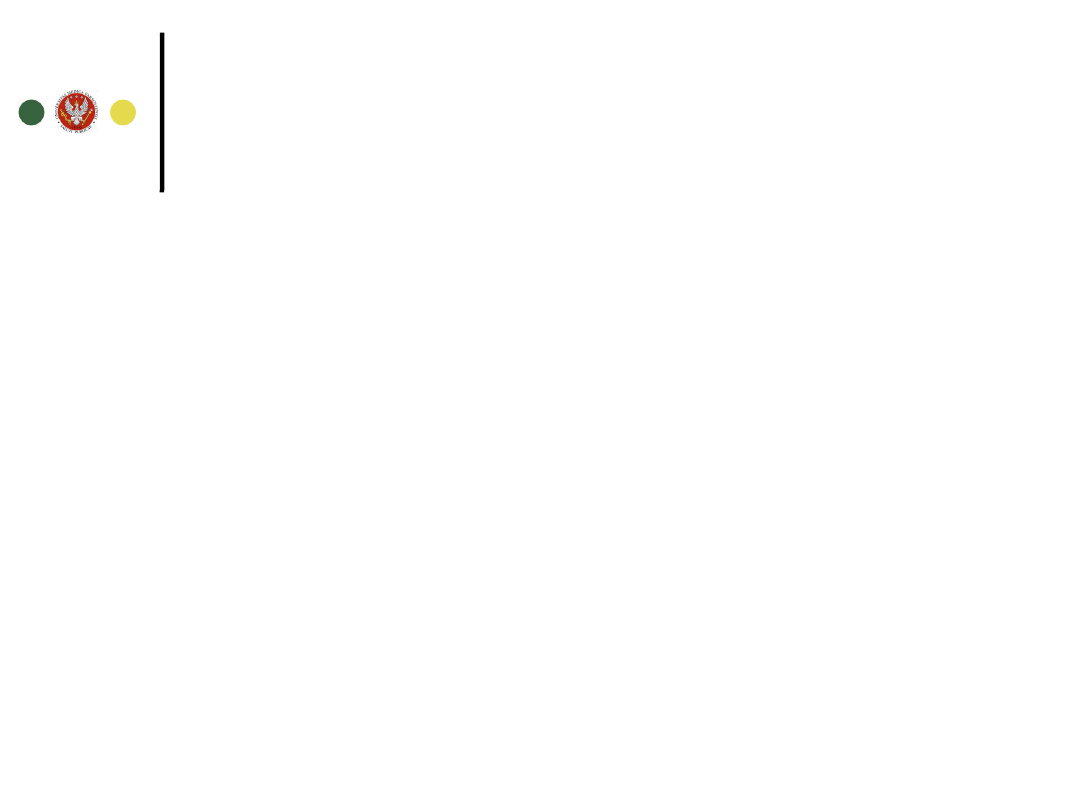
Jaką chorobę możemy rozpoznać u
chorego?
Ostra białaczka szpikowa
Przewlekła białaczka szpikowa
Nadpłytkowość samoistna
Czerwienica Prawdziwa
Odczynowa granulocytoza/trombocytoza
RBC WBC
PLTS
+++ Granulocytosis
Inc
+/-
+
95,00%-
+
Nrml Granulocytosis
Inc
+/-
+
50,00%-
+
PBSz
Nrml Granulocytosis
Inc
+/-
+
-
+
+/-
Mielofibroza
#ARG!NeutropeniaDec +++
++
50,00%-
+/-
Nrml Granulocytosis
Nrml/ Inc
-
-
-
-
-
Nrml Nrml/
Granulocytosis Inc
-
-
-
-
?
włóknienie
szpiku
Speno
megalia
J AK2
mutacja
BCR-
ABL
Ryzyko
zakrzepicy/
trombozy
Czerwienica
prawdziwa
Nadpłytkowość
samoistna
Odczynowa
granulocytoza
Odczynowa
nadpłytkowość
PBSz - patogeneza
Mutacja zachodząca w KKM, w wyniku której powstaje
białaczkowa komórka macierzysta, polega na zamianie części
ramion długich chromosomów 9 i 22 (jest to tzw.
zrównoważona translokacja)
Tak zmieniony chromosom 22 nosi nazwę chromosomu
Filadelfia (Ph’)
Gen c-abl koduje jedno z białek czynnych przy proliferacji
komórki należące do cytoplazmatycznych kinaz tyrozynowych
Najczęściej gen ten jest przeniesiony w sąsiedztwo części genu
bcr na chromosomie 22; gen bcr (od breakpoint cluster region)
stosunkowo często ulega przełamaniu - stąd jego nazwa
Powstaje na chromosomie 22 gen fuzyjny – onkogen o nazwie
bcr-abl, charakterystyczny dla PBSz
Klon nowotworowy pod wpływem bcr-abl wykazuje: nadmierną
proliferację, niezależność od zewnątrzkomórkowych czynników
wzrostu i cytokin, zahamowanie apoptozy
PBSz - patogeneza
Translokacja t(9:22)(q34:q11) powoduje w
komórce dodatkowo zmniejszoną stabilność
DNA i podatność na następne mutacje;
pojawienie się takiej dodatkowej mutacji w
jednej z komórek PBSz powoduje powstanie
bardziej złośliwego podklonu i klinicznie
przejawia się przejściem choroby w fazę
przyspieszoną, a następnie kryzę blastyczną
PBSz - Rozpoznanie
Obecność chromosomu Filadelfia w badaniu kariotypu met.
GTG lub FISH (95%chorych)
Obecność genu fuzyjnego BCR-ABL wykrywanego met. RT-
PCR (100% chorych)
FAG = 0 lub wartości bliskie 0
Hiperleukocytoza ze względną limfopenią
Wybitnie bogatokomórkowy szpik z dominacją linii
granulocytowej (90%)
PBSz – Jaka to faza choroby?
Faza przewlekła
Faza przyspieszona (akceleracji)
Kryza blastyczna

PBSz – Jaka to faza choroby?
Faza przewlekła - BCR/ABL
Faza przyspieszona (akceleracji)
10-19% blastów we krwi obwodowej lub szpiku,
bazofilia >20%,
małopłytkowość <100 000/mm³,
nadpłytkowość >1000 000/mm³,
dodatkowe aberracje chromosomowe (klonalna ewolucja cytogenetyczna),
powiększenie śledziony lub zwiększenie leukocytozy oporne na leczenie.
Kryza blastyczna
>20% blastów,
pozaszpikowe nacieki białaczkowe
Obecność co najmniej jednego z wymienionych objawów jest konieczna do rozpoznania fazy
przełomu blastycznego CML.
PBSz – Jaka to faza choroby?
Faza przewlekła – przeciętnie 3-5 lat (u około 25%
chorych rocznie przechodzi w następną fazę)
Faza przyspieszona (akceleracji) – występuje tylko u
około 50% chorych i trwa kilka miesięcy i następnie
przechodzi w kryzę blastyczną – narastające objawy
ogólne
Kryza blastyczna – jako zejście fazy przyspieszonej lub
(nagle) fazy przewlekłej, wyjątkowo jako
„pierwotna”K.B.; w ciągu kilku tygodni doprowadza
do zgonu – przebiegiem odpowiada ostrej białaczce;
kryza może być mieloblastyczna (częściej) lub
limfoblastyczna (rzadziej)
PBSz – Jak leczyć tego chorego?
Obserwacja
Hydroksymocznik
Przeszczepienie allogenicznych komórek
krwiotwórczych
Interferon alfa
Inhibitory kinazy tyrozynowej (imatynib, dazatynib,
nilotynib)
Chemioterapia skojarzona
Co jest wskazane w tym wypadku?
PBSz – Jak wyleczyć tego chorego?
Obserwacja
Hydroksymocznik
Przeszczepienie allogenicznych komórek
krwiotwórczych
Interferon alfa
Inhibitory kinazy tyrozynowej (imatynib, dazatynib,
nilotynib)
Chemioterapia skojarzona
Pierwotna mielofibroza
Pierwotna mielofibroza
(OMF-osteomyelofibrosis)
(OMF-osteomyelofibrosis)
Klonalny nowotwór mieloproliferacyjny –
nadmierna proliferacja głównie megakariocytów i
prekursorów granulocytów w szpiku kostnym
Wzmożona produkcja cytokin stymulujących
włóknienie szpiku (Il-8, TGF-β, PDGF, FGF)
Hematopoeza pozaszpikowa
Nieefektywna hematopoeza
Organomegalia
Pierwotna mielofibroza
Pierwotna mielofibroza
(OMF-osteomyelofibrosis)
(OMF-osteomyelofibrosis)
Zapadalność 0,5-1,5/ 100 000 na rok
Mediana wieku rozpoznania 7 dekada
Występowanie u młodych dorosłych i
dzieci – sporadyczne
Mutacja JAK2 V617F – u około 50%
chorych
Mediana przeżycia 3-7 lat
Pierwotna mielofibroza
Pierwotna mielofibroza
(OMF-osteomyelofibrosis)
(OMF-osteomyelofibrosis)
OMF- objawy:
Utrata masy ciała
Stany podgorączkowe
Poty nocne
Osłabienie
Ból w lewym górnym kwadrancie brzucha
Splenomegalia: u >90%
Hepatomegalia u >50%
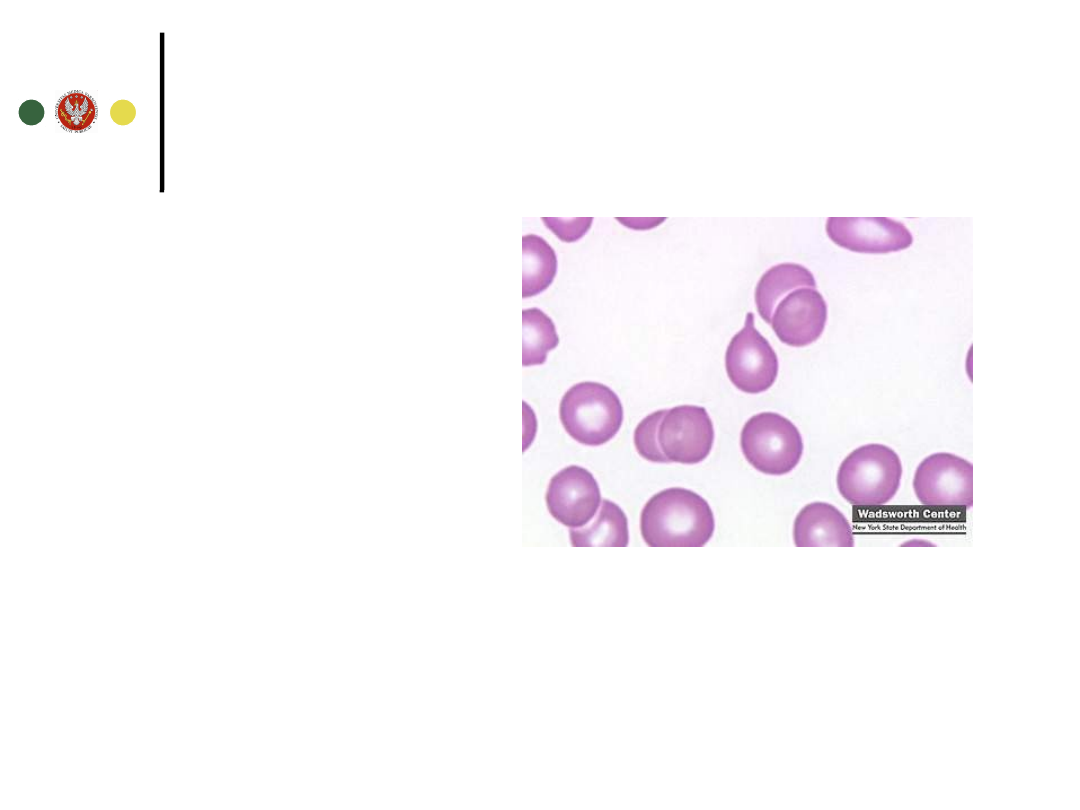
Niedokrwistość i postępująca pancytopenia
Morfologia+rozmaz mikroskopowy
LDH
Oznaczenie BCR-ABL oraz Jak2 V617F
Mielogram i trepanobiopsja szpiku (+ewent.
badanie cytogenetyczne)
OMF- badania diagnostyczne:
Duże
Proliferacja megakariocytów z cechami atypii oraz włóknienie
retykulinowe lub kolagenowe w szpiku
Wykluczenie PRV, CML BCR-ABL+ oraz innych nowotworów układu
krwiotwórczego
Obecność JAK 2 V617F lub innych markerów klonalności (jeśli brak
– wykluczenie przyczyn wtórnego włóknienia – np. infekcji,
uszkodzenia toksycznego, chorób autommunologicznych,
przerzutów do szpiku, HCL i in.)
Małe:
Wzrost liczby WBC i RBC
Wzrost aktywności LDH
Niedokrwistość
Splenomegalia
Rozpoznanie: 3 duże i 2 małe
OMF- kryteria rozpoznania:
RBC – niedokrwistość
WBC i PLT – wszelkie opcje
możliwe
Rozmaz: obecne prekursory
ukł białokrwinkowego
(mielocyty, mieloblasty)
oraz erytroblasty, Dakrocyty
Trepanobiopsja!!!!! –
rozległe włóknienie szpiku.
Możliwość „suchych biopsji”
OMF- badania laboratoryjne:
"Puste biopsje" + pancytopenia +
splenomegalia – duże prawdopodobieństwo
pierwotnej mielofbrozy, wskazanie do
trepanobiopsji.
Nie ma rozpoznania OMF bez wykonania
trepanobiopsji

•
Faza komórkowa – początkowy okres, rozrost wszystkich linii
komórkowych, szczególnie dużo megakariocytów (może być
nadpłytkowość), leukocytoza z leukoerytroblastycznym
obrazem rozmazu krwi obwodowej, początki splenomegalii
• Faza włóknienia – coraz mniej komórek, coraz więcej
włókien; pogłębiająca się anemizacja; tendencja do
pancytopenii, wyraźna organomegalia; nie daje się w sposób
klasyczny pobrać szpiku - tzw. "puste biopsje”)
• Faza sklerotyzacji – przebudowa struktury kostnej (ogniska
osteosklerotyczne), złamania patologiczne, pogłębiająca się
pancytopenia, znaczna organomegalia; całkowite przeżycie –
4-5 lat; zgony z powodu wylewów do OUN i krwotoków oraz
infekcji; transformacja w postać ostrą podobną do OBSz w
około 25% przypadków
OMF- fazy choroby:
32
•
Substytucja KKCz i KKP
•
HU – tylko w fazie komórkowej (1-2g/d).
•
Glikokortykosterydy przewlekle (hamowanie
włóknienia) – wątpliwa skuteczność
•
Splenektomia – rzadko stosowana, w przypadkach
ucisku na narządy sąsiednie, powtarzających się
zawałów śledziony.
•
Napromienianie śledziony (częste powikłania, mało
skuteczne)
•
Talidomid/lenalidomid (postulowane działanie
immunomodulujace wydzielanie cytokin)
OMF- leczenie:
33
·Transplantacja allogenicznych komórek
krwiotwórczych:
- jedyna metoda prowadząca do wyleczenia
- jedynie dla młodszych chorych w dobrym
stanie ogólnym
- wysoki odsetek powikłań śmiertelnych po
przygotowaniu mieloablacyjnym, wyniki
znacznie lepsze po RIC
OMF- leczenie:
34
Nadpłytkowo
Nadpłytkowo
ść
ść
samoistna
samoistna
•
Choroba nowotworowa układu krwiotwórczego
spowodowana rozrostem linii megakariocytowej,
prowadząca do wzrostu liczby płytek krwi.
Przebieg powolny, bardzo rzadko transformacja w
OBSz albo MDS (ok. 5%) lub mielofibrozę wtórną
(5-8%)
•
U ok. 40-50% chorych stwierdza się mutację JAK2
V617F
•
Mediana wieku przy rozpoznaniu: 7 dekada
•
Zapadalność 1-2/ 100 000 na rok
Nadpłytkowo
Nadpłytkowo
ść
ść
samoistna
samoistna
ET
ET
– kryteria rozpoznania
– kryteria rozpoznania
Liczba płytek krwi ≥ 450 G/l
Zwiększenie liczby dojrzałych megakariocytow
w szpiku kostnym, bez odchyleń w linii
czerwonokrwinkowej i granulocytarnej
Wykluczenie: czerwienicy prawdziwej,
pierwotnej mielofibrozy, PBSz BCR-ABL+,
zespołu mielodysplastycznego i innych
nowotworów mieloidalnych
Wykrycie JAK 2 V617F lub innego markera
klonalności o podobnym charakterze – w
przypadku nie wykrycia – wyklucznie
nadpłytkowości odczynowej
Rozpoznanie: wszystkie powyższe
ET – obraz kliniczny:
Bez objawów klinicznych
Bóle głowy, parestezje, zawroty głowy,
TIA, zaburzenia widzenia
(mikrozatorowość)
Powikłania zakrzepowo-zatorowe: udar;
zawał serca, zakrzepica żył głębokich,
zesp. Budd-Chiari
Krwawienia: defekt funkcjonalny PLT
(głównie u chorych z wysoką liczbą PLT >
1 500 G/l)
Spleno- i hepatomegalia
Erytromelalgia
PLT > 1 500 G/l lub wysokie ryzyko
zakrzepicy(jak w PV):
Hydroksymocznik (cel: PLT< 600 G/l)
ASA (przy PLT < 1 500 G/l)
PLT < 1 500 G/l oraz niskie i pośrednie
ryzyko zakrzepicy:
ASA
ET – leczenie:
Anagrelide (wybiórczo hamuje dojrzewanie
megakariocytów i płytkotworzenie) przy
nietolerancji lub nieskuteczności HU – ryzyko
włóknienia szpiku oraz ostrej niewydolności
serca i zaburzeń rytmu u ludzi starszych; wraz
z ASA ryzyko powikłań krwotocznych
Transplantacja allogenicznych komórek
krwiotwórczych – przypadki kazuistyczne
Interferon alfa – w przypadku nietolerancji
innych leków; w ciąży
Trombocytafereza – doraźne obniżenie liczby
PLT w przypadku bardzo wysokich wartości
ET – leczenie:
Przypadek kliniczny 2
71 letni mężczyzna z policytemią – wzrostem
liczby krążących erytrocytów
Htc – 60% (norma 38-50%)
Historia – O co zapytamy?
Papierosy
Alkohol
Cukrzyca
Infekcje
Wywiad rodzinny
Swędzenie
Choroby serca
Zakrzepica
Zmiana rytmu wypróżnień
Podróże
Leki
Badanie przedmiotowe
Szczupły mężczyzna o zaczerwienionej
twarzy
Bez zmian osłuchowych nad płucami i
sercem
Prawidłowe RR i HR
Wyczuwalna śledziona
Jak wpływa to na dalszą diagnostykę?
Morfologia krwi
WBC: 15,7 G/l
LYM 1,0 G/l
MID 0,7 G/l
GRAN 14 G/l
Ht 60
Hgb 19 g%
MCV 79 fL
PLT 485 G/l
Która z wartości może mieć wpływ na dalszą
diagnostykę?
Jakie dodatkowe badania byłyby
wskazane?
RTG kl. Piersiowej?
Biopsja szpiku?
Saturacja O2?
Poziom Erytropoetyny?
Obecność mutacji JAK 2 w limfocytach?
Jakie rozpoznania są prawdopodobne?
Policytemia związana z paleniem
Policytemia związana z diuretykami
Czerwienica prawdziwa
Czerwienica związana z nowotworem nerki
Czerwienica związana z nowotworem wątroby
Czerwienica prawdziwa
Polycythemia vera (PV)
•
Choroba nowotworowa układu
krwiotwórczego spowodowana mutacją komórki
prekursorowej mielopoezy; jest rozrostem o
wieloletnim przebiegu i rzadko przekształcającym
się w ostrą białaczkę szpikową
•
U niemal wszystkich chorych wykrywa się
somatyczną mutację kinazy Janus 2 (JAK 2 V617F)
•
Klon nowotworowy, zachowuje zdolność do
różnicowania i dojrzewania, prowadzi do
nadprodukcji przede wszystkim krwinek
czerwonych, ale mogą być również nadmiernie
wytwarzane neutrofile i płytki krwi
Czerwienica prawdziwa
Przebiega w 3 fazach:
Prodromalnej (niewielki wzrost liczby erytrocytów)
Policytemicznej (wzrost masy krwinek czerwonych)
Wyczerpania (zwłóknienie szpiku kostnego,
nieefektywna hematopoeza, hematopoeza
pozaszpikowa, hipersplenizm
Należy wyeliminować inne przyczyny poligobulii, np.:
czerwienica rzekoma - odwodnienie;
Wszelkie przyczyny prowadzące do niedotlenienia
tkanek i wzrostu wydzielania Epo - np. POCHP i inne
choroby
Autonomiczne wydzielanie Epo (rak jasnokomórkowy
nerki, rak wątrobowokomórkowy, torbielowatość
nerek, transplantacja nerki)
Czerwienica rodzinna (nadmierna ekspresja
receptora EPO)
Stosowanie sterydów anabolicznych, zesp.Cushinga i
in
.
48
Morfologia krwi obwodowej
Oznaczenie mutacji JAK 2 V617F
Trepanobiopsja szpiku, mielogram, hodowla
komórkowa
Stężenie erytropoetyny w surowicy
Gazometria tętnicza, rtg klp, usg j.b, ewnt.
inne badania do diagnostyki różnicowej
Czerwienica prawdziwa - diagnostyka
Duże:
Hb> 18,5 g% (M); > 16,5 (K) lub udowodnione
zwiększenie masy krążących erytrocytów
Obecność mutacji JAK 2 V617F lub innej
czynnościowo podobnej (np. w exonie 12)
Małe:
Nadmiernie komórkowy szpik kostny w stosunku do
wieku z zaznaczoną proliferacją linii
czerwonokrwinkowej, granuocytarnej i
płytkotwórczej
Stężenie EPO w surowicy poniżej normy
Spontaniczny wzrost kolonii erytroidalnych w hodowli
Rozpoznanie: 2 duże + 1 małe lub pierwsze duże i dwa
małe
Czerwienica prawdziwa - rozpoznanie
Czerwienica prawdziwa – jakie
powikłania?
Zakrzepica Tętnicza?
Zakrzepica Żylna?
Ostra białaczka szpikowa?
Osteomielofibroza?
Ostra białaczka limfoblastyczna?
Dna?
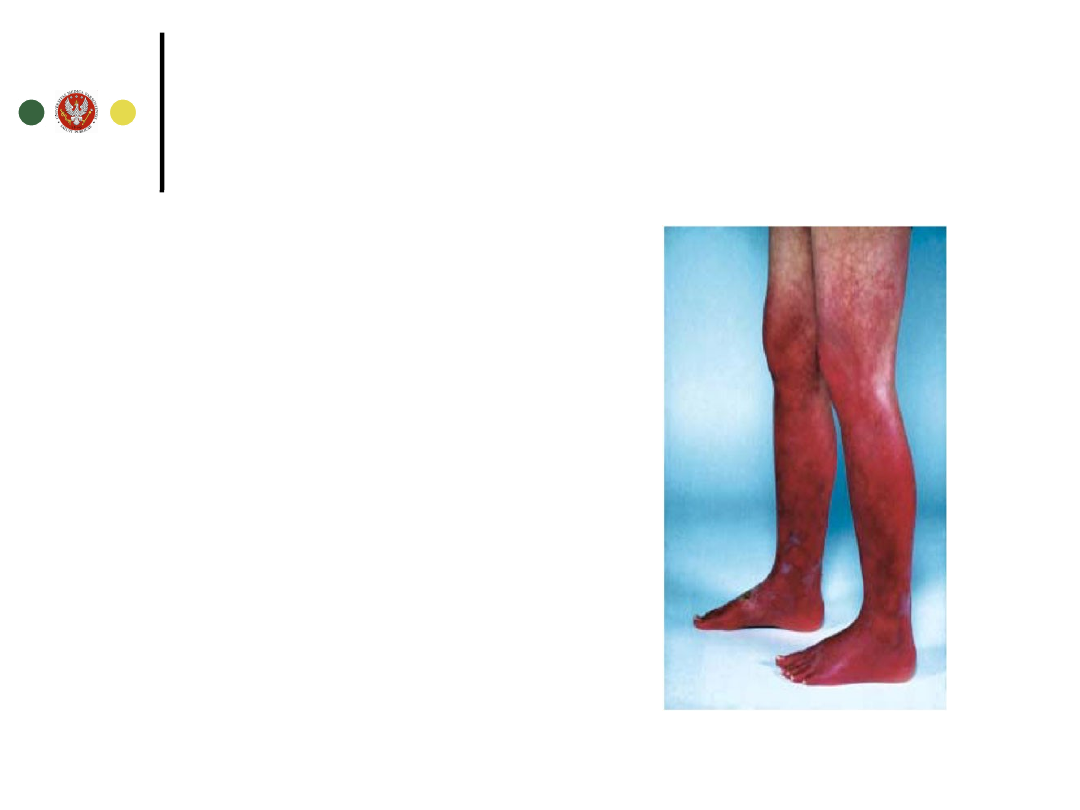
Są konsekwencją poliglobulii, splenomegalii lub
powikłań zakrzepowo-zatorowych
Zaczerwienienie skóry, przekrwienie spojówek
Dyskomfort w lewym górnym kwadrancie brzucha -
splenomegalia
Zespół nadlepkości: bóle i zawroty głowy, zaburzenia
widzenia, szum w uszach, splątanie
Świąd skóry, zwłaszcza po kąpieli, erytromelalgia
Dna moczanowa
Nadciśnienie tętnicze, żylna choroba zakrzepowo-
zatorowa (w tym zesp.Budd-Chiari), udar niedokrwienny,
zawał m. sercowego
Czerwienica
prawdziwa objawy
Czerwienica prawdziwa
Zaburzenia zakrzepowo-
zatorowe: przyczyna
śmierci 30-40%
pacjentów
Ok. 15-20% -
transformacja do
MDS/AML
Możliwe powikłania
krwotoczne (głównie
przy towarzyszącej
nadpłytkowości)
Czerwienica prawdziwa –
jak leczyć tego chorego?
Upust krwi
Upust krwi i
hydroxymocznik
Chlorambucil
Radioaktywny fosfor
Anagrelid
Interferon
Leczenie
Leczenie
PV
PV
•
Chorzy niskiego ryzyka zakrzepicy:
•
Krwioupusty- Ht < 45% M; < 42% K
•
Chorzy wysokiego ryzyka zakrzepicy oraz w przypadku
narastającej leukocytozy, nadpłytkowości i
organomegalii:
•
Leczenie cytostatyczne– hydroksymocznik,
chlorambucyl lub pipobroman (ryzyko wtórnej
białaczki)
•
Leczenie interferonem – u chorych nie tolerujących
cytostatyków; w okresie ciąży
Chorzy pośredniego ryzyka zakrzepicy:
Decyzja co doleczenia zindywidualizowana, raczej
jedynie ASA
Leczenie
Leczenie
PV
PV
U wszystkich chorych kwas acyetylosalicylowy
75-100 mg/d (nie przy plt>1 500 G/l);
Allopurinol przy hyperurykemii;
Substytucja żelaza jeśli pozahematologiczne
objawy niedoboru
Nadpłytkowość oporna na cytostatyki – ewentualnie anagrelid
Transplantacja allogenicznych komórek krwiotwórczych –do
rozważenia u chorych w okresie mielofibrozy lub z powodu
transformacji do ostrej białaczki/MDS
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
Wyszukiwarka
Podobne podstrony:
Marcinkowski, Bajek, Galewska (2010) Aktywność studentów w Internecie
Wykład 3 powtórzenie 2010 studenci (1)
Wykład 5 2010 studenci
Wykład 5 2010 studenci ppt
transplantologia tx studenci 2010
Wykład 3 powtórzenie 2010 studenci
Zagadnienia dla studentow I roku WF na egzamin z biochemii 2010, Wychowanie Fizyczne (materiały i no
gis woiągi notatki dla studentów 2010 11, GIS-Geograficzne Systemy Informacji
slajdy dla studentów 2010
slajdy dla studentów 2010
STUDENCI WiP 2010, WIP, Sem.III, FIZY1
INFORMATOR STUDENCKI 2010 2011 (2), STUDIA, WZR I st 2008-2011 zarządzanie jakością, specjalność ZJi
Foresight Technologiczny 2010 11 logistyka STUDENCI
2010 11 zima plan zajec dla studentow v roku
Zagadnienia z fizyki dla studentow I roku, ZiIP PP 2010-2011, Semestr I, Fizyka techniczna
Zadanie-podatki-dla studentów 2010, Ogrodnictwo, Semestr V, Ekonomika, Ekonomika z chomika ;)
Etyka w biznesie, ^ Turystyka i Rekreacja GWSH Katowice, 3 semestr, podstawy przedsiebiorcz, gwsh Mt
więcej podobnych podstron