BADANIA UKŁADU
HEMOSTATYCZNEGO
EDYTA ZDZIŁOWSKA
KLINIKA HEMATOLOGII CM UJ

HEMOSTAZA
-
zespół procesów fizjologicznych
zapewniających płynność krwi krążącej, szczelność
łożyska naczyniowego oraz sprawne hamowanie
krwawienia przy przerwaniu ciągłości ściany naczyń
krwionośnych.
HEMOSTAZA PIERWOTNA
–
powstanie czopów
hemostatycznych zasklepiających światło
uszkodzonego naczynia. Biorą w niej udział płytki
krwi.
HEMOSTAZA WTÓRNA
–
to powstanie sieci fibryny
wzmacniającej czop płytkowy dzięki działaniu czynnika
tkankowego aktywującego krzepnięcie krwi.
GŁÓWNE ELEMENTY TO
:
ściany naczyń, płytki krwi,
układ krzepnięcia i fibrynolizy oraz endogenne inhibitory
krzepnięcia.

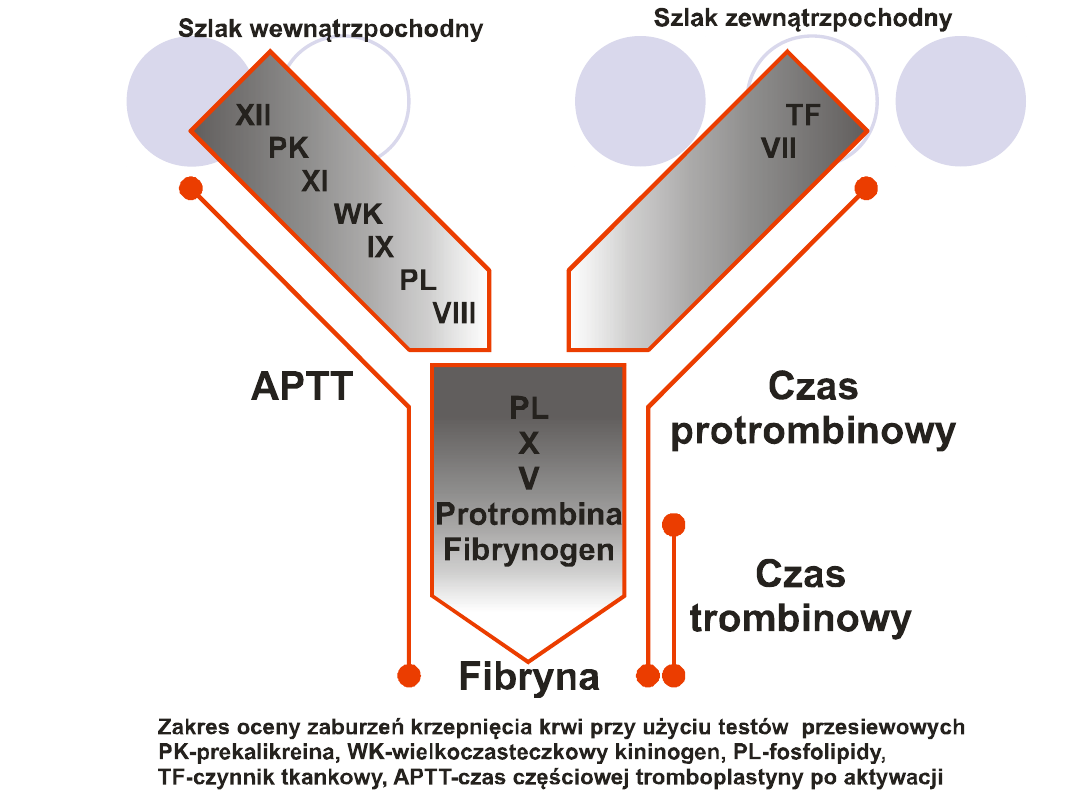
UKŁAD KRZEPNIĘCIA
ZADANIE -
zamiana rozpuszczalnego białka –
fibrynogenu w
nierozpuszczalną fibrynę.
CZYNNIKI BIORĄCE UDZIAŁ W TYM PROCESIE
TO:
12 białek osocza,1 białko integralne błon komórkowych,
fosfolipidy błon komórkowych, jony wapniowe, jony cynku
oraz pośrednio witaminy K
PODZIAŁ BIAŁKOWYCH CZYNNIKÓW
KRZEPNIECIA:
Czynniki kontaktu- XI, XII, prekalikreina, wielocząsteczkowy
kininogen
Czynniki zespołu protrombiny- II, VII, IX, X
Czynniki wrażliwe na trombinę- I, V, VIII, XIII.
Cz.XIa
HMWK
PL
Ca+++ Zn++
Cz.IXa
cz.VIIIa
PL
Ca++
TENAZA
PROTROMBINAZA
Cz.XIa
HMWK
PL
Ca+++ Zn++
T
TM
PL
Ca++
Cz.Xa
Cz.Va
PL
Ca++
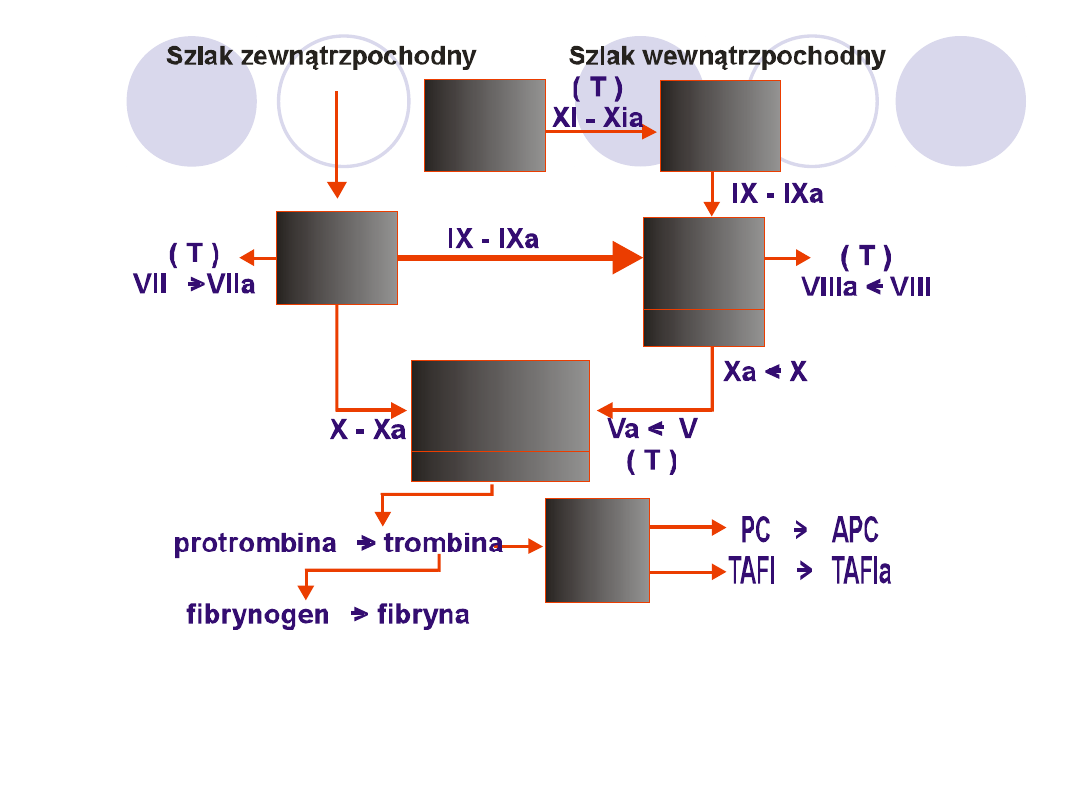
Kompleksowe reakcje aktywacji układu krzepnięcia.
PK-prekallikreina, HMWK-wielkoczasteczkowy kinionogen,
PL - fofsfolipid, Ca++ - jony wapnia, Zn++ - jony cynku,
TM-trombomodulina, PC-białko C, APC-aktywne białko C,
TAFI - inhibitor fibrynolizy aktywowany przez trombinę,
TAFIa - aktywna forma TAFI, T - trombina
Cz.VIIa
TF
PL
Ca++

W warunkach fizjologicznych ścisłe powiązania miedzy
szlakami krzepnięcia a jego aktywację można przedstawić
jako proces 4 kompleksów enzymatycznych:
1.
Tenazy zewnątrzpochodnej- TF-VIIa-X,
2.
Kompleksu aktywacyjnego cz.IX,
3.
Tenazy wewnątrzpochodnej- VIIIa-IXa-X,
4.
Protrombinazy-Xa-Va-II.
Ostatnia faza krzepnięcia to: przejście fibrynogenu w sieć
fibryny. Trombina odszczepia od fibrynogenu 2 pary
fibrynopeptydów A i B, pozostały monomer fibryny
polimeryzuje i tworzy sieć fibrynową stabilizujący się przy
udziale cz.XIII a-katalizuje powstanie wiązań krzyżowych
miedzy monomerami.
INHIBITORY KRZEPNIĘCIA
AT III
–
białko inaktywujące proteazy serynowe ,łączy
się z trombiną (1:1) i unieczynniają oraz cz. IXa, Xa, XIa,
XIIa. Heparyna zwiększa ok.1000 razy szybkość działania
ATIII.
Ochrona krążenia przed działaniem aktywnych
enzymatycznie cz.krzepnięcia oraz ograniczaniu
wykrzepiania przy uszkodzeniu naczynia. Niedobór ATIII
może być wrodzony- 3-8% ze zwiększonym ryzykiem
żylnych incydentów zakrzepowych lub nabyty-
spowodowany ostra zakrzepicą, DIC, schorzeniami
wątroby, doustnymi lekami antykoncepcyjnymi, HTZ

TFPI-
inhibitor szlaku czynnika tkankowego- białko w
osoczu związane z lipoproteinami, w obecności cz. Xa wiąże
i inaktywuje kompleks TF-VIIa.
Wrodzony niedobór –bardzo rzadki-obserwowany u rodziny
ze skłonnością do zakrzepic
Podwyższenie inhibitora w: hipercholesterolemii, ciąży,
rozsiewie nowotworowym, po injekcjach heparyny.
W DIC- brak korelacji z innymi badaniami stąd jego
przydatność diagnostyczna bardzo mała.

BIAŁKO C-S –
witamino-K zależny inhibitor inaktywujący cz.Va , VIIIa przy
udziale kofaktora b.S, fosolipidów i jonów wapnia.Układ
aktywowany trombiną związaną z endotelialną
trombomoduliną przekształca się w aktywowane białko
C(APC)
Wrodzony niedobór b.C u homozygot objawia się w
pierwszych dniach życia jako plamica piorunująca z
zakrzepicą naczyń mózgowych. U heterozygot nawracające
zakrzepice żylne i zatorowość płucna.
Niedobór b.C i S towarzyszy DIC, zakrzepicy żył głębokich,
ciężkiemu uszkodzeniu wątroby, jako następstwo terapii L-
asparaginazą, warfaryną
.
HC II –
heparynowy kofaktor- wiąże i inaktywuje trombinę,
silniej w obecności heparyny.
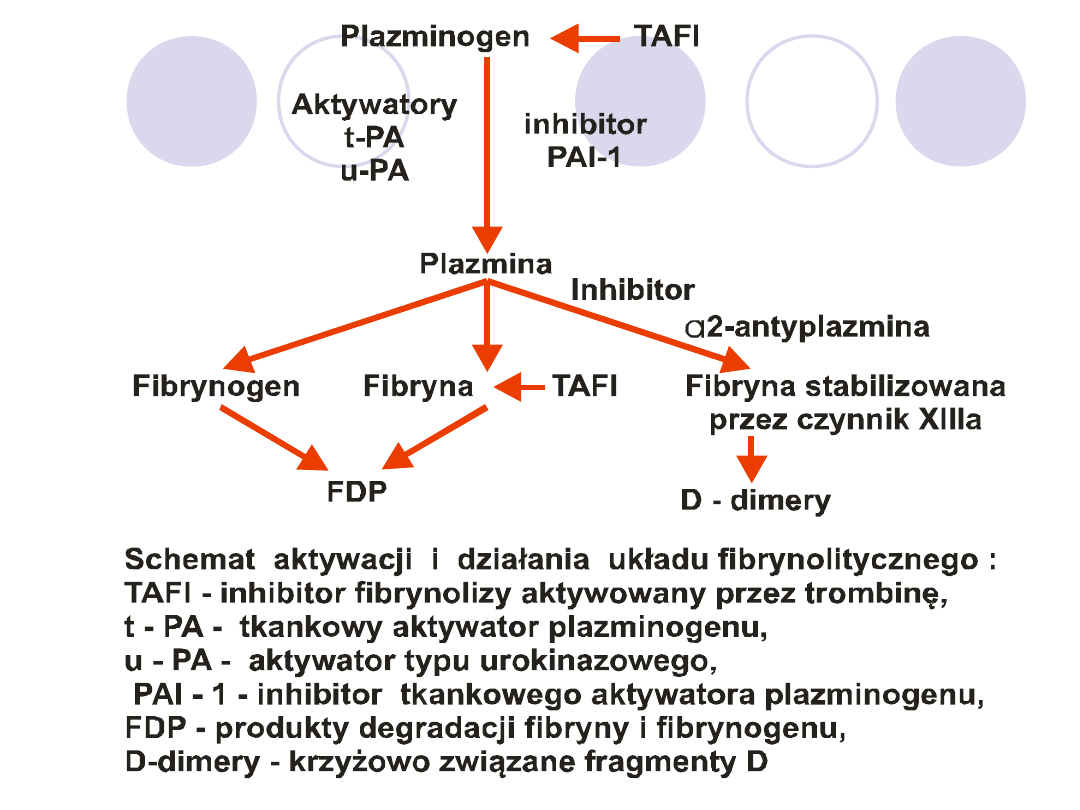
FIBRYNOLIZA
ZADANIE:
rozpuszczenie śródnaczyniowych złogów
fibryny i utrzymanie drożności naczyń.
GŁÓWNE SKŁADNIKI:
plazminogen (nieczynny
enzym), aktywatory przekształcające plazminogen w
plazminę, inhibitory –samej plazminy jak i jej aktywatorów.
AKTYWATORY:
wytwarzane jako jednołańcuchowe
prekursory
t-PA-
w komórkach naczyń,
u-PA-
w wielu
komórkach i narządach(nerki).

INHIBITORY:
PAI-1-
wiąże i inaktywuje t-PA lub u-PA
,
PAI-2-
szybciej inaktywuje u-PA niż t-PA,
L2-antyplazmina-
łączy się z plazminą w sposób
stechiometryczny. Po jej wyczerpaniu plazminę
inaktywuje
L-2 makroglobulina.
TAFI
-
inhibitor fibrynolizy aktywowany przez trombinę
związaną z trombomoduliną- odszczepia od fibryny C-
końcowe reszty lizyny co ułatwia wiązanie się plazminogenu
i t-PA z fibryną.

Aktywacja plazminogenu pod wpływem t-PA ma głównie
znaczenie w rozpuszczeniu fibryny i utrzymaniu drożności
naczyń,
Plazmina tworząca się przy udziale u-PA uczynnia
prometaloproteinazy macierzy pozakomórkowej
degradujące składniki macierzy stąd ważna rola w
przebudowie tkanek i migracji komórek –angiogenezie,
gojeniu się ran, wzroście nowotworów i tworzeniu
przerzutów.
FDP
-
powstają przy trawieniu fibryny i fibrynogenu.
Wielocząsteczkowe produkty degradacji oznaczane jako
X,Y,D,E.
D-dimer
powstaje przy trawieniu usieciowionej fibryny
przez plazminę (połączone wiązaniem krzyżowym)
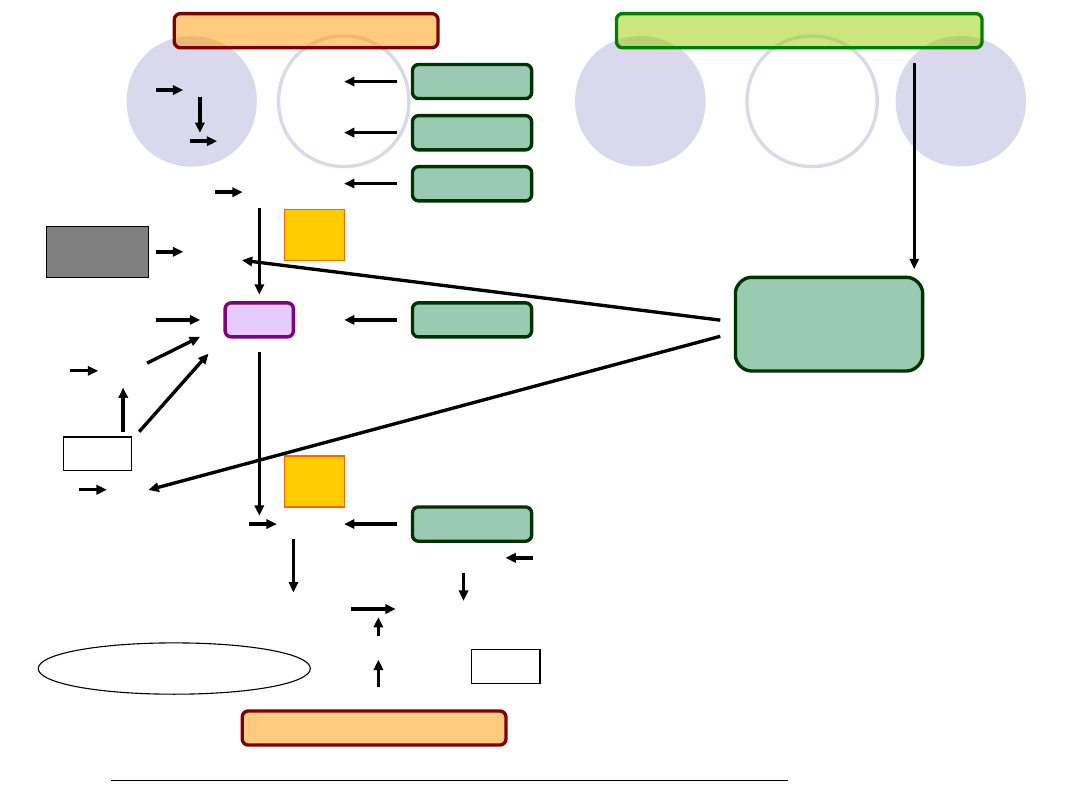
Rys. Współdziałanie układów: krzepnięcia, antykoagulacyjnego i fibrynolizy
XII XIIa
XI XIa
IX IXa
AT III
AT III
AT III
Białko C
EPCR
Biąłko S
Trombina
Czynnik V
Trombomodulina
UKŁAD KRZEPNIĘCIA
UKŁAD ANTYKOAGULACYJNY
PL Ca
++
VIII/vW
VIIIa
AT III
Xa
X
VII
VIIa
TFPI
FT
V
Va
PL Ca
++
Aktywne
białko C
AT III
II
IIa
Fibrynogen Fibryna
XIIIa
XIII
Plazmina
Plazminog
en
UKŁAD FIBRYNOLIZY
TAFI
Aktywatory/inhibitory

PŁYTKI KRWI
Najmniejsze (2-4m, objętość 8-12 fl)) bezjądrzaste
elementy morfotyczne krwi powstające z cytoplazmy
megakariocytów. Otoczone są trójwarstwową błoną
lipoproteinową z podbłonowymi elementami kurczliwymi-
aktyną, miozyną i kalmoduliną, które pozwalają na zmianę
kształtu płytki w procesach agregacji i adhezji.Błona
płytkowa jest miejscem interakcji składników osocza i
uszkodzonych komórek śródbłonka. Zawiera fosfolipidy,
cholesterol, glikolipidy i glikoproteiny.

Do ziarnistości płytkowych zaliczamy
:
ziarnistości gęste
-
uwalniające ADP,ATP,GDP,GTP
,
serotoninę, j.Ca
2+
,j.Mg
2+
,katecholaminy, P-selektyna
ziarnistości alfa-
białka swoiste płytek: PF
4
,
-tromboglobulinę,
białka adhezyjne :selektyna P,
cz. vWF, trombospondyna,
fibronektyna,
cz.krzepnięcia i fibrynolizy: cz.V , XI,
fibrynogen, kininogen HMWK,białko S,
t-PA, PAI-1
Lizosomy
-
kwaśne hydrolazy
,
Peroksysomy
-
katalaza.
RECEPTORY PŁYTEK KRWI
GP Ia/IIa
(CD49b/CD29b)
Receptor dla kolagenu
GPIb(alfa,beta), GP
IX,GP V
(CD 42c,42b,42a,42d)
Receptor dla vWF-
upośledzenie funkcji-choroba
vWF
Niedobór GPV,IX-upośledzenie
adhezji-z.Bernarda-Souliera
GP IIb/IIIa (CD41,61)
Kompleks łączący się z
fibrynogenem również vWF,
trombospondynę,fibronektynę
. Brak lub dysfunkcja
powoduje skazę krwotoczną
(trombastenia Glanzmana)
GPIII b (CD36)
Receptor trombospondyny
GP IIa/Ic (CD29,49e)
Receptor fibronektyny
GP Ic/IIa (CD49f/CD29)
Receptor lamaliny
GP53 ( CD63)
Uwalniana z ziarnistości
glikoproteina łącznie z P-
selektyną odp. za adhezję
płytka/monocyt,
płytka/leukocyt lub
płytka/limfocyt

ADHEZJA
Poprzez swoje receptory mogą się łączyć do różnych białek
eksponowanych na powierzchni leukocytów, monocytów,
limfocytów, uszkodzonego nabłonka lub do powierzchni
podśródbłonkowej głównie kolagenu, fibronektyny,
trombospondyny.
Adhezja do podścieliska przez tzw. siły ścinania ( duża
rola cz.vWF- pomost pomiędzy płytką a ścianą naczynia)
Łączenie płytek między sobą tylko przez fibrynogen-
odpowiedni receptor płytkowy( kompleks IIIa/II b,
CD61/CD41)

AKTYWACJA
powoduje zmianę kształtu z dyskowatego na sferyczny, a
do pojedynczej warstwy płytek przyłącza się następna.
Powstają agregaty płytkowe z udziałem fibrynogenu i
fibrynonektyny.
Różni agoniści tj.kolagen, ADP, adrenalina, serotonina,
czynnik aktywujący płytki PAF, tromboksan A2(TX2), łączą
się ze swoistymi receptorami płytki .
Jednoczesna fosforylacja białek wewnątrzpłytkowych,
przemieszczanie j.wapniowych z kanalików gęstych do
cytoplazmy, zmniejszenie stężenia cAMP, pobudzenie
przemiany kwasu arachidonowego oraz uwolnienie
substancji w ziarnistościach powodują dalszą aktywację.

AGREGACJA
I faza-
odwracalna- za pośrednictwem ADP-zależnego
tworzenia kompleksu CD61/CD41 oraz fibrynogenu
następuje łączenie się płytek ale brak jeszcze uwolnienia
zawartości ziarnistości.
II faza
–
nieodwracalna- degranulacja, uwolnieni TXA
2
,
stabilizacja konglomeratu płytkowego przez siatkę fibryny.
Ostatnia faza to retrakcja skrzepu i ostateczna
stabilizacja.

UDZIAŁ PŁYTEK W HEMOSTAZIE:
tworzą pierwotny czop hemostatyczny,
udostępniają fosfolipidy do reakcji krzepnięcia-
fosfatydyloserynę i fosfatydyloetanolaminę.
Umożliwia to tworzenie się kompleksu tenazy i
protrombinazy.

PODSTAWOWE TESTY I BADANIA
Do badań pobiera się zawsze krew żylną .
Antykoagulant- cytrynian sodu 3,2%( 9:1)
Nie należy używać szklanych probówek, próbek
podejrzanych o skrzepnięcie lub hemolizę.
Do badań stosuje się osocze cytrynianowe
(2000 obr/10 min.) w ciągu 2-4 h od pobrania
Można korzystać z osoczy mrożonych (kilkanaście dni w
-20
o
C lub tygodni w -70
o
C)
Osocza standardowe (mieszanina od 10-20 zdrowych
dawców), osocza referencyjne liofilizowane
.

BŁĘDY PRZEDANALITYCZNE
Zbyt duża ilość cytrynianu,
Hemoliza,
Hiperbilirubinema,
Lipemia,
Niewłaściwe przechowywanie próbki(>4 godzin,
nieopowiednia temp.)
Obecność heparyny.

CZAS KRWAWIENIA
-
doświadczalne zranienie skóry ↔ ustanie wypływu krwi
- hemostaza pierwotna
- nie zależy od czynników krzepnięcia (poza vWF)
- przedłużony w skazach płytkowych, naczyniowych, czasem
w chorobie von Willebranda
- metoda Ivy: opaska sfingomanometru (40 mmHg), nacięcie
na skórze przyśrodkowej powierzchni przedramienia (dł. 10
mm, gł. 2,5 mm), norma: 2-10 min
- metoda Duke’a (opuszka palca, płatek ucha)
- Wartości referencyjne- 3-8 min
.

Czas krzepnięcia
to czas spontanicznego krzepnięcia krwi w probówkach w
temp. 37C bezpośrednio po wynaczynieniu.
- postępowanie- 1ml krwi żylnej do szklanej probówki
włączając stoper. Probówkę umieszcza się w łaźni wodnej i
co 30 sek. przechyla się aż do chwili gdy swobodny
przepływ krwi zostanie zatrzymany.
NORMA : 4 - 10 min.

Czas rekalcynacji
czas od momentu dodania do osocza cytrynianowego
CaCl2 do chwili powstania skrzepu w temp. 37 C. Jest
równoważny czasowi krzepnięcia.
postępowanie - do probówki 0,2 ml osocza + 0,2 ml
ogrzanego CaCl2, włączamy stoper do chwili powstania
skrzepu.
NORMA: 60-180 sek.

Czas częściowej tromboplastyny po aktywacji
(APTT - activated partial thromboplastin time)
czas krzepnięcia osocza cytrynianowego po dodaniu
kefaliny, kaolinu oraz chlorku wapnia.
Kaolin zwiększa dostępność czynnika kontaktu- XII, skraca
czas krzepnięcia a wyniki są jednolite uniezależnione od
niejednakowego uczynnienia przez szkło probówki. Zamiast
kaolinu aktywatorem może być celit lub kwas elagowy
Kefalina jest fosfolipidowym odczynnikiem otrzymanym z
mózgów króliczych, działa jak czynnik płytkowy 3 ,
uniezależnia pomiar od zmian płytkowych .

Jest próbą krzepnięcia zapoczątkowanego przez układ
wewnątrzpochodny –aktywacji protrombiny ( cz.
V,VIII,IX,X,XI,XII)z wyłączeniem wpływu krwinek płytkowych.
Zależy więc od osoczowych czynników oraz stężenia
protombiny i fibrynogenu. Mierzy czas od uczynnienia
czynników kontaktu do powstania fibryny. Większa czułość
niż czas krzepnięcia.
Jest czuły na niedobory także czynnika V,X, protrombiny i
fibrynogenu natomiast nie wrażliwy na niedobór cz.VII.
NORMA: 22-55 sek.
Wydłużenie:
-hemofilia A (VIII),B (IX)oraz II,V,X,XI,XII,
- obecność heparyny,
- krążący antykoagulant, antykoagulant toczniowy
-obecność produktów degradacji fibryny i fibrynogenu

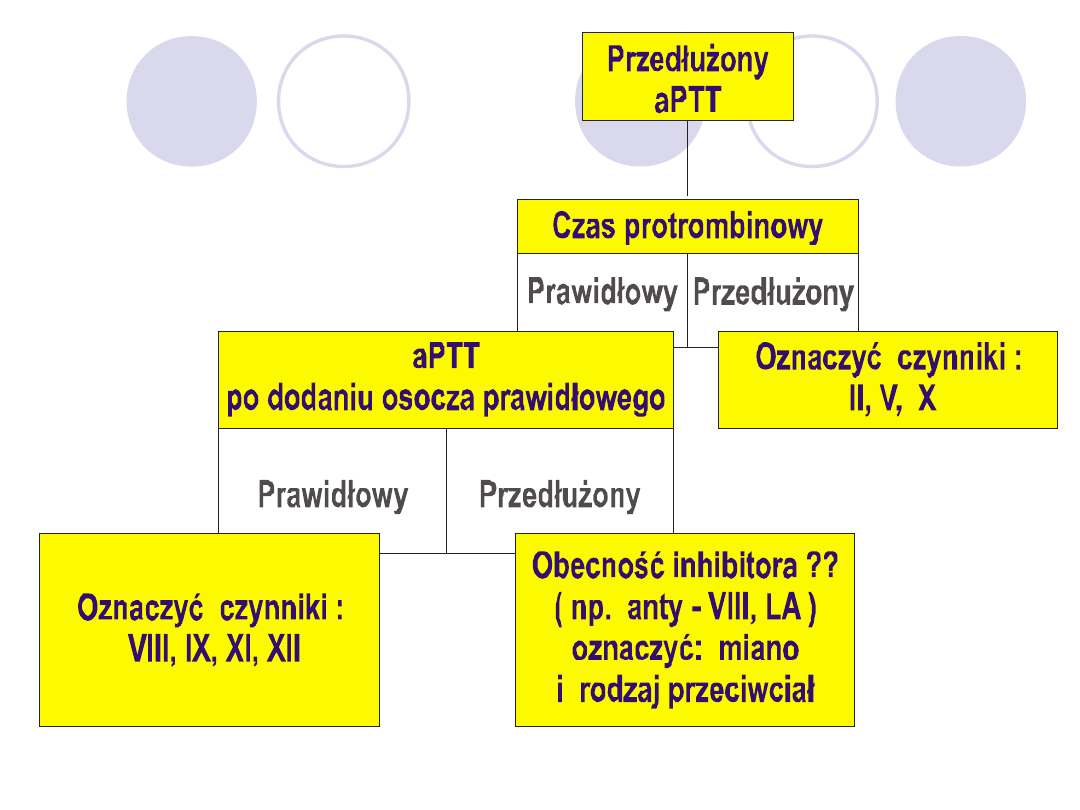
Aby odróżnić prosty niedobór cz.krzepnięcia od
powodowanych obecnością krążącego antykoagulantu
porównuje się aPTT osocza badanego z osoczem
prawidłowym i mieszaniny dwóch równych objętości osocza
badanego z osoczem prawidłowym po inkubacji przez 60
min.w temp.37 stopni.
O prawidłowej korekcji aPTT wnioskuje się gdy różnica
miedzy aPTT osocza badanego i mieszaniny jest większa niż
50% różnicy między aPTT badanego i kontrolnego.
Inaczej stwierdza się brak korekcji i rozpoznaje się obecność
krążącego antykoagulanta.

Przykład
aPTT kontrolnego= 35 sek,
aPTT badanego= 60 sek
aPTT mieszaniny= 42sek.
Korekcja prawidłowa ponieważ 60-42=18 60-35=25.
25:2=12,5, 18>12,5.
Stąd przyczyna przedłużenia jest niedobór czynników
krzepnięcia.
Jeśli aPTT mieszaniny= 52sek-to brak korekcji, ponieważ 60-
52=8, 60-35=25,25:2=12,5 8<12,5.
Stąd przyczyna przedłużenia aPTT jest krążący
antykoagulant.
APTT
zalecany jest jako test kontroli leczenia heparyną
niefrakcjonowaną (lek przeciwzakrzepowy stosowany u
pacjentów z żylną i tętniczą zakrzepicą oraz w profilaktyce
ch.zakrzepowo-zatorowej).
Heparyna tworzy kompleks z ATIII i inaktywuje trombinę i
cz.Xa. Terapeutyczne stężenie heparyny we krwi odpowiada
1,5-2,5 krotne przedłużenie aPTT w porównaniu do wartości
przed leczeniem.
Heparyny drobnocząsteczkowe(frakcjonowane) monitoruje
się nie jest aPTT przydatne można oznaczać aktywność
anty-Xa.
Czas protrombinowy ( czas Quicka ,
tromboplastynowy
)
czas krzepnięcia osocza cytrynianowego po dodaniu chlorku
wapniowego z dodatkiem tkankowej tromboplastyny ( cz.III)
,który w kontakcie z cz.VII uczynnia układ krzepnięcia. Nie
zależy od PLT i innych cz.krzepnięcia poza-II,V,VII,X i
fibrynogenem.
Tromboplastyna uzyskiwana z tkanek zwierzęcych lub
metodami inżynierii genetycznej.
NORMA : 12-16 sek. ( 80-120%)
czas osocza kontrolnego x100
Wskaźnik%=
czas osocza badanego
czas osocza badanego
Współczynnik.=
czas osocza kontrolnego
INR
( international normalized ratio)
Czas osocza badanego
ISI
INR = [
]
Czas osocza prawidłowego
Norma : 0,9-1,4

Wydłużenie:
-niedobory II,V,VII,X,
-niedobory witaminy K,
-leczenie antykogulantami,
-choroby miąższu wątroby,
-w hipo- i afibrynogemii,
-DIC,
-obecność heparyny i FDP,
-różne: leczenie salicylanami, białaczka,
mocznica,ch.Addisona-Biermera itd.
Skrócenie
PT nie jest patologią a świadczy o niewłaściwym
przechowywaniu próbek.

Wskazania do wykonania testów PT, a PTT :
przedoperacyjnym badaniu pacjentów ze zwiększonym
ryzykiem krwawień
ocenie aktywności biologicznej czynników krzepnięcia toru
zewnątrzpochodnego (w przypadku PT ) czynników: II, V, VII,
X i fibrynogenu, lub wewnątrzpochodnego (w przypadku
APTT) czynnika VIII, IX, XI, XIII, fibrynogenu;
monitorowaniu leczenia doustnymi antykoagulantami (w
przypadku PT) lub terapii przeciwzakrzepowej, np. z
zastosowaniem heparyny i hirudyny (w przypadku APTT);
ocenie funkcji wątroby;
wykrywaniu inhibitorów układu wewnątrzpochodnego
powstających wtórnie do różnych schorzeń – szczególnie
inhibitorów FVIII lub antykoagulanów toczenia (w przypadku
APTT).

IZOLOWANE
PRZEDŁUŻENIE APTT
błąd pobrania (heparyna)
leczenie HNF
hemofilia A, B, C (FVIII, FIX,
FXI)
czasem choroba von
Willebranda
anomalia Hagemana (FXII)
niedobór WK, prekalikreiny
antykoagulant toczniowy
IZOLOWANE
PRZEDŁUŻENIE INR
leczenie doustnymi lekami
przeciwkrzepliwymi
(antagoniści wit. K)
niedobór wit. K
hypoprokonwertynemia (FVII)
choroby wątroby
PRZEDŁUŻENIE APTT oraz INR
niedobór FV, FX, niedobór protrombiny (FII)
znaczny niedobór czynników zależnych od wit. K
zaburzenia po masywnych przetoczeniach krwi
niedobór fibrynogenu, dysfibrynogenemia (też
przedłużony TT!)
DIC (też TT)
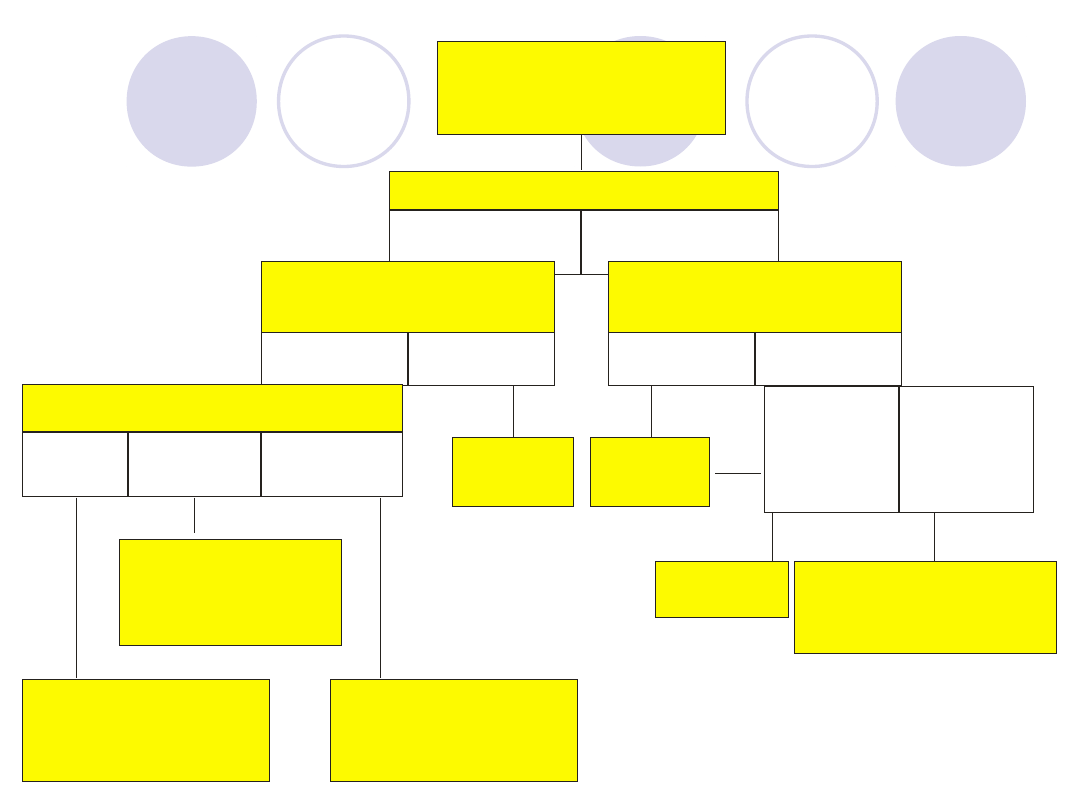
Disfibrynogenemia
Zaburzenia
polimeryzacji
Hipofibrynogenemia
wrodzona
lub nabyta
Hipofibrynogenemia
Białko ostrej fazy
Zaburzenia
wrodzone
Zaburzenia syntezy
wątrobowej
Zwiększona degradacja
Czas trombinowy
Przedłużony
Przedłużony
Prawidłowy
Prawidłowy
Prawidłowe Obniżone
Obniżony Prawidłowy Podwyższony
Obecność
heparyny
Obecność
inhibitora
Czas batroksobinowy
(reptylazowy)
Czynniki II, V, VII, X
Fibrynogen
Niedobór
czynnika
pojedynczego
Niedobór
kilku
czynników
Przedłużony
czas protrombinowy

Markery generacji trombiny
oznaczanie fragmentów protrombiny 1+2 (F1+2)
powstających z cząsteczki protrombiny pod wpływem
aktywnego czynnika X. U osób leczonych antykoagulantami
zaobserwowano obniżenie poziomu F1+2. Uważa się, że to
oznaczenie może służyć jako uzupełnienie rutynowego
badania PT; i mogłoby wskazywać prawdziwy stopień
supresji układu krzepnięcia.okres półtrwania F1+2-90 min.
Oznaczenie kompleksów trombiny z antytrombiną (TAT)-
marker krótkotrwałej generacji trombiny
.
Oznaczanie fibrynopeptydu A(FPA)-16-aa fragment
łańcucha alfa fibrynogenu odszczepiany przez trombinę.
Wszystkie oznaczenia u osoczu ubogopłytkowym metodami
immunoenzymatycznymi

Markery generacji trombiny oznacza się głównie w celach
badawczych.
Wzrost ich stężenia : w zakrzepicy żylnej, ostrych
incydentach sercowo-naczyniowych, DIC, po dużych
urazach i operacjach.
F1+2-wykazuje silnie ujemna korelację z INR do wartości 4-
może być przydatny do monitorowania leczenia doustnymi
antykoagulantami
.

Próba ogólna układu fibrynolizy
-
pomiar czasu upłynnienia czasu skrzepu krwi. Czas jest
wypadkową czynności wszystkich czynników
uczestniczących w fibrynolizie.
Wyk.: 2 ml krwi do probówki w 37 C, oznacza się czas
upłynnienia skrzepu . Obserwować co 30, 60 min.
NORMA: ponad 24h
W hiperfibrynolizie upłynnienie jest obserwowane po 3-6
h, niekiedy kilkanaście min.

Oznaczenie zużycia protrombiny
w czasie krzepnięcia zużywają się cz.I,V,VIII,XIII również
trombina prawie w całości przechodzi w protrombinę.
Prawidłowo w surowicy stwierdza się mniej niż 10%
protrombiny w odniesieniu do 100 % aktywności w osoczu.
im więcej protrombiny w surowicy tym mniejsze jej zużycie
w procesie krzepnięcia i tym większe upośledzenie
aktywności jednego lub kilku czynników
( V,VIII,IX,X lub płytkowego 3 )

Czas lizy skrzepu euglobulin
ogólna próba układu fibrynolitycznego-miara aktywności
aktywatorów fibrynolizy
.
czas upłynnienia skrzepu wytrąconego przez roz.kwaśnym
r-rem o słabej sile jonowej (pH 5,2-5,9)
Wytrącona euglobulinowa frakcja zawiera min.
plazminogen, plazminę,aktywatory plazminogenu oraz
fibrynogen i inne czynniki krzepnięcia VIII,XII,XIII.
Aktywność fibrynolityczna jest duża ponieważ euglobuliny
są prawie całkowicie pozbawione antyplazmin. NORMA: 2-
4h.
Obniżenie: choroby wątroby,
stany uczynnienia fibrynolizy,
Wzrost: choroby nerek,
Osoby leczone inhibitorami fibrynolizy
.

FDP (PDF)
oznaczenie produktów degradacji fibrynogenu i fibryny.
Niezbędne przy np.ostrej lub przewlekłej postaci
rozsianego krzepnięcia śródnaczyniowego.
Metodami półilościowymi-
cząsteczki lateksu opłaszczone poliklonalnymi Ab
specyficznymi dla fragmentów D i E ludzkiego
fibrynogenu aglutynują w obecności produktów
degradacji fibrynogenu. Stopień rozcieńczenia przy
którym pojawia się aglutynacja jest określony w teście.
NORMA w surowicy <1 mg/ml
Ilościowymi- immunochemicznymi
.

Wzrost stężenia:
-rozsiane krzepnięcie śródnaczyniowe z hiperfibrynolizą,
-pierwotna hiperfibrynoliza,
-leczenie lekami trombolitycznymi ,
-zakrzepica żyl,
-zawał m.sercowego,
zator tętnicy płucnej,
przewlekłe ch.watroby,
nowotwory złośliwe.
Obniżenie:
-po przeszczepie nerek,
-niektóre ch.nerek,
-rak pęcherza moczowego,
-skaza krwotoczna.

Stężenie D-dimerów
ważne jest odróżnienie produktów degradacji fibrynogenu
(hiperfibrynoliza) i fibryny (trwanie zakrzepu!).
FDP-DD jest to swoisty produkt degradacji fibryny. Jest
skutkiem trawienia przez plazminę fibryny związanej
krzyżowo przez cz. XIII w czasie stabilizacji polinerów
fibryny. Ma znaczenie w rozpoznawaniu DIC i zakrzepicy żył
głębokich. Zalecane badanie po zabiegach operacyjnych, u
chorych leżących.
Metodami półilościowymi
-cząsteczki lateksu opłaszczone monoklonalnymi Ab
przeciwko D-dimerom aglutynują w obecności produktów
degradacji fibryny . Surowica: 500g/ml mocz 20g/ml
(0,5g/ml) Wzrost stężenia D-dimeru
-

CZAS TROMBINOWY
ocena ostatniej fazy krzepnięcia. Zmiany fibrynogenu w
fibrynę, z wyłączeniem etapu stabilizacji skrzepu przez cz.
XIII.
Przydatny gdy stwierdza się przedłużenie cz.
protrombinowego i aPTT.
Mierzy się czas krzepnięcia po dodaniu stałej ilości
trombiny.
Przedłużony:
– hipofibrynogenemia, dysfibrogenemia,
-
obecność inhibitorów: heparyny, FDP
NORMA:16-21 sek.

FIBRYNOGEN
białko syntetyzowane w wątrobie,
białko ostrej fazy,
kofaktor agregacji płytek – fibrynogen przez wiązanie
glikoproteiny IIb i IIIa bierze udział w tworzeniu czopu
płytkowego.
Substrat dla tombiny, po odszczepieniu fibrynopeptydów A i
B,cząstki fibrynogenu polimeryzują i tworzą fibrynę ,która
pod wpływem cz.XIII ulega usieciowieniu - wzmacnia on
czop płytkowy i przymocowuje go do ściany naczynia. Te
mechanizmy powodują zatrzymanie krwawienia. Fibrynogen
pośrednio uczestniczy w gojeniu się ran, ponieważ na
powierzchni utworzonej z niego fibryny współdziałają
komórki biorące udział w tym procesie, tj. erytrocyty, płytki
krwi, makrofagi i fibroblasty.
Fibrynogen odgrywa istotną rolę w regulacji lepkości krwi.

Najczęściej stosowaną metodą do oznaczania fibrynogenu
jest zmodyfikowany pomiar czasu trombinowego opisany w
1957 roku przez Claussa-metoda koagulometyczną .
Jest to czas krzepnięcia rozcieńczonego osocza po dodaniu
wysokich stężeń trombiny jest odwrotnie proporcjonalnie do
stężenia fibrynogenu. Badanie to jest więc miarą ostatniego
etapu wspólnej drogi w kaskadzie krzepnięcia.
Wartości prawidłowe stężenia fibrynogenu wynoszą: 1,8-3,5
g/l.
Także metodami turbidymetrycznymi lub
nefelometrycznymi (bardziej wiarygodne w dgn.DIC) oraz
immunochemicznymi stosowanymi w dgn dysfibrynogenemii
lub celach badawczych.

Stężenie fibrynogenu -
podwyższone:
-
fizjologicznie podczas miesiączki i w ciąży,
-
stany zapalne jako białka ostrej fazy (ostre stany
gorączkowe, choroby zakaźne, duże zabiegi operacyjne,
urazy). Brak wzrostu fibrynogenu w tych stanach może
sugerować wzmożoną fibrynolizę lub zespół wykrzepiania
sródnaczyniowego (DIC).
-
w przebiegu chorób nerek (zespół nerczycowy, kłębkowe
zapalenie nerek, zespół hemolityczno-mocznicowy),
-
kolagenozy (toczeń ruminiowaty, zapalenie guzkowe
okołotetniczne), w nocnej napadowej hemoglobinurii,
-
choroby nowotworowe , plamica zakrzepowa małopłytkowa.
-
Ponadto fibrynogen jest czynnikiem ryzyka choroby
niedokrwiennej serca.

Stężenie fibrynogenu -
obniżone:
wrodzone niedobory fibrynogenu, afibrynogenemia,
hipofibrynogenemia
choroby wątroby (piorunujące zapalenie wątroby, marskość
wątroby, martwica wątroby)\
zespół rozsianego wykrzepiania śródnaczyniowego (DIC),
skazy fibrynolityczne (pokrwotoczne, pourazowe,
pooparzeniowe, ostra białaczka promielocytowa,
nowotwory) ,
monoukleoza zakaźna,
leki: fenobarbital, streptokinaza, urokinaza, L-asparaginaza.

Białko C
Aktywność białka C metodą:
-koagulometryczną -na podstawie aPTT- roz.osocze mieszane
jest z osoczem niedoborowym b.C. Po dodaniu jadu daboi
będącego silnym aktywatorem b.C następuje przedłużenie
czasu wytrącenia skrzepu proporcjonalne do zawartości
białka C w próbce.
- amidolityczną-spektofotometryczny pomiar szybkości
rozszcepiania substratu chromogennego przez aktywowane
wcześniej b.C
Prawidłowa aktywność-70-140%
Stężenie białka C-immunochemicznymi.prawidłowe 3-6 mg/l
Oznaczane przy podejrzeniu wrodzonej trombofilii.

Białko S
Aktywność na podstawie przedłużenia PT i aPTT w próbce
osocza rozcieńczonej i mieszanej z osoczem z niedoborem
b.S, po dodaniu aktywowanego b.S.
Stężenie zaś wolnej frakcji metodami immunochemicznymi
po wcześniejszym wytrąceniu frakcji związanej lub stosując
przeciwciała. Duże problemy ze standaryzacją metody
ponieważ może wystapić reaktywność z nieaktywnymi
postaciami b.S.

OPORNOŚĆ NA AKTYWNE BIAŁKO C
APC hamuje powstanie trombiny przez wybiórcze trawienie
cz.V,VIII.Zmiany w budowie bądź czynności tych czynników
lub zmniejszenie ich stężenia mogą prowadzić do oporności
na APC;
1.zmodyfikowane ozn. aPTT w obecności APC bądź
bez APC
-dodanie APC do osocza powoduje co najmniej 2-krotne
przedłużenie aPTT-wynik jako wspłóczynnik wrażliwości na
APC(APC:SR) czyli stosunek aPTT po dodaniu APC do aPTT
bez podania APC.Jeśli nie rozciencza się próbki badanej
osoczem pozbawionym cz.V to fałszywie dodatnie wyniki
uzyskuje się u chorych leczonych doustnymi
antykoagulantami lub heparyną i z niedoborem cz.II,VIII i
białka S.
2.oznaczenie PT ,czasu krzepnięcia po dodaniu jadu żmii
Russella lub cz.Xa w obecności APC z użyciem osocza
pozbawionego cz.V
Brak oporności –APC:SR>2

Oporność stwierdza się u osób:
-z czynnikiem V Leiden- odróżnienie homo- od
heterozygoty,
-cz.V Cambrige(Arg 306Thr) lub cz.V Hong Kong(Arg
306Gly)
-zespołem antyfofolipidowym(APS)
-zwiększoną aktywnością cz.VIII,
-kobiet w ciąży lub stosujących doustne środki
antykoncepcyjne.
OPORNOŚĆ NA APC (BEZ WZGLĘDU NA PRZYCZYNĘ)
ZWIĘKSZA RYZYKO WYSTĄPIENIA ŻCHZZ.

OZNACZANIE AT III
metody oznaczenia aktywności oparte na hamowaniu
trombiny i cz.Xa przez ATII w próbce po dodaniu heparyny.
Resztkowa aktywność trombiny oznacza się:
met.amylolityczną- spektrofotometryczny pomiar szybkości
rozszczepiania substratu chromogennego do barwnego
produktu
met.koagulometryczną- mierząc szybkość tworzenia
skrzepu po dodaniu fibrynogenu.
Stężenie ATII-oznacza się metodami immunochemicznymi.
NORMA:aktywność –80-120% normy
Stężenie –0,19-0,31 g/l

INNE CZYNNIKI KRZEPNIĘCIA
Oznaczenie cz.II,V,VII,VIII,IX,X,XI,XII w osoczu
ubogopłytkowym.
Aktywność można oznaczać metodami
1.
Koagulometrycznymi- stanowią modyfikację czasu
PT(V,VII,X) oraz aPTT(VIII,IX,XI),
2.
Spektofotometrycznymi z użyciem substratów
chromogennych- uważane za czulsze ponieważ pozwalają
na duże rozc. Co ogranicza wpływ innych czynników i
antykoagulantów
3.
Metodami immunochemicznymi.
NORMY:
V,VII,IX,X,XI,XII- 70-130% normy,
VIII -50-150%
Wykonuje się w przypadku wyjaśnienia przyczyny
nieprawidłowego PT lub aPTT. W hemofiliach do
monitorowania leczenia koncentratami cz.krzepnięcia

Oznaczenie cz.vW
Badanie efektu stabilizującego cz.VIII-modyfikacja aPTT
wykonanego w próbce osocza zmieszanego z osoczem z
niedoborem cz.VIII oraz badanie wiązania vWF z cz.VIII
Badanie agregacji pod wpływem rystocetyny w jednym lub
kilku stężeniach(kofaktor rystocetynyvWF:RC)
Stężenie cz.vWF metodami immunochemicznymi
Multimeryczne formy cz.vWF za pomocą elektroforezy i
swoistych metod detekcji (autoradiografii)
Norma:stężenia i aktywność-70-150%
Indukowana rystocetyną agregacja <20%

OZNACZENIE PRZECIWCIAŁ
PRZECIWPŁYTKOWYCH METODĄ
IMMUNOENZYMATYCZNĄ
Immunogenność krwinek płytkowych związana jest z
glikoproteinami na błonie komórkowej swoistych dla płytek
ale także wspólne z antygenami erytrocytów –ABH,
Lewis,P,Li oraz limfocytami –HLA kl lI głównie locus A i B.
Dlatego też obecne w krążeniu chorego alloprzeciwciała
przeciwko antygenom erytrocytów czy limfocytów mogą
niszczyć także płytki krwi.
Obecnie uznanych jest 9 układów antygenowych płytek od
HPA-1 do 9.
W wyniku procesów autoimmunizacyjnych glikoproteiny
błonowe mogą być przyczyną powstawania autoprzeciwciał.
Najczęściej spotykane są przeciwciała skierowane do
glikoproteiny:
IIb /IIIA, Ib /IX, IV, Ia/IIa.

ZESTAWY IMMUNOENZYMATYCZNE
GTI-PAKPLUS- test wykrywający przeciwciała p/antygenom
HLA kl.I oraz Ib/IX, IV także niektórych IIb/IIIa i Ia/Iia z
możliwością rozróżnienia przeciwciał p/HPA-1 lub HPA-3a,1b lub
3b,5a,5b
GTI –PAKAUTO-test wykrywający przeciwciała przeciwko
glikoproteinom płytkowym IIb/IIIa i Ia/IIa rozróżniający
przeciwciał krążące i opłaszczone na płytkach.
ZASADA-surowica lub eluat umieszczane są w studzienkach
płytki których dno opłaszczone glikoproteinami płytkowymi i
antygenami HLA kl.I. Obecne w surowicy przeciwciała p/płytkom
i antygenom HLA kl.I zostają związane w studzienkach,
natomiast inne są wypłukiwane. Następnie dodaje się r-r
poliwalentnej surowicy koziej p/immunoglobulinom człowieka
sprzężonej z fosfatazą alkaliczną.Po inkubacji surowica zoztaje
odpłukana po czym dodany zostaje z-r substancji barwnej-
fosforan paranitrofenylu(PNPP).Po kolejnej inkubacji barwa
zostaje zatrzymana r-rem wodorotlenku sodu.Pomiar
intensywności zabarwienia bada się za pomoca
spektrofotometru przy długości fali 405 lub 410 nm(ref,490nm)
Próbki zanieczyszczone bakteryjnie, lipemiczne, zhemolizowane
mogą prowadzić do zafałszowania wyniku testu.

-można obserwować zjawisko agregacji pod wpływem różnych
aktywatorów.
Agregację można badać:
a) w osoczu bogatopłytkowym metodą turbidymetryczną
Działa na zasadzie luminometru mierzącego natężenie wiązki
światła widzialnego.
Niezagregowane płytki dają największe zmętnienie.
-aktywatory : arachidonian sodu, ADP, kolagen,
epinefryna,rystocetyna.
Pełny proces agregacji- dwufazowa krzywa odpowiadająca
fazom agregacji
b) metodą impendacyjną w pełnej krwi – polega na pomiarze
oporności elektrycznej.
AGREGOMETR

Zastosowanie agregometrii
:
-w diagnostyce wrodzonych i nabytych defektów płytek krwi,
-w diagnostyce choroby von Willebranda,
-ocenie reakcji na leki antyagregacyjne.

PFA-100® -
Analizator do oceny prawidłowości procesu hemostazy
pierwotnej. Uznany w 1998 roku za standard - pozwala na
szybkie wykrycie dysfunkcji płytek, zarówno dziedzicznych,
jak i nabytych czy wywołanych przez leki o działaniu
przeciwpłytkowym
.
w warunkach in-vitro szybko i łatwo mierzy czas utworzenia
się czopu płytkowego, wykorzystując do tego rzeczywiste
środowisko hemodynamiczne. Do pomiaru potrzebna jest
niewielka ilość krwi pełnej (800 µl) pobranej na cytrynian
dopuszczalne probówki Sarstedt-Monovette i BD Vacutainer)
i jednorazowe wkłady testowe:

Wkład testowy Kolagen/Epinefryna służy do wykrywania
dysfunkcji trombocytów wywołanej czynnikami
wewnątrzpłytkowymi, niedoborem czynnika von
Willebranda, lub defektów spowodowanych przyjmowaniem
leków przeciwpłytkowych. Ze względu na wysoką
predykcyjną wartość testu (ponad 90%) w wykrywaniu
defektów hemostazy pierwotnej – jest idealnym narzędziem
do testowania wzmożonej tendencji krwawienia przed
jakimkolwiek zabiegiem operacyjnym.
COL/EPI: 85 – 165 sek.

Wkład testowy Kolagen/ADP znakomicie sprawdza się w
potwierdzaniu lub wykluczaniu efektu zażywania leków
przeciwpłykowych na bazie kwasu acetylosalicylowego
(ASS). Zredukowany czas okluzji poniżej 73 sek. wskazuje
na wzmożoną aktywność płytek i powinien być wskazaniem
do wtórnej terapii lekowej u pacjentów przygotowywanych
do zabiegów kardiologicznych i implantacji stentów, by
zapobiec zakrzepicy. COL/ADP: 71 – 118 sek.
Liczba płytek powinna wynosić powyżej 100 000 kom. / µL,
wartość hematokrytu powyżej 30%. Test powinien być
przeprowadzony najwcześniej po 15 minutach od pobrania,
nie później niż do 2 godz. po pobraniu krwi.

Pomiar okuzji za pomoca PFA-100 może nie wykryć:
- łagodnych trombocytopatii,
-łagodnych defektów czynnika von Willebranda,
-różnicować pomiędzy chorobą von Willebranda a defektami
płytek krwi.

Agregometr do szybkiej oceny funkcji płytek-
PLATELETWORK’S
- pozwala na procentową ocenę
agregacji w świeżych próbkach krwi pobranej podczas
interwencyjnych zabiegów kardiologicznych Pozawala na
równoczesne pomiary przyłóżkowe liczby płytek i ocenę
funkcji agregacyjnej.

Metoda cytometrii przepływowej
Ocena struktury i funkcji płytek,
Wykrywa aktywację wykorzystując reakcję specyficznych
przeciwciał monoklonalnych z białkami błonowymi.
CD62(P-selektyny),kompleksu GP IIb/IIIa
Pomiar RNA płytkowego-pomocne w różnicowaniu
małopłytkowości.
Ograniczenie-tylko wyspecjalizowane ośrodki posiadające
cytometr przepływowy.

Badania wykonywane w przypadku
podejrzenia małopłytkowości poheparynowej
-spowodowana występowaniem obecnością IgG
skierowanych przeciwko neoepitopom powstałym w
kompleksie heparyna-cz.płytkowy 4.
-znaczenie ma wczesne rozpoznanie.Na początku terapii
zaleca się monitorowanie liczby płytek.
-przeciwciała wykrywa się: metodami czynnościowymi lub
immunoenzymatyczną(ELISA)

TROMBOELASTOGRAFIA
Metoda pozwalająca na graficzne przedstawienie dynamiki
procesów krzepnięcia i fibrynolizy oraz właściwości
fizycznych tworzącego się skrzepu.
Badanie wykonuje się w pełnej krwi cytrynianowej po
dodaniu jonów wapnia. Ocenie poddaje się parametry
odpowiadające różnym etapom krzepnięcia i
fibrynolizy(R,K,alfa,MA,Ly30
PrzedłużonyR-niedobory czynników osoczowych,
Zmniejszony kąt alfa- niedobór fibrynogenu,
Obniżenie amplitudy MA-zaburzenia płytek
Ly 30-procesy fibrynolizy.

Wskazania:
-ocena hemostazy w czasie zabiegów kardiochirurgicznych,
-w chirurgii wątroby,
-dgn stanów nadkrzepliwości i zaburzeń hemostazy w ciąży,
-monitorowanie pacjentów z wrodzonymi skazami
krwotocznymi,
-ocenie reakcji na rekombinowany aktywowany cz.VII i leki
antyfibrynolityczne

FIBRINTIMER II
Półautomatyczny koagulometr skierowany do małych pracowni
i małej ilości wykonywanych badań
Oznaczenie metodą optyczno-mechaniczną
Możliwość oznaczenia PT, aPTT, TT, fibrynogenu,
cz.krzepnięcia,ATIII,
Brak automatyzacji,oznaczenie stężenia czynników na
podstawie krzywej wykreślanej dla każdego nowego
zestawu odczynników

SYSMEX CS-2000i / 2100i

najnowszy model analizatora koagulologicznego
przeznaczony do laboratoriów o średniej wydajności(ok..180
oznaczeń (PT+APTT)/h)
system detekcji HIL (Hemolysis – Icterus - Lipemia)
weryfikujący jakość próbki jeszcze przed analizą, w celu
wykrycia ewentualnych substancji interferujących
jednoczesne wykorzystanie 5 długości fal przy testach
wykrzepialnych – pozwalający na automatyczny wybór
najlepszej długości fali dla konkretnej próbki.
automatyczna rejestracja czynności konserwacyjnych wraz z
dostarczaniem dokumentacji potwierdzającej ich wykonanie
automatyzacja funkcji: analiz powtórnych, powtórek z
rozcieńczeniem, zlecenia kaskady testów
wprowadzenie funkcji wielokrotnych rozcieńczeń (Multi-
Dilution Analysis = MDA)
rozbudowany systemem kalibracji
łatwość integracji z laboratoryjną siecią informatyczną

Koagulometr BCS® XP

W pełni automatyczny system zapewnia bardzo wydajną analizę
testów wykonywanych w pracowni hemostazy – zarówno tych
rutynowych, jak i specjalistycznych - do 380 testów na godzinę
(PT).
pozwala na dopasowanie pracy analizatora do potrzeb
użytkownika – wykonywanie oznaczeń typu „reflex-testing”,
definiowalne profile, badania cito.
Możliwość wykonywania wszystkich technikami pomiaru
jednocześnie: wykrzepiania, chromogenną, immunologiczną;
Zastosowanie pomiaru przy kilku długościach fali pozwala na
oznaczanie próbek lipemicznych, żółtaczkowych czy
hemolizowanych;
Poszerzony zakres rozcieńczeń (do 1:1200) i szeroki zakres
pomiarowy dla testów specjalistycznych
Autoasystent – opcja oprogramowania pozwalająca na
sprawniejsze zarządzanie próbkami wymagającymi ponownego
pomiaru, rozcieńczenia, przekalkulowania lub zlecenia
dodatkowych testów
Automatyczne monitorowanie poziomu odczynników;
Kontrola jakości uwzględniająca karty Levey-Jeningsa i reguły
Westgarda;
Możliwość zapamiętania krzywych kalibracyjnych dla 6 różnych
serii tego samego odczynnika .

Testy rutynowe
PT
APTT
Fibrynogen pochodny i wg Clauss’a)
TT
Czas batroksobinowy
AT III
D-Dimer
Tendencje do krwawień
Czynnik II - Czynnik XI
Czynnik XIII
Czynnik von Willebranda – Antygen
Badania przesiewowe w kierunku trombofilii - ProC® Global
Czynnik V-Leiden
Białko C
Białko S
Antykoagulanty tocznia
Czynnik XII

Fibrynoliza
Plazminogen
Alfa-2 antyplazmina
PAI
C1-inhibitor
Komplement
Monitorowanie
Heparyna
Heparyna niskocząsteczkowa
Hirudyna

Sysmex®CA-560 kompaktowy analizator
koagulometryczny
w pełni automatyczny koagulometr, przeznaczony do
laboratoriów o małym i średnim obciążeniu, wydajność ok. 20-
30 próbek /h
oznaczenia metodami wykrzepiania, chromogennymi i
immunologicznymi – oferując duży wybór testów przy czym aż 5
parametrów może być wykonywanych jednocześnie np. PT,
APTT, fibrynogen, TT, AT III lub D-Dimer.
Oznaczanie fibrynogenu metodami: ilościową, jak i wyliczenia z
PT;
Testy rutynowe
Fibrynogen (pochodny i wg Claussa)
APTT
TT
Czas batroksobinowy
AT III
D-Dimer Czynnik VIII
Trombofilia
Białko C
Antykoagulanty tocznia
Heparyna niskocząsteczkowa

BADANIA MORFOLOGII I FUNKCJI PŁYTEK KRWI
-
liczba płytek, objętość płytek
-rozmaz, mikroskop elektronowy
-czas krwawienia (Ivy, Duke)
-czas życia płytek krwi (metody izotopowe)
-PFA-100 (adhezja/agregacja płytek)
-agregacja pod wpływem ADP, kolagenu, rystocetyny,
adrenaliny, kw. arachidonowego

MAŁOPŁYTKOWOŚĆ RZEKOMA
(PSEUDOTROMBOCYTOPENIA)
-
artefakt laboratoryjny
-nieistotna klinicznie
-aglutynacja płytek
in vitro
w obecności EDTA
-ok. 0,2% zdrowych osób (przeciwciała przeciw epitopom
błony płytek, aktywne w wyniku obniżenia stężenia jonów
Ca
2+
w osoczu)
-infekcje, leki
-pobrać krew na cytrynian lub heparynę + ocenić rozmaz krwi
obwodowej

ZABURZENIA WYDZIELANIA ZIARNISTOŚCI
PŁYTKOWYCH
Choroba puli magazynowej (wrodzony brak ziarnistości α
/zespół szarych płytek/ lub ziarnistości gęstych)
Zaburzenia wydzielania ziarnistości do krwi
łagodna skaza płytkowa
brak drugiej fazy agregacji pod wpływem ADP/adrenaliny,
osłabienie agregacji wywołanej kolagenem
prednizon, desmopresyna, KKPł
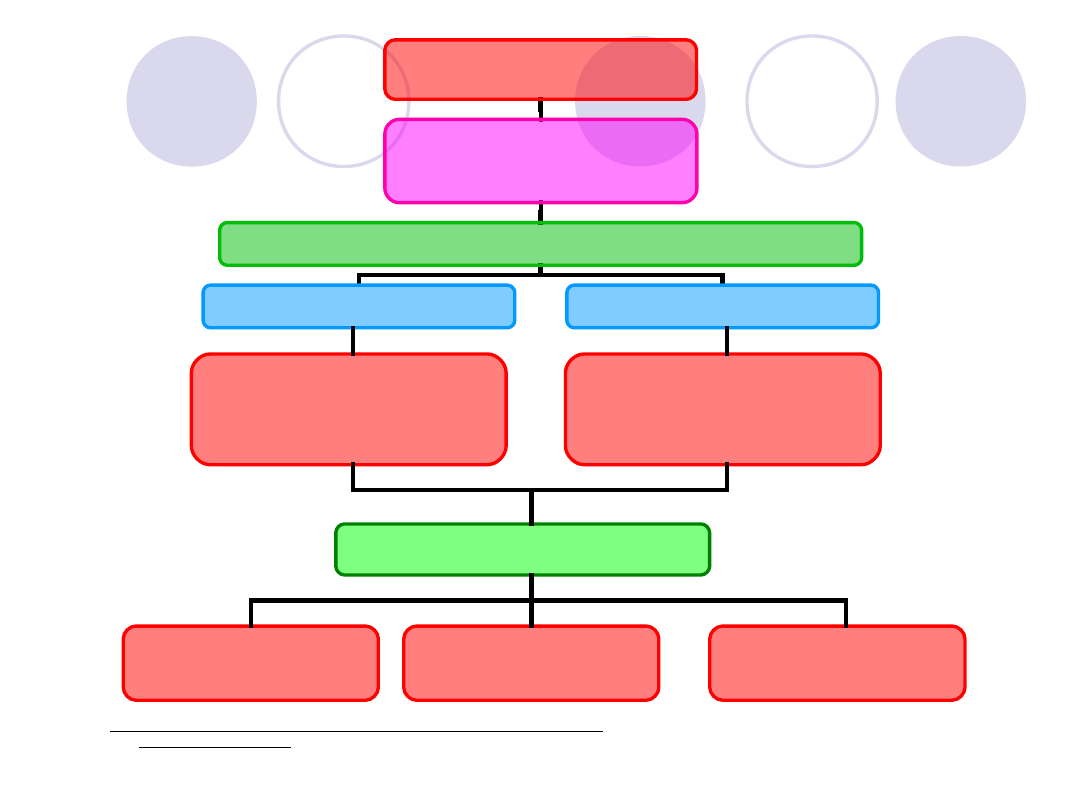
Rys. Schemat postępowania diagnostycznego w przypadku niewyjaśnionej
skłonności dokrwawień
WYWIAD RODZINNY
cechy krwawienia
BADANIE FIZYKALNE
choroby współistniejące
cechy krwawienia
Badania
podstawowe
-
przesiewowe
Naczyniowo
-
płytkowe
Osoczowe
Badania uzupełniające
naczyniowe,płytkowe,osoczowe
Naczynia krwionośne
_ testy czynnościowe
i badania specjalistyczne
Płytki - agregacja
Reakcja uwalniania
Badania cytometryczne
Czynniki krzepnięcia
Inhibitory (przeciwciała)
Krzepnięcie
APTT
PT
Fibrynogen(czas trombinowy)
Liczba płytek
Rozmaz krwi obwodowej
Czas okluzji i inne
Czas krwawienia

SKAZY
NACZYNIOWE
rzadkie, raczej łagodne
krwawienia: wybroczyny, krwotoczne wykwity skórne i na
błonach śluzowych, krwawienia z dziąseł, siniaczenie
(rzadziej: inne krwawienia śluzówkowe)
prawidłowa funkcja płytek, sprawny układ osoczowy
czas krwawienia może był przedłużony
„kruchość naczyń”: test Rumpela i Leedego (objaw
opaskowy) – może być dodatni też w małopłytkowości
(historyczne znaczenie)
biopsja skóry
obraz kliniczny, prawidłowe badania układu płytkowego
i
osoczowego

I.WRODZONE
1.
naczyniakowatość krwotoczna,
2.
zespół Marfana,
3.
zespół Ehlersa-Danlosa
4.
Wrodzona łamliwość kości
II.NABYTE
plamica Schonleina-Henocha(alergiczna)
plamica polekowa,
plamica w przebiegu zakażeń,
plamica ortostatyczna,
plamica starcza,
plamica w nadczynności kory nadnerczy,
gnilec.

Płytkowe skazy krwotoczne
Skazy związane z nieprawidłową liczba płytek
Małopłytkowości:
1. spowodowane zmniejszonym wytwarzaniem płytek
-niedokrwistość alpastyczna,
-nacieczenie szpiku( białaczki, przerzuty
nowotworowe),
-zakażenia wirusowe,
-niedobory wit.B12 i/lub kw.foliowego

Wrodzone uwarunkowane zmniejszonym
wytwarzaniem płytek
Wrodzona hipoplazja magakariocytowa,
Współistniejąca z brakiem kości promieniowej
N.Fanconiego,
Dziedziczna związana z zaburzeniem dojrzewania
megakariocytów
Zespoły z mutacją genu MYH 19 na chromosomie 22q12-13
Zespół Alporta,
Zespół Wiskotta-Aldricha

Nabyte uwarunkowane zmniejszonym
wytwarzaniem płytek
-
niedokrwistość alpastyczna,
-nacieczenie szpiku( białaczki, przerzuty nowotworowe),
-zakażenia wirusowe,
-niedobory wit.B12 i/lub kw.foliowego
Małopłytkowość cykliczna,
Promeniowanie jonizujące,
Po leczeniu mielosupresyjnym,
Wskutek alkoholizmu,
Nocna napadowa hemoglobinuria

2. spowodowane nadmiernym niszczeniem płytek
Immunologiczne:
- samoistna plamica małopłytkowa,
-małopłytkowość poprzetoczeniowa,
-małopłytkowość polekowa(w tym poherparynowa HIT),
-w przebiegu chorób autoimmunologicznych -toczeń
rumieniowaty układowy,
-w przebiegu chłoniaków złośliwych,
Po przeszczepieniu szpiku,
Po leczeniu surowicą antylimfocytową

Nieimmunologiczne
-zakażenia,
DIC,
Zespół hemolityczno-mocznicowy
Zakrzepowa plamica małopłytkowa.
Uwarunkowane nieprawidłowym rozmieszczeniem płytek
-hipersplenizm

Nadpłytkowości
1.Pierwotne-
Nadpłykowość samoistne,
Inne zespoły mieloproliferacyjne (czerwienica
prawdziwa,zwłóknienie szpiku,przewlekła białaczka
szpikowa)
2.Wtórne-
Zapalenia ostre i przewlekłe,
Choroba nowotworowa,
Niedobór żelaza,
Po krwotokach,
Po splenektomii i innych zabiegach chirurgicznych

Skazy związane z zaburzeniem czynności
płytek
Wrodzone
:
Zaburzenia błony płytkowej:
-zaburzenia adhezji –ch.Bernarda-Souliera,
-zaburzenia agregacji- trombastenia Glanzmanna,
-zaburzenia w zakresie uwalniania substancji wewnątrzpłytkowych-
ch.puli magazynowej (niedobór ziarnistości gęstych); zespół
szarych płytek(niedobór alfa ziarnistością)
-defekt receptora kolagenu (Ia/IIa)
-zaburzenia prokoagulacyjnej aktywności płytek- zespół Scotta-
zaburzenie transportufosftydyloseryny z wewnetrznej do
zewnętrznej warstwy błony płytkowej,zmniejszona ilość miejsc
wiążących cz.Va i Xa,zmniejszona generacja trombiny na
powierzchni płytek

-
zaburzenia sekrecji lub transdukcji sygnału
:
zaburzenia w zakresie uwalniania substancji
wewnątrzpłytkowych- ch.puli magazynowej (niedobór
ziarnistości gęstych); zespół szarych płytek(niedobór alfa
ziarnistością)
-defekt receptora kolagenu (Ia/IIa)
-zaburzenia prokoagulacyjnej aktywności płytek- zespół Scotta
-zaburzenie transportu fosftydyloseryny z wewnetrznej do
zewnętrznej warstwy błony płytkowej,zmniejszona ilość
miejsc wiążących cz.Va i Xa,zmniejszona generacja
trombiny na powierzchni płytek
-skaza krwotoczna płytkowa Quebec

Nabyte:
-polekowe,
-mocznica,
-przewlekłe zespoły mieloproliferacyjne,
-gammapatie monoklonalne,
-choroby wątroby,
-DIC,
-krążenie pozaustrojowe.

Zespół Bernarda i Souliera
-
wrodzony defekt syntezy kompleksu glikoprotein Ib/IX/V
(receptor dla vWF) ciężka skaza skórno-śluzówkowa
-długi czas krwawienia, niewielka małopłytkowość, duże
płytki, brak agregacji pod wpływem rystocetyny
Trombastenia Glanzmanna
-wrodzony defekt syntezy kompleksu glikoprotein IIb/IIIa
(receptor dla fibrynogenu, vWF, trombospondyny,
fibronektyny)
skaza skórno-śluzówkowa od wczesnego dzieciństwa,
- długi czas krwawienia, liczba płytek w/n, brak agregacji pod
wpływem ADP, kolagenu, kw. arachidonowego, adrenaliny,
agregacja pod wpływem ristocetyny PRAWIDŁOWA

Osoczowe skazy krwotoczne
I.WRODZONE
1.
Hemofilia A,B
2.
Choroba von Willebranda,
3.
Niedobór czynników-I,II,V,VII,X,XI,XIII.
II.NABYTE
Niedobór witaminy K,
Choroby watroby,
DIC,
Pierwotne uczynnienie fibrynolizy,
Krążące antykoagulanty.

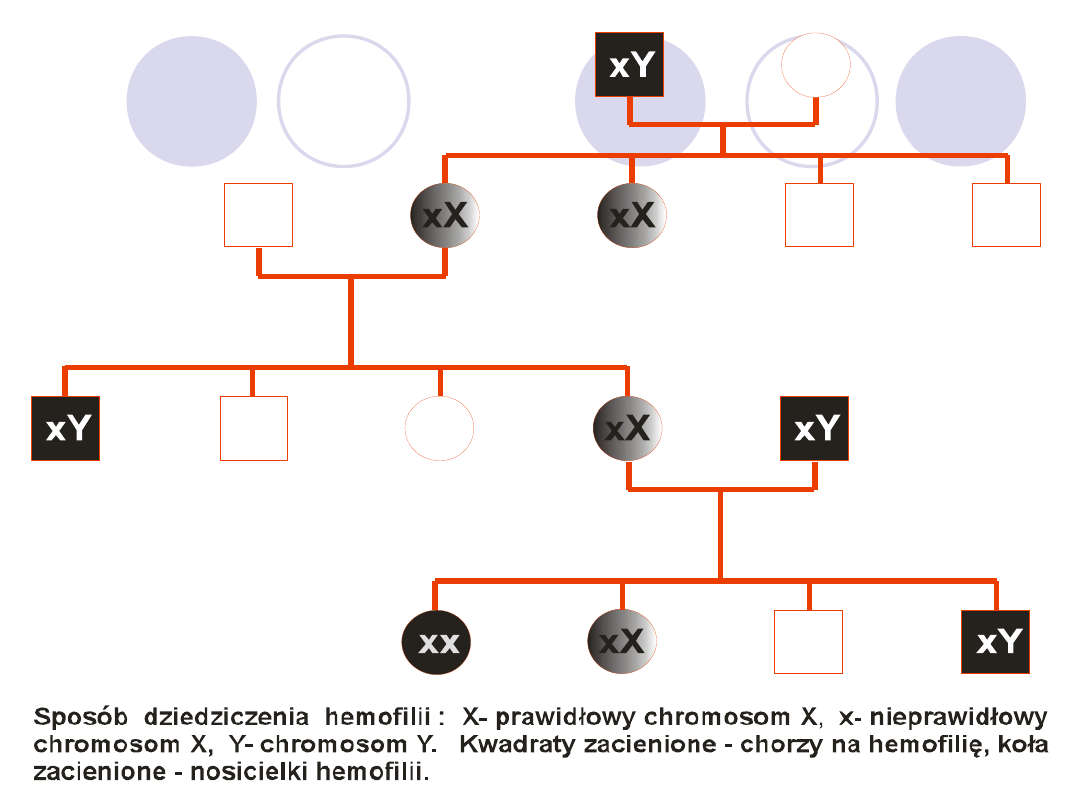
HEMOFILIA A
Wykrywana przeciętnie u 1 na 50-10tys. męskich
noworodków niezależnie od rasy
Brak syntezy cz.VIII lub jej zmniejszenie lub synteza
nieprawidłowego białka,
Gen czynnika zlokalizowany na chromosomie X,
Sprzężony z płcią- ujawnia się u mężczyzn, kobiety są
nosicielkami
U kobiet nosicielek aktywność zmniejszona do połowy gdy
spadnie poniżej 25% normy-skłonność do krwawień
W badaniach laboratoryjnych,przedłużone aPTT,
zmniejszona aktywność cz.VIII

MECHANIZM ZABURZEŃ
-
zewnątrzpochodny szlak krzepnięcia nie jest w stanie
zapewnić prawidłowej hemostazy stąd skłonność do
krwawień
-
cz.X a jest szybko inaktywowany przez AT i TFPI- za mała
ilość wygenerowanej trombiny aby przekształcić fibrynogen w
fibrynę
-
Ale ta ilość trombiny wystarcza do aktywacji czVIII który
wzmacnia aktywację cz.Xa i następuje wyrzut trombiny co
umożliwia utworzenie stabilnej fibryny.
-
Ponadto cz.Xa szybko jest włączany w kompleks
protrombinazy i unika szybkiej inaktwyacji przez AT i TFPI.
-
Wykryto ok.1500 różnych mutacji w genie cz.VIII,niektóre
predysponują do najgroźniejszego powikłania jakim jest
wytworzenie przeciwciał p/cz.VIII

a)
ciężka- aktywność cz.VIII poniżej 1%,ok.50-60 %
przypadków
nawracające samoistne krwawienia śródstawowe-
zniszenie stwów, mięśni i kalectwo; samoistne wylewy do
narządów wewnętrznych, krwotki z p.pokarmowego,
krwiomocz, krwawienia śródczaszkowe. Bez uzupełnienia
niedoborowego czynnika chory może się wykrwawić.
b) umiarkowana- aktywność cz.VIII-1-5% normy,ok.25-30%
przypadków -wylewy sporadyczne ,ale krwawienia
pourazowe są równie poważne
c) łagodna- aktywność cz.VIII 6-50% normy ,ok.15-20%
przypadków charakteryzuje się krwawieniami w
następstwie dużych urazów i po operacjach
chirurgicznych.
3 postacie: ciężka, umiarkowana i łagodna
XY
XY
XY
XY
XX
XY
XX

HEMOFILIA B
Wrodzony niedobór cz.IX
Sposób dziedziczenia, obraz kliniczny oraz
postacie choroby podobne jak w hemofilii A
Rozpoznanie musi być potwierdzone oznaczeniem
aktywności cz.IX

Choroba von Willebranda
Najczęstsza łagodna lub umiarkowana skaza krwotoczna
skórnośluzówkowa (1%ogólnej populacji) dziedziczona
autosomalnie ,
Podłożem molekularnym choroby jest defekt/niedobór
osoczowej glikoproteiny-cz.vW prowadzący do zaburzeń
hemostazy w postaci skazy lub zakrzepicy
Czynnik von Willebranda jako multimer ma podwójną
funkcję- uczestniczy w adhezji i agregacji płytek oraz tworzy
kompleks z cz.VIII chroniąc go przed degradacją -zaburzenia
na poziomie hemostazy wtórnej jaki pierwotnej.
Objawy- krwawienia z nosa, dziąseł, siniaki,u kobiet
przedłużąjące się krwawienia miesięczne, krwawienia po
ekstrakcji zębów i zabiegach chirurgicznych. W cięższych
postaciach krwawienia dostawowe, pourazowe krwawienia
domięśniowe.
Objawy klasycznej, wrodzonej ch.vW łagodnieją z wiekiem,
natomiast nasilające się objawy w wieku starszym to
postać nabyta rozwijająca się.
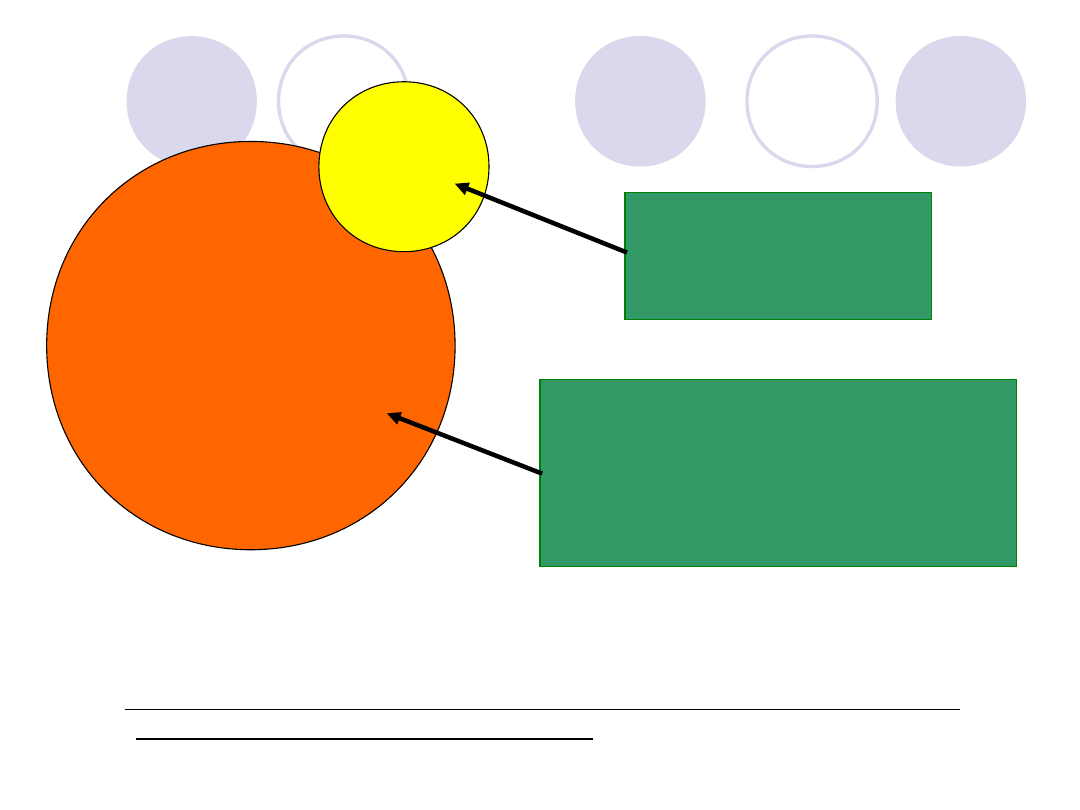
Rys. Kompleks czynnika von Willebranda i czynnika VIII oraz podstawowe
badania laboratoryjne obu czynników
Czynnik
von Willebranda
Czynnik
VIII
VIII:C i VIII:Ag
vWF:Ag
vWF:R-Co
Inne specyficzne
badania

Choroba jest heterogenna zarówno pod względem
genetycznym jak i klinicznym
Różnicowanie typów opiera się na analizie obrazu
klinicznego, wywiadzie rodzinnym i analizie laboratoryjnej.

TY
P
SKAZA KRWOTOCZNA
WYNIKI
1
Łagodna lub
umiarkowana
(niedobór ilościowy)
vWf:Ag,R:Cof,VIII:c-<50%
Rozkład multimerów prawidłowy
2A
Łagodna lub
umiarkowana
(niedobór jakościowy)
najczęstsza postać
vWf:Ag,R:Cof,VIII:c-zmniejszone
Brak dużych i pośrednich
multimerów
2B
Łagodna lub
umiarkowana
vWf:Ag,R:Cof,VIII:c-zmniejszone
Brak dużych multimerów,
małopłytkowość,RIPA zwiększony
2M
Łagodna lub nasilona
vWf:Ag,R:Cof,VIII:c-zmniejszone
Obniżone R;Cof pomimo obecności
dużych i pośrednich multimerów,
2N
Łagodna lub nasilona
zwiększonyobniżone VIII:c,
prawidłowy rozkład multimerów
3
Ciężka
(niedobór ilościowy)
vWf:Ag,R:Cof,VIII:c- znacznie
zmniejszone(<5%)
Brak lub śladowa ilość multimerów

DIC- zespół rozsianego wykrzepiania
wewnątrznaczyniowego
.
Zespół wtórny do wielu różnych stanów
klinicznych
Aktywacja krzepnięcia z wytworzeniem dużej
ilości fibryny która wiąże płytki i formuje
zakrzepy blokujące przepływ krwi w drobnych
naczyniach krwionośnych stąd niedokrwienne
uszkodzenie wielu narządów,
Dochodzi do zużycia płytek,fibrynogenu i innych
cz.krzepnięcia co objawia się skazą krwotoczną
Częstość DIC-1:1000 hospitalizowanych chorych

Czynniki inicjujące DIC
:
Nadmierne wytwarzanie trombiny w szlaku zależnym od
czynnika tkankowego i aktywnego cz.VII oraz upośledzenie
funkcjonowania endogennych inhibitorów krzepnięcia-
ATIII,białka C,TFPI.
Wzrost stężenia inhibitora aktywatora plazminogenu typu 1-
hamuje aktywność układu fibrynolizy co upośledza zdolność
organizmu do rozpuszczania mikrozakrzepów
Interakcje między czynnikami krzepnięcia i mediatorami
reakcji zapalnej. Na komórkach śródbłonka,
kom.jednojądrzastych, płytkach, fibroblastach,komórkach
mięśni gładkich znajdują się przezbłonowe receptory
aktywowane przez proteazy (PAR)

MECHANIZM
Wytworzenie dużej ilości trombiny przekształcającej
fibrynogen w fibrynę, która tworzy zakrzepy w świetle
drobnych naczyń. Tam uwięzione zostają płytki krwi. Jeśli
aktywacja krzepnięcia nie zostanie w porę zahamowana
zakrzepy blokują dopływ krwi do narządów i powoduje ich
niewydolność. Wytworzenie wielu zakrzepów wyczerpuje
czynniki krzepnięcia i pytki co objawia się skazą krwotoczną
.Powstające produkty degradacji fibryny i fibrynogenu
zaburzają mechanizm hemostazy w wyniku : działania
antykoagulacyjnego, hamowania funkcji płytek, działania
cytotoksycznego na śródbłonek i zwiększenia
przepuszczalności ściany naczyń włosowatych.

Choroby i stany kliniczne ,w których może
rozwinąć się DIC
:
-zakażenia-bakteryjne(posocznice),riketsjowe, wirusowe,
pierwoniakowe
-powikłania ciąży, porodu(przedwczesne oddzielenie łożyska)
-nowotwory-guzy lite, mielo- i limfoproliferacje,
-hemoliza wewnątrznaczyniowa-po przetoczeniu krwi
niezgodnej,nocna napadowa hemoglobinuria,
-ch.naczyń krwionośnych-naczyniaki,tętniaki,zator tętnicy
płucnej
-rozległe uszkodzenie tkanek-oparzenia,wstrząsy,
Może być uruchamiane na różnych drogach-uszkodzenie
śródbłonka,proteaz aktywujących krzepnięcie,zwolnienie
przepływu krwi. Podstawowe znaczenie mają cytokiny IL-
1,TNF,IL-6

Nie ma jednego testu laboratoryjnego ,którego
wynik pozwoliłby jednoznacznie potwierdzić lub
wykluczyć rozpoznanie DIC. Warunkiem koniecznym
jest wykrycie choroby ,w przebiegu której doszło do
uogólnionej aktywacji krzepnięcia.
DIC o ostrym przebiegu zawsze stanowi
bezpośrednie zagrożenie życia a rokowanie zależy
od możliwości wyleczenia choroby podstawowej.
PAMIETAĆ !

Diagnostyka DIC obejmuje:
badanie przesiewowe: aPTT , PT, TT, PLT
testy potwierdzające : FDP, D-dimery
testy uzupełniające:
w zakresie ukł. krzepnięcia- AT III,cz.V, cz.VIII, czas lizy
euglobulin ,kompleksy TAT,PAP, fragment 1+2 protrombiny,
w zakresie funkcji płytek: tromboksan, cz.płytkowy 4,TG.

Ostry DIC:
Gwałtowny przebieg,
małopłytkowość(>100-0pkt.,<100>50-1pkt.,<50
-2pkt.)
przedłużone aPTT, PT,TT
Zmniejszenie stężenia fibrynogenu,
Zmniejszenie innych cz.krzepnięcia,
Zwiększenie stężenia D-dimerów
Przewlekły DIC
Liczba płytek nieznacznie zmniejszona, dynamika
zmian
płytek
zwiększone stężenie F1+2,TAT(testy bardzo czułe
lecz mało swoiste)
Dynamika zmian PT,FDP
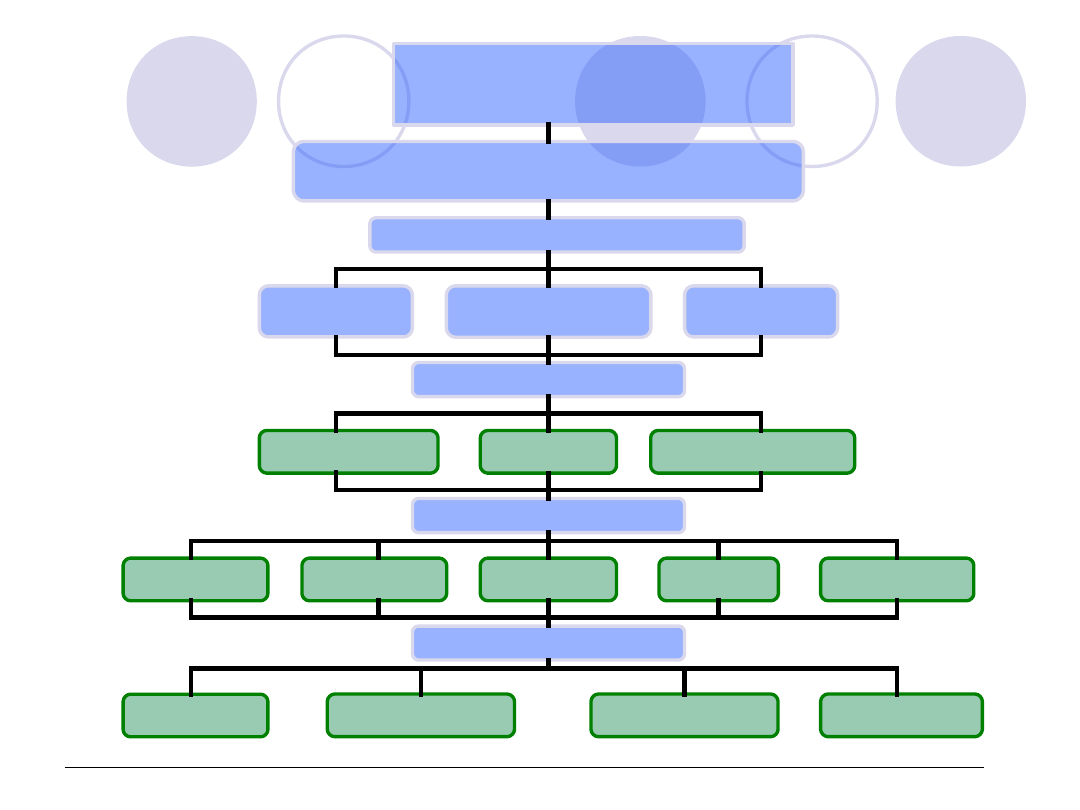
Rys. Schemat postępowania diagnostycznego w przypadku podejrzenia zespołu wykrzepiania śródnaczyniowego (DIC)
Wywiad
Przyczyny DIC
Cechy skazy (skórne,narządowe)
Choroby współistniejące
Badania fizykalne- cechy i objawy skazy, lokalizacja Objawy
choroby podstawowej
Układ krzepnięcia
APTT, PT, TT
Badania podstawowe - przesiewowe
Fibrynogen
Morfologia
PLT
Badania uzupełniające
D-dimery
Antytrombina
Monomery fibryny
Badania 2-go rzutu
TAT i PAP
FPA i FPB
Czynnik V i VIII
F1 + 2
Plazmoniogen
Badania rzadkie
b-beta 15 – 42
b-beta 1 – 118
Beta-
tromboglobulina
TXA
2
PF 4

Leczenie
Leczenie choroby podstawowej .
W sepsie –odpowiednia antybiotykoterapia , zwalczenie wstrząsu, wyrównanie
kwasicy, wyrównanie zaburzeń wodno-elektrolitowych.
W ch. nowotworowych -chemio- i radioterapia, ew. leczenie operacyjne.
Wyrównanie zaburzeń hemostazy
-leczenie substytucyjne (kkcz, kkp, osocze świeżo mrożone, krioprecypitat,
koncentrat fibrynogenu, koncentrat czynników zespołu protrombiny,
rekombinowanego cz.VII a)
-heparyna,
-koncentraty inhibitorów krzepnięcia,
-ihibitory fibrynolizy

TROMBOFILIE( nadkrzepliwość
)
To wrodzone lub nabyte zaburzenia mechanizmów
hemostazy ,które prowadzą do zakrzepów. Do
zakrzepicy dochodzi w wyniku zachwiania
równowagi pomiędzy naturalnymi układami
antykoagulacyjnymi a czynnikami sprzyjającymi
aktywacji krzepnięcia. Najczęstszym objawem
tych defektów jest żylna choroba zakrzepowo-
zatorowa ale także rzadko zakrzepy tętnicze.

TROMBOFILIE WRODZONE
I. Niedobory inhibitorów krzepnięcia
-niedobór ATIII- znanych jest ok.125 zaburzeń w genie
antytrombiny(delecje, mutacje typu zmiany sensu, mutacje
nonsensowne) dziedziczenie autosomalne dominujące.
-niedobór białka C -ok.160 mutacji genu białka C
dziedziczonych autosomalnie dominująco.
-niedobór białka S- ok.130 mautacji.
II. Zwiększona aktywność czynników krzepnięcia
-poilmorfizm genu cz.V(Arg506Glu)-czynnik V Leiden ,
-mutacje G20210A genu protrombiny,
-zwiększona zawartość w osoczu czynnika VIII(>150%),
Dysfibrynogeniemia
III.Inne
-hipercholecysteinemia,
-niedobór plazminogenu.

TROMBOFILIA NABYTA-ZESPÓŁ
ANTYFOSFOLIPIDOWY
-niezapalna układowa choroba tkanki łącznej,
charakteryzująca się współwystępowaniem zakrzepicy
naczyniowej lub powikłań położniczych oraz krążących
przeciwciał antyfosfolipidowych.
Kryteria kliniczne
Zakrzepica naczyń: Jeden lub więcej epizodów zakrzepicy
w naczyniach tętniczych, żylnych (z wyjątkiem zakrzepicy
żył powierzchownych) lub włosowatych w obrębie
jakiejkolwiek tkanki lub narządu, potwierdzony badaniem
obrazowym, dopplerowskim lub histologicznym. W obrazie
histopatologicznym zmianom zakrzepowym nie powinno
towarzyszyć zapalenie ściany naczynia.

Niepowodzenie położnicze
co najmniej jedno obumarcie morfologicznie prawidłowego
płodu po 10. tygodniu ciąży (prawidłowa morfologia płodu
udokumentowana za pomocą ultrasonografii lub badania
bezpośredniego) lub
co najmniej jeden przedwczesny poród morfologicznie
prawidłowego płodu przed 34. tygodniem ciąży w związku
ze stanem przedrzucawkowym, rzucawką lub ciężką
niewydolnością łożyska lub
co najmniej trzy samoistne poronienia o niewyjaśnionej
przyczynie przed 10. tygodniem ciąży, z wykluczeniem
przyczyn związanych ze zmianami anatomicznymi lub
zaburzeniami hormonalnymi u matki oraz
chromosomalnymi u obojga rodziców

Kryteria laboratoryjne
Obecność antykoagulantu toczniowego(LA) w osoczu wykrytego co
najmniej 2-krotnie w odstępie minimum 12 tygodni, metodami zaleconymi
przez International Society on Thrombosis and Haemostasis
Przeciwciała antykardiolipinowe (ACA)w klasie IgG lub IgM w średnim lub
dużym stężeniu (tzn. >40 GPL lub MPL, lub <99. centyla) wykryte co
najmniej 2-krotnie w odstępie minimum 12 tygodni standaryzowaną
metodą ELISA
Przeciwciała przeciw β2-glikoproteinie I obecne w surowicy lub osoczu (w
mianie >99. centyla) wykryte co najmniej 2-krotnie w odstępie minimum
12 tygodni standaryzowaną metodą ELISA
Zespół antyfosfolipidowy rozpoznaje się, gdy jest spełnione co najmniej 1
kryterium kliniczne i 1 kryterium laboratoryjne. Kryteriów nie należy
stosować, jeżeli objawy kliniczne choroby wystąpiły w okresie <12 tygodni
lub >5 lat od momentu wykrycia przeciwciał antyfosfolipidowych (APLA).
Badania laboratoryjne
wydłużenie APTT
małopłytkowość
Hemoliza śródnaczyniowa

Powikłania
Zakrzepica żylna lub tętnicza
zatorowość płucna
zawał serca, kardiomiopatia
małopłytkowść
niedokrwistość hemolityczna
zespół hemolityczno-mocznicowy
krwawienia
białkomocz
nadciśnienie naczynionerkowe
owrzodzenia skóry
udar mózgu
upośledzenie wzroku i słuchu
kurcze mięśniowe
napady migreny
poronienie
stan przedrzucawkowy
wewnątrzmaciczne opóźnienie wzrostu - IUGR

Zaburzenia hemostazy w chorobach wątroby
Wątroba to miejsce syntezy :
a) osoczowych cz.krzepnięcia ( zależne od wit. K- II, VII, IX, X
oraz niezależne od wit. K- I, V, VIII, XI, XIII, fibrynogen)
b) inhibitorów krzepnięcia( AT III, białko C, b. S, b. Z)
c) czynników fibrynolizy ( plazminogen, PAI-1, TAFI, alfa 2-
antyplazmina)
d) czynnik wzrostu płytek -trombopoetyna.

ZABURZENIA DOTYCZACE CZYNNIKÓW KRZEPNIĘCIA
-niewydolność wątroby wiąże się ze spadkiem cz.krzepnięcia-
II, V, VII, IX, X, XII, XIII.
-proporcjonalnie do stopnia uszkodzenia hepatocytów spada
stężenia cz.V,
-w osterj niewydolności jako pierwsze obniżeniu ulegają cz.o
krótkim okresie półtrwania-VII,V,IX,X,II
-niewydolność prowadzi do spadku wit.K co zaburza syntezę
cz.krzepnięcia
-w ch.wątroby przebiegających ze stanem zapalnym wzrasta
stężenie cz.vW i cz.VIII
- w przewlekłych zapaleniach wątroby, żółtaczce
cholestatycznej czy raku wątroby obserwuje się podwyższone
stężenie fibrynogenu. Ale jest to białko nieczynne
funkcjonalnie ze wzg. na nieprawidłowo uformowane
łańcuchy.
W schyłkowych stadiach marskości obserwujemy obniżony
poziom fibrynogenu.
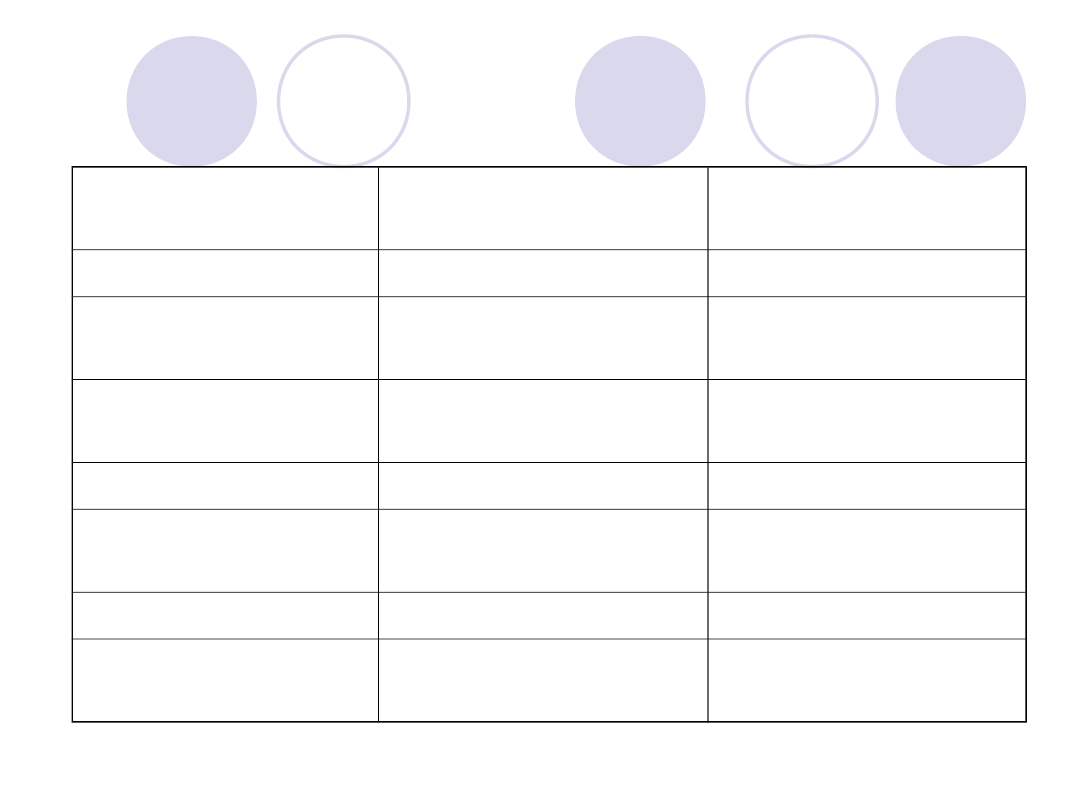
HEMOSTAZA U NOWORODKÓW I KOBIET W
CIĄŻY
BADANIE
NOWORODKI
KOBIETY W
CIĄŻY
PLT
N
N
STĘŻENIE
FIBRYNOGENU
ZWIĘKSZONE
N
STĘŻENIE
PROTROMBINY
NIEZNACZNIE
ZWIĘKSZONE
OBNIŻONE
STĘŻENIE CZ.V
N
N
STĘŻENIE CZ.VII i
X
ZWIĘKSZONE
OBNIŻONE
STĘŻENIE CZ.VIII
ZWIĘKSZONE
N
STĘŻENIE CZ.IX
NIEZNACZNIE
ZWIĘKSZONE
OBNIŻONE
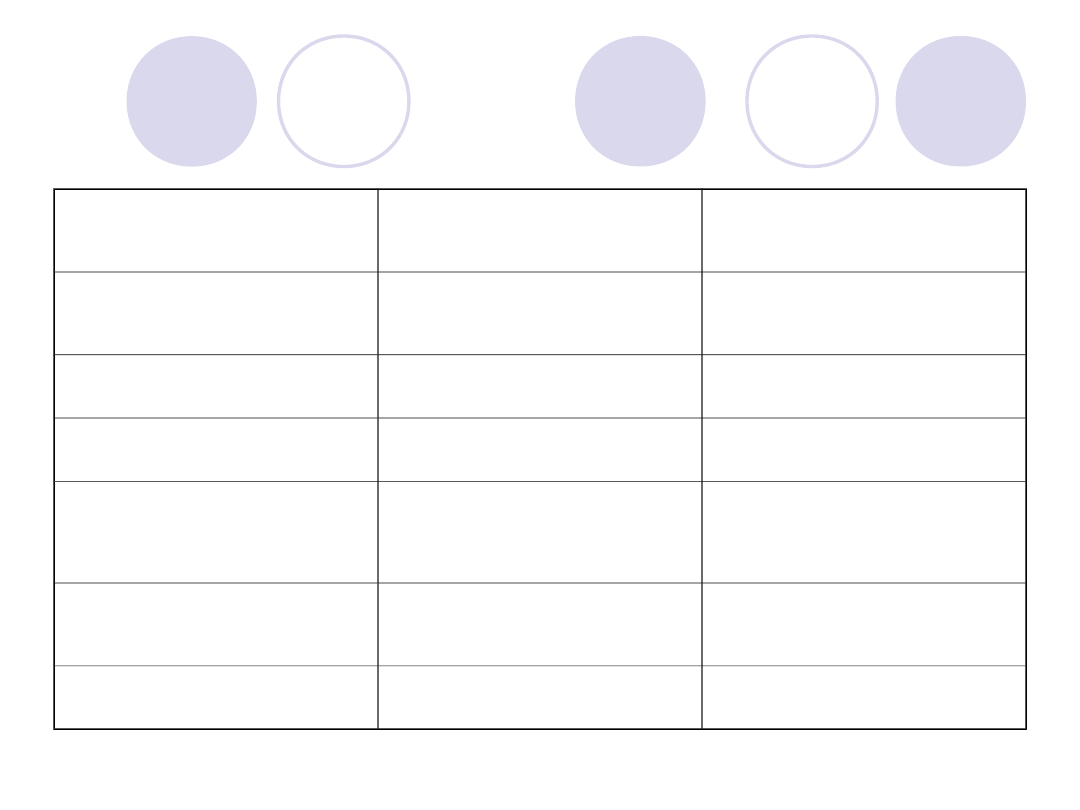
STĘŻENIE CZ.XI
NIEZNACZNIE
ZMNIEJSZONE
ZMNIEJSZONE
STĘŻENIE CZ.XII
NIEZNACZNIE
ZWIĘKSZONE
ZMNIEJSZONE
STĘŻENIE CZ.XIII
ZMNIEJSZONE
ZMNIEJSZONE
STĘŻENIE AT III
ZMNIEJSZONE
ZMNIEJSZONE
STĘŻENIE
PLAZMINOGENU
ZWIĘKSZONE
ZMNIEJSZONE
CZAS
TROMBINOWY
N
PRZEDŁUŻONY
FDP
N lub ZWIĘKSZONE N lub ZWIĘKSZONE

Przykład 1
PT INR=1,0
aPTT=80 sek,
TT=15 sek,
Fibrynogen= 2,0 g/l,
PLT= 145 000/ul

aPTT kontrolnego= 35 sek,
aPTT badanego= 80 sek
aPTT mieszaniny= 62sek.
Korekcja nieprawidłowa ponieważ 80-62=18 80-
35=45. 18<22,5.
Stąd przyczyna przedłużenia aPTT jest krążący
antykoagulant

Przykład 2
PT INR=1,2
aPTT=100 sek,
TT=14 sek,
Fibrynogen= 1.9 g/l
PLT=160000/ul

aPTT kontrolnego= 35 sek,
aPTT badanego= 100 sek
aPTT mieszaniny= 62sek.
Korekcja prawidłowa ponieważ 100-62=38 100-
35=65. 38>32,5.
Stąd przyczyna przedłużenia aPTT jest brak
czynników osoczowych

Cz.VIII =80%,
czIX =45%
Cz.X=110%

HEMOFILIA TYPU B-postać łagodna

Przykład 3
PT INR=3,5
aPTT=25sek,
TT=16 sek,
Fibrynogen =1,8 g/l
PLT =180000/ul

cz. II = 100%,
cz. X =110%,
cz. V =10%.

NIEDOBÓR CZ.V-
Hipoprokonwertynemia
W Polsce zarejestrowanych jest ok.135 chorych

Przykład 4
PT INR=1,2
aPTT= 50sek,
TT=15 sek,
Fibrynogen =1,9 g/l

aPTT kontrolnego= 35 sek,
aPTT badanego= 50 sek
aPTT mieszaniny= 42sek.
Korekcja prawidłowa ponieważ 50-42=8 50-
35=15. 8>7,5.
Stąd przyczyna przedłużenia aPTT jest brak
czynników osoczowych

FVIII =80%,
FIX =85%
FX=110%
F XI=100%
FXII=2%

ANOMALIA HAGEMANA
Pacjentka diagnozowana z powodu przedłużenia
APTT oraz nadmiernego krwawienia po operacji
migdałków.

Przykład 5
PT INR=1,6
aPTT= 45sek,
TT=15,8 sek,
Fibrynogen =1,8 g/l
VWF:RCo 49,4%
FXI 72,2%
FXII 87,3%
FVIII 52,4%
FIX 75,4%
FX 32%

ŁAGODNY NIEDOBÓR CZYNNIKA X

Przykład 6
Czas krwawienia=25 min,
PT INR=1,0
aPTT= 42sek,
TT=15,8 sek,
PLT =250000/ul
vWF:RCo <10%
FVIII < 10%
vWF:Ag < 1%
FXII 87,3%
FIX 78,4%
Czas okluzji KOL/EPI-250 sek

CHOROBA VON WILLEBRANDA TYP III
Ciężka postać-pacjentka cierpi na przedłużające się krwawienia
miesięczne, znaczna anemizacja

Przykład 7
Czas krwawienia=21min,
PT INR=1,0
aPTT= 42sek,
TT=15,8 sek,
vWF:RCo 20%
FVIII 75%
vWF:Ag 45%
RIPA-dodatni
PFA-100 Col/Epi > 300 s, Col/ADP > 270 s
PLT 10000/ul

CHOROBA VON WILLEBRANDA TYP IIB
Rozpoznanie na podstawie wywiadu rodzinnego,objawów klinicznych i
testów laboratoryjnych.
Rzadki typ choroby von Willebranda odznacza się tendencją do
małopłytkowości (u podłoża choroby leży zwiększone powinowactwo
czynnika vWF do płytkowej glikoproteiny Ib). W stanach, które wiążą się ze
wzrostem aktywności vWF w osoczu (stres, choroba, stan zapalny, silny
wysiłek) może pojawić się lub pogłębić małopłytkowość
.

Przykład 8
PT -1,0 INR
aPTT= 42sek,
TT=18 sek,
vWF:RCo 58%
FVIII 62%
FX 34%
F II 103%
FV 82%
RIPA- ujemny
PLT 370000/ul
Agregacja płytek pod wpływem ADP 88%, kolagenu 80%,
epinefryny 98, ristocetyny 85% i arachidonianu sodu 85%

CHOROBA VW typ 1 oraz niedoboru
FX.

Przykład 9
PT INR=1,1
aPTT= 36sek,
fibrynogen=3,6g/l,
vWF:RCo 28%
FVIII 33%
vWF:Ag 44%
FXII 55%
FIX 65%
FXI 84%
PLT 218000/ul

CHOROBA VON WILLEBRANDA
Pacjentka w trakcie diagnostyki .
Nie ma przeciwwskazań do hormonalnej terapii zastępczej. Stosowanie HTZ
często łagodzi przebieg choroby i jest zalecane u pacjentek z chorobą von
Willebranda.

Przykład 10
Czas krwawienia=35 min.
PT INR=1,2
aPTT=40sek,
Plt =130 tys, MPV >30 um
Test agregacji- brak pod wpływem rystocetyny

Cytometria przepływowa
– upośledzenie kompleksu Ib/IX/V

ZESPÓŁ BERNARDA-SOULIERA

Przykład 11
PT -1,9 INR
aPTT= 72sek,
TT=29 sek,
Fibrynogen=1,0g/l,
FDP 10 mg/ml
D-dimery 800 ug/ml
PLT 10000/ul

ROZSIANE KRZEPNIĘCIE ŚRÓDNACZYNIOWE

FACTOR V-LEIDEN RESULTS:
Mutation analyzed: 1691G>A
Factor 5 Mutation Interpretation: PRESENT
Factor 5 Mutation genotype: G/A
The patient is a heterozygote (one copy of the gene positive) for
the
Factor V 1691G>A (Leiden) mutation.
Zakrzepica żył głębokich jest spowodowana spowolnieniem przepływu krwi
w naczyniach żylnych. Jest to wieloczynnikowa choroba wywołana
interakcją między czynnikami genetycznymi (np. czynnik V - mutacja typu
Leiden, mutacja genu protrombiny) i środowiskowymi (np. siedzący tryb
życia, otyłośc, palenie tytoniu, zawał serca, udar mózgu, niektóre
zaburzenia składu krwi oraz wiek > 60 roku życia).
Mutacja czynnika V jest dziedziczona autosomalnie dominująco, polega na
zastąpieniu argininy przez glutaminę w pozycji 506 łańcucha ciężkiego
(R506Q).
Pacjenci z mutacją typu Leiden posiadają białkowy czynnik V niewrażliwy
na inaktywację przy udziale białka C. W takiej sytuacji białkowy czynnik V
ma podwyższoną aktywność i może powodować zwiększone ryzyko
powstania zmian zakrzepowo-zatorowych w organiźmie.

Pacjent z dodatnim wywiadem rodzinnym w kierunku skazy
płytkowej o typie zaburzenia czynności płytek krwi (u matki i
dwóch sióstr znaczne osłabienie agregacji po epinefrynie – do 10-
30%).
Wywiad krwotoczny: w przeszłości krwawienia z nosa, niewielkie
krwawienia z dziąseł. Po usunięciu zęba przedłużone krwawienie.
Nie przechodził zabiegów chirurgicznych.
INR 1,0
APTT 31,9 s
fibrynogen 1,8g/l
FVIII 80% R:Cof 70%
PLT 192000/ul
PFA-100: Col/EPI 169 s , Col/ADP 111 s .
Agregacja pod wpływem kw. arachidonowego, ADP, kolagenu,
ristocetyny prawidłowa, zredukowana do 10% pod wpływem
epinefryny
W rozmazie krwi obwodowej obecne duże płytki.
Wskazana dalsza diagnostyka w kierunku wrodzonych defektów
płytkowych w Instytucie Hematologii i Transfuzjologii w
Warszawie.

Dgn: Podejrzenie hipofibrynogenemii
Wyniki badań poniżej – dalsza diagnostyka w toku. Obniżenie aktywności
fibrynogenu może mieć u pacjenta charakter wrodzony lub wtórny do
istniejącej patologii wątorby. Dzis pobrano dalsze badania.
Wskazana diagnostyka siostry z uwagi na krwawienie po ekstrakcji zęba –
bardzo proszę o wykonanie u siostry badań: morfologia, APTT, PT, TT (czas
trombinowy) oraz aktywność fibrynogenu.

Dgn:
Wrodzona hipofibrynogenemia
Na podstawie wyników badań oraz wywiadu rodzinnego (obniżenie aktywności
fibrynogenu u siostry) można u Pacjenta rozpoznać wrodzony, łagodny niedobór
fibrynogenu. Jest to przyczyną zwiększenia wartości czasu APTT i PT w dostarczonych
przez pacjenta badaniach. Wyniki mogą jednak różnić się w zależności od
laboratorium.
Hipofibrynogenemia może również powodować zwiększoną tendencję do zakrzepicy.
W okresach przedłużonego unierochomienia oraz w innych sytuacjach sprzyjających
zakrzepicy wskazana (przy braku objawów krwotocznych) profilaktyka heparyną
drobnocząsteczkową.
Wskazane oznaczenie aktywności fibrynogenu u braci oraz ojca.
INR 1,5
APTT 40 sek
fibr 1,2 g/l (n. 1,8-3,5)
TT 25 sek
l

Dgn:
Zespół antyfosfolipidowy
Pacjent skierowany z powodu przedłużenia APTT (60 s) i PT (76 s)
oraz umiarkowanej małopłytkowości.
Wywiad krwotoczny ujemny (liczne ekstrakcje zębów bez powikłań –
ostatnia ok. 12 lat temu, operacji nie przechodził, krwawienia z nosa
w młodości – niewielkie, po wysiłku, z dziąseł – bez krwawień,
siniaczenie – nie, krew w moczu, stolcu – nie, 3 tygodnie temu
niewielkie plamienie koło cewnika po wysiłku).
Wywiad rodzinny – u matki częste krwawienia z nosa.
Wywiad w kierunku epizodów zakrzepowych ujemny.
INR 1,4
APTT 69,5 s
fibr 3,4 g/l TT 17,4 s PLT 53000/ul (na cytrynian 48000/ul)
Test korekcji APTT zdrowym osoczem – dodatni dla APTT
FII 55,6%
FVII 48,5%
FIX 100,1%
FX 73,5%
FVIII 140,2%
vWF:RCo 80,6%
vWFAg 129,72%
Antykoagulant tocznia dodatni
Ig antykardiolipinowe 85,83 GPL (IgG), 3,84 MPL (IgM)

Dgn:
choroba Rendu-Oslera-Webera (wrodzona
naczyniakowatość krwotoczna)1:50 000)
Objawy kliniczne: na wargach obecne punkcikowane
wybroczyny/naczyniaki od ok. 4 lat (ilość zmienna),okresowo
również na dziąsłach, krwioplucie. Od tego czasu również
krwawienia z nosa i plamienia z dziąseł przy szczotkowaniu zębów,
dłuższe krwawienia po skaleczeniach. Nie miesiączkuje (po
histerektomii), wcześniej obfite miesiączki. Operacje
(histerektomia, usunięcie jajnika, porody i cięcie cesarskie bez
powikłań, ekstrakcje zębów bez powikłań).
Badania podstawoe układu hemostazy w normie. Badania w
kierunku małopłytkowości, choroby von Willebranda, hemofilii C,
trombocytopatii ujemne.
Całość obrazu klinicznego wskazuje na naczyniakowatość
wrodzoną.
INR 0,9
APTT 26,1 s
fibr 6,4 g/l
FXI 107,6 %
vWF:RCo 169,5% PLT 176000/ul
Agregacja płytek prawidłowa pod wpływem arachidonianu sodu,
ADP, epinefryny, kolagenu, rystocetyny

NORMY
aPTT-25-35 sek
PT 0,9-1,4,
TT-12-16 sek,
Fibrynogen -1,8-3,5g/l
PLT 150-450 000
Czas krwawienia 2-7 min
D-dimery < 350ug/dl
FDP <800 ng/ml
AT III 75-130%
F V-70-130%
FVIII-50-150%
FIX-70-130%
F X-70-130%
FXII-70-130%
vW Ag-50-150%
FvW-50-150%
Czas okluzji kol/epinef-85-165 sek,
kol/ADP- 71-118 sek
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
- Slide 91
- Slide 92
- Slide 93
- Slide 94
- Slide 95
- Slide 96
- Slide 97
- Slide 98
- Slide 99
- Slide 100
- Slide 101
- Slide 102
- Slide 103
- Slide 104
- Slide 105
- Slide 106
- Slide 107
- Slide 108
- Slide 109
- Slide 110
- Slide 111
- Slide 112
- Slide 113
- Slide 114
- Slide 115
- Slide 116
- Slide 117
- Slide 118
- Slide 119
- Slide 120
- Slide 121
- Slide 122
- Slide 123
- Slide 124
- Slide 125
- Slide 126
- Slide 127
- Slide 128
- Slide 129
- Slide 130
- Slide 131
- Slide 132
- Slide 133
- Slide 134
- Slide 135
- Slide 136
- Slide 137
- Slide 138
- Slide 139
- Slide 140
- Slide 141
- Slide 142
- Slide 143
- Slide 144
- Slide 145
- Slide 146
- Slide 147
- Slide 148
- Slide 149
- Slide 150
- Slide 151
- Slide 152
- Slide 153
- Slide 154
- Slide 155
- Slide 156
- Slide 157
- Slide 158
- Slide 159
- Slide 160
Wyszukiwarka
Podobne podstrony:
Badanie układu kostno stawowego i mięśniowego (1)
Ananatomia i fizjologia badania ukladu oddechowego u dzieci
BADANIE UKLADU REGULACJI CIAGLE Nieznany (2)
METODY BADANIA UKŁADU LIMFATYCZNEGO
Badanie układu napędowego z silnikiem bezszczotkowym z magnesami trwałymi
METODY BADANIA UKŁADU LIMFATYCZNEGO, Mieszanka Mareckiego
Badanie układu sterowania z regulatorem PID
Badanie układu z elementami nieliniowymy
Badanie układu zapłonowego
Sprawozdanie 2, całkuj-różnik wymuszsin, Badanie układu całkująco - różniczkującego przy wymuszeniu
Badanie układu oddechowego(1)
BADANIE UKŁADU CZASOWEGOU5
Badanie ukladu nerwowego id 781 Nieznany
Badanie układu pokarmowego
więcej podobnych podstron