GENETYKA WYKŁAD 12
choroby genetyczne
Chorobę można definiować w różny sposób. To zaburzenie równowagi wewnętrznej organizmu.
Choroba może wystąpić gdy zaburzone jest współdziałanie genotypu z objawami fenotypu.
choroby zależne od środowiska - to np. zatrucie, złamania, urazy - są niezależne od genotypu
choroby zależne od genów:
od genotypu zależą zaburzenia ilości chromosomów:
zespół EDWARDSA (trisomia chromosomu 18)
- niedorozwój umysłowy
- niski wzrost, zniekształcona czaszka, liczne zmiany wewnętrzne
- do 4 miesiąca życia następuje zgon, 30% dzieci umiera już w pierwszym miesiącu
zespół PATAUA (trisomia chromosomu 13)
- szczelina w tęczówce, ślepota
- głuchota, znieksztłcenie uszu
- wodonercze; przepuklina pępkowa
- ślepota, wady serca, rozszczep warg i podniebienia
- śmierć do 3 roku życia
zespół DOWNA (trisomia 21 chromosomu)
- mutacja autosomalna
- mongolizm, małpia bruzda na dłoni, obniżone napięcie mięśni
- wady serca, upośledzenie umysłowe
- przyspieszone starzenie
- wiele zmian wewnętrznych
zespół TURNERA ( X0)
- monosomia chromosomu X - występuje tylko u ♀
- niepłodność, niedorozwój umysłowy i somatyczny, wady serca
- szeroka płetwiasta szyja, zmiany barwnikowe skóry, 4 i 5 palec ręki znacznie krótsze
~ czasem defekt mozaikowy - tylko niektóre komórki chore
zespół KLINEFELTERA ( XXY)
- tylko u ♂
- 47 chromosomów
- hermafrodyta, słabo rozwinięte narządy płciowe, niedorozwój umysłowy, somatyczny
- mają ciałko Barra ( kolejny X w formie euchromatyny, wynika z nondysjunkcji chromosomów)
~ zarówno niedobór jak i nadmiar chromosomów jest szkodliwy
choroby zależne od środowiska i genotypu: (oba czynniki tak samo ważne)
rozedma płuc
płaskostopie
rozszczep podniebienia
zajęcza warga (wrodzone rozszczepienie wargi górnej, któremu często towarzyszy rozszczepienie podniebienia.)
wady serca
hipercholesterolemia (to wada metaboliczna, podwyższone powyżej normy stężenie cholesterolu w osoczu krwi
miażdżyca
cukrzyca
łuszczyca
zapalenia płuc, oskrzeli
choroby psychiczne
choroby genetyczne mogą być recesywne - ujawniają się wtedy w homozygocie recesywnej
heterozygoty są zwykle źródłem choroby, nosicielami
choroby dominujące występują rzadko
choroby genetyczne wynikające z błędów w metabolizmie:
~ mogą dotyczyć przemian aminokwasów, węglowodanów, lipidów)
- zaburzenia w szlaku metabolicznym przekształcającym fenyloalaninę do aminokwasów endogennych:
ketokwasy białko melanina
fenyloalanina tyrozyna
tyroksyna i trójjodotyronina (hormony tarczycy)
kwas
homogentyzynowy
CO2 i H2O
schemat przemian fenyloalaniny w organizmie człowieka
alkaptonuria
- zaburzenia przemian kw. homogentyzynowego w CO2 i H2O; prowadzi do gromadzenia się kw. homogentyzynowego w organizmie, który blokuje enzymy i zatruwa mózg
~ tyrozyna po kilku przemianach przekształca się w kwas homogentyzynowy, który nie ulega dalszym przemianom z pwodu braku odpowiedniej oksydazy
- „choroba niebieskich pieluszek” - nienormalne zabarwienie moczu u dzieci
- uszkodzenia w szkielecie ( w chrząstkach stawów) (kwas łączy się z kolagenem)
- brak produktu melaniny - prowadzi do albinizmu
fenyloketonuria
- blok metaboliczny uniemożliwiający przekształcenie fenyloalaniny w tyrozynę
- duże stężenie fenyloalaniny jest toksyczne => upośledzenie umysłowe
- niedostatek tyrozyny ( a zatem i melaniny) - jasna skóra skłonna do schorzeń, jasne, mysie włosy
- nadmiar ketokwasów fenylooctowego i fenylopropionowego - specyficzny mysi zapach potu i moczu
- leczenie: dieta uboga w fenyloalaninę, podawanie tyrozyny
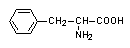
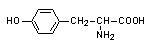
fenyloalanina hydroksylaza tyrozyna
inne choroby metaboliczne:
galaktozemia
~ galaktoza jest jednym z podstawowych cukrów w organizmie, brak oksydazy galaktozy (brak urydylotransferazygalaktozo-1-fosforanowej) prowadzi do nierozkładania tego związku do glukozy
~ nadmiar w mięśniach - trujący
choroba glikogenowa
~ wrodzone zaburzenia metaboliczne, polegające na gromadzeniu nieprawidłowych ilości glikogenu w tkankach (brak zdolności metabolizmu glikogenu)
~ glikogen może gromadzić się w różnych organach, np. trzustce (=> powiększenie organu; brak enzymu kwaśnej maltazy)
choroba fafizm
~ brak enzymu dehydrogenazy glukozo-6-fosforanowej (gen odpowiedzialny jest na chromosomie X - choroba sprzężona z daltonizmem i hemofilią)
inne:
choroby związane z metabolizmem lipidów. bloki metaboliczne w ich metabolizmie powodują choroby w układzie nerwowym (OUN)
np. choroba Tay'a - Sachsa:
- .śmierć do 4 roku życia
- mutacja enzymu β-heksoaminidazy A lub B
- nagromadzenie gangliozydów w organizmie
np. choroba Niemana - Picka:
- gromadzenie sfingomieliny i cholesterolu w tkankach
- brak enzymu lipidazy sfingomielinowej (sfingomielinazy)
- leczenie nieznane
choroby związane z metabolizmem mukopolisacharydów:
spichrzyce - brak degradacji mukopolisacharydów w lizosomach
np. zespół Sanfilippo - MPS III (mukopolisacharydoza typu III )
np. zespół Hurler'a - MPS II
np. zespół Huntera - MPS II
(* MPS - oznacza brak specyficznych lizosomalnych enzymów danego typu, np. typu II albo III)
choroby związane z nieprawidłowym metabolizmem pierwiastków:
- hemochromatoza - nagromadzenie Cu lub Fe w wątrobie, trzustce, sercu; powiększenie organów, zaburzenia pracy organów
- zespół Wernickiego - Korsakowa - brak witaminy B1 i B2; defekt związanej z tym tranketolazy, która bierze udział w syntezie witamin
~ otępienie, luki w pamięci
mukowiscydoza: *
~ powoduje zaburzenia funkcjonowania kanalików chlorkowych błon komórkowych
~ w układzie oddechowym gromadzi się zbyt dużo śluzu - co blokuje powierzchnię oddechowaą, jest znakomitą pożywką dla bakterii
~ zaburzenia pracy przewodu pokarmowego
~ „choroba słonego potu” - dużo Cl w pocie
* - nie zaliczam mukowiscydozy do chorób zw. z wadliwym metabolizmem pierwiastków, bo chodzi tu o zatrzymywanie Cl- w nadmiarze w stosunku do dużego wydalania Na+ z komórki - ale nie jest to METABOLIZM tych pierwiastków
hemoglobinopatie
mutacja w pozycji 146 łańcucha białka: zamiana kwasu glutaminowego -> walinę
stanowi to w tropikach biologiczne przystosowanie - chorzy na nią nie zapadają na malarię
~ np. hemoglobinopatia S - niedokrwistość sierpatokrwinkowa (=skaza sierpowata)
choroby warunkowane czynnikiem dominującym:
- psychoza maniakalno - depresyjna (ujawnia się już w heterozygocie)
- pląsawica Huntingtona
~ objawy w 40. roku życia
~ stopniowa degeneracja układu nerwowego, drżenie kończyn, niekontrolowanie mięśni
- achondroplazja - brak wzrostu kości długich i karłowatość
- amelia - brak kończyn górnych i dolnych
- „rybia łuska” - nadmierne rogowacenie skóry, mutacje w genach kodujących białka strukturalne
- porfiria
- hipercholesterolemia (podwyższone powyżej normy stężenie cholesterolu w osoczu krwi - nieprawidłowość metaboliczna)
choroby mitochondrialne:
DNAmit to 16 569 pz bez intronów, 37 genów w tym:
~ 22 kodujące tRNA
~ 13 kodujących mRNA
~ 2 kodujące rRNA
choroby powodują mutacje punktowe lub delecje
- mutacja w pozycji 11778 G≡C => A=T => 340 pozycja aminokwasu Arg -> His
~ efekt dotyczy 4 podjednostek dehydrogenazy NADH
~ zapadają na nią mężczyźni w wyniku interakcji z chromosomem X
~ ślepota
- zespół LHON
zanik nerwu wzrokowego, dotyka obustronnie ♂ w 10 - 30 roku życia, całkowita ślepota w przeciągu 8 tygodni
- zespół KSS
~ spowodowany przez dużą delecję (2-8kbp)
~ paraliż zewnętrznych mięśni oka, zmiany w siatkówce; wady serca
- zespół (fenotyp) MELAS
~ ślepota; wady serca
fenotypy MERFF i NARP - dotyczą chorób mitochondrialnych (DNAmit)
LECZENIE CHORÓB GENETYCZNYCH:
- klasycznie przez terapię genową
~ można leczyć ( => leczyć) i uzdrowić (=> uzdrowić na zawsze)
uzdrowić można np. przez usunięcie wadliwego organu (polipy okrężnicy)
- leczenie to przedłużenie życia i złagodzenie skutków (np. przy fenyloketonurii - odpowiednia dieta może zapobiec poważnym zmianom w organizmie)
dieta eliminująca (eliminuje dany składnik z pożywienia - fenyloalanina, galaktoza w glaktozemii)
dieta uzupełniająca (np. o wiatminę C - szkorbut)
- zastąpienie produktów brakujących/wadliwych genów: podawanie insuliny w cukrzycy
- hamowanie konkurencyjne: głównie enzymu; obniża się aktywność danego enzymu; gdy jest go mało - indukuje się aktywność
- zastąpienie chorego organu (torbielowatość nerek =. nowotwory)
poradnictwo genetyczne:
- terapia genowa - naprawa genu, zamiana genu, zmiana ekspresji genu
- Anderson, 1990r. - udana terapia genowa:
do faga T4 wprowadzono gen Ada (deaminaza adenozyny)
- gen CD4 w przypadku wirusa HIV - syntezuje się oligonukleotydy antysensowne, które mogą blokować ekspresję genów i uniemożliwiać chorobę
- terapia genowa dotyczy też nowotworów
- diagnostyka presymptomatyczna: jeżeli jedno dziecko rodzi się z wadą i cała rodzina choruje, to prowadzi się diagnostykę ciąży w 8-11 tyg. (izolacja trofoblastów z płynu owodniowego, możliwość uzyskania 10 μg DNA, amplifikuje PCR i bada wady)
diagnostyka ta dotyczy kilku technik:
amniocenteza - pobranie komórek w płynu owodniowego
fetoskopia - endoskopia, uwidacznianie płodu
badania kosmówki, pobieranie krwi, skóry
wypłukiwanie embrionu
NOWOTWORY:
- przyczyna zgonów
- to komórka, która traci zdolność samokontroli nad wzrostem i podziałami
- powstaje w sposób ciągły, w 3 etapach
przyczyny:
- mutacje (spontaniczne, indukowane)
- wirusy: zmiany w DNA (np. SV40 do 7 chromosomu; RNA - ptasia i mysia białaczka)
wirus atakuje, gdy osobnik osłabiony
cechy:
- lekceważenie praw wzrostu (autonomia komórki)
- utrata organizacji, celowości, funkcji komórki
- utrata zdolności do przylegania (choroba błon komórkowych)
- beztlenowy metabolizm (nadmierna produkcja kwasu mlekowego)
- niestabilność genetyczna (zmiana kariotypu chromosomów)
cytogenetyka - określenie, czy nowotwór jest złośliwy
kolchicyna, kolcemid - zatrzymanie komórki w metafazie
5
Wyszukiwarka