
Cancer Risk According to Type and Location of ATM
Mutation in Ataxia-Telangiectasia Families
E. Cavaciuti,
1,2
A. Lauge´,
3
N. Janin,
4
† K. Ossian,
4
J. Hall,
5
D. Stoppa-Lyonnet,
3
and N. Andrieu
1,2
*
1
Inserm Emi 00-06, Evry, France
2
Institut Curie, Service de Biostatistiques, Paris, France
3
Institut Curie, Service de Ge´ne´tique Oncologique, Paris, France
4
Institut Gustave Roussy, Villejuif, France
5
International Agency for Research on Cancer, DNA Repair Group, Lyon, France
Epidemiological studies have indicated that ataxia-telangiectasia (AT) heterozygotes in AT families have an increased risk of
cancer, particularly of breast cancer (BC). However, in BC case– control studies, no significant differences were found in the
frequency of ATM mutations between patients and controls. In such studies missense mutations were found more frequently
than truncating mutations, suggesting that the cancer risk depends on mutation type. To investigate this possibility, we assessed
the risk of BC according to the type and position of the ATM truncating mutation in extended AT families. DNA or RNA that
had been isolated from blood or buccal cells of AT children and their relatives was screened for ATM germ-line mutations using
restriction endonuclease fingerprinting, the protein truncation test, fluorescence-assisted mismatch analysis, and direct
sequencing. The standardized incidence ratio of cancer associated with ATM heterozygosity status and type of mutation was
estimated. We tested for genotype–phenotype correlations by simulations, permuting mutations among parental branches. No
significant difference was found in the relative risk of breast cancer or any other type of cancer based on mutation type.
However, the occurrence of BC may be associated with truncating mutations in certain binding domains of the ATM protein
(e.g., P53/BRCA1,
-adaptin, and FAT domains; P ⫽ 0.006). In this limited sample set, the presence of missense or truncating
ATM mutations was not associated with different cancer risks. The risk of BC appeared to be associated with the alteration
of binding domains rather than with the length of the predicted ATM protein.
©
2004 Wiley-Liss, Inc.
INTRODUCTION
Ataxia-telangiectasia (AT) is a rare autosomal
recessive disorder characterized by progressive
neuronal degeneration, immunological deficiency,
radiosensitivity, and increased risk of cancer. Epi-
demiological studies on AT families have shown
that AT heterozygotes (HetATs) have an increased
risk of developing cancer, particularly breast cancer
(BC) in female relatives. Since the ATM gene was
isolated (Savitsky et al., 1995a), BC cases and con-
trols have been screened for ATM mutations in
order to evaluate the public health implications.
Indeed, although AT is a rare disease, it is esti-
mated that 0.5%–1% of those in the general popu-
lation are AT mutation carriers. Thus, any in-
creased risk of cancer associated with ATM carrier
status is of public health relevance. Many of the
first case– control studies failed to find significant
differences between cases and controls in the fre-
quency of ATM mutations (Vorechovsky et al.,
1996a, 1996b; FitzGerald et al., 1997; Chen et al.,
1998). However, it was noted that missense muta-
tions were detected more frequently than truncat-
ing mutations than would have been expected
based on the profiles found in AT families. This
discrepancy between the findings of familial stud-
ies and those of case– control studies suggests that
the two types of ATM heterozygosity (i.e., the pres-
ence of truncating or missense mutations) confer
different cancer risks (McConville et al., 1996;
Gatti et al., 1999; Meyn, 1999), with missense mu-
tations associated with an increased risk of cancer
and truncating mutations leading to an AT pheno-
type. Thus, the increased risk of cancer observed in
relatives of AT children could have resulted from
factors associated with being a relative, particularly
being a mother, of an AT child rather than as a
direct consequence of ATM heterozygosity, as had
been suggested by Olsen et al. (2001). The pres-
†Present address: De´partement de Ge´ne´tique Humaine, CHU
Sart Tilman, Lie`ge, Belgique.
Supported by: Institut Gustave Roussy, Villejuif and Institut
Curie, Paris; Ligue Nationale Contre le Cancer; Comite´ des Hauts-
de-Seine de la Ligue Contre le Cancer; Service de Radioprotection
d’Electricite´ de France; INSERM.
*Correspondence to: Dr. Nadine Andrieu, Inserm Emi 00-06/
Service de Biostatistiques, Institut Curie, 26 rue d’Ulm, 75248 Paris
Cedex 05, France. E-mail: nadine.andrieu@curie.net
Received 5 April 2004; Accepted 15 July 2004
DOI 10.1002/gcc.20101
Published online 23 September 2004 in
Wiley InterScience (www.interscience.wiley.com).
GENES, CHROMOSOMES & CANCER 42:1–9 (2005)
RESEARCH ARTICLE
©
2004 Wiley-Liss, Inc.

ence of missense mutations was associated with a
16-fold increase in BC risk in two AT families
(Stankovic et al., 1998). One of these mutations
(T7271G/Val2424Gly) was associated with BC in
an Australian BC family (Chenevix-Trench et al.,
2002), although this association was not confirmed
in recent studies (Bernstein et al., 2003; Szabo et
al., 2004). Moreover, an abnormally high number of
spontaneous tumors, including a substantial num-
ber of mammary tumors, were observed in het-
erozygous knock-in mice (Atm-
⌬SRI) carrying a
mutation synonymous to that of the human in-
frame ATM deletion 7636del9 (Spring et al., 2002).
The difference in cancer risk associated with mu-
tation type could result from truncating mutations
leading to the synthesis of either no ATM protein
or a small amount of a shortened, possibly unstable
form, yet missense mutations leading to synthesis
of a normal level of abnormally functioning ATM
protein (McConville et al., 1996; Gatti et al., 1999;
Meyn, 1999). A truncated protein would have no or
little effect on the activity of the ATM protein
produced by the normal allele in ATM heterozy-
gotes (HetATM) and therefore would have little or
no effect on the overall function of ATM. In con-
trast, the protein encoded by a missense-mutated
allele could act as a dominant-negative inhibitor of
the ATM protein produced by the normal allele.
This would result in an alteration in the activity of
the DNA damage detection and repair system, in
which ATM is involved, and therefore in an in-
crease in cancer risk (Chenevix-Trench et al., 2002;
Scott et al., 2002). Indeed, the 7636del9 mutant
protein of the knock-in mice displayed dominant–
negative activity in cellular assays (Spring et al.,
2002). The results of in vitro studies using cells
heterozygous for ATM mutations also supported
the idea that a dominant–negative interaction oc-
curs between the normal and mutated forms of the
ATM protein. Bakkenist and Kastan (2003)
showed that ATM protein is held inactive in dimer
or multimer forms and that cellular irradiation in-
duces rapid intermolecular autophosphorylation,
causing dimer/multimer dissociation and initiating
ATM kinase activity. Thus, the presence of a de-
fective ATM protein may inhibit activation of the
normal protein. Fernet et al. (2004) observed that
cell lines carrying missense mutations were on av-
erage more radiosensitive than those carrying trun-
cating mutations. This finding is also consistent
with dominant–negative interactions involving
missense mutations and influencing HetATM cell
survival. None of the previous AT family studies
that assessed the risk of BC considered differences
associated with the presence of the two types of
ATM mutation. Furthermore, little is known about
the frequency or type of ATM mutation associated
with increased risk of cancer at other sites (Swift et
al., 1976; Swift et al., 1991; Swift and Su, 1999;
Angele and Hall, 2000; Geoffroy-Perez et al., 2001).
In the present study, we assessed the risk of
cancer among 34 families with AT children resi-
dent in France associated, first, with each of the
two types of ATM mutations (i.e., truncating or
missense mutations and in-frame deletions), and
second, for the truncating mutations, with the pre-
dicted length of the truncated protein. The stabil-
ity and therefore activity of a truncated ATM pro-
tein may differ according to its length and thus
influence the ability of the cell to repair DNA
damage and the risk of developing cancer.
MATERIALS AND METHODS
A family study of AT children was carried out in
France between June 1994 and February 1997. The
main design features of this study and the genotyping
of the ATM locus were described previously (Janin et
al., 1999) and are briefly summarized.
AT children were identified by pediatricians or
cytogeneticists. They were eligible for inclusion in
the study if they and their family were living in
France. Demographic data and data on the occur-
rence of cancer at any site were collected by trained
interviewers from first- (parents and siblings), second-
(uncles, aunts, and grandparents), and third- (great-
uncles and -aunts, great-grandparents, and cousins)
degree relatives of the AT children. A blood or buccal
cell sample was taken from each AT child and any
siblings, with parental agreement. A blood sample
was taken from each of the other family members
over age 18 who accepted participation in the study
and gave informed consent.
The proportion of cancers that were confirmed
differed according to cancer site. For example, 18
of the 29 BC and all the ovarian cancers were
confirmed by medical records, and there was com-
plete agreement between the case reports and the
pathology records. The proportion of prostate, co-
lon, pancreatic, and liver cancers confirmed was
lower (44%, 33%, 17%, and 0%, respectively). On
average, only 26% of the cancers at these sites were
confirmed from pathology reports.
The methods used for the identification of ATM
germ-line mutations were modified as new tech-
niques became available during the study period
(conducted between 1996 and 2003). For AT pa-
tients from 24 of the 34 families, eight overlapping
cDNA fragments covering the whole coding se-
2
CAVACIUTI ET AL.

quence (62 coding exons) were obtained using
RNA isolated from the corresponding lymphoblas-
toid cell line. Analysis of fragment size was used to
screen for the presence of any large partial dele-
tions of the ATM gene. Smaller mutations were
detected by restriction endonuclease fingerprinting
(REF), the protein truncation test (PTT), fluores-
cence assisted mismatch assays (FAMA), or, more
recently, direct sequencing. When a variant pattern
was observed by REF, PTT, or FAMA, the cDNA
was sequenced in both directions using DyeDeoxy
Terminator Cycle sequencing kits (Applied Biosys-
tems, Inc., Foster City, CA) using an ABI377 DNA
sequencer or ABI PRISM 3100 genetic analyzer
(Applied Biosystems, Inc.). When an abnormal
transcript was detected, the genomic DNA was
analyzed to identify the origin of the splicing de-
fect. Since the end of 2003, the 62 coding exons
and the intron– exon boundaries have been ana-
lyzed by sequencing using the BigDye Terminator
Cycle Sequencing V1.1 Ready Reaction Kit (Ap-
plied Biosystems, Inc.). SeqScape software was
used for sequence analysis (Lauge et al., unpub-
lished data).
The parental origin of each mutation was deter-
mined by identifying the mutations in DNA sam-
ples from both the mother and father. Lympho-
blastoid cell lines were unavailable for the AT
children of 9 families, and in these families, ATM
screening was performed on the parents. When no
mutation was found, ATM heterozygosity was de-
termined from the haplotypes. The haplotypes
were established from the segregation pattern in
the extended families by use of markers closely
linked to ATM (Janin et al., 1999). This method of
diagnosis assumes that a minimal number of ge-
netic recombination events have occurred. When
ATM heterozygosity status could not be deter-
mined, the probability that the individual shared
an ATM mutation with his or her closest HetATM
relative was calculated. As a result, four classes of
relatives were defined: relatives that were obligate
HetATM, relatives with a 0.5 probability of being
an ATM carrier (50% HetATM), relatives with a
0.25 probability of being an ATM carrier (25%
HetATM), and relatives that were not carriers
(non-HetATM). Siblings of AT children for whom
no DNA samples were available had a 0.66 proba-
bility of being HetATM: there were few such sib-
lings (six girls), all of whom were very young and
thus accounted for very few person-years in the
study population. To limit the number of classes,
these relatives were included in the 50% HetATM
category. Similarly, 15 subjects for whom the prob-
ability of being heterozygotes was 0.17 or less were
included in the non-HetATM group. Four men
with a 0.33 probability of being HetATM were
included in the 25% HetATM group. None of
these individuals was diagnosed with cancer.
Relatives of AT children were considered at risk
from birth until their age at interview or age at
death (for subjects unaffected by cancer) or until
their age at diagnosis (for subjects with cancer).
Nonmelanoma skin cancers were not considered
pathological, and thus these subjects were consid-
ered at risk until age at interview or age at death or
until development of cancer at another site. When
the age at diagnosis of cancer was unknown, age at
death was used. The expected number of cancers
per 5-year age category was calculated from the
French age-, sex- and period-specific (1978 –1982,
1983–1987,
1988 –1992)
estimated
incidences
(Benhamou et al., 1990; De Vathaire et al., 1996;
Me´ne´goz and Che´rie´-Challine, 1999), using the
PYRS program (Coleman et al., 1986). The esti-
mated incidences for the period 1978 –1982 were
used to calculate the expected number of cancers
in the period before 1978 and the estimated inci-
dences for the period 1988 –1992 were used to
calculate the expected number of cancers in the
period after 1992. The standardized incidence ratio
(SIR) of cancer associated with HetATM status
was estimated from the ratio between the observed
number of cases (O) and the expected number of
cases (E) in the AT families. We also calculated the
relative risk (RR) weighed on the a priori probabil-
ity of being HetATM (RR
w
; Thompson and Eas-
ton, 2002). Two-sided 95% confidence intervals
(CIs) for RR estimates, heterogeneity, and trend
tests were based on the approximation of the Pois-
son distribution (Breslow and Day, 1987; Thomp-
son and Easton, 2002).
The association of the location of truncating mu-
tations in the ATM gene and the risk of cancer was
investigated by dividing the ATM protein arbi-
trarily into 15 sections, 14 of which contained 200
amino acids (aa) and one of which contained 256 aa.
The significance of the association of the location
of the truncating mutations with BC risk was esti-
mated by simulation, permuting mutations among
parental branches as previously described (Gayther
et al., 1995; Thompson and Easton, 2001). Parental
branches with truncating mutations were randomly
permuted 10,000 times within the 15 sections. The
resulting deviance statistics were calculated under
the null hypothesis of no genotype–phenotype cor-
relation and were compared with the deviance sta-
tistic calculated from this data set. The significance
3
CANCER RISK ACCORDING TO ATM MUTATION

level was calculated as the proportion of the ran-
dom deviance statistics that were at least as large as
the deviance observed. The number of relatives
with cancer was calculated by counting the number
of cancers occurring either in obligate-HetATM
relatives or in all relatives (excluding the non-
HetATM), weighting and not weighting on the a
priori probability of being HetATM.
RESULTS
Thirty-four of the 35 families contacted agreed to
participate in this study. The 34 families included
1,423 relatives of AT patients (mean of 42 relatives
per family, SD
⫽ 19, and a total of 64,492 person-
years) with information available concerning demo-
graphic characteristics and the occurrence of any can-
cer.
DNA
samples
were
collected
from
401
individuals, which allowed us to classify 412 other
individuals as either obligate-HetATM or non-
HetATM. The mean ages of the groups of relatives
for each ATM heterozygote status were similar (data
not shown), and the overall mean age was 45.4 years.
The ATM mutation was not assessed in 15 of the
69 (maternal and paternal) branches from the 34
AT families (one family had AT children who were
cousins and was considered to have three branches)
because no lymphoblastoid cell line was available
from the AT child or from one of its parents. In the
54 branches tested, 45 mutations were detected
(83.3%), of which 34 were distinct (Table 1): 29
TABLE 1. ATM Mutations Detected Among the 34 Families (69 Analyzed Parental Branches)
Nucleotide change (genomic origin identified)
Amino acid change
Number of parental branches
Truncated protein
73 del4 and 73 del7 (IVS04-2 A
⬎G)
ter31 and ter32
a
2
118 del4
ter42
1
137 del4
ter54
1
381 delA
ter128
2
497 del22
b
ter169
1
1066 del170
b
ter372
1
1563 delAG
ter564
4
2734 C
⬎T
Gln912stop
1
2838 ins65 (IVS20–495 del4)
ter969
2
3275 C
⬎A
Ser1092stop
1
3711 del5
ter1243
2
3753 delTAT
⬎insGA
ter1255
2
3802 delG
ter1268
1
4661 insA
ter1560
1
5644 C
⬎T
Arg1882stop
1
5791 delG
⬎insCCT
ter1936
1
5932 G
⬎T
Glu1978stop
1
6004 C
⬎T
Gln2002stop
1
6007 del89 (6095 G
⬎A)
ter2007
1
6100 C
⬎T
Arg2034stop
1
6452 ins24 (IVS46-1 del6)
ter2159
1
7327 C
⬎T
Arg2443stop
1
7630 del11 (IVS53-2 A
⬎C)
ter2566
2
7792 C
⬎T
Arg2598stop
1
7789 del139 (IVS55
⫹ 5 delG)
ter2599
2
8011 del13 (IVS56-2 A
⬎C)
ter2678
1
8030 delA
ter2681
1
8140 C
⬎T
Gln2714stop
1
8395 del10
ter2802
1
Missense mutation or in-frame deletion
7462 T
⬎C
Cys2488Arg
1
7875 TG
⬎GC
Asp-Ala2625Glu-Pro
1
8489 T
⬎G
Val2830Gly
1
3403 del174 (3576 G
⬎A)
del58
2
3874 del120 (IVS28
⫹ 1 G⬎A)
del40
1
Nucleotide numbering according to Savitsky et al.,1995b.
a
IVS04-2 A
⬎G : homozygote genomic mutation that leads to two different proteins in the same individual.
b
No genomic origin identified.
4
CAVACIUTI ET AL.

resulted in premature termination codons at vari-
ous positions in the ATM gene; two were in-frame
deletions [3874 del 120 (IVS 28
⫹1 G3A) and 3403
del174 (3576 G
3A), leading to the deletion of 40
and 58 aa, respectively, within a region of the ATM
protein containing no identified functional do-
mains; in both proteins, the PI-3 kinase domain
remained intact, encoded by the in-frame mutated
allele]; and three were missense mutations, all in or
around the region encoding the PI-3 kinase domain
of the ATM protein. Only the sequence variants
that were predicted to lead to an amino acid sub-
stitution in the functional domains of the ATM
protein were considered missense mutations. Vari-
ants were found in the FAT-binding and PI-3 ki-
nase domains, which were rare events. Other vari-
ants were considered “unclassified variants,” and in
the analyses, branches with such unclassified vari-
ants were combined with branches with no de-
tected mutation.
The frequencies of the parental branches in
which (i) a truncating mutation or (ii) a missense
mutation or an in-frame deletion segregated were
86.7% (39 of 45) and 13.3% (6 of 45), respectively.
The 39 branches with a truncating mutation in-
cluded 809 relatives (396 women), the six branches
with a missense or an in-frame type mutation in-
cluded 146 relatives (70 women), and branches for
which no ATM mutation was found included 238
relatives (118 women). The RR of cancer was
assessed by SIR (Table 2). In addition to exclud-
ing nonmelanoma skin cancer, we excluded liver
cancer from the other cancer sites examined be-
cause we were not able to verify these cancers
histologically and therefore were unable to dis-
tinguish primary hepatic cancer from metastasis
originating from a cancer at another site. The RR
of
BC
associated
with
being
an
obligate
HetATM was 3.96 (95% CI: 1.81–7.53) and was
similar for all types of mutation: 3.68 for individ-
uals carrying truncating mutations, 3.70 for those
carrying missense or in-frame mutations, and
4.76 for those with undetected mutations. For
the group with uncertain ATM heterozygosity
TABLE 2. Relative Risk of Cancer According to Mutation Type and HetATM Status
a
Type of mutation
HetATM status
O
E
SIR (95% CI)
Weighted SIR (95% CI)
Breast (among female relatives)
All types
Obligate-HetATM
9
2.27
3.96 (1.81–7.53)
2.43 (1.32–4.09)
25%
⫹ 50% HetATM
8
8.37
0.96 (0.41–1.88)
Truncating mutation
Obligate HetATM
5
1.36
3.68 (1.18–8.58)
1.93 (0.78–3.95)
25%
⫹ 50% HetATM
4
5.86
0.68 (0.18–1.75)
Missense or in-frame mutation
Obligate-HetATM
1
0.27
3.70 (0.05–20.6)
1.96 (0.03–10.9)
25%
⫹ 50% HetATM
0
0.50
—
No detected mutation
Obligate-HetATM
2
0.42
4.76 (0.53–17.2)
3.25 (0.59–9.89)
25%
⫹ 50% HetATM
1
1.09
0.92 (0.01–5.10)
Non-HetATM
11
8.21
1.34 (0.67–2.40)
All sites except breast, liver, and nonmelanoma skin (both male and female relatives)
All types
Obligate-HetATM
13
14.39
0.90 (0.48–1.54)
1.07 (0.75–1.49)
25%
⫹ 50% HetATM
47
46.11
1.02 (0.75–1.36)
Truncating mutation
Obligate-HetATM
9
8.53
1.06 (0.48–2.00)
1.30 (0.85–1.90)
25%
⫹ 50% HetATM
34
30.55
1.11 (0.77–1.56)
Missense or in frame mutation
Obligate-HetATM
0
1.25
—
1.10 (0.21–3.28)
25%
⫹ 50% HetATM
6
3.14
1.91 (0.70–4.16)
No detected mutation
Obligate-HetATM
2
2.22
0.90 (0.10–3.25)
0.61 (0.13–1.76)
25%
⫹ 50% HetATM
3
7.22
0.42 (0.08–1.21)
Non-HetATM
33
43.59
0.76 (0.52–1.06)
a
O, observed number of cases; E, expected number of cases.
5
CANCER RISK ACCORDING TO ATM MUTATION
(i.e., both the 50% and 25% HetATM groups
combined), the RR of BC was not increased
either for the entire group or for the mutation-
type subgroups. The RR of cancer at other sites
was not increased for any HetATM status or for
any type of mutation.
The RR was 8.43 (95% CI: 2.73–19.8) for all
obligate-HetATM female relatives under 45 years
and was 2.39 (95% CI: 0.64 – 6.10) for female rela-
tives
ⱖ 45 years. However, the RRs of the two
groups were not significantly different (results pre-
viously published; Geoffroy-Perez et al., 2001).
The RR was then weighted (RR
w
) on the a priori
probability of being a heterozygote: the RR
w
for
female relatives under 45 years (6.32, 95% CI:
1.94 –15.2) was significantly higher (P
⫽ 0.001)
than that for female relatives
ⱖ 45 years (0.68, 95%
CI: 0.08 –2.46; data not shown).
The RR of cancer also was estimated on the
basis of the presence of a truncating mutation in
the different sections of the ATM gene. BC risk was
significantly associated with the location of the
truncating mutation (P
⫽ 0.006). In other words,
the truncating mutations in individuals with BC
were not randomly scattered throughout the ATM
gene. We used the RR
w
to be able to exploit the
majority of the available information on cancer oc-
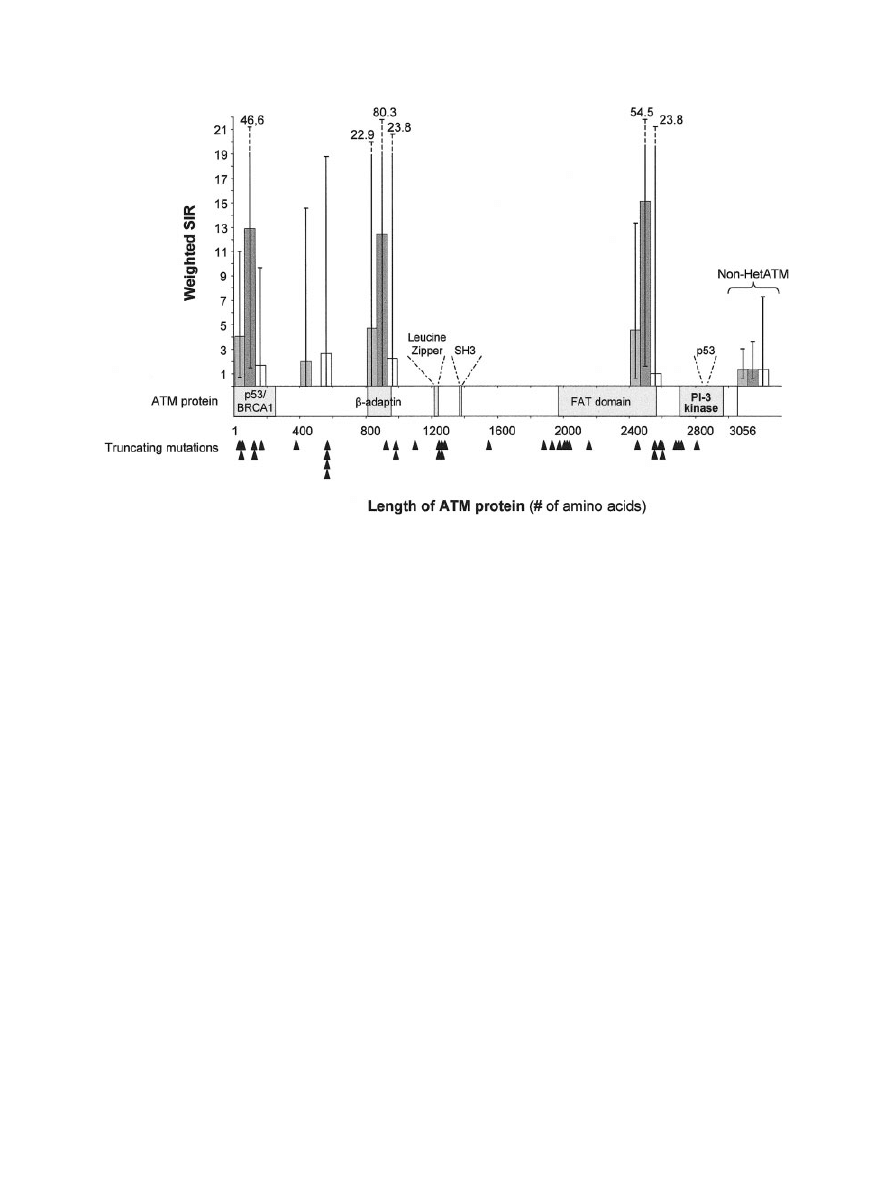
currence in the families (Fig. 1 shows the results
for BC for women as a group and for women sub-
divided into two age classes). The RR
w
of BC was
particularly high for mutations in four sections of
the ATM protein. Interestingly, the three highest
values corresponded to domains of the ATM pro-
tein that were previously characterized: the N ter-
minal, 200 aa, which is part of the P53/BRCA1
binding domain; the 800th–1,000th aa, part of the
-adaptin binding domain; and the 2,400th–
2,600th aa, at the end of the FAT-binding domain
(Stankovic et al., 2002). We were unable to identify
any significant trend or difference in estimated RR
among these four sections. However, it should be
noted that the power of this study for detecting
differences between sections was very low because
of the small sample sizes. The point estimates for
the four sections were all similar, around 4. The
point estimates were higher for female relatives
under 45 years of age than for those 45 years and
older for all sections.
The results for cancer risk at other sites for all
relatives and for all relatives by gender are shown
Figure 1.
Relative risk of breast cancer according to the predicted
length of ATM proteins resulting from truncating mutations. Relative
risks and 95% confidence intervals were estimated by dividing the ATM
protein arbitrarily into 15 sections [14 contained 200 amino acids (aa)
and 1 contained 256 aa] and are shown as dashed for women
ⱖ 45
years old, white for women
⬍ 45 years old, and gray for all women.
Black arrowheads indicate truncating mutations. For comparative pur-
poses, the relative risks for the three age groups of female relatives in
the non-HetATM group are shown.
6
CAVACIUTI ET AL.
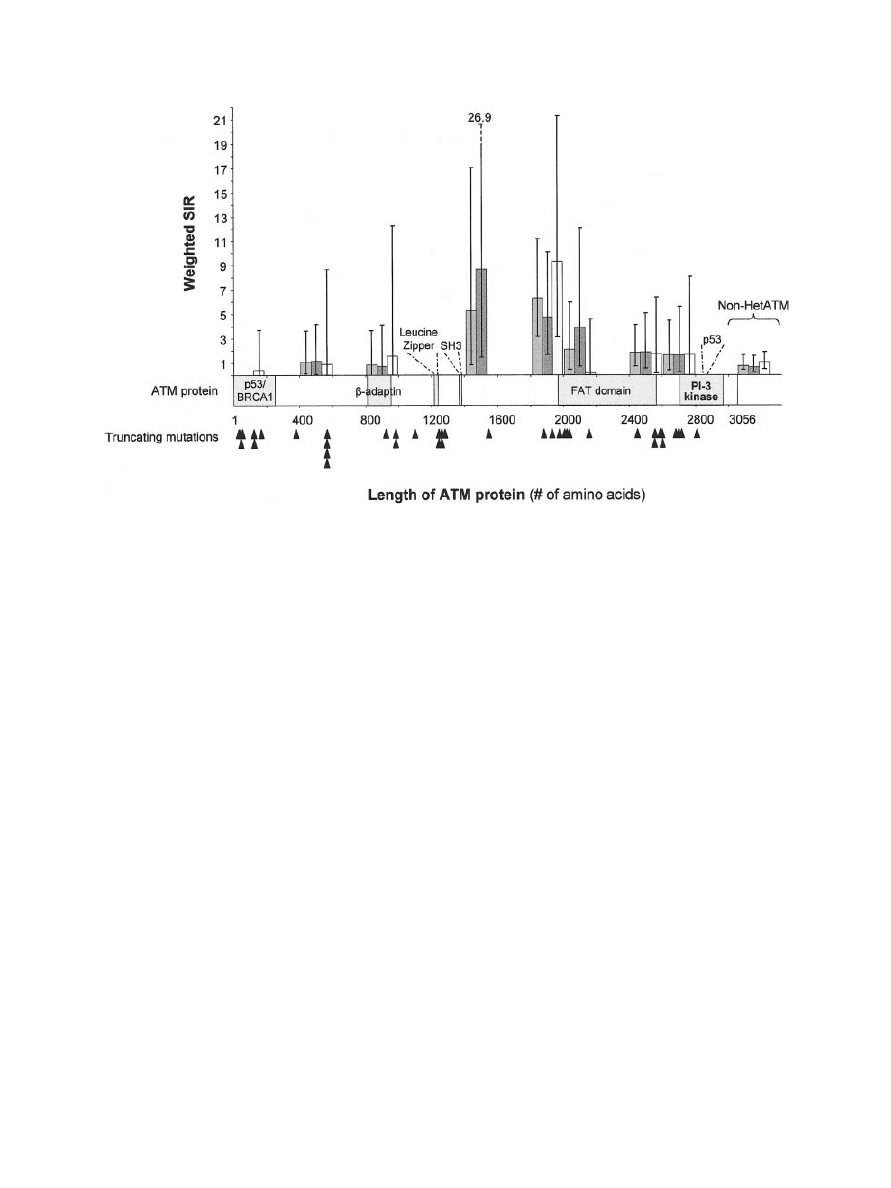
in Figure 2. The site of cancer other than BC was
not associated with the location of the mutation
(P
⫽ 0.19). In other words, the mutations in indi-
viduals with cancers at sites other than the breast
were randomly scattered through the ATM gene,
even when only cancers of the digestive tract were
considered. The point estimate of the RR
w
for
cancer with truncating mutations in the first half of
the protein was not increased. For truncating mu-
tations resulting in an ATM protein truncated in
the middle (between the 1,400th and 1,600th aa or
between the 1,800th and 2,000th aa), the RR
w
point estimate of developing a cancer at a site other
than the breast was high [5.31, 95% CI: 0.83–17.05
(not significant), and 6.24, 95% CI: 3.39 –11.19,
respectively]. The most frequent cancers associ-
ated with these two sections were cancers of the
digestive tract (colorectal and stomach, 6 of the 16
declared cancers) and lung cancer (5 of the 16).
The increased risk for the section between the
1,400th and 1,600th aa was observed only for male
relatives (only cancers of the digestive tract), but
that for the section between the 1,800th and
2,000th aa was observed similarly for both women
and men and for all cancer types. These regions of
the ATM protein do not contain any known func-
tional domains. The point estimates for RR
w
were
lowest for truncating mutations in the part of the
gene encoding the C-terminal fragment of the pro-
tein. The estimated RR
w
did not differ signifi-
cantly according to the location of a truncating
mutation; indeed, we were unable to identify any
significant trend or difference in the RR estimates
between the different sections of the ATM gene for
cancers at sites other than the breast.
DISCUSSION
The RR of cancer (at breast or other sites) did
not differ significantly according to the type of
ATM mutation carried by the AT family members.
Even though the power of our study for detecting
differences between subgroups was limited, the
RR point estimates gave no indication that there
were different risks for BC or for cancer at other
sites associated with either truncating or missense
and in-frame mutations. However, as there were
few missense and in-frame mutations considered in
this study, our estimate is imprecise. The cancer
risk at other sites associated with truncating muta-
tions was not significantly related to the position of
the truncating mutation, although the point esti-
mates indicated that the associated risk was higher
Figure 2.
Relative risk of cancer at any site according to the pre-
dicted length of ATM proteins resulting from truncating mutations.
Relative risks and 95% confidence intervals were estimated by dividing
the ATM protein arbitrarily into 15 sections [14 contained 200 amino
acids (aa) and 1 contained 256 aa] and are shown as dashed for men,
white for women, and gray for all individuals. Black arrowheads indicate
truncating mutations. For comparative purposes, the relative risks for
the three groups of relatives in the non-HetATM group are shown.
7
CANCER RISK ACCORDING TO ATM MUTATION

for mutations in the middle region of the protein.
Conversely, BC risk was associated with the posi-
tion of some truncating mutations: mutations in
certain binding domains of the ATM protein (i.e.,
P53/BRCA1,
-adaptin, and FAT-binding do-
mains) led to a significantly increased risk of BC.
One possible explanation is that ATM mRNA car-
rying mutations in such domains is preferentially
eliminated, resulting in haploinsufficiency and
therefore in increased cancer risk. An alternative
possibility is that this increased risk may be asso-
ciated with the absence of certain binding domains
or with the presence of altered binding domains in
a truncated ATM protein produced in cells carrying
such mutations. The mechanism by which the pro-
duction of a truncated protein affects the ability of
the normal ATM protein to function is unknown.
The transcription and translation of such ATM vari-
ants will have to be analyzed in order to understand
the underlying molecular mechanisms.
In our study, no ATM mutations were detected
in about 13% of the tested branches. In this cate-
gory, the BC risk was slightly higher than the risk
observed in other mutation categories, but the RR
estimates did not differ significantly. It is difficult
to attribute this slightly increased risk to the pos-
sible presence of a particular type of mutation. A
variety of techniques were used for mutation
screening with varying sensitivity of detection for
certain classes of sequence variants, and possible
differences in the stability of ATM transcripts, de-
pending on the mutation type, also may influence
the detection sensitivity. Extreme scenarios in
which missed mutations are either all truncating or
all missense and in-frame lead to similar overall
conclusions: that there are no indications of differ-
ent risks for BC or cancer at other sites according to
the type of ATM mutation present.
Poor sensitivity of a self-reported family history of
cancer (i.e., underreporting) may have led to an un-
derestimation of the relative risk of cancer. However,
most BC cases were verified, and BC has been re-
ported with great accuracy in numerous other studies
(e.g., Theis et al., 1994). We were unable to verify
many of the cancers at other sites, such as the colon.
However, a recent study showed that a family history
of cancer was reported with a high positive predictive
value (Ziogas and Anton-Culver, 2003). We were un-
able to verify the disease status of relatives declared
unaffected as France has no national cancer registry.
However, it has been estimated that about 98% of
reports of a negative family history are correct (Aitken
et al., 1995; Ziogas and Anton-Culver, 2003). Calcu-
lation of the expected number of cancer cases is
known to be sensitive to the reference population
used. The incidence of several cancers, including BC,
has been increasing in Western European countries
for a number of decades. In this study, we used the
estimated incidence of cancer for 1978 –1982 as the
reference population for the calculation of the ex-
pected number of cancer cases among relatives at risk
before
1978
(mostly
grandmothers/grandfathers,
great-aunts/great-uncles,
and
great-grandmothers/
great-grandfathers of AT children), possibly leading
to overestimating this number. Inversely, the ex-
pected number of cancers among relatives at-risk
after 1992 (mostly mothers/fathers and aunts/uncles)
may be underestimated because we used the esti-
mated incidences of cancer for 1988 –1992 as the
reference population. This may have biased the RR
estimates but would not affect the comparisons be-
tween risks associated with mutation type.
The estimates of weighted relative risk of BC
were lower than the estimates of relative risk of
BC among those in the obligate-HetATM group.
This is because there was no trend in the asso-
ciation between BC risk and HetATM status and
no increased risk for relatives with uncertain
HetATM status. This lack of a trend may have
been a result of bias from having a higher pro-
portion of known heterozygotes among BC cases
than among unaffected family members. Indeed,
ATM heterozygosity was uncertain in 29% of BC
cases, whereas ATM heterozygosity was uncer-
tain in 44% of unaffected family members.
Therefore, the slight and not significantly in-
creased risk of BC of non-HetATM individuals
may have reflected this possible bias, suggesting
that obligate-HetATM BC risk was not substan-
tially overestimated. It should be noted that Ol-
sen et al. (2001) also found no gradient of BC
incidence above background levels between
mothers, sisters, aunts, and cousins without a
bias in the proportions of known HetAT status
because the status was defined a priori according
to the relationship with the AT child.
In conclusion, our findings do not support the
hypothesis that the two types of ATM mutation are
associated with different risks of cancer among AT
families (McConville et al., 1996; Gatti et al., 1999;
Meyn, 1999). However the risk of BC may be
associated with the location of truncating muta-
tions. As our study was limited by sample size,
more powerful studies, using data sets pooled from
international sources, are needed to confirm our
observations.
8
CAVACIUTI ET AL.

ACKNOWLEDGMENTS
We are very grateful to the physicians who
helped us contact families with AT children—J.-O.
Bay, C. Billard, M.-T. Boguais, J.-P. Gout, B. Le-
heup, N. Philip, J.-P. Pollet, M.-F. Croquette, C.
Griscelli, and M. Debre´—and to the participating
families. We also thank Josyane Le Calvez for tech-
nical assistance and Alex Edelman & Associates for
the linguistic revision of the manuscript.
REFERENCES
Aitken J, Bain C, Ward M, Siskind V, MacLennan R. 1995. How
accurate is self-reported family history of colorectal cancer? Am J
Epidemiol 141:863– 871.
Angele S, Hall J. 2000. The ATM gene and breast cancer: is it really
a risk factor? Mutat Res 462:167–178.
Bakkenist CJ, Kastan MB. 2003. DNA damage activates ATM
through intermolecular autophosphorylation and dimer dissocia-
tion. Nature 421:499 –506.
Benhamou E, Laplanche A, Wartelle M, Gignoux M, Me´ne´goz F,
Robillard J, Schaffer P, Schraub S, Flamant R. 1990. Statistiques
de sante´. Paris: INSERM.
Bernstein JL, Bernstein L, Thompson WD, Lynch CF, Malone KE,
Teitelbaum SL, Olsen JH, Anton-Culver H, Boice JD, Rosen-
stein BS, Borresen-Dale AL, Gatti RA, Concannon P, Haile RW.
2003. ATM variants 7271T
⬎G and IVS10–6T⬎G among women
with unilateral and bilateral breast cancer. Br J Cancer 89:1513–
1516.
Breslow NE, Day NE. 1987. Statistical Methods in Cancer Re-
search. IARC: Lyon.
Chen J, Birkholtz GG, Lindblom P, Rubio C, Lindblom A. 1998.
The role of ataxia-telangiectasia heterozygotes in familial breast
cancer. Cancer Res 58:1376 –1379.
Chenevix-Trench G, Spurdle AB, Gatei M, Kelly H, Marsh A, Chen
X, Donn K, Cummings M, Nyholt D, Jenkins MA, Scott C, Pupo
GM, Dork T, Bendix R, Kirk J, Tucker K, McCredie MR, Hopper
JL, Sambrook J, Mann GJ, Khanna KK. 2002. Dominant negative
ATM mutations in breast cancer families. J Natl Cancer Inst
94:205–215.
Coleman M, Douglas A, Hermon C, Peto J. 1986. Cohort study
analysis with a FORTRAN computer program. Int J Epidemiol
15:134 –137.
De Vathaire F, Koscielny S, Rezvani A, Laplanche A, Este`ve J,
Ferlay J. 1996. Statistiques de sante´. Paris: INSERM.
Fernet M, Moullan N, Lauge A, Stoppa-Lyonnet D, Hall J. 2004.
Cellular responses to ionising radiation of AT heterozygotes: dif-
ferences between missense and truncating mutation carriers. Br J
Cancer 90:866 – 873.
FitzGerald MG, Bean JM, Hegde SR, Unsal H, MacDonald DJ,
Harkin DP, Finkelstein DM, Isselbacher KJ, Haber DA. 1997.
Heterozygous ATM mutations do not contribute to early onset of
breast cancer. Nat Genet 15:307–310.
Gatti RA, Tward A, Concannon P. 1999. Cancer risk in ATM
heterozygotes: a model of phenotypic and mechanistic differences
between missense and truncating mutations. Mol Genet Metab
68:419 – 423.
Gayther SA, Warren W, Mazoyer S, Russell PA, Harrington PA,
Chiano M, Seal S, Hamoudi R, van Rensburg EJ, Dunning AM, et
al. 1995. Germline mutations of the BRCA1 gene in breast and
ovarian cancer families provide evidence for a genotype-pheno-
type correlation. Nat Genet 11:428 – 433.
Geoffroy-Perez B, Janin N, Ossian K, Lauge A, Croquette MF,
Griscelli C, Debre M, Bressac-de-Paillerets B, Aurias A, Stoppa-
Lyonnet D, Andrieu N. 2001. Cancer risk in heterozygotes for
ataxia-telangiectasia. Int J Cancer 93:288 –293.
Janin N, Andrieu N, Ossian K, Lauge A, Croquette MF, Griscelli C,
Debre M, Bressac-de-Paillerets B, Aurias A, Stoppa-Lyonnet D.
1999. Breast cancer risk in ataxia telangiectasia (AT) heterozy-
gotes: haplotype study in French AT families. Br J Cancer 80:
1042–1045.
McConville CM, Stankovic T, Byrd PJ, McGuire GM, Yao QY,
Lennox GG, Taylor MR. 1996. Mutations associated with variant
phenotypes in ataxia-telangiectasia. Am J Hum Genet 59:320 –
330.
Me´ne´goz F, Che´rie´-Challine L. 1999. Le cancer en France: inci-
dence et mortalite´. Paris: Ministe`re de l’emploi et de la solidarite´.
Meyn MS. 1999. Ataxia-telangiectasia, cancer and the pathobiology
of the ATM gene. Clin Genet 55:289 –304.
Olsen JH, Hahnemann JM, Borresen-Dale AL, Brondum-Nielsen
K, Hammarstrom L, Kleinerman R, Kaariainen H, Lonnqvist T,
Sankila R, Seersholm N, Tretli S, Yuen J, Boice JD Jr, Tucker M.
2001. Cancer in patients with ataxia-telangiectasia and in their
relatives in the nordic countries. J Natl Cancer Inst 93:121–127.
Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L,
Tagle DA, Smith S, Uziel T, Sfez S. 1995a. A single ataxia
telangiectasia gene with a product similar to PI-3 kinase. Science
268:1749 –1753.
Savitsky K, Sfez S, Tagle DA, Ziv Y, Sartiel A, Collins FS, Shiloh Y,
Rotman G. 1995b. The complete sequence of the coding region of
the ATM gene reveals similarity to cell cycle regulators in differ-
ent species. Hum Mol Genet 4:2025–2032.
Scott SP, Bendix R, Chen P, Clark R, Dork T, Lavin MF. 2002.
Missense mutations but not allelic variants alter the function of
ATM by dominant interference in patients with breast cancer.
Proc Natl Acad Sci USA 99:925–930.
Spring K, Ahangari F, Scott SP, Waring P, Purdie DM, Chen PC,
Hourigan K, Ramsay J, McKinnon PJ, Swift M, Lavin MF. 2002.
Mice heterozygous for mutation in Atm, the gene involved in
ataxia-telangiectasia, have heightened susceptibility to cancer.
Nat Genet 32:185–190.
Stankovic T, Kidd AM, Sutcliffe A, McGuire GM, Robinson P,
Weber P, Bedenham T, Bradwell AR, Easton DF, Lennox GG,
Haites N, Byrd PJ, Taylor AM. 1998. ATM mutations and phe-
notypes in ataxia-telangiectasia families in the British Isles: ex-
pression of mutant ATM and the risk of leukemia, lymphoma,
and breast cancer. Am J Hum Genet 62:334 –345.
Stankovic T, Stewart GS, Byrd PJ, Fegan C, Moss PAH, Taylor AM.
2002. ATM mutations in sporadic lymphoid tumors. Leuk Lym-
phoma 43:1563–1571.
Swift M, Morrell D, Massey RB, Chase CL. 1991. Incidence of
cancer in 161 families affected by ataxia-telangiectasia. N Engl
J Med 325:1831–1836.
Swift M, Su Y. 1999. Link between breast cancer and ATM gene is
strong. BMJ 318:400.
Swift M, Sholman L, Perry M, Chase C. 1976. Malignant neoplasms
in the families of patients with ataxia-telangiectasia. Cancer Res
36:209 –215.
Szabo CI, Schutte M, Broeks A, Houwing-Duistermaat JJ, Thor-
stenson YR, Durocher F, Oldenburg RA, Wasielewski M, Odefrey
F, Thompson D, Floore AN, Kraan J, Klijn JG, van den Ouweland
AM, Wagner TM, Devilee P, Simard J, ’t Veer LJ, Goldgar DE,
Meijers-Heijboer H. 2004. Are ATM mutations 7271T
3G and
IVS10 – 6T
3G really high-risk breast cancer-susceptibility al-
leles? Cancer Res 64:840 – 843.
Theis B, Boyd N, Lockwood G, Tritchler D. 1994. Accuracy of
family cancer history in breast cancer patients. Eur J Cancer Prev
3:321–327.
Thompson D, Easton D. 2001. Variation in cancer risks, by mutation
position, in BRCA2 mutation carriers. Am J Hum Genet 68:410 –
419.
Thompson D, Easton DF. 2002. Cancer Incidence in BRCA1 mu-
tation carriers. J Natl Cancer Inst 94:1358 –1365.
Vorechovsky I, Luo L, Lindblom A, Negrini M, Webster AD, Croce
CM, Hammarstrom L. 1996a. ATM mutations in cancer families.
Cancer Res 56:4130 – 4133.
Vorechovsky I, Rasio D, Luo L, Monaco C, Hammarstrom L, Web-
ster AD, Zaloudik J, Barbanti-Brodani G, James M, Russo G, et al.
1996b. The ATM gene and susceptibility to breast cancer: anal-
ysis of 38 breast tumors reveals no evidence for mutation. Cancer
Res 56:2726 –2732.
Ziogas A, Anton-Culver H. 2003. Validation of family history data in
cancer family registries. Am J Prev Med 24:190 –198.
9
CANCER RISK ACCORDING TO ATM MUTATION
Wyszukiwarka
Podobne podstrony:
Guide to Selection and Use of D Nieznany
LUNGA Approaches to paganism and uses of the pre Christian past
Niqaab according to Quran and Sunnah
Microstructures and stability of retained austenite in TRIP steels
feminism and formation of ethnic identity in greek culture
Causes and control of filamentous growth in aerobic granular sludge sequencing batch reactors
article expenditure patterns and timing of patent protection in a competitive R&D environment
Herbs Of The Field And Herbs Of The Garden In Byzantine Medicinal Pharmacy
Microstructures and stability of retained austenite in TRIP steels
feminism and formation of ethnic identity in greek culture
Dialectic Beahvioral Therapy Has an Impact on Self Concept Clarity and Facets of Self Esteem in Wome
Modeling And Simulation Of ATM Networks
The Name and Nature of Translation Studies In James S Holmes
FIDE Trainers Surveys 2012 08 31 Uwe Bönsch The recognition, fostering and development of chess tale
Variation in NSSI Identification and Features of Latent Classes in a College Population of Emerging
Wójcik, Marcin; Suliborski, Andrzej The Origin And Development Of Social Geography In Poland, With
article expenditure patterns and timing of patent protection in a competitive R&D environment
więcej podobnych podstron