Analizowane surowce i produkty w technice materiałów wiążących i budowlanych:
-surowce i kamienie wapienne, dolomity, kruszywa
-półprodukty i produkty
-kontrola jakości wody stosowanej w procesie technologicznym
-kontrola procesów spalania i składu gazów spalinowych
Cel i zakres badań:
1.Kontrola składu surowców, kontrola jakości półproduktów i produktów
Kontrola i nadzór nad przebiegiem procesu technologicznego
-analiza fazowa
-analiza chemiczna: metody normowe obejmujące oznaczanie składy główne (straty prażenia SiO2, CaO, MgO, Na2O, K20, Fe2O3, Al2O3, TiO2, oraz anionów SO3, Cl-, S2-
2.Badanie wpływu małych i śladowych ilości pierwiastków (głównie metali), na przebieg procesów fizyko-chemicznych (np. procesy hydratacji, samorozpadu spieku samorozpadowego)
3.Analiza zawartości metali śladowych (Pb, Cd, Zn, Cr, Mn, Tl, As, Be, Mo, Sb, Sn, V) w surowcach (np. kamień wapienny) i produktach przemysłu materiałów budowlanych (cementy) otrzymanych z zastosowaniem technik ekologicznych
4.Badania zawartości substancji toksycznych (metale ciężkie, chrom VI, siarczki, cyjanki) w wyciągach wodnych otrzymywanych w wyniku ługowania cementu oraz spoiw cementowych lub żużlowo-alkalicznych immobilizowanych metalami ciężkimi
5.Badania twardości wody stosowanej do celów przemysłowych- kontrola wydajności determinacji H20 na jonitach, analiza H20
6.Analiza gazów, kontrola zawartości CO2, CO w gazach spalinowych
7.Analiyka środowiska-badanie wpływu działalności przemysłowej na poziom stężenia środowiska natur.:
-tereny sąsiadujące z obiektami przemysłowymi, gleby, powietrze
-wody, ścieki
-powietrze
-flora i fauna, biologiczne aspekty zanieczyszczeń (liście, drzewa iglaste, owoce)
Chrom VI w cemencie dostaje się 3 drogami:
-z surowców
-z wymurówki pieca
-z mielenia
Analityka:
1.Analiza jakościowa
2.Analiza ilościowa
3.Analiza strukturalna
4.Analiza rozmieszczenia
5.Analiza specjacyjna- oznaczenie form fiz. i chem. pierwiastka
-szczegółowa
-grupowa
-frakcjowanie
Metoda analityczna -srtategiczna koncepcja uzyskiwania optymalnych informacji o obiekcie badań przy założonej zasadzie pomiaru
Zasada pomiaru-wykorzystanie zjawisk fizykochem. w celu uzyskania informacji analitycznych ( kodowanie i dekodowanie)
Próbka-podzbiór populacji podlegający bezpośrednio badaniu ze względu na daną cechę w celu wyciągnięcia wniosków wniosków kształtowaniu się wartości tej cechy w populacji
Próbka reprezentatywna-struktura nie różni się pod względem badanej cechy istotnie od struktuy populacji generalnej
Próbka laboratoryjna-przygotowana z próbki ogólnej reprezentująca wł. Partii produkty, przeznaczona do proadzenia analiz
Próbka analityczna-część produktu wydzielona z próbki labol. Przeznaczona w całości do jednego oznaczenia lub wykorzystania bezpośrednio do badania lub obserwacji
Postępowanie analityczne określone jest przepisami roboczymi:
1.Pobieranie próbek
2.Przygotowanie łącznie z niezbędnymi do tego odczynnikami, materiałami pomocniczymi, sprzętem
3.Układu pomiarowego pomiarowego uwzględnieniem stosowania przyrządówi zmiennych parametrów pomiaru
4.Wzorcowanie
5.Zakresu stosowania (oznaczalności i zakres zawartości)
6.Selektywności/specyficzności
7.Błedy systematyczne i przypadkowe
8.Wartości ślepej i granic oznaczalności
9.Zapotrzebowanie czasu i kosztów jednej analizy
Kryteria doboru metody analitycznej:
1.Rodzaj próbki
2.Przybliżony skład
3.Rodaj wyznaczanej informacji (oznaczenie skł. Głównego i śladowego)
4.Wymagana dokładność i precyzja
5.Czas trwania oznaczenia, pracochłonność
6.Ilość wykonywanych analiz i ich częstotliwość
7.Posiadana ilość próbki, stopień zanieczyszczenia lub konieczność jej zabezpieczenia
8.Posiadana aparatura, możliwość jej nabycia i koszty zakupu aparatury
9.Wpływ matrycy
10.Dostępność odczynników i ich trwałość
11.Koszt analizy
12.Osobiste przyzwyczajenia i wprawa
13.Normy krajowe, EWG, systemy jakości, badania kredytowe, ciągła automatyczna kontrola procesów przemysłowych
Kryterium podziału metod analitycznych:
1.Metody klasyczne (wagowe i objętościowe)
Metody instrumentalne
2.Analiza śladowa <10-2 IUPAC:
-ślady 10-2-10-8%,
-mikroślady 10-8-10-11%,
-monoślady 10-11-10-14%,
-pikoślady 10-14-10-17%
3.Metody bezwzględne (nie wymagają kalibracji np. met. Wagowa, miareczkowanie, elektrograwimetria, kubmetria)
Metody względne
CEMENT:
Skł. Główne: SiO2, CaO, MgO, Al2O3, Fe2O3, Na2O, K2O, SO3
Akcesoryczne: TiO, MnO, P2O5, V2O5, BaO, Cr, S
Śladowe: CrIII, CrVI, Tl, As, Pb, Te
Metody klasyczne
Wagowe: SiO2 SO3 Ba2+
Miareczkowe głównie kompleksometryczne: SiO2, Fe2O3, Al2O3, CaO, MgO, SO3, Cl-
Metody instrumentalne:
-ASA atomowa spektometria absorpcyjna Na2O K2O
-ICPAES plazmowa spektometria emisyjna
-Płomieniowa spektometria emisyjna
-spektofotometria absorpcyjna (kolorymetria) SiO2, TiO2, Fe2O3, MnO2, P2O5,
-potencjometryczne (chlorki, fluorki, borany)
-Tubimetryczne (chlorki)
-Woltamperometryczne TiO2 Fe2O3 MnO2
Metody roztwarzania w przemyśle cementowym:
1.wymywanie (leaching)
DEV-kruszymy i ciągu kilkunastu godzin wytrącamy zawiesinę z wody i oznaczamy zawartość subst. Toksycznych
TANK-kostkę betonu moczymy w r-rze i badamy po czasie ile substancji toksycznych przeszło do roztworu.
2.roztwarzanie w kwasach
3.stapianie
4.spalanie (paliwa) As Cd Cr Pb Ni Tl Zn
5.wykorzystująca lotność substancji w wys. temp. Hg Tl (lotne składniki wyłapujemy na specjalnych katalizatorach np. Hg na wacie złotej)
Roztwarzanie surowców mineralnych i krzemianowych
I.Sposoby tradycyjne:
1.w kwasach
2.przez stapianie
3.przez spiekanie
II.Sposoby nowoczesne
1.roztw. w kwasach pod ciśnieniem
2.roztw.w kwasach przy użyciu energii mikrofalowej
Roztwarzanie w kwasach:
1.kwasy nieutleniające: HCl HBr HF H2SO4-rozc. HClO4-rozc.
2.kwasy utleniające: HNO3 H2SO4-stęż. HClO4-stęż.
3.kwasy kompleksujące: HCl HF - tworzący chloro i fluoro kompleksy co na ogół ułatwia proces rozpuszczania ale kompleksy mogą być lotne.
Rozkład HF:
Obecność H2SO4, HNO3 lub HClO4 zapobiega hydrolizie SiF4
H2SO4 zapobiega stracie TiF4, ZrF4, NbF4, TaF4
HNO3 lub HClO4 -bardzo odpowiednie w obecności Ba, Sr, Ca, Pb
Zastosowanie: Oznaczenie z naważek Fe(II), metali alkalicznych Ti, Mn, P oraz śladów metali.
Materiał naczyń: Pt, teflon, polietylen, polipropylen
Stapianie
Dobór topnika, wysoka temperatura, duże stężenie odczynników
Krzemiany-topniki alkaliczne:
-wodorotlenki NaOH KOH
-węglany Na2CO3, K2CO3 +ew.NaNO3
-nadtlenki Na2O2
-borany Na2B4O7, LiBO2, Li2B4O7
W nowoczesnych instrumentalnych met. anal.
-czas oznaczenia-poniżej minuty.
Ograniczenia tradycyjnych metod roztwarzania:
-czas 40 min do 24h
-niecałkowity rozkład niektórych próbek
-zanieczyszczenie próbki spowodowane dużą ilością stosowanych odczynników
Zastosowanie: oznaczenie głównych składników do analizy śladów.
Rozkład HF lub miesz. kwasów pod ciśnieniem
Szybki i bezpieczny sposób roztwarzania próbek nieorg. w HF, HCl i miesz. silnych kwasów min. takich jak HF+HNO3, HF+H2SO4 lub rozpuszczanie próbek organicznych w alkaliach lub kwasach utleniających: -temp 110-250°C, -ciśnienie 100-300bar, -czas 30-120min
Nadmiar kwasu wiąże się w kompleks z kwasem barowym.
Budowa:
-naczynie teflonowe o V=20-200ml
-autoklaw w postaci bomby w płaszczu metalowym
-korpus zakręcany kołpakiem z zaworem
-urządzenie grzewcze: piecyki, łaźnia wodna, piaskowe, olejowe bloki grzewcze o kontrolowanej i regulowanej temp.
Zalety:
-tańsze naczynia teflonowe, chemicznie odporne
-możliwość zastosowanie silnych kwasów np. HF, woda królewska
-energiczny przebieg reakcji chemicznych umożliwiający całkowity rozkład substancji trudno roztwarzanych
-skrócony czas roztwarzania
-hermetyzacja operacji
-zatrzymywane subst łatwolotne
-możliwość oznaczenia SiO2
Zastosowanie:
-szybki rozkład szkieł, skał, minerałów, azotków w autoklawie przy pomocy HF, HCl, H2SO4 i innych silnych kwasów
-łatwy rozkład sub organicznych w autoklawach w obecności alkaliów lub HNO3 lub innych czynników o właściwościach utleniających.
Roztwarzanie przy użyciu mikrofal
Rozkład mieszaniną kwasów (HF, HF+HNO3) w hermetycznie zamkniętym naczyniu teflonowym w kuchni mikrofalowej. Wysokie ciśnienie do 150atm i wysokie temp.
Zalety:
-czas 15-30min
-brak kontaminacji (zamknięte naczynie, mała ilość odczynników)
-brak strat spowodowanych lotnością niektórych substancji
-odprowadzenie par kwasów, co zapobiega zanieczyszczeniu atm laboratorium
-doskonały sposób mineralizacji organicznej
Typy materiałów:
1.substancje odbijające promieniowanie mikrofalowe
2.przeżroczyste dla promieniowania mikrofalowego
3.adsorbujące, rozpraszające promieniowanie mikrofalowe
Wytrącanie osadów:
-Rozdzielanie
-Wykrywanie
-Oznaczanie
Klasyczne metody analizy ilościowej
-Metody wagowe
-Metody miareczkowe
Metody wagowe
-Określenie masy oznaczanej substancji (analitu), przeprowadzonej w trudno rozpuszczalny związek za pomocą odpowiedniego odczynnika strącającego.
-Zastosowanie do oznaczania makroskładników
*Praktycznie wszystkie metale i niemetale
-Zalety: duża dokładność, błąd < 0.1 %
-Wady: długi czas wykonania
Warunki jakie musi spełniać osad w analizie wagowej:
-Praktycznie nierozpuszczalny w wodzie, niski iloczyn rozpuszczalności < 1·10-5 M
-Stały, określony skład chemiczny
-Postać umożliwiająca szybkie i łatwe sączenie
-Duża masa cząsteczkowa, której składnik oznaczany masa stanowi niewielką część, pożądane strącanie za pomocą związków organicznych
-np.Hydroksychinolinian glinu Al(C9H6ON)3, m Al /3(C9H6ON) = 0.06
M C9H6ON = 144.16 g/mol
M Al = 26.98 g/mol
Roztwór, układ wieloskładnikowy jednofazowy, o cząsteczkowej dyspersji. Składnik roztworu będący w przewadze ilościowej nosi nazwę rozpuszczalnika, pozostałe składniki są rozpatrywane jako substancje rozpuszczone.
Roztwór nasycony, roztwór substancji pozostający w równowadze z osadem tej substancji
Roztwór przesycony, roztwór substancji o stężeniu większym od stężenia roztworu nasyconego w danej temperaturze. Roztwór przesycony można otrzymać przez ostrożne oziębianie roztworu nasyconego w wyższej temperaturze.
Roztwór nienasycony roztwór, w którym stężenie substancji rozpuszczonej jest mniejsze niż wynika to z jej rozpuszczalności w danych warunkach
Iloczyn rozpuszczalności, stała równowagi reakcji rozpuszczania związku jonowego:
AmBn ↔ mA(aq)n+ + nB(aq)m-
równa iloczynowi stężeń solwatowanych jonów podniesionych do potęgi równej odpowiednim współczynnikom stechiometrycznym z równania reakcji rozpuszczania:
L = [A(aq)n+]m[B(aq)m-]n.
-Przekroczenie iloczynu rozpuszczalności dla jonów An+ i Bm- spowoduje wzrost stężenia niezdysocjowanego AmBn i wytrącenie się osadu.
-Iloczyn rozpuszczalności zależy od temperatury, ciśnienia.
-Roztwór nienasycony: L < [A(aq)n+]m[B(aq)m-]n
-Roztwór nasycony: L = [A(aq)n+]m[B(aq)m-]n.
-Roztwór przesycony: L > [A(aq)n+]m[B(aq)m-]n.
Czynniki wpływające na rozpuszczalność osadów:
Efekt wspólnego jonu -jony wspólne wprowadzone do roztworów nasyconych powodują zmniejszenie rozpuszczalności trudno rozpuszczalnych osadów
-Dodatek elektrolitu o wspólnym jonie w ilości dwukrotnie większej od ilości potrzebnej do wytrącenia powoduje zmniejszenie rozpuszczalności osadu
-Większy nadmiar powoduje wzrost rozpuszczalności osadu
Efekt solny zwiększenie rozpuszczalności trudno rozpuszczalnego osadu pod wpływem soli, nie mających jonu wspólnego z osadem.
Wpływ temperatury wzrost temperatury powoduje wzrost rozpuszczalności osadów.
Krystalizacja, proces tworzenia się i wzrostu kryształu z cieczy przechłodzonej, roztworu przesyconego lub przesyconej pary (fazy gazowej).
Przyczyny:
-Ochłodzenie roztworu
-Odparowanie rozpuszczalnika
-Dodanie odczynnika powodującego powstanie nowego związku o małym iloczynie rozpuszczalności
Rozpoczęcie krystalizacji:
-Powstanie zalążków nowej fazy (zarodki krystalizacji, nukleacja),
--nukleacja homogeniczna (bez udziału faz obcych)
--nukleacja heterogeniczna (tworzenie się zarodków na cząstkach zanieczyszczeń, ścianach naczynia)
*Stężenie roztworu iloczyn rozpuszczalności liczba zarodków krystalizacyjnych
*Przyspieszanie tworzenia zarodków
--Pocieranie ścianek naczynia tworzenie ostrych krawędzi sprzyjających adsorpcji i tworzeniu zarodków
--Szczepienie roztworu
*Wielkość kryształów zależy od:
--Szybkości tworzenia zarodków krystalizacji
--Szybkości wzrostu pojedynczego kryształu
Szybkość tworzenia zarodków > szybkość narastania kryształu osad drobnokrystaliczny
Szybkość tworzenia zarodków < szybkość narastania kryształu osad grubokrystaliczny
Rozpuszczalność kryształów zależy od ich wielkości: małe kryształy lepiej rozpuszczalne od dużych; rozpuszczalność dużych kryształów jest w przybliżeniu stała
Typy osadów
-Krystaliczne
*Drobnokrystaliczne (BaSO4)
*Grubokrystaliczne (MgNH4PO4)
-Koloidowe
*Postać osadu
--Serowate (AgCl)
--Galaretowate (Fe(OH)3, Al(OH)3)
*Powinowactwo fazy rozproszonej do rozpraszającej
--Liofilowe - cząstki koloidu solwatowane przez fazę rozpraszającą
---Roztwory wodne: Hydrofilowe (SiO2 nH2O)
--Liofobowe - cząstki koloidu nie solwatowane przez fazę rozpraszającą
---Roztwory wodne: Hydrofobowe (AgCl, As2S3)
Koagulacja
Roztwór koloidalny zol
Zol→ żel, osad↓
Zol↔ żel
Koagulacja - tworzenie i wzrost agregatów prowadzący do wydzielenia się fazy stałej
-Odwracalna
-Nieodwracalna
Koagulacja polega na zmniejszeniu stopnia dyspersji układów koloidalnych a więc na łączeniu się cząstek fazy rozproszonej w większe zespoły w wyniku obniżania ładunku elektrycznego powierzchni cząstki koloidalnej.
Czynniki wywołujące koagulację:
-dodatek elektrolitu,
-dodatek koloidu o ładunku przeciwnym, co powoduje rozładowanie cząstek,
-naświetlanie radiochemiczne (np. promieniowanie b powoduje koagulację zoli dodatnich),
-działanie mechaniczne (mieszanie, wytrząsanie),
-ogrzewanie (np. ścinanie się białka),
-dehydratacja lub desolwatacja przez dodanie środków odwadniających, np. alkoholu lub acetonu,
-odparowanie lub wymrażanie ośrodka dyspersyjnego.
Wysalanie - koagulacja koloidów liofilowych - wydzielanie fazy rozproszonej w obecności dużych stężeń elektrolitów.
-Elektrolit powoduje kompresję elektrycznej warstwy podwójnej wokół cząstki obniżając w jej pobliżu ładunek elektryczny.
-Stosuje się sole o jonach ulegających silnej hydratacji, łatwo rozpuszczalne i wyższej wartościowości, np. MgSO4, ale także (NH)2SO4 i Na2SO4.
---Zdolność wysalająca zależy od charakteru zarówno kationu jak i anionu a także od ich promieni jonowych.
UWAGA - ilość elektrolitu nie może być zbyt duża, ponieważ może zajść nie tylko zobojętnienie ładunku cząstek koloidalnych, ale także adsorpcja nadmiarowych jonów odpychanie cząstek naładowanych petyzacja
Wytrącanie osadów krystalicznych
*Cel uzyskanie osadu grubokrystalicznego konieczne jak najmniejsze przesycenie roztworu
*Wytrącanie
-Z roztworów rozcieńczonych
-Przy pomocy rozcieńczonego odczynnika strącającego
-W podwyższonej temperaturze
-Odczynnik strącający dodawany stopniowo, kroplami
-Z roztworu mieszanego
-W obecności substancji zwiększających rozpuszczalność osadu
-Stosować dojrzewanie osadu
---Rekrystalizacja (osad drobnokrystaliczny osad grubokrystaliczny; zanieczyszczony mniej zanieczyszczony)
----Oczyszczenie osadu od współstrąconych zanieczyszczeń
----Polepszenie właściwości sączenia
Wytrącanie osadów koloidalnych
Wytrącanie:
-Roztwory stężone (w porównaniu z stosowanymi do wytrącania osadów krystalicznych), które następnie rozcieńcza się, celem zmniejszenia adsorpcji zanieczyszczeń
-W podwyższonej temperaturze
-W obecności elektrolitów
-Odsączenie po opadnięciu osadu na dno zlewki
Współstrącanie, współwytrącanie, zjawisko wydzielania się na strąconym osadzie analitycznym innych, zanieczyszczających go substancji, mimo nieprzekroczenia ich iloczynów rozpuszczalności.
Przyczyną współstrącania może być:
-Adsorpcja powierzchniowa jonów,
-okluzja,
-powstawanie kryształów mieszanych
-Tworzenie nowych związków między cząsteczkami osadu a jonami w roztworze.
-Strącanie następcze
-Mechaniczne zatrzymywanie
Współstrącanie jest nie tylko procesem przeszkadzającym w otrzymaniu czystego osadu analitycznego ale pełni pozytywną rolę przy oddzielaniu śladowych ilości substancji w procesie oddzielania śladów z zastosowaniem nośników.
Adsorpcja
Adsorpcja, zjawisko gromadzenia się jakiejś substancji (adsorbatu) na powierzchni ciała stałego lub cieczy(adsorbentu). Adsorpcja przez osady krystaliczne z układu wieloskładnikowego jest zawsze selektywna
W pierwszej kolejności adsorbowane są:
-Jony wspólne z osadem
-Jeżeli w roztworze mamy kilka soli o jonie wspólnym z osadem adsorpcji ulegnie przede wszystkim sól trudniej rozpuszczalna
-Adsorpcja elektrolitów obcych ma charakter selektywny, adsorbowane są jony które tworzą z jonami osadu związki najtrudniej rozpuszczalne
-Związki słabo zdysocjowane są silnie adsorbowane(H2S).
Adsorpcja wymienna
Jony z powierzchni adsorbenta wymieniane są na jony o tym samym ładunku znajdującymi się w roztworze, im trudniej rozpuszczalny związek może powstać z jonów obecnych w roztworze tym łatwiej może zachodzić adsorpcja wymienna.
Adsorpcja
(AxBy)osad + nCr-r ↔ [(AxBy)C]+ Q
Desorpcja
-Podwyższenie temperatury
-Zmniejszenie stosunku powierzchni osadu do jego objętości
Zmniejszenie stężenia jonów adsorbowanych
Okluzja
Zjawisko zamykania obcych jonów pochodzących z roztworu lub cząsteczek rozpuszczalnika przez kryształy szybko rosnące w tym roztworze.
--okluzja jonów chlorkowych w kryształach siarczanu baru rosnących w stężonym roztworze chlorku baru.
--Zaokludowane jony można usunąć przez przekrystalizowanie osadu (osad analityczny) w podwyższonej temperaturze.
Mechaniczne zatrzymywanie
--Przypadkowe zatrzymanie przez tworzący się osad innych faz, np. wody, kurzu
--Proces celowego wychwytywania małych ilości innych faz poprzez dodanie substancji stałej do fazy ciekłej
Tworzenie kryształów mieszanych
Roztwory stałe, kryształy mieszane,
--substancje jednorodne chemicznie, składające się z kryształów utworzonych z co najmniej dwóch odrębnych rodzajów chemicznych. Węzły sieci krystalicznej roztworu stałego obsadzone są w sposób przypadkowy przez różne kationy (lub aniony).
--Tworzone przez związki izomorficzne (jony o tej samej wartościowości i podobnej objętości)
--nBaSO4·mKMnO4 różowy
Wytrącanie następcze -Polega na wytrącaniu na powierzchni osadu innej substancji zwykle o jonie wspólnym z osadem, zachodzącym w czasie pozostawania osadu w kontakcie z roztworem macierzystym.
Analiza miareczkowa
Analiza miareczkowa (objętościowa) metoda analityczna ilościowa wykorzystująca pomiar objętości roztworu mianowanego zużytego w reakcji z oznaczanym składnikiem
Miareczkowanie czynność w analizie miareczkowej polegająca na dodawaniu z biurety roztworu mianowanego (titranta) do roztworu substancji oznaczanej aż do osiągnięcia punktu końcowego miareczkowania.
Roztwór mianowany, titrant, -roztwór odczynnika chemicznego o znanym stężeniu dodawany z biurety do roztworu oznaczanej substancji podczas miareczkowania.
Miano stężenie titranta (mol/L, M)
Substancje podstawowe
*Substancje podstawowe, substancje wzorcowe
*Pełnią rolę wzorców w analizie miareczkowej
--Substancje stałe
--Jednorodne
--Bardzo czyste (suma domieszek < 0.01 - 0.02%)
--Łatwe do wysuszenia i przechowywania
--Trudno lotne
--Niehigroskopijne
--Reagujące ściśle stechiometrycznie z roztworem, który ma być mianowany.
*Służą do:
--Przygotowania roztworów mianowanych
--Nastawiania miana roztworów mianowanych
Reakcja chemiczna może stać się podstawą oznaczenia miareczkowego:
*Przebiegająca stechiometrycznie
*Przebiegająca szybko
*W której dodawany odczynnik nie reaguje z innymi substancjami obecnymi w roztworze
*Umożliwiająca detekcję punktu równoważnikowego
*W której biorą udział związki chemiczne tworzące trwałe roztwory
Ilość analitu oznaczanego metodami miareczkowymi:
0.001 g - 0.1 g
Dokładność: 0.1 - 0.2 %
--Zależy od dokładności przygotowania roztworu mianowanego
--Zależy od dokładności pomiaru objętości cieczy
Roztwory wzorcowe
1.Przenieść ilościowo naważkę (np. 1,0000 g) substancji podstawowej do kolby miarowej 1L i rozpuścić wodzie, uzupełnić do kreski
--Roztwór wzorcowy pierwotny
2.Przenieść naważkę (np. 1,00g) odczynnika do kolby miarowej 1L i rozpuścić w wodzie, uzupełnić do kreski. Następnie zmianować tak przygotowany roztwór wykorzystując:
a)naważkę substancji podstawowej
b)Inny mianowany roztwór
--Roztwór wzorcowy wtórny
Klasyfikacja metod miareczkowych
Kryteria
*Typ reakcji zachodzącej podczas miareczkowania
*Sposób prowadzenia miareczkowania
-Bezpośrednie
-Pośrednie
--Odwrotne
--Podstawieniowe
*Sposób detekcji punktu końcowego
-Wizualna
---Zmiana barwy
---Pojawienie się osadu
-Instrumentalna
---Zmiana pH
---Absorbancji
---Przewodnictwa
---SEM ogniwa
Klasyfikacja metod miareczkowych - typ reakcji
*Reakcje kwas - zasada
--Alkacymetryczne
---Alkalimetria (titrant: zasada)
---Acydymetria (titrant: kwas)
*Reakcje kompleksowania
--Kompleksometria
*Reakcje redoks
--Redoksymetria (redoksometria)
--Manganometria (KMnO4)
--Cerometria (Ce(SO4)4)
--Chromianometria (K2Cr2O7)
--Jodometria (I2)
*Reakcje strąceniowe
--Stąceniowe
--Argentometryczne (AgNO3)
Sposób prowadzenia miareczkowania
Miareczkowanie bezpośrednie
--Oznaczana substancja reaguje bezpośrednio z dodawanym titrantem
----Używany jest jeden titrant
Miareczkowanie pośrednie
-Oznaczana substancja nie reaguje bezpośrednio z titrantem, lecz z inną substancją, którą się miareczkuje
-Miareczkowanie odwrotne (odmiareczkowanie nadmiaru)
--do badanego roztworu dodaje się odmierzoną ilość roztworu mianowanego w nadmiarze i nadmiar odmiareczkowuje za pomocą drugiego titranta
--Używane dwa titranty
Miareczkowanie podstawieniowe (substytucyjne)
-Oznaczany składnik A uwalnia z odpowiedniego związku równoważną ilość składnika B, który jest następnie odmiareczkowywany za pomocą titranta.
PR i PK
Punkt równoważnikowy (PR) - punkt miareczkowania, w którym cała zawartość oznaczanego składnika przereagowała z dodawanym odczynnikiem
Jak sprawdzić kiedy to ma miejsce?
-Zmiana barwy
-Wytrącenie osadu
-Zmiana pH roztworu
-Zmiana przewodnictwa roztworu, zmiana SEM ogniwa
Punkt końcowy (PK) - wyznaczony doświadczalnie moment, w którym uznajemy że cała ilość oznaczanego składnika roztworu przereagowała z dodawanym odczynnikiem
PR = PK, błąd oznaczenia = 0
Wskaźniki PK miareczkowania
Wskaźniki - odczynniki, które zmieniają barwę w PR reakcji titranta z oznaczanym składnikiem.
-Zmiana barwy (wskaźniki dwubarwne)
-Zanik barwy pierwotny (wskaźniki jednobarwne)
-Zabarwienie bezbarwnego pierwotnie roztworu
Klasyfikacja wskaźników
-Wskaźniki kwasowo-zasadowe
-Wskaźniki redoks
-Wskaźniki kompleksometryczne
Miareczkowanie alkacymetryczne
-Reakcja zobojętniania
-Krzywa miareczkowania
-Jedno i wieloprotonowy kwas i zasada
-Zasada doboru wskaźnika -zmiana barwy wskaźnika powinna być wewnątrz skoku miareczkowania
Wskaźniki to słabe kwasy lub zasady organiczne, których cząsteczki mają inną barwę niż jony
Kompleksometria dział analizy miareczkowej, w którym w czasie miareczkowania następuje utworzenie rozpuszczalnych
i słabo zdysocjowanych kompleksów.
-Kompleksy niechelatowe
-Kompleksy chelatowe
Kompleksony - kwasy aminokarboksylowe
Wskaźniki kompleksometryczne
Wskaźniki redoks: Błękit wariaminowy, 3,3'-dimetylonaftydyna
W czasie miareczkowania następuje zmiana potencjału na skutek związania kationu w kompleks
Miareczkowanie Cu2+ przy pomocy EDTA; zmiana potencjału Cu2+/Cu+ na skutek wyczerpania Cu2+
Metalowskaźniki:
I grupa - związki bezbarwne tworzące z kationami metali barwne kompleksy, np. kwas salicylowy, tiocyjanian amonu
II grupa - związki, które reagując z kationem powodują zmętnienie, np. kwas szczawiowy
III grupa - wskaźniki metalochromowe
Wskaźniki metalochromowe barwniki organiczne zdolne do tworzenia kompleksów
z metalami przy czym reakcji towarzyszy zmiana zabarwienia
Wymagania stawiane wskaźnikom metalochromowym:
-Wysoka czułość reakcji - mała ilość wolnych kationów tuż przed PR musi dawać intensywnie zabarwiony kompleks
-Reakcja barwna musi być specyficzna i selektywna
Kompleks MIn musi być trwały, ale trwałość M-In musi być mniejsza od trwałości M-EDTA
M + In M-In
M-In + EDTA M-EDTA + In
-Reakcje tworzenia kompleksów muszą zachodzić szybko
-Różnica barw M-In i M-EDTA musi być wyraźna
Wskaźniki metalochromowe:
Mureksyd, fiolet pirokatechinowy, kalces (oznaczanie Ca2+ obok Mg2+, zmiana barwy z czerwonej na niebieską)
Błędy w analizie miareczkowej:
-Błąd miareczkowania - różnica między objętością titrantu zużytą do osiągnięcia PK a objętością potrzebną do osiągnięcia PR miareczkowania
Vtitrant błąd dodatni, c
Vtitrant błąd ujemny, c
-Błąd cechowania - źle skalibrowane naczynia miarowe
-Błąd temperaturowy - t ≠20°C
-Błędy przy sporządzaniu roztworów
-Błędy związane z pomiarem objętości
--Błąd kropli, vkropli = 0,03 mL
--Błąd spływu
--Błąd odczytu
Analiza gazów
Kontrola i automatyzacja przemysłowych procesów technologicznych
-spalanie gazów
-oczyszczanie gazów
Kontrola stężenia gazów szkodliwych dla zdrowia
Kontrola składu wybuchowych mieszanin gazowych
Analizatory gazów
1.chemiczne
2.Fizykochemiczne
3.fizyczne-ze względu na rodzaj: -telekonduktometryczne , -telekonduktometryczne ze spalaniem katalitycznym, -magnetyczne
4.przetworniki półprzewodnikowe: -typu tlenkowego TGS, -jonoselektywne tranzystory typu MOS-ISFET, -chemiczne czułe tranzystory typu MOS-CSFET
5.Spektograf mas
6.Chromatografy gazowe z detektorami
-termokondyktometryczne (TCD)
-termochemicznym
-płomieniowym (FDP)
-płomieniowo-jonizacyjnym (FID)
-wychwytu elektrolitów (ECD)
-termojonowym (NPD)
-spektometrem mas (GC-MS)
-spektometrem IR (GC-IR)
-spektometrem AAS (GC-AAS)
-spektometrem AES (GC-AES)
Analizatory chemiczne
-z mieszaniny gazowej absorbujemy skł. w r-r pochłaniającym i stężenie tego składnika wyznaczamy z pomiaru obj. mieszaniny gazowej przed i po adsorpcji. Względnie mamy dwa rodzaje zaliczane do analizatorów chemicznych:
1.spalać oznaczony składnik i jego stężenie oznaczamy na podstawie objętości przed i po spaleniu
2.utleniony składnik i (np. CO → CO2) i z objętości CO2 oznaczamy CO
Do pomiaru służą aparaty Orsata
-CO2- 50% r-r KOH
-O2- 40% r-r KOH+pirogalol
-CO- 25% r-r NH4Cl+20% Cu2Cl2
-H2 i CH4- spalanie H2 290oC i CH4 850-900oC
-biureta pomiarowa
-zbiornik poziomujący
Analizatory fizykochemiczne:
-met. AAS
-met. polarograficzna
absorbujemy selektywnie z miesz. gaz. składniki (najczęściej w śladowych ilościach), ale stężenie wyznaczamy z pomiaru własności fizycznych r-r pochłaniającego.
Analizatory fizyczne:
Stężenie oznaczanego skł. gazowego wyznaczamy mierząc jego niektóre właściwości fizyczne jak ρ, p, przewodnictwo cieplne, zdolność do spalania w obecności katalizatorów, właściwości magnetyczne, optyczne, pochłaniania promieniowania, przy czym właściwości te różnią się znacznie od właściwości innych składników mieszaniny gazowej.
Analizatory termokonduktometryczne = cieplnoprzewodnościowe, katarometry
Zasada pomiaru: wykorzystanie różnic przewodności cieplnej gazów
Wsp. przewodnictwa gazów:
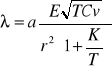
r-czynny płomień cząstki gazowej
k-stała uwzględniająca działanie elektrostatycznych sił międzycząsteczkowych
E-współ. Wzajemnej energii przy zderzeniu
1.Przewodnictwo ciepne gazów zależy od prom. cząstki gazowej i różnią się bardzo znaczenie między sobą: r ↓ przew.↑
2.przew. nie zależą od ciśnienia
3.przew. cieplne znacznie zmienia się z temp. T, λT=λo(1+βl)
4.przew. ciepne gazów w mieszaninie ma wł. addytywne
Przew. Względne λ/λpow λpow=1
Zasada pomiaru skł. miesz. 2 gazów wykorzystuje wł. addytowe przewodnictwa cieplego
λm= λ 1m1+ λ 2m2
m1 m2- cząstkowe stężenia składników gazów (względna molowa zaw. Skł. w r-r.)
Mieszanina 2 gazów
![]()
p1 p2- zaw. gazu w %
(nie wszystkie mieszaniny spełniają zasadę addytywności)
Pomiar w warunkach przemysłowych:
W warunkach przemysłowych porównujemy z przewodnictwem cieplnym gazu porównawczego i mierzymy temp. czujnika nagrzewanego do odpowiedniej temp. I chłodzonego przepływającym gazem. Im wyższe przewodnictwo analizowanego gazu tym szybciej chłodzi się grzejniki, powstaje różnica temperatur.
Analizatory z katalitycznym spalaniem
Zasada pomiaru - wykorzystanie efektu reakcji spalania gazu:
CO+1/2O2=CO2 + 67700 kcal/mol
H2+1/2O2=H2O + 5800
Wartość temp. mieszaniny wyższa od temp. zapłonu (ok. 350oC)
Spalanie katalityczne:
1.aktywny katalitycznie drut platynowy, który jest jednocześnie czynnikiem pomiarowym
2.reakcja zachodzi na katalizatorze rozdrobnionym a efekt cieplny reakcji mierzy się za pomocą oddalonego czujnika.
Dokładność oznaczenia mała 10%,
Tania, prosta metoda
Ograniczenia- przyczyny błędów:
-wpływ temperatury otoczenia
-wahania w napięciu zasilającym mostek
-zmiany objętości przepływu
-spadek aktywności analizatora
-inne składniki mieszanin gazów
Analizatory optyczne
Na podczerwień
Prom. IR pochłaniają wszystkie gazy z wyjątkiem gazów pierwiastkowych pierwiastkowych dwuatomowych drobinach (O2, N2) oraz jednoatomowe gazy szlachetne.
Met. umożliwia analizę gazów przem. I oznacznie CO, CO2, H2O,
H2S, SO2, NH3, HCN,
-dobra selektywność
-wysoka czułość
-duża uniwersalność
-możliwość pomiaru ciągłego ciągłego małym opóźnieniem
Analizy magnetyczne
O2 i niektóre inne gazy wykazują właściwości paramagnetyczne w odróżnieniu od większości gazów, które są diamagnetyczne ( molekularny moment magnetyczny=0)
Siły pola magnetycznego działają na sub. Paramagnetyczne zgodnie z kierunkami sił pola. Na diamagnetyczne odwrotnie.
Przetworniki Półprzewodnkowe
Typu tlenkowego w analizie gazowej
Budowa: warstwy tlenków SnO2, ZnO, Fe2O3, Cr2O3, CoO, MnO2.
Zasada pomiaru: znaczna wrażliwość struktur półprzewodnikowych na obecność śladowych ilości gazów, które wpływają na zmianę stanów powierzchniowych gazów. Zaadsorbowane na powierzchni półprzewodnika cząstki gazu mogą być wytwarzać ładunek powierzchniowy. Zmiany ładunków powierzchniowych powodują zmiany przewodnictwa przetwornika, które można zmierzyć.
Zastosowanie: do ciągłego pomiaru.
Spektometria mas
-Analiza b.skomplikowanych mieszanin gazów.
-Wstępna jonizacja mieszaniny gazowej jonizacji wolnych atomów.
-Rozdzielenie w separatorze stosuje się jako kryterium:
m/z = masa jonu/ładunku
-Otrzymujemy widmo masowe tzw. korpuskularne
-Ponadto oznaczamy polimery, metale, ciała stałe.
-jako metoda samodzielna lub detektor w mtalach sprzężonych
Spektrometria jonowa
MS-informacja o strukturze i składze ilościowym i jakościowym badanych próbek.
Rejestracja widm masowych przedstawiających rozkład naładowanych cząsteczek lub jonów w zależności od ich masy a dokładniej m/z
Cząstki naładowane: wolne atomy: fragmenty cząstek
-układ wpr. próbki
-komora jonizacyjna i przyspieszająca
-analizator (separator) jonów
-detektor jonów elektryczny/fotograficzny
-wzmacniacz prądu
-rejestrator widm MS
Metody wzbudzenia (jonizacji):
1.zderzenie elektronów (bombardowanie elekt.) -agresywny sposób, powoduje też fragmentyzacje sub. org.
2.przyłączenie elektronu-stosujemy elektrony termiczne
3.jonizacja za pomocą pola
4.jonizacja chemiczna
5.dejonizacja w polu
6.fotojonizacja
Separator jonów decyduje o czułości i selektywności.
Obecnie stosowany jest separator kwadropolowy -stosować można bezpośrednio lub pośrednio (jako czujnik) np.: analiza DTA- uzyskamy informacje o składzie i stężeniu wydzielających się gazów.
Zastosowanie: Budownictwo:
-identyfikacja polimerów-odporność termiczna
-badanie surowców
-badanie farb, klejów, żywic- analiza składników lotnych
-badanie katalizatorów- analiza produktów
-ceramika- warunki syntezy
-paliwa-produktów spalania
Uzyskane techniką TGA-MS informacje umożliwiają:
-wybór i dobór odpowiednich materiałów i surowców
-przewidywanie właściwości
-poprawa jakości produktów
Chromatografia gazowa:
Klasyfikacja:
1.wg. stanu skupienia fazy ruchomej:
-Chromatografia gazowa
-chromatografia cieczowa
-chromatografia fluidalna
2.wg. stanu skupienia fazy stacjonarnej
-gaz-ciecz (GLC)
-ciecz-ciecz (LLC)
-gaz-ciało stałe (GSC)
-ciecz-ciało stałe (LSC)
Ciało stałe jako faza stacjonarna: adsorbent (ch.adsorpcyjna), wymieniacz jonowy (ch.jonowymienna)
Ciało stałe jako sito molekularne (ch. Sitowa)
Ciecz osadzona na nośniku jako faza stacjonarna (ch.podziałowa)
Chromatografia gazowa:
A)gaz-ciało stałe (GSC)- adsorbcyjna chrom. Gazowa
B)gaz-ciecz (GLC)- podziałowa chromat. gazowa, kapilarna chrom. gazowa
Adsorbery:
-węglowe: węgiel aktywny, węgiel grafityzowany, karboksyle, węglowe sita molekularne
-krzemionkowe: żel krzemionkowy, krzemionka porowata
-sita molekularne: zeolity, molekularne sita organiczne
-polimery porowate
-tlenki glinu, MgO
Nośniki ciekłych faz stacjonarnych
-ziemie okrzemkowe
-cegły szamotowe
-teflon sproszkowany
-wąskie szklane kapilary
Ciecze stosowane w podziałowej CG:
-oleje silikonowe
-poliglikole
-estry złożone
-węglowodory nasycone
Rozdzielanie:
Na skutek różnicy objętości składników w średnicy ruchomej
ciało stałe: prawo izotermy Langumira
ciecze: prawo Henrego, prawo podziału Nersta
po wyjściu z kolumny rozdzielone składniki trafiają do defektora.
Schemat:
1.urządzenie do regulowania przepływu gazu inertnego
2.dozownik gazu analizowanego: -manualne, -kran sześciodrożny
3.kolumna rozdziałowa znajdująca się w termostacie
4.detektor
Czułość analizy zależy od rodzaju detektora:
-konduktometryczny (katarometr)
-płomieniowo-jonizacyjny:
a)wprowadzany gaz do płomienia; na skutek jonizacji zmiana przewodnictwa między okładkami elektrod→ zmiana przewodnictwa płomienia; nie można oznaczyć CO i CO2
b)detektor płomieniowy termiczny-efekt cieplny
c)detektor płomieniowo-fotometryczny - zw. siarki i fosforu; po spaleniu związki te mają zdolność luminescencji mierzonej spektrometrem
d)detektor wychwytu elektronów
Zalety:
-wysoka czułość
-idealna selektywność
-wysoka rozdzielczość
-uniwersalność
-analiza wieloskładnikowa
-czas analizy krotki-on line
-doskonałe zautomatyzowanie procesów rozdziału i detekcji
Zastosowanie:
-Przemysł organiczny
-przemysł petrochemiczny
-przeróbka węgla i gazu ziemnego
-Przemysł koksowniczy i gazowy
-przem. Spożywczy
-przem. Szklarski
-przem. Farmaceutyczny
-analiza gazów wylotowych
-Badanie zanieczyszczeń środowiska: wody, gleby, powietrza
Analiza jakości szkła:
-intensyfikacja procesów klarowania
-kontrola technologicznego wytopu szkła
-poprawa jakości szkła
-zapewnienie określonych właściwości
-wyjaśnienie mechanizmu szeregu procesów przebiegających w układzie gaz-masa
Gazy: O2, H2, N2, CO, CO2, SO2
Zawartość gazów zależy od:
-składu zestawu
-warunków wytopu, atm. gazowej
-fizykochemicznego oddziaływania gazów z masą szklaną
-rozp. gazów w szkle i tworzenia zw. chem. z komponentami szkła (O2, CO2, SO2, H2O)
Analiza nastawiona jest na rozwiązanie problemów:
-składu pęcherzyków gazowych, w których gaz uwalnia się mechanicznie
-składniki gazów rozpuszczonych w masie szklanej, która ze szkła wydobywa się drogą wysokotemperaturowej ekstrakcji próżniowej.
Metody rozdzielania
I.Rozdzielanie ukł. jednoskładnikowych.
1.Rozdzielanie na podstawie przemiany materii:
a)chemiczne:
-strącanie,
-współstrącanie śladów na nośniku,
-rozdzielanie wewnętrzne,maskowanie,
-wymiana jonowa(chromatografia jonowymienna).
b)rozdzielanie elektrochemiczne:
-elektroliza,
-elektroliza wewnętrzna,
-amalgmacja.
2.Rozdzielanie bez przemiany materii:
a)rozdzielanie na podstawie efektów kinetycznych:
-dializa,
-osmoza,odwrócona osmoza,
-elektrodializa,
-elektroforeza.
b)rozdzielanie na podstawie zmian stanu:
-destylacja,
-krystalizacja,
-sublimacja.
c)rozdzielanie na podstawie równego podziału i adsorpcji:
-ekstrakcja,
-chromatografia gazowa,
-chromatografia cieczowa.
Rozdzielanie śladów:
Strącanie i współstrącanie.
Mechanizm:
1.Izomorfizm.
2.Adsorpcja.
3.Okluzja.
Niewielkie ilości pierwiastka strąca się na kolektorach. Potem oddzielamy to na sączku i w ten sposób pozbywamy się głównego składnika, który pozostaje w roztworze.
Rozdzielanie wewnętrzne tzw. maskowanie:
Bardzo przydatne w analizie cementu. Rozdzielanie przy pomocy kompleksu EDTA. Tworzą się kompleksy pierwiastków (Fe, Al, Ti, Ca, Mg) z EDTA.
Elektrograwimetria:
Na elektrodzie stałej-oznaczanie pierwiastków śladowych.
Elektroda rtęciowa-oznaczanie śladów. Dużo większe możliwości oznaczania pierwiastków niż w przypadku elektrody stałej.
Zjawiska wykorzystywane przy rozdzielaniu:
1.Wykorzystanie cienkich warstw(filtry).
2.Wykorzystanie lotności pierwiastków (mat.budowlane, skały)
3.Ekstrakcja(metoda wykorzystuje różnicę rozpuszczalności analizowanych substancji-dwie niemieszające się ciecze).
4.Wymiana jonowa(podział jonitów:kationity-anionity,stałe-cienkie,nieorganiczne-organiczne,naturalne-półsyntetyczne-syntetyczne).
Nowoczesne metody rozdzielania:
Dializa-oddzielanie cząsteczek, przede wszystkim koloidalnych od cząsteczek rzeczywistych na zasadzie dyfuzji przez membramy.
Elektrodializa-oddzielanie cząsteczek, przede wszystkim koloidalnych od cząsteczek rzeczywistych na zasadzie dyfuzji przez membramy w polu elektrycznym.
Elektroforeza-rozdzielanie naładowanych cząstek na podstawie ich różnych ruchliwości w polu elektrycznym.
Elektroforeza kapilarna-mamy 2 elektrody. Pole elektryczne o dużym napięciu(100-500 V/cm)powoduję ruch ładunków do katody-ujemne a do anody-dodatnie. Pozwala rozdzielać sub. organiczne i nieorg. z dużą szybkością.
Zalety tej metody:
-b.niewielka próbka,
-wysoka rozdzielczość,
-duża szybkość rozdziału,
-można zastosować różne typy detektorów,
-wszechstronność zastosowań,
-analiza próbek z roztworów wodnych,
-ekonomia.
Zastosowanie:
-analiza wody,
-przemysł farmaceutyczny(jakość leków),
-analiza materiałów biologicznych a także in-vivo,
-przemysł spożywczy(kontrola jakości),
-chemia sądowa(DNA, toksykologia, analiza śladów-zbrodnia, określenie czasu zgonu).
Błędy w analizie chemicznej:
1.Błędy przypadkowe-niewielkie, niepływające na wynik średni, są przyczyną nieznacznych różnic pomiędzy wynikami uzyskanymi a wynikiem średnim. Błędy elementarne, powstające w toku poszczególnych operacji postępowania analitycznego. Spowodowane są: niedokładnością przyrządów, niedoskonałością zmysłów, wahaniami w sieci zasilającej.
2.Błędy systematyczne-mają charakter stały, jednokierunkowy, mają ściśle określoną przyczynę(niesprawność przyrządów, ich niewłaściwe użytkowanie, niewystarczająca czystość odczynników).
a)instrumentalne-wady przyrządów(zła kalibracja) i zanieczyszczenie odczynników,
b)metodyczne-związane ze specyfiką metody analitycznej i stosowanego układu chemicznego(nieilościowy przebieg reakcji chem., współstrącanie, reakcje uboczne),
c)grube-powstające na skutek niedopatrzenia lub z winy niedostatecznie starannego pracownika(niewłaściwe pobieranie i przechowywanie próbek, matematyczny błąd obliczeniowy).
Mediana-wynik środkowy, gdy jest liczba wyników nieparzysta. W przypadku, gdy jest liczba wyników parzysta obliczamy średnią z dwóch środkowych wyników.
Moda-wynik najczęściej występujący w danej próbie.
Oszacowanie odchylenia standardowego σ:
σ-odchylenie standardowe, reprezentuje błąd bezwzględny dla xi któremu odpowiada punkt przegięcia na krzywej Gaussa.
Przedział ufności-reprezentuje granice, w których z dużym prawdopodobieństwem znajduje się poprawny wynik o ile nie pojawią się błędy przypadkowe.
Testy statystyczne:
Precyzja-dobra zgodność równoległych wyników oznaczeń. Miara odtwarzalności i powtarzalności wyników.
Powtarzalność-precyzja uzyskiwana przez tego samego analityka w konkretnym laboratorium.
Odtwarzalność-precyzja uzyskiwana przez różnych analityków z różnych laboratoriów przy zachowaniu tej samej procedury analitycznej.
Wiarygodność analizy -otrzymywanie dokładnych i precyzyjnych wyników, niepewność związana z danym wynikiem powinna być mała.
Czułość-stosunek sygnału pomierzonego do szumu.
Systemy jakości:
Jakość-ogół właściwości oraz cech i charakterystyk produktów lub usług(np.wykonanie analizy i uzyskanie wyniku) spełniających określone oczekiwania i wymagania.
Jakość metody analitycznej-charakterystyki oraz parametry charakterystyczne dla metody analitycznej, sprzętu oraz wyników pomiarów w odniesieniu do ich przydatności do spełnienia stawianych wymagań.
Dla metody analitycznej charakterystyka obejmuje takie parametry jak:
-dokładnośc i precyzja,
-odtwarzalność,
-specyficzność,
-czułość,
-zakres liniowości,
granice wykrywalności i oznaczalności.
System jakości-struktura organizacyjna, zasoby, odpowiedzialności, zbiór procedur, których wprowadzenie i przestrzeganie ma zapewnić, że działanie laboratorium analitycznego spełnia warunki ustanawiane przez zleceniodawców. Jest to struktura organizacyjna pozwalająca na zarządzanie jakością.
Zarządzanie jakością-ten aspekt ogólnej funkcji zarządzania, który określa i wdraża politykę w dziedzinie jakości.
Polityka w dziedzinie jakości-wszystkie zamierzenia i kierunki działania przedsiębiorstwa dotyczące jakości, formułowane przez naczelne kierownictwo.
Program jakości-dokument zawierający konkretne procedury dotyczące jakości, rozdzielenie środków, pieniędzy i chronologię działań.
Zapewnienie jakości-całość planowanych i systematycznych działań koniecznych do uzyskania dostatecznej pewności, że produkt czy usługa spełniają wymagania jakościowe.
Obejmuje:
-zarządzanie jakością,
-program jakości,
-nadzór nad jakością(ciągłe kontrolowanie i weryfikacja stanu procedur, metod, warunków, procesów i usług oraz analiza uzyskanych wyników).
Sprawdzanie jakości-systematyczne i niezależne badanie pozwalające określić czy działania i procedury w zakresie jakości odpowiadają zaplanowanym przedsięwzięciom i w jakim stopniu wymagania jakościowe są spełnione.
Sterowanie jakością- metody i działania w celu spełnienia wymagań jakościowych, wykonywane przez personel laboratorium. Podstawą sterowania jakością w laboratorium jest stosowanie tzw.dobrej praktyki laboratoryjnej.
Dobra praktyka laboratoryjna(GLP)-zestaw sformalizowanych reguł dotyczących całości działania laboratorium i wykonywanych w nim poszczególnych czynności. Sprawdzanie sprawności organizacji i personelu.
Program zapewnienia jakości-zdefiniowany system obejmujący personel niezależny od prowadzonych badań powołan do zapewnienia jakości zgodnie z GLP.
-wymagania związane z pomieszczeniem i środowiskiem,
-wykwalifikowany personel,
-odpowiednie wyposarzenie, mat.referencyjne, gospodarka odpadami,
-wymagania dla odczynników i instrumentów,
-systemy sprawdzające, testy i mat.referencyjne,
-wykonywanie badań,
-przedstawianie wyników badań,
-przechowywanie i archiwizacja materiałów i zapisów.
Charakterystyka procedur analitycznych-dokumenty opisujące procedury opisywanych testów i innych czynności laboratoryjnych.
Akredytacja laboratorium-formalne uznanie, potwierdzone określonym dokumentem, że laboratorium jest kompetentne do wykonywania określonych badań, czyli posiada określony system zarządzania jakością, odpowiednią aparaturę, stosuje sprawdzone procedury analityczne, ma wykwalifikowany personel i prowadzi pełną dokumentacje wykonywanych czynności.
Etapy związane z uzyskaniem akredytacji:
-wystąpienie z wnioskiem o akredytacje zawierającym księgę jakości, instrukcji zapewnienie jakości, charakterystykę procedur badawczych,
-centralne biuro jakości wybiera zespół inspekcyjny,którego zadaniem jest sprawdzenie czy dostarczone informacje są adekwatne,
-po udzieleniu akredytacji kontrola z zewnątrz odbywa się co roku.
Akredytacja udzielana jest na okres 4 lat i po tym okresie procedura certyfikacyjna jest powtarzana.
Walidacja-kompleksowa procedura sprawdzająca czy dana metoda analityczna jest wolna od błędów nie tylko w ramach kalibracji,ale przede wszystkim wynikających z interferencji przy analizie próbek rzeczywistych.
$ KADI $ PRODACTIONS
ALL RIGHTS RESERVED ®
1
Wyszukiwarka
Podobne podstrony:
Instrukcja G, Poniedziałek - Materiały wiążące i betony, 05. (03.11.2011) Ćw G - Badania surowców ce
Sprawozdanie nr 1 CECHY TECHNICZNE MATERIAfLOW BUDOWLANYCH, Budownictwo studia pł, sprawka maater
badania techniczne materiałów z tworzyw sztucznych, Materiały budowlane z Materiałoznastwem
cechy techniczne materialow budowlanych
Badanie cech technicznych c, Materiały Budowlane
Materiały budowlane - Wybrane cechy techniczne materiałów, Wybrane cechy techniczne wyrobów/materi
Badanie wybranych?ch technicznych materiałów budowlanych
Cechy techniczne materiałów budowlanych
analiza kondycji ekonomicznej hurtowni materiałów budowlanyc
Niektóre materiały i wyroby budowlane, Podłogi drewniane-cechy techniczne, Techniczne
Instrukcja B, Poniedziałek - Materiały wiążące i betony, 04. (27.10.2011) Ćw B - Badanie właściwości
Obl, Poniedziałek - Materiały wiążące i betony, 02. (17.10.2011) Ćw D - Badanie właściwości zapraw b
analizy surowcow materialow i innych nakladow, Analiza i inne
Sprawozdanie nr 1 CECHY TECHNICZNE MATERIALOW BUDOWLANYCH, budownictwo, materiały budowlane
1 Cechy techniczne materiałów budowlanych
Badania techniczne materiałów kamiennych, Materiały budowlane z Materiałoznastwem
Pytania dzial produkcyjny C3, Materiały Dietetyka, PWSZ (Nina nevermind), Wyposażenie techniczne (Ni
Instrukcja F, Poniedziałek - Materiały wiążące i betony, 10. (08.12.2011) Ćw F - Badanie właściwości
więcej podobnych podstron