STAN GAZOWY
Cząsteczki charakteryzują się tym, że maja duża wartość energii kinetycznej ruchu postępowego. Przy dużych odległościach międzycząsteczkowych i małych siłach oddziaływania międzycząstecz, substancje zajmują objętość i przyjmują kształt naczynia, w którym się znajdują. Cząsteczki tworzące gaz znajdują się w ustawicznym ruchu, zderzają się ze sobą i wymieniają energie.
PRAWA GAZU DOSKONALEGO
- Cząsteczki gazu doskonałego mają objętość własną równą zeru, masa ich skupiona jest w punkcie matematycznym
- cząsteczki znajdują się w ciągłym ruchu postępowym
- zderzenia między cząsteczkami są sprężyste
- między kolejnymi zderzeniami cząsteczki gazu doskonałego poruszają się ruchem prostoliniowym
- zderzenia cząstek ze ściankami naczynia są rejestrowane makroskopowo jako ciśnienie
- ruch cząstek jest chaotyczny
- cząsteczki gazu doskonałego są chemicznie obojętne.
1. Prawo Boyle'a-Mariotte'a
pV=const
2. Prawo Gay-Lussaca
Dla p=const, stała masa gazu ogrzana o 1K, zwiększy swoją objętość o α
Vo w temp 273,16K (0 0C)
Vt obj gazu w temp t, to
Vt=Vo+αVot=Vo(1+αt)
α=1/273,16=0,00366
Vt=273,16K+t/273,16K
Stała masa gazu dla V1w t
V2 w t2
V1/V2=273,16+t1/273,16+t2
V1/V2=T1/T2→V1/T1=V2/T2
V = const·T
Izochora dla gazu doskonalego
Gaz w stałej objętości V ogrzany o 1K zwiększa swoje ciśnienie o 1/273,16 ciśnienia jakie ma 00C. Pt=Po(1+(t/273,16)
P/T=const
Równanie stanu gazu doskonałego
V=f(P,T) dV=(δV/δT)ttdp+(δV/δT)pdT
Z p.Boyle'a (δV/δT)t=-k1/p2
Z p.Gay-Lussaca (δV/δT)p=k2
dV=(k1/p2)dp+k2dT
k1/p=V , k2=V/T → k1=pV
dV=-V(dp/p)+V(dT/T)
(dV/V)+(dP/P)=dT/T
Całkujemy; lnV+lnp=LnT+C'
pV=CT→ pV/T=C
C - zależy od masy gazu dla 1 mola; R-stala gazowa
pV=RT; pV=nRT- dla n moli gazu r-nie Clapeyrona
KINETYCZNA TEORIA GAZÓW
1. Gaz stanowi zbiór jednakowych kulistych, doskonale sprężystych cząstek
2. Cząsteczki gazu znajdują się w stanie ruchu postępowego
3. Cząsteczka jedna nie działa na druga dopóki się nie zderza
4. Ruch cząsteczek jest całkowicie bezwładny, prostoliniowy pomiędzy zderzeniami: cząsteczki maja różne prędkości.
ROWNANIE WAN DER WAALSA
Gazy rzeczywiste
(p+p')=p+a/V2
(p+a/V2)(V-b)=RT
(p+a/V2T)(V-b)=RT Równanie Berthelota
(p+a/T(V+C)2)(V-b)RT Równanie Clausiusa
Prawo Daltona
P mieszaniny gazów =∑Pi
Pcalk=Pa+Pb+Pc
PaV=naRT...
(Pa+Pb+Pc)V=(na+nb+nc)RT
n=na+nb+nc
(pa+pb+pc)V=nRT
Pa/p=na/n=Xa; Pa=PXa
PRAWO FICHA
Dm=-Ds.(dc/dx)dt
Ilość gazu dm dyfundująca
W czasie dt z jednej warstwy do drugiej odległej o dx jest wprost proporcjonalna do powierzchni s i gradientu stężenia - dc/dx
Dc/dt=D(d2C/dx2)
D - wspł. dyfuzji zależy od rodzaju gazu
D=1/3Uλ;λ- średnia droga swobodna,
U - średnia prędkość
RÓW VAN DER WAALSA a stan krytyczny zachowanie się gazu rzeczywistego poddawanego sprężeniu w różnych temp, można przedstawić za pomocą r-nia Van Der Waalsa. Po rozwinięciu i przekształceniu otrzymujemy
r-nie 3 stopnia względem V
V3=(b+RT/p)V2+(2/p)V-ab/V=0
R - nie to powinno mieć 3 pierwiastki tzn. spełniające je wartości V. Przez przekształcenie p=(RT/V-b)-a/V2 Graficznie otrzymany dla temp niżej od krytycznej krzywej 3 stopnia maja 3 pierwiastki wartości
(V-V1)(V-V2)(V-V3)=0
(V-Vk)3
TERMODYNAMIKA CHEMICZNA
I ZASADA TERMODYNAMIKI
Wielkości ekstensywne i intensywne
Ekstens: zależne od ilości substancji i rosnące ze wzrostem jej masy (V,U,H,S...) Wielkości te są addytywne (dodają się)
Intens: nie zależą od ilości substancji (P,t(T),d,c(stęż))
RÓWNANIE I ZAS TERODYNAMIKI
ΔU=ΔQ+Δ(-W)
Procesy termodynamiczne
Praca rozszerzania się
DW=-pdV
∫dW=-∫pdV= -pΔV
gdzie ΔV=V2-V1
a) proces izochoryczny: V=const,
Jeżeli dV=0 to ΔU=ΔQ
DW=0 dU=dQ
b) proces izobaryczny: p=const
W=pΔV; W jest dodatnie dla sprezenia ,ujemne dla rozproszenia
1a) Rozszerzenie V (sprężanie) dla gazu doskonałego
Tego dla p=const, V1>V2
V1=RT1/p,V2=RT2/p
W=±(pΔV)=±{p[(RT1/p)-RT2/p]}=±RΔT, gdzie
T1-T2=ΔT, T2-T1=ΔT
2a) Rozszerzenie V ( sprężanie) izotermiczne T=const
WT=∫(RT/V)dV=RTlnV2/V1
Gdy T=const to objętość molowa jest odwrotnie proporcjonalna do ciśnienia (i od stężenia C=1/V)
WT=RTlnV2/V1=RTlnp1/p2=RTlnC1/C2
3a) Rozszerzenie V (sprężanie) izotermiczne i izobaryczne: T=const, p= const
Jeżeli w układzie zachodzi reakcja to możliwe jest izobaryczna zmiana objętości
ν1-liczba moli przed reakcja
ν2 -liczba moli po reakcji
V1=ν1(RT/p) V2=ν2(RT/p)
Wp, t=ΔνRT
PRAWO JOULE'A
Jeżeli przy t=const zmieni się p lub V gazu doskonałego to mimo tego jego U pozostanie bez zmian (δU/δV)t=(δU/δp)t=0
Jeżeli ΔU=0,to Q=W
Q=W=RTlnV2/V1=RTlnp1/p2=RTlnC1/C2
Dla gazu rzeczywistego następuje zmiana energii wewnętrznej
ENTALPIĘ (H) wyrażamy wzorem:
![]()
Gdzie:
U - energia wewnętrzna układu.
p - ciśnienie układu.
v - objętość układu.
W procesach izobarycznych ciepło równa się energii wewnętrznej ΔU = q, a w przemianie izochorycznej entalpii układu ΔH = q. W przemianach izotermicznych, jak sama nazwa wskazuje ΔU i ΔH są równe zero(powyższe zależności przyjmuje się tylko dla gazów doskonałych).
ENTALPIA I ENERGIA wewnętrzna są funkcjami stanu. O czym mówi prawo Hessa: W przemianach izochorycznych i izobarycznych, w których nie występuje praca nie objętościowa, efekty cieplne odpowiednio ΔU i ΔH, nie zależą od drogi przemian a tylko od stanu początkowego i końcowego.
CIEPŁO MOLOWE to ilość ciepła potrzebna do ogrzania jednego mola substancji o jedne stopień. Rozróżniamy ciepło molowe w stałej objętości Cv i w stałym ciśnieniu Cp. Termodynamika wykazuje:
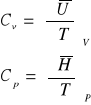
Dla gazów doskonałych jednoatomowych wynoszą one Cv = 3/2R i Cp = 5/2R.
CIEPŁO ROZPUSZCZANIA (QN) jest to ilość ciepła wydzielana lub pochłonięta podczas rozpuszczenia l mola substancji w n molach rozpuszczalnika. Ta ogólna definicja zawiera w sobie pojęcia pochodne.
PEŁNE CIEPŁO ROZPUSZCZANIA (QNAS) występuje wówczas, gdy do rozpuszczenia l mola substancji użyjemy takiej ilości rozpuszczalnika, że powstanie roztwór nasycony.
PIERWSZE CIEPŁO ROZPUSZCZANIA (QAQ) towarzyszy rozpuszczeniu l mola substancji w nieskończonej ilości rozpuszczalnika (powstaje roztwór nieskończenie
rozcieńczony).
CZĄSTKOWE CIEPŁO ROZPUSZCZANIA towarzyszy rozpuszczeniu l mola substancji w tak dużej ilości roztworu o określonym stężeniu, że stężenie to nie ulegnie zmianie. Jeżeli ten roztwór był praktycznie nasycony to mówimy wówczas o OSTATNIM CIEPLE ROZPUSZCZANIA. Z trzech ostatnich definicji wynika, że mają one charakter pojęć granicznych.
ENTALPIA: ZAWARTOŚĆ (pojemność)
1. Q=U+W, δQ=dU+pdV+Vdp, p=const
2. Q=dU+pdV
δQ=dU+pdV, ΔQ=ΔU+pdV=ΔH - zmiana entalpii
CIEPLO MOLOWE
Rozpatrzymy ciało jednorodne, które podczas zmiany stanu termodynamicznego nie ulega przemianie chemicznej i nie zmienia stanu skupienia
δQ/δT=(dU/dT)+(dW/dT)
Wlk.δQ/δT (dla 1 mola można zapisać)δQ/δT=C jest ilością ciepła potrzebna do ogrzania 1 mola o 10K.Rozniczka δQ/δT zależy od sposobu zmian(od drogi procesu)i nie jest rozniczka zupelna dla V=const δW=0 i → Cr=(δQ/dT)v=(dU/dT)v, a jeżeli p= const, to dW=pdV i
Cp=(dQ/dT)p=(dU/dT)p+p(dV/dT)p
Oznacza, ze Cp=(dH/dT)p
Cv - ciepło molowe w stałej objętości
Cp - ciepło mol w stałym ciśnieniu
Cp>Cv
Dla gazu doskonałego stosując prawo Joule'a otrzymamy (dU/dT)p=(dU/dT)v,
P(dV/dT)p=pd/dT(RT/p)p=R
Cp-Cv=R
Zależność ciepła mol od temperatury
Adiabata gazu doskonałego:
δQ=0,dU=dW, dU=-pdV
dU=CpdT, to dT=pdV/Cv
pV=RT a Cp-Cv=R,
różniczkujemy i
dT=Vdp/Cp, dzielimy
Cp/Cv=(VdP/pdV)
Cp/Cv=K, całkujemy
PVK=const, a wstawiając
P=RT/V mamy TVK-1=const
II ZASADA TERMODYNAMIKI
W każdej przemianie samorzutnej biegnącej w układzie izolowanym (ΔU=0) entropia układu wzrasta, osiągając w stanie równowagi układu wartość maksymalna.
O samorzutności przebiegu reakcji decydują;
1. Dążenie układu do osiągnięcia równowagi energetycznej
2. Dążenie układu do osiągnięcia maksimum prawdopodobieństwa termodynamicznego. Zrealizowanie wszystkich możliwych kombinacji obsadzeń poziomów energetycznych układu przez elementy tego układu, czyli osiągniecie maks entropii
- dla procesów przebiegających w warunkach izotermiczno-izochorycznych energii swobodna to ΔF=ΔU-TΔS
w warunkach izotermiczno-izobarycznych entalpia swobodna to ΔG=ΔH-TΔS
III ZASADA TERMODYNAMIKI
Entropia substancji o strukturze doskonałego kryształu w temp zera bezwzględnego jest równa zeru St=∫(cp/T)dT
PLYNY
1.GAZY (jak wyżej)
2. CIECZE
We wszystkich cieczach znajdujących się w ruchu podczas przesuwania się jednych warstw względem drugich powstają siły tarcia na skutek istnienia sil przyciągania miedzy cząsteczkami cieczy. Od strony poruszającej się szybciej działa na warstwę poruszająca się wolniej siła przyśpieszenia, a od strony warstwy poruszającej, na warstwę poruszającą się szybciej siła hamująca. Siły te nazywamy siłami tarcia.
CIECZ DOSKONALA
Ciecz nieściśliwa pozbawiona lepkości, w takiej cieczy nie występują siły tarcia wewnętrznego a wprawiona w ruch może się poruszać bez konca..
LEPKOŚĆ
Lepkość bezwzględna (dynamiczna)
Jest liczbowo równa sile przypadającej na jednostkę powierzchni, potrzebnej do utrzymania jednostkowej różnicy
Prędkości miedzy warstwami cieczy odległymi od siebie o jednostkę długości nwz=nb/nw
Płynność: γ=1/n
Lepkość kinematyczna:γ=n/d
RUCHY CIECZY W WISKOZYMETRZE OSTWALDA
- laminarny, czyli warstwowy (warstwy cieczy jakby ślizgały się po sobie)
- turbulentny, czyli burzliwy (podczas przepływu cieczy powstają w niej zawirowania)
- pośredni czyli nie ustalony
METODY POMIARU LEPKOŚCI BEZWZGLĘDNEJ CIECZY:
Metoda oparta na wzorze Poiseuille'a
Lepkość cieczy można oznaczyć, mierząc czas przepływu objętości cieczy V przez kapilarę o promieniu R .Do pomiaru stosuje się wiskozymetr Oswalda (ma on kształt U-rurki. W jego lewej części występuje rozszerzenie, które przechodzi przez kapilarę. U góry i na dole rozszerzenia na rurce wiskozymetru znajdują się kreski a i b. Do prawej części rurki wlewa się taka ilość cieczy ,aby jej poziom znajdował się poniżej kreski b. Zasysa się gruszka do lewego ramienia powyżej kreski a i mierzymy czas przepływu cieczy pomiędzy a i b.nx=nwdxtx/dwtw
Metoda oparta na wzorze Stokesa
Siła oporu działająca na kulisty przedmiot poruszający się w cieczy lepkiej jest wprost proporcjonalna do jego prędkości, współczynnika lepkości cieczy i jego promienia. Kula opadająca w cieczy w pierwszej chwili porusza się ruchem przyspieszonym,w miarę wzrostu prędkości opadania wzrasta siła tarcia R, która po pewnym czasie równoważy działając na kulkę siła ciężkości F, co powoduje, że kulka spada ruchem jednostajnym ze stałą prędkością n=2r2(d-dc)gt/9l
Stała kulki n=K(d-dc)t
NAPIECIE POWIERZCHNIOWE
Na powierzchni kropel cieczy (ogólnie cieczy)
Jest zgromadzona pewna energia E zwana energia powierzchniowa
dE=γds.;γ - napięcie powierzchniowe[N/m]
Jeżeli T rośnie to γ maleje. Jeżeli T- T krytyczna to γ=0
Wg Eotrosa molowa energia powierzchniowa cieczy jest prostoliniowa funkcja temp
Iloczyn Vmγ1/ 4zostal nazwany przez Baczynskiego i Sugdena PARNACHORA i oznaczane jest często literka p.Wartosc p jest addytywne i konstytutywne
REFRACHORA F=-Plog(nd20-1)
NAPIĘCIE POWIERZCHNIOWE - jest to zmniejszenie potencjału termodynamicznego układu towarzyszące zmniejszeniu jego powierzchni o jednostkę 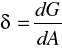
. , lub siła styczna do powierzchni potrzebna do rozerwania błonki powierzchniowej na długości jednostkowej. Jednostką napięcia powierzchniowego jest J/m2 lub N/m.
RÓWNANIE IZOTERMY ADSORPCJI GIBBSA.
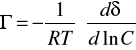
gdzie:
R - stała gazowa
T - temperatura
C - stężenie substancji rozpuszczonej.
γ - napięcie powierzchniowe roztworu.
METODY POMIARU NAPIĘCIA POWIERZCHNIOWEGO.
Metoda Kapilarnego wzniesienia. Polega na oznaczenia wzniesienia słupa cieszy w kapilarze oraz do pomiaru gęstości cieszy. γx/γw=hxdx/hwdw
Metoda stalagmometryczna (kropelkowa). Pomiar napięcia powierzchniowego, odbywa się przez wyznaczenia wielkości kropli odrywającej się od kapilary. Ciężar tej kropli będzie się równał napięciu powierzchniowemu.
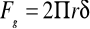
(r - promień kapilary a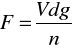
, gdzie V objętość roztworu, d - jego gęstość, g -przyspieszenie grawitacyjne, n - liczba kropel na objętość V). Pomiar ten przeprowadza się najczęściej metodą względną, przyjmując za ciecz odniesienia wodę.Metoda dorywania piersi cienia. Pomiar sprowadza się do pomiaru przy pomocy wagi torsyjnej, siły oderwania pierścienia od powierzchni cieczy.
Metoda pęcherzykowa (maksymalnego ciśnienia baniek). Metoda ta może służyć do pomiaru napięcia miedzy fazami ciecz-gaz i ciecz-ciecz. Polega ona na mierzeniu ciśnienia potrzebnego do wyciśnięcia z kapilary, pęcherzyka gazu lub kropelki cieczy.
Metoda Wilhemlmy'ego (płytkowa). Polega na mierzeniu siły potrzebnej do oderwania płytki od jednej z faz.
Metoda profilu leżącej kropli. Polega na określeniu kształtu kropel lub pęcherzyków w stanie spoczynku.
ROZPUSZCZALNOSC:
a. nieorg. C2H5/H20
b. praktycznie nie rozp Hg/H2O
c. org. butanol/H2O
Wyłączając szczegółowe przypadki, skład mieszaniny różni się od składu jej pary
a) Prawo Daltona dla pary głosi ze ciśnienie ogólnie p=p1+p2 a liczby moli n1 i n2 składników n1/n2=p1/p2
b) Równanie Gibbsa-Duhema N1dlnp1+N2dlnp2=0 gdzie N2=1-N1. Ta zależność słuszna nie jest dla cieczy wzajemnie nierozp.
Ciecze wzajemnie nierozpuszczalne:
P=p10+p20 wiec temp wrzenia mieszaniny jest zawsze niższa niż temp wrzenia składników 1 lub 2 w stanie czystym
Skład pary n1/n2=p10/p20 skład pary nie zależy od składników mieszaniny ciekłej. Temp wrzenia mieszaniny ciekłej jest stała tak długo dopóki nie zniknie jedna z warstw
Ciecze o nieograniczonej wzajemnej rozp:
Typ I Wg prawa Raulta
P1=p10N1 p2...
P=p10N1+P20N2
N1/n2=N1p10/N2p20
Takie zależności odpowiadają układom doskonałym. Dość dobrze stosuje =a się do tego typu zależności takie jak H2O+D2O
Typ II
TypIII
PRZEMIANY FAZOWE I ROWNOWAGI FAZOWE
Reguła faz Gibbsa:
z=s-f+2
z - liczba stopni swobody
s - liczba składników
f - liczba faz
2 - liczba uwzględniająca dwa parametry stanu ciśnienie i temperaturę, gdy z=1 zmiana jednego parametru:cisnienia lub temp. ,nie powoduje zmiany równowagi fazowej; z=0, zmiana jakiegokolwiek parametru narusza stan równowagi fazowej
PRAWO PODZIALU NERNSTA Jeżeli do układu złożonego z dwóch niemieszających się cieczy (benzen(A) i woda(R))wprowadzimy trzeci(kw.octowy(B))to po ustaleniu sie rownowagi stosunek stezenkw do benzenu i wodyK=lnabA/abR
Jeżeli roztwory są rozcieńczone to K=CbI/CbIIK=K1+2K2k12Cw-izoterma podziału
EKSTRAKCJAV roztworu zawierające a gramów substancji, którą trzeba wydzielić
Działamy ekstrahentem w ilość v1, a1 pozostało a-a1 przeszło do ekstrahenta
An=a[V/kV1+V]n
ADSORPCJA (zjawisko gromadzenia się subst na granicy faz w warstwach powierzchniowych
-pochłanianie powierzchni
-adsorbat substancja adsorbowana
-adsorbent-substancja adsorbująca na swojej powierzchni adsorbat
Adsorpcja gazów wprost proporcjonalna do p,a odwrotnie do Tto<T1...
Empiryczne równanie Freudlicha A=kpn
Rownanie Gibbsa a=C/RT
Teoria adsorpcji Langumira:
1. Powierzchnia stałych adsorbentów ma centra aktywne
2. Proces adsorbcji ma charakter dynamiczny
3. Każde centrum adsorbuje tylko 1 cz czyli adsorbent pokrywa się warstwa monomolekularna]
4. Ciepło adsorbcji jest identyczne, dla kazdego centrum
Jeżeli z oznacza pow adsor zajęta przez cz zaadsorb to (1-z) oznacza powierzchnie wolna
Va=kap(1-z)
Vd=kdz
A=αp/1+βp- izoterma adsorpcji Langumuira
Wyróżniamy adsorpcje fizyczna i chemiczna
PRAWO PODZIALU RÓWNOMIERNEGO ENERGII
Każdy rodzaj ruchu można rozłożyć na niezależne ruch składowe, których liczba jest zwana liczba stopni swobodnych
Biorąc pod uwagę przypadkowy ruch to każdy stopień swobody przypada przeciętnie taki sam udział w ogólnej ilości energii niezależnie od tego czy jest to mobr, mpost, mdrg, Dla cząstek o m stopniach swobody otrzymujemy K=Cp/Cv=m+2/m
CIEPŁO WŁAŚCIWE CIAŁ STAŁYCH
Reguła Dulonga i Petita
Ciepło atomowe wszystkich prostych ciał jest jednakowe i wynosi ok. 6,4
Reguła Koppa-Newmanna
Ciepło mol ciał stałych związków chem = sumie ciepeł atomowych pierwiastków składowych.
Elementy termochemii
Stan standardowy: założono, że wartość funkcji termodynamicznej dla 1 mola subs prostej w 25C i pod ciśnieniem 101325Pa są równe 0
Standardowe ciepło tworzenia zw chem. jest równe ilości energii wymieniony na sposób ciepła przez układ w którym zachodzi synteza 1 mola związku chem z subst prostych w warunkach standard gdy V=const
Standardowa entalpia tworzenia zw chem dotyczy przemiany jak wyżej, gdy p=const
Prawo Hessa:
Pozwala obliczyć ciepło dowolnej reakcji ze stosunkowo minimalnej liczby danych wyjściowych
Ciepło reakcji jest również suma algebraiczna ciepeł tworzenia reagentów.
PROCESY ODWRACALNE I NIEODWRACALNE
Entropia: Jeśli prowadzi proces odwracalny tak ze Q jest ciepłem przemiany przy T=const, to zmiana entropii przemiany wyraża się równaniem:
ΔS=Qodw/T [J/K]
ROWNOWAGA CHEMICZNA
- podejście kinematyczne
V1
dA+dB↔lL+mM
V2
V1=k1[A]d[B]b
V2=k2[L]l[M]m
K=k1/k2=[L]l[Mm]/[A]d[B]b
K - stała równowagi chem.
K=Ze-Eakt/RT
- podejście termodynamiczne
dA+bB=lL+mM
Wartość entalpii swobodnej jest funkcja aktywności tej subst.
G=Go+Rtlna
Gdy zachodzi reakcja przy T,p=const
ΔG=-RTlnK
1ΔG=ΔGo+RTln(allamm/aadabb)
lub
2-ΔG=RTlnK-RTln2
-ΔG=RTln(k/2)
R-nania te noszą nazwę izotermy Van't Hoffa z r-nania 1 wynika, że przy T, p=const zmiana ΔG towarzysząca reakcji chemicznej zależy od:
-rodz reakcji i subst reagujących, co opisuje stała równowagi K
-od stosunku aktywności produktów do aktyw substratów, co wyraża wlk Z
Podstawowe znaczenie równań Van't Hoffa
Polega na tym, że umożliwia ono przewidywanie kierunku przebiegu reakcji.
Dla przemian samorzutnych ΔG<0(ujemne) oznacza to, że ΔG
Proces samorzutny musi maleć.
Gdy ΔG>0 (dodatnie)to w danych warunkach nie może przebiegać samorzutnie
Wpływ temp na stała równowagi chem.
T=d(ΔGo)/dT=
ΔGo-RT2[d(lnk)/dT
Równanie Gibbsa-Helmholtza dla warunków standard ma postać:
ΔGo=ΔHo+T[d(ΔGo)/dT]
Porównując te dwa równania mamy:RT2[d(lnk)/dT]=ΔHo
Równanie to zwane jako równaniem izobary Van't Hoffa ujmujące wpływ temp na stałą równowagi reakcji
D(lnk)/dT=ΔH/RT2
Otrzymane równanie stosuje się do każdego szczególnego przypadku: całkujemy, przechodzimy do log i
Log(k1/k2)=ΔH/2,303R
[T2-T1/T2-T1]
Wykorzystanie izobary:
1. K1wyzn w T1 (dla reakcji)
K2 wyznaczamy w T2
Obliczyć można ΔH
Jeżeli znane sa te wlk w danej temp można wyzn Kx,Tx
2 a. Dla reakcji endotermicznych (ΔH dodatnie) logk rośnie ze wzrostem temp, oznacza to, że wzrost temp podnosi wydajność reakcji endotermicznych
b. dla reakcji egzotermicznych (ujemne) logk spada ze wzrostem temp. Obniżanie temp sprzyja wzrostowi wydajności takich reakcji
KINETYKA CHEMICZNA
SZYBKOŚĆ REAKCJI
V=dc/dt=dx/dt
c - stężenie substatur
t - czas reakcji
x - stężenie produktu
Wg uznania, warunków, wygody, można rejestrować albo ubytek substratu albo przyrost stężenia produktu. Z kinetycznego punktu widzenia reakcje można podzielic na jedno i wieloczasteczkowe: nie ma tu formalnych ograniczen,ale prawdopodobieństwo jednoczesnego odpowiedniego zderzenia tych trzech lub więcej cząstek jest bardzo mało prawdopodobne.
Znakomita większość reakcji to reakcje dwu lub jednocząsteczkowe. Reakcje trójcząsteczkowe czas często okazują się wieloetapowymi
SZYBKOŚĆ REAKCJI (v) jest miarą ilości zużytego substratu lub utworzonego produktu w czasie. Dla większości reakcji chemicznych prowadzonych w zamkniętym układzie można wyrazić ją wzorem:
![]()
Jest to postać różniczkowa. Szybkość rekcji można też wyrazić za pomocą równania kinetycznego danej reakcji:
![]()
[A], [B], [C] są stężeniami reagentów a współczynniki a, b, c są wyznaczane eksperymentalnie (dla reakcji prostych w przybliżeniu równają się współczynnikom stechiometrycznym)
Współczynnik proporcjonalności k w równaniu kinematycznym przyjęło nazywać się STAŁĄ SZYBKOŚCI reakcji. Jej wartość nie zależy od stężenia substratów i produktów oraz czas, zaś jest zależnością temperatury oraz stężenia i rodzajów katalizatorów.
Sumę wykładników z równania kinematycznego nazywa się RZĄDEM REKCJI.
![]()
n1L1+n2L2+...= n1'M+n2'M+...V=kCL1n1CL2n2
Wykładniki n1,n2 wskazują na rząd reakcji względem danego składnika, zatem można uwzględnić rzędowość reakcji wzgl wybranego składnika
- rzędu pierwszego:V=kC1
- drugiego V=kC1C2 lub V=kC12
- trzeciego V=K1C13 lub...
- zerowego V=k lub V=kC0
Równania kinetyczne
dx/dt=k(a-x)0 Rzędowość zerowa
a - stężenie początkowe substratu
x - ubytek substratu lub stężenie produktu po czasie t
(a-x)- stężenie substratu, który nie wszedł jeszcze w reakcje
k - stała szybkości reakcji
CZĄSTECZKOWOŚĆ REAKCJI określa ilość cząsteczek reagujących ze sobą w elementarnym akcie reakcji. Rząd reakcji jest równy liczbowo jej cząsteczkowości, gdy reakcje zalicza się do prostych.
OKRES (CZAS) POŁOWICZNYM reakcji (τ1/2) to czas, w którym stężenie danego substratu mierzone w czasie t = 0, zmniejszy się do połowy początkowej wartości.
Dla reakcji I rzędu
T1/2=ln2/k=0,69/k
Dla reakcji II rzędu przyjmując, że stężenia obu substratu były jednakowe -dc/dt=kc2
Z czego otrzymujemy -dc/c2=kdt
1/c=kt+const
gdy t=0,c=0 to wygodniej zapisać const=1/a i a-c/ac=kt
x/a(a-x)=kt
Przykładem reakcji dwu cząsteczek jest zmydlanie tłuszczów
CH3COOC2H5+OH−=CH3COO- +C2H5OH
Reakcje można wyśledzić monitorując (przez miareczkowanie) próbki tzn oznaczając w nich OH- (zasadę)
Jeżeli początkowe stężenie estru = a a zasady = b a≠b to r-nie kinetyczne da się zapisać
Dx/dt=k(a-x)(b-x),po scałkowaniu
(1/a-b)ln[b(a-x)/a(b-x)] ostatnie równanie można wykorzystać do graficznego wyznaczania stałej szybkości reakcji.
Reakcje złożone:
I produkty końcowe większości reakcji chemicznych (szczególnie organicznych są wynikiem jednego lub większej liczby różnych biegnących równolegle lub następujących po sobie procesow(etapow)
II Produkty pośrednie mogą być nieobecne w zapisach równań stechiometrycznych reakcji
III zdarza się często ze znaleziona doświadczalnie szybkość reakcji nie pasuje do tych obliczonych na podstawie równań stechiometrycznych
IV Na szybkość reakcji maja wpływ również takie czynniki chemiczne (katalizatory) Teoria zderzeń aktywnych
OGNIWEM GALWANICZNYM - nazywa się układ, w którym energia elektryczna powstaje w wyniku reakcji chemicznej. Podstawowymi jego częściami są dwa półogniwa połączone ze sobą tak, aby była możliwość wymiany ładunków elektrycznych pomiędzy nimi.
PÓŁOGNIWO - składa się z elektrody zanurzonej w roztworze zawierającym odpowiedni rodzaj jonów.
SIŁA ELEKTROMAGNETYCZNA SEM - jest różnicą potencjałów na elektrodach ogniwa niepracującego. (tzn. otwartego, I 0)
limI0 U = E
E = U + IR
WZÓR NESRTA, na sem ogniwa:
![]()
POTENCJAŁ STANDARDOWY PÓŁOGNIWA, jest to potencjał, gdy aktywność odpowiedniego jonu, biorącego udział w reakcji zachodzącej na elektrodzie jest równa jeden. Jest to wartość względna, wyznaczano względem standardowego półogniwa wodorowego, dla którego przyjęto wartość potencjału równą zero.
ELEKTRODY CHINHYDROWE, zaliczamy do półogniw redoks. Chinon ulega redukcji do hydrochinonu. W reakcji tej biorą udział dwa elektrony i dwa kationy wodorowe. Właśnie dlatego ogniwo zawierające to półogniwo o wykorzystuję się do pomiaru pH danego roztworu. Potencjał takiego układu określa przekształcony wzór Nersta:
![]()
MIARECZKOWANIE POTENCJOMETRYCZNE wykonuję się niemal identycznie jak miareczkowanie objętościowe z tą różnicą, że zamiast obserwowanie zmiany barwy wskaźnika mierzy się SEM ogniwa napełnionego tego roztworem. Taki typ miareczkowania przeprowadza się, gdy nie jest możliwe miareczkowanie objętościowe z powodów np.: mętności badanego roztworu, lub braku wskaźnika.
KONDUKTOMETRYCZNE
Odwrotnością oporu właściwego nazywamy PRZEWODNICTWEM WŁAŚCIWYM lub konduktywności przewodnika.
![]()
Przewodnictwo właściwe jest funkcja temperatury, a dla roztworów elektrolitów zależy także od ich lepkości i wartości dielektrycznej. W dużym stopniu uzależnione jest też w przypadku roztworów od ich stężenia. Dlatego wprowadza się pojęcia PRZEWODNICTWA MOLOWEGO I RÓWNOWAŻNIKOWEGO. Definicje tych wielkości są następujące:
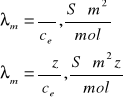
Gdzie ![]()
- przewodnictwo molowe,![]()
- przewodnictwo równoważnikowe, z - sumaryczna wartość jonów jednakowego znaku, powstających podczas dysocjacji jednej cząsteczki, ce - stężenie molowe elektrolitu.
RUCHLIWOŚĆ JONU (kationu lub anionu), jest to stosunek prędkości jonu do natężenia pola elektrycznego, w którym znajduje się jon. Ruchliwość jonu nie zależy od natężenia pola elektrycznego.
MIARECZKOWANIE KONDUKTOMETRYCZNE, polega miareczkowaniu kwasu zasadą (lub na odwrót) lub mieszaniny kwasów. Bardzo ruchliwe jony wodorowe, są zastępowane mniej ruchliwymi kationami z wodorotlenku. Powoduje to spadek przewodnictwa. Po osiągnięciu punktu zobojętnienia, rośnie stężenie jonów hydroksylowych, które także są bardzo ruchliwe. Przewodnictwo zwiększa też jednocześnie coraz większa ilość jonów w roztworze. Przy miareczkowaniu słabych kwasów, nie zauważa się początkowego spadku przewodnictwa gdyż powstaje mocny elektrolit. Pomimo tego po osiągnięciu punktu równoważnikowego, stężenie jonów OH— i ciągłe zwiększanie się sumy wszystkich jonów, powoduje powstanie wyraźnego załamania w punkcie zobojętnienia na krzywej miareczkowania. Przy miareczkowaniu mieszaniny mocny - słaby kwas, najpierw zostaje zneutralizowany mocny kwas.
KOLOIDY
POJĘCIE I KLASYFIKACJA KOLOIDÓW Stan koloidalny jest stanem skupienia materii równie powszechnym jak stan gazowy, ciekły lub stały. Cechą charakterystyczną stanu koloidalnego jest niski stopień rozdrobnienia. Układy koloidalne lub krótko - koloidy, są to układy dyspersyjne, najczęściej dwuskładnikowe, o wyglądzie układów fizycznie jednorodnych, chociaż w rzeczywistości oba składniki nie są ze sobą zmieszane cząsteczkowo.
Składnik tworzący fazę ciągłą układu nazywamy ośrodkiem dyspersyjnym lub rozpraszającym, drugi zaś fazą rozproszoną lub składnikiem rozproszonym. Faza rozproszona składa się z cząstek koloidalnych o wymiarach od 1 do 100 nm, a nawet do 500 nm. Należy tu jednak zaznaczyć, że do układów koloidalnych zaliczamy nie tylko te, które mają wszystkie trzy wymiary "koloidalne", lecz także i te, w których dwa a tylko nawet jeden wymiar jest koloidalny, czyli ma wartość od 1 do 500nm. W związku z tym układy koloidalne można podzielić na układy z cząstkami trójwymiarowymi, układy z cząstkami blaszkowatymi i układy z cząstkami nitkowatymi. Jeżeli cząstki fazy rozproszonej mają jednakową wielkość, układ nazywamy mono lub izo-dyspersyjnym, jeżeli różną - polidyspersyjnym. Układy koloidalne, które spotykamy w przyrodzie czy laboratorium mają jednak najczęściej charakter polidyspersyjny.
Układy dyspersyjne o wymiarach cząstek większych od 500 nm nazywamy układami mechanicznymi (zawiesinami lub suspensjami, w których cząstki ulegają sedymentacji), zaś układy o wymiarach cząstek rozproszonych mniejszych od 1nm układami o rozdrobnieniu cząsteczkowym (roztworami rzeczywistymi).
Stan rozproszenia koloidalnego jest bardzo rozpowszechniony, zarówno w świecie przyrody ożywionej (różnorodne białka, pektyny, węglowodany) i nieożywionej (gliny, mgły, pył wulkaniczny), jak również wśród związków otrzymanych sztucznie w laboratorium chemicznym (mydła, niektóre barwniki, siarka koloidalna, tlenki metali itd.). Oprócz tego w przyrodzie występuje dużo związków, których cząsteczki mają wymiary charakterystyczne dla układów koloidalnych, zwane są one eukoloidami (np. skrobia, celuloza, kauczuk, keratyna, kolagen glikogen itd.), znane są również syntetyczne eukoloidy, jak polistyreny i inne tworzywa sztuczne. Podczas rozpuszczania eukoloidów powstają samorzutnie układy koloidalne.
Najbardziej rozpowszechnione są układy koloidalne o ciekłym ośrodku dyspersyjnym, zwane roztworami koloidalnymi, liozolami lub zolami. Jeżeli ośrodek dyspersyjny jest wodą, zwane są hydrozolami, jeżeli alkoholem alkozolami, jeżeli benzenem - benzenozolami itd. Ogólnie, jeżeli ośrodek dyspersyjny jest cieczą organiczną, układy koloidalne nazywa się organozolami, jeżeli zaś gazem gazozolami (w przypadku powietrza - aerozolami).
KOLOIDY LIOFOBOWE I LIOFILOWEKoloidy w zależności od powinowactwa do rozpuszczalnika (ośrodka rozpraszającego) dzielimy na liofilowe (duże powinowactwo względem rozpuszczalnika - emulsoidy), np. białka, tanina, żelatyna, Fe(OH)3, Al(OH)3 i liofobowe sunspensoidy, np. zole metali, wodorotlenków pewnych metali, sole metaliczne, np. As2S3, AgCl. Zjawisko łączenia się cząstek fazy rozproszonej z ośrodkiem dyspersyjnym nazywamy ogólnie solwatacją a w przypadku ośrodka wodnego - hydratacją.
W wyniku solwatacji koloidów liofilowych cząsteczki fazy rozproszonej ulegają stabilizacji. Natomiast koloidy liofobowe nie ulegają solwatacji albo też ulegają tylko nieznacznie a czynnikiem stabilizującym je jest głównie ładunek elektryczny.
METODY OTRZYMYWANIA KOLOIDÓW
W celu uzyskania rozdrobnienia koloidalnego stosowane są dwa rodzaje metod:
METODY DYSPERSYJNE, polegające na rozdrabnianiu cząstek o wymiarach większych od 500nm aż do uzyskania wymiarów charakterystycznych dla roztworów koloidalnych. Należą tu głównie: rozdrabnianie mechaniczne (rozcieranie), metoda Brediga, polegająca na rozpyleniu głównie czystych metali w łuku Volty, peptyzacja (polega na działaniu roztworem odpowiedniego elektrolitu na świeżo wytrącony trudno rozpuszczalny osad), rozpylanie za pomocą ultradźwięków, rozpylanie katodowe, termiczne itd.
METODY KONDENSACYJNE, polegające na łączeniu cząsteczek lub jonów w większe zespoły aż do osiągnięcia rozdrobnienia koloidalnego. Najważniejsze z tego typu metod to zmniejszenie rozpuszczalności, redukcja, czasem utlenienie, hydroliza, polimeryzacja, metoda zarodnikowa i inne. METODY OCZYSZCZANIA KOLOIDÓWOczyszczanie koloidów od domieszek substancji tworzących roztwory rzeczywiste (ciała krystaliczne i elektrolity) ma duże znaczenie w zapewnieniu trwałości zolu. Do najczęściej stosowanych do tego celu metod należą: dializa, elektroliza, ultrafiltracja, elektrodekantacja i adsorpcja wymienna na jonitach. Jedną z łatwiejszych metod jest dializa. W celu przeprowadzenia dializy woreczek z błony półprzepuszczalnej (kolodium, celofan lub naturalna błona zwierzęca) napełniamy czystym rozpuszczalnikiem (najczęściej wodą) i umieszczamy w naczyniu z zolem. Substancje o rozdrobnieniu cząsteczkowym przechodzą przez membranę do wewnętrznej cieczy i mogą być w ten sposób usunięte z koloidu. Zmieniając stale ciecz wewnętrzną możemy koloid doprowadzić do żądanej czystości. Tak działające urządzenie do doczyszczania koloidów zwane jest dializatorem. Elektrodializa jest połączeniem dializy z elektrolizą a jej przewaga nad dializą polega głównie na większej szybkości oczyszczania (dializę należy prowadzić co najmniej przez kilkanaście dni, podczas gdy elektrodializa już po upływie 2-3 dni daje czysty zol).
WŁAŚCIWOŚCI UKŁADÓW KOLOIDALNYCH
WŁAŚCIWOŚCI MECHANICZNE Jedną z najbardziej charakterystycznych cech układów koloidalnych są ruchy Browna. Zjawisko polega na ciągłych chaotycznych ruchach postępowych, obrotowych i drgających, fazy rozproszonej w ośrodku ciekłym lub gazowym. Ruchy Browna można zaobserwować przypatrując się np. cząstkom kurzu oświetlonym cienką wiązką światła w zaciemnionym pomieszczeniu.
WŁAŚCIWOŚCI OPTYCZNE Jedną z najbardziej charakterystycznych cech układów koloidalnych jest efekt Tyndalla. Polega on na tym, że jeżeli przez roztwór koloidalny przepuszczamy wiązkę światła, to wskutek uginania się promieni na cząstkach fazy rozproszonej, mniejszych od długości fali, światło staje się widoczne w postaci smugi świetlnej. Intensywność tego efektu jest tym większa im większa jest różnica między współczynnikami załamania fazy rozproszonej i ośrodka dyspersyjnego. Efekt Tyndalla został wykorzystany w konstrukcji ultramikroskopu, który ma duże zastosowanie w różnorodnych badaniach koloidów, np. liczenie cząsteczek, obserwacja ruchów Browna, pomiar szybkości koagulacji i inne. Kształty geometryczne cząstek fazy rozproszonej o wymiarach odpowiadających rozdrobnieniom koloidalnym możemy obserwować jedynie w mikroskopie elektronowym. ABSORPCJA ŚWIATŁA. Niektóre układy koloidalne mają silniejszą absorpcję aniżeli rozproszenie światła. Pomiar absorpcji światła jest jedną z metod badania układów koloidalnych, pozwala on na oznaczanie stężenia fazy rozproszonej i na śledzenie przebiegu koagulacji. Barwa układów koloidalnych uwarunkowana jest zarówno przez absorpcję, jak i przez rozproszenie światła. Zależy ona od wielkości, kształtu i stopnia agregacji cząstek fazy rozproszonej. W świetle rozproszonym może być ona inna niż w świetle przechodzącym. Ten sam układ koloidalny może mieć różną barwę w zależności od stopnia rozproszenia. WŁAŚCIWOŚCI ELEKTRYCZNE. Na skutek adsorpcji jonów elektrolitu z roztworu na powierzchni cząstki koloidalnej powstaje ładunek elektryczny. W wyniku tej adsorpcji tworzy się podwójna warstwa elektryczna złożona z powłoki wewnętrznej, czyli adsorpcyjnej, przylegającej mocno do powierzchni zewnętrznej, będącej warstwą jonów przeciwnego znaku. Zależnie od tego jakie jony są adsorbowane na powierzchni, cząstka może być naładowana albo ujemnie albo dodatnio. Jednak znak ładunku elektrycznego nie jest ich cechą charakterystyczną. Ta sama bowiem cząstka koloidalna może mieć ładunek dodatni lub ujemny, zależnie od środowiska. Na przykład koloidalny jodek srebra AgJ w roztworze zawierającym jony srebra jest naładowany dodatnio, w roztworze zaś zawierającym jony jodkowe ujemne (patrz rysunek).
Wyszukiwarka