Technologiczne kryteria klasyfikacji surowców naturalnych
Wyliczanie wielkości zasobów.
Do obliczenia wielkości zasobów w złożach stosować metodę objętościową, statystyczną lub bilansu materiałowego. Metoda objętościowa polega na obliczeniu objętości pustych przestrzeni w skale zbiornikowej. Objętość ta odpowiada objętości występujących w skale zbiornikowej nagromadzeń węglowodorów; przelicza się ją zwykle na objętość, jaką węglowodory zajmowałyby w warunkach normalnych. Objętość węglowodorów w złożu nazywa się zasobami geologicznymi, a ta część zasobów która po zastosowaniu określonych metod eksploatacji złoża można wydobyć na powierzchnie nazywa się zasobami wydobywanymi. Definiuje się także zasoby bilansowe, czyli zasoby które opłaca się eksploatować.
Klasyfikacja zasobów
Złoża gazu ziemnego i ropy naftowej występują w różnych warunkach geologicznych i na różnych (często bardzo dużych) głębokościach. Określenie granic i objętości zbiornika złożowego bywa więc trudne i wymaga wielu kosztownych prac rozpoznawczych (wiertniczych, geofizycznych).
W zależności od stopnia rozpoznania i udokumentowania zasobów podejmuje się decyzję dotyczące zagospodarowania i rozpoczęcia eksploatacji złóż. Z tego względu najbardziej przydatnym sposobem klasyfikacji jest ich podział na kategorie obejmujące złoża rozpoznane i udokumentowane w podobnym stopniu. W
W krajach zachodnich zasoby dzieli się na przemysłowe (kategorie A,B,C1 i C2) i prognostyczne (D1 i D2). Warunkiem zaliczenia zasobów do kategorii A lub B jest określenie geologicznej budowy oraz wielkości złoża. Konieczne jest ponadto uzyskanie gazu lub ropy z co najmniej dwóch lub trzech otworów dowierconych do tego samego poziomu w danym złożu. Należy również określić wydajność poziomu, statyczne ciśnienie złożowe, dozwolone natężenia wydobycia z otworu wiertniczego, jakość ropy lub gazu.
Wybrane procesy technologiczne pozyskiwania paliw i surowców dla przemysłu chemicznego
Podstawowe surowce naturalne (węgiel, ropa naftowa, gaz ziemny)
Klasyfikacja rop naftowych ze względu na gęstość, zawartość siarki oraz zawartość składników grupowych. Podstawowe właściwości fizykochemiczne rop naftowych
Gęstość określonej ropy zależy istotnie od struktury jej składu węglowodorowego. Gęstość węglowodorów rośnie bowiem zazwyczaj wzrostem wartości stosunku liczby atomów węgla do atomów wodoru w ich cząsteczkach. Ropy zawierające dużo parafin i naftenów charakteryzują się niską wartością stosunku C/H. Ponieważ wodór jest lżejszy od węgla, więc takie ropy mają gęstość mniejszą od gęstości rop aromatycznych. Gęstość, a zwłaszcza stosunek H/C, umożliwia więc określenie w sposób przybliżony stopnia parafinowości lub aromatyczności rop naftowych, jak też wydzielonych z nich frakcji. W typowych ropach wartość H/C wynosi 1,8.
Zawartość siarki należy do kryterium klasyfikacji chemicznych rop, co umożliwia ocenienie jej przydatności dla danej rafinerii. Ocena ta dotyczy technologicznych możliwości przeróbki konkretnej ropy na określone produkty z zachowaniem ich zadowalającej wydajności i pożądanej jakości. Bywa niekiedy , że rafineria nie może przerabiać określonej ropy ze względu na brak odpowiedniego wyposażenia technologicznego np. z braku instalacji do odsiarczania produktów.
Ze względu na zawartości siarki ropy dzielimy na niskosiarkowe (wartość %), średniosiarkowe (wartość %) oraz wysokosiarkowe (wartość%)
Zawartość składników grupowych
Systematyka technologiczna węgli (typy węgli, składniki węgli, asortyment węgli). Podstawowe właściwości fizykochemiczne węgli.
Systematyka technologiczna porządkuje węgle wg ich przydatności do różnych celów z punktu widzenia:
-typu węgla (zaliczenie do typów w zależności od zawartości części lotnych, ciepła spalania i tzw. spiekalności
-stopnia czystości (zawartość siarki i popiołu, wilgotności)
-asortymentu tj. klasy ziarnistości (węgle grube, kęsy, kostka, orzech, węgle średnie, węgle miałowe)
Te trzy elementy charakterystyki technologicznej węgla są brane pod uwagę przy podejmowaniu decyzji o przeznaczeniu węgla na cele opałowe lub skierowaniu go jako surowca do przemysłu koksochemicznego czy do instalacji przeróbki chemicznej. Rozróżniamy 4 podstawowe procesy chemicznej przeróbki węgla:
*1 - Rozkład termiczny bez dostępu powietrza:
-niskotemperaturowy (<600 °C) - półkoksowanie
-wysokotemperaturowy (> 800 °C) - koksowanie
*2 - Zgazowanie parą wodną i tlenem (wytwarzanie gazu syntezowego)
*3 - Upłynnianie bezpośrednie polegające na wytworzeniu ciekłych frakcji węglowodorowych metodą katalitycznego uwodornienia lub pirolizy węgla
*4 - Wytwarzanie karbidu - surowca do produkcji acetylenu
Podział węgli ze względu na stopień uwęglenia (stopień metamorfizmu):
-Torf (55-60% C, 30-40% SNO)
-Węgle brunatne (60-75% C 20-30% SNO)
-Węgle kamienne (75-92% C 2-20% SNO) - te z kolei dzielimy na : płomienne, gazowo-płomienne, gazowe, gazowo-koksowe, koksowe, chude
-Węgle antracytowe i antracyt (92-98% C 1-4% SNO)
Klasyfikacja gazów ziemnych ze względu na skład, składniki węglowodorowe i nie węglowodorowe gazów ziemnych. Podstawowe właściwości fizykochemiczne gazów ziemnych. Liczba Wobbego, definicja i sens fizyczny
Gaz ziemny to surowiec mineralny znajdujący się w skorupie ziemskiej w fazie gazowej. Gaz ten spotyka się w złożach w postaci rozproszonej w wodach podziemnych lub ropie naftowej
Gazy ziemne oprócz węglowodorów stanowiących ich podstawową masę zawierają często także i inne składniki, jak CO2, CO, H2S, N2, H2 i gazy szlachetne. Składniki węglowodorowe to głównie metan i etan oraz w znacznie mniejszych ilościach wyższe alkany - propan, butany oraz pentany i heksany.
W gazie ziemnym z różnych złóż różna jest zawartość poszczególnych węglowodorów. Gaz, który składa się wyłącznie z metanu i etanu (>90%) nazywa się gazem suchym. Typowymi gazami suchymi są gazy błotne, kopalniane (towarzyszące węglowi kamiennemu) oraz gaz ziemny pochodzący ze złóż typowo gazowych. Natomiast gaz, który oprócz metanu zawiera węglowodory o większej od niego masie molowej jest nazywany gazem kondensatowym lub mokrym (nazwy nieprecyzyjne). Towarzyszy on na ogół złożom ropy naftowej
Liczba Wobbego jest to iloraz ciepła spalania (Q) i pierwiastka kwadratowego z gęstości względnej gazu (d) odniesionej do powietrza. Wartość liczby Wobbego jest podstawą do podziału paliw gazowych na podgrupy.
Wyróżnia się:
dolną Liczbę Wobbego - gdy za wartość kaloryczną przyjmuje się jego wartość opałową;
górną Liczbę Wobbego - gdy za wartość kaloryczną przyjmuje się jego ciepło spalania.
Przerób ropy naftowej; destylacja, procesy pogłębionej przeróbki naftowej
W procesach przeróbki ropy naftowej i frakcji węglowodorowych z niej otrzymywanych wytwarza się:
*gaz płynny
*paliwa silnikowe (benzyny, oleje napędowe, paliwa odrzutowe)
*oleje smarowe i smary
*oleje opałowe
*stałe węglowodory naftowe (parafiny, cerezyny, wazeliny)
*asfalty drogowe i przemysłowe
*surowce węglowodorowe do syntez organicznych
Przygotowanie ropy do przeróbki
Ropa wydobyta ze złoża zawsze zawiera zanieczyszczenia mechaniczne oraz pewną ilość porwanego gazu ziemnego i wody z rozpuszczonymi w niej solami. Woda (solanka) występuje w ropie w postaci emulsji. Zawodnienie ropy ogromnie utrudnia prowadzenie jej destylacji. Konieczne jest bowiem ogrzewanie nie tylko ropy, ale i zawartego w niej balastu wodnego. Ponadto para wodna zakłóca proces. Głębokie odsolenie ropy jest konieczne, gdyż zmniejsza korozję aparatury w instalacjach destylacji ropy i instalacjach dalszej przeróbki różnych frakcji naftowych.
Metody rozbijania emulsji:
-mechaniczne (filtrowanie i obróbka ultradźwiękami)
-chemiczne (dodawanie różnych deemulgatorów)
-termiczne (podgrzewanie i odstawanie wody, przemywanie gorącą wodą)
-elektryczne (działanie pola elektrycznego)
Ropę odsoloną do zawartości stoli poniżej 5mg/dm3 można poddawać rozdzielaniu przez destylację I rektyfikację.
Instalacje destylacji rurowo-wieżowej (DRW)
Przemysłowe procesy destylacji ropy naftowej polegają na łączeniu destylacji prowadzonej z jednokrotnym lub wielokrotnym odparowaniem z rektyfikacją faz parowej i ciekłej. W procesach tych rozdziela się ropę na frakcję, mogące stanowić składniki produktów handlowych lub półprodukt - surowiec w różnych procesach rafineryjnych.
W instalacjach destylacji ropy głównymi aparatami są piece rurowe i kolumny rektyfikacyjne zwane wieżami. Stąd powszechnie używa się potocznego określenia instalacja destylacji rurowo-wieżowej (DRW).
Nowoczesne instalacje DRW pracują w sposób ciągły i zazwyczaj są dwustopniowe. Pierwszy stopień stanowi destylacja pod ciśnieniem atmosferycznym, drugi pod ciśnieniem zmniejszonym. Istnieją jednak również rafinerie, w których ropę rozdziela się, prowadząc tylko destylacje atmosferyczną
Frakcja |
Odbiór |
Zasadnicze |
Zakres temperatury wrzenia |
|
- |
- |
- |
0,1 Mpa |
8-9 kPa |
Gaz suchy i płynny |
S+DA |
C1-C4 |
<20 |
- |
Benzyna lekka |
S+DA |
C5-C9 |
40-150 |
- |
Benzyna ciężka |
DA |
C9-C12 |
150-190 |
- |
Nafta |
DA |
C10-C16 |
190-240 |
- |
Lekki olej napędowy |
DA |
C15-C22 |
240-350 |
- |
Destylaty próżniowe |
- |
- |
- |
- |
lekki |
DP |
>C20 |
>350 |
220-250 |
średni |
DP |
|
|
250-300 |
ciężki |
DP |
|
|
310-380 |
Gudron |
DP |
- |
- |
- |
Aparatura instalacji DRW
W skład podstawowej aparatury instalacji DRW wchodzą piece rurowe, kolumny (wieże) frakcjonujące oraz wymienniki ciepła, chłodnice i pompy.
Schemat technologiczny i parametry pracy dwustopniowej
Część atmosferyczna DRW
*1 Odsolona ropa tłoczona jest przez wymienniki ciepła, w których jest ogrzewana ciepłem frakcji odprowadzanych z kolumn atmosferycznej i próżniowej i jest wprowadzana do kolumny wstępnej
*2 Tam odbywa się tzw. stabilizacja ropy tj. oddzielenie gazów i oddestylowanie z ropy benzyny lekkiej.
*3 Ropę pozbawioną najlżejszych frakcji ogrzewa się następnie w piecu do temp. ok. 350 st. C i wprowadza do kolumny atmosferycznej.
*4 Pozostałość(mazut) gromadzi się w zbiorniku. Jako frakcję szczytową odbiera się benzynę lekką. Po skropleniu i oddzieleniu gazu benzyna ta jest kierowana do zbiorników. Frakcje boczne ochładza się w wymiennikach ciepła i także odprowadza do zbiorników magazynowych.
b) Część próżniowa instalacji DRW
*1 Mazut ze zbiornika ogrzewa sięw piecu do temp 420 st. C i wprowadza do kolumny próżniowej, w której rozdestylowuje się go na frakcje olejowe.
*2 Pozostałość z kolumny (gudron) kieruje się przez wymiennik i chłodnicę do ogrzewanego parą zbiornika.
*3 Przeprowadzenie destylacji mazutu z parą wodną
Pogłębiona przeróbka destylatów ropy naftowej
Pogłębiona przeróbka ma na celu zwiększenie ilości produktów lekkich. Produkty destylacji atmosferycznej przerabia się w następujący sposób:
Frakcja benzynowa - poddawana jest procesowi reformingu otrzymując reformat, z którego wydzielane są węglowodory aromatyczne oraz benzyna o wysokiej liczbie oktanowej. Wodór powstały w tym procesie kierowany jest do instalacji hydrorafinacji i hydroodsiarczania oleju napędowego.
Frakcja olejowa (z destylacji i instalacji krakingu katalitycznego)- poddawana jest hydroodsiarczania, powstały siarkowodór kierowany jest do instalacji otrzymania siarki metodą Clausa.
Destylaty próżniowe - poddaje się reakcjom krakingu katalitycznego (otrzymując gaz, benzynę o liczbie oktanowej ~80, oraz oleje napędowe i opałowe. Cięższe frakcje oleju próżniowego poddawane są rafinacji i odparafinowaniu otrzymując oleje smarowe i parafinę, oraz procesowi koksowaniu z otrzymaniem koksu naftowego.
Gudron - poddawany jest procesowi oksydacji w celu otrzymania asfaltu
Procesy przemysłu rafineryjnego do pozyskiwania paliw: hydrorafinacja, reforming, hydrokraking, kraking katalityczny
Reforming
Głównym produktem reformingu są komponenty benzyn silnikowych zwane reformatami, które charakteryzują się wysoką liczbą oktanową, co wynika z dużej zawartości węglowodorów aromatycznych i izoparafinowych. Reformaty są wciąż zdecydowanie najważniejszym źródłem tych węglowodorów. Ich wydzielanie z reformatów stanowi jednak specyficzny problem optymalizacyjny, gdyż:
-węglowodory aromatyczne nadające wysoką oktanowość reformatowi są jednocześnie niezwykle ważnymi surowcami w syntezach organicznych
-zawartość benzenu oraz sumy jednopierścieniowych węglowodorów aromatycznych w benzynach jest od kilku lat ostro limitowana ze względów ekologicznych.
Istota pracy instalacji reformingu polega na przekształceniu średnich i ciężkich benzyn z DRW, niekiedy z dodatkiem benzyn tzw. Wtórnego pochodzenia (np. hydrokraking lub kraking katalityczny), w benzyny o charakterze nasyconym i LO > 90. W stosunku do surowca uzyskuje się wzrost liczby oktanowej o 30-40 jednostek.
Podczas katalitycznego reformingu zawarte w benzynach węglowodory ulegają różnym reakcjom chemicznym przyspieszanym i ukierunkowywanym przez dwufunkcyjny katalizator zawierający platynę osadzoną na tlenku glinu o odpowiedniej budowie sieci krystalicznej. Tlenek ten posiada właściwości kwasowe, które intensyfikuje dodatek chloru jako promotora. Najważniejsze reakcje chemiczne przebiegające w procesie katalitycznego reformingu benzyn:
Odwodornienie sześcioczłonowych naftenów do węglowodorów aromatycznych
Cykloheksan -(-H2)-> Cykloheksen -(-2H2)-> Benzen + 3H2
Reakcji sprzyjają wysoka temperatura (>520K) i małe ciśnienie cząstkowe wodoru sprzyjają osiągnięciu wyższego stopnia przereagowania.
Dehydroizomeryzacja pięcioczłonowych naftenów
Metylocyklopentan cykloheksan Benzen + 3H2
Podobnie jak wyżej duża temperatura i niskie ciśnienie cząstkowe.
Dehydrocyklizacja parafin
Heksan -(-H2)-> cykloheksan -(-3H2)-> Benzen + 3H2
Pierwsze stadium polega na pośrednim powstawaniu olefin, które zostają zaadsorbowane na dwóch sąsiednich centrach katalizatora.Następnie pierścień zostaje zamknięty I następuje natychmiastowe odwodornienie.
Izomeryzacja
Podczas reformingu przebiegają trzy typy reakcji izomeryzacji węglowodorów:
*izomeryzacja n-parafin do izoparafin
*izomeryzacja zawartych w surowcu i tworzących się w procesie węglowodorów alkiloaromatycznych
*izomeryzacja pięcioczłonowych cykloolefin do sześcioczłonowych
5) Hydrokrakowanie
Reakcje te prowadzą do węglowodorów nasyconych o mniejszej masie molowej, często o budowie bardziej rozgałęzionej.
Surowiec do procesu reformingu poddaje się rektyfikacji i kieruje się frakcje wrzącą w temperaturze powyżej 85 °C, która poddaje się następnie hydrorafinacji. Ze względu na wrażliwość katalizatora na związki siarki, frakcje tę należy wcześniej poddać procesowi odsiarczania. Z surowca należy również usunąć związki azotu, gdyż podczas procesu uwodorniają się one do amoniaku, który osłabia kwasowy charakter nośnika.
Reformowanie prowadzi się w temp. 480-525 °C i pod ciśnieniem 0.7-3.0 MPa. Bardzo ważne jest utrzymanie intensywności cyrkulacji gazu wodorowego dobranej odpowiednio do strumienia masy zasilającego surowca. Zapobiega to zakoksowaniu i dezaktywacji katalizatora.
W praktyce przemysłowej ostrość procesu reformingu ocenia się na podstawie dwóch parametrów: molowego stosunku wodoru do surowca i szybkości objętościowej.
Oprócz reformatu otrzymuje się także gaz wodorowy oraz gaz płynny.
Niskociśnieniowe instalacje reformingu z cykliczną regeneracją katalizatora
Instalacja wyposażona jest w ciąg trzech reaktorów oraz dodatkowy tzw. Swing reaktor, którym można zastąpić dowolny z zasadniczych reaktorów. W każdym momencie pracy instalacji jeden z tych czterech reaktorów znajduje się w cyklu regeneracji katalizatora. Polega on na włączeniu do pracy reaktora ze świeżo zregenerowanym katalizatorem z jednoczesnym rozpoczęciem cyklu regeneracji w kolejnym reaktorze. Instalacja jako całość pracuje w sposób ciągły, lecz poszczególne reaktory -periodycznie.
Podstawowym produktem instalacji jest wysokooktanowy reformat LOB=95-103 (bez dodatku tetraetyloołowiu) lub wydzielany z niego koncentrat węglowodorów aromatycznych.
Instalacje reformingu z ciągłą cyrkulacją i regeneracją katalizatora
Jest to generacja instalacji technologicznych pozwalających prowadzić stabilnie proces reformingu z utrzymywaniem wysokiej temperatury (do 540 °C) i niskiego ciśnienia (0,3-1,0 MPa) w reaktorach. Ciągły dopływ świeżego zregenerowanego katalizatora zapewnia przy tym możliwość utrzymywania tych zaostrzonych parametrów technologicznych w okresach czasu ograniczonych jedynie koniecznością przeprowadzenia prac konserwacyjno-remontowych. Wymiane katalizatora prowadzi się bez zatrzymania instalacji. Instalacje z ciągłą regeneracją są oferowane przez dwie firmy - UOP i IFP. Obydwa procesy różnią się znacznie.
Zalety i wady procesów UOP i IFP.
Ciągłe procesy reformingu UOP i IFP prowadzi się pod niskim ciśnieniem. Z tego względu konieczne było zastosowanie specjalnych rozwiązań konstrukcyjnych aparatury (pieców, wymienników ciepła, reaktorów). Trudność polegała na takim zaprojektowaniu tych aparatów, aby spadek ciśnienia był w nich minimalny. Drugim trudnym problemem technologicznym jest odzyskiwanie reformatu unoszonego z gazem wodorowym oddzielającym się w separatorze od cieczy pod stosunkowo niskim ciśnieniem. Stosuje się wtedy tzw. Recontacting pod wyższym ciśnieniem.
Zalety procesów reforming z ciągłą regeneracja katalizatora uwidoczniają się wyraźnie przy ich porównaniu z procesem klasycznym. W procesie ciągłym uzyskuje się większą o ok. 2% obj. Wydajnośćreformatu (C5+) oraz większą wydajność wodoru.
Kraking katalityczny
Z samego rozdestylowania ropy można, zależnie od jej jakości otrzymać 15-20% benzyn. Dodatkowae ilości benzyn wytwarza się z destylatów próżniowych w wyniku poddania ich procesi katalitycznego krakingu.
Surowcem procesu są destylaty próżniowe z DRW lub mazut, będące złożoną mieszaniną węglowodorów. Z tego względu reakcje zachodzące podczas krakowania są skomplikowane i wielokierunkowe. Przebieg tych reakcji zależy od składu surowca i warunków prowadzenia procesu. Jednocześnie z reakcjami rozkładu cząsteczek węglowodorów zachodzą reakcje wtórne: kondensacja, cyklizacja oraz izomeryzacja. Na przebieg tych reakcji zasadniczy wpływ ma temperatura i obecność katalizatora.
Kraking katalityczny prowadzi się w temp. 380-440 °C, w których nawet bez użycia katalizatora zachodzą ze znaczną szybkością następujące reakcje termiczne:
-rozerwanie wiązań C-C w cząsteczkach parafin z wytworzeniem olefin o mniejszej masie molowej
-odwodornienie naftenów prowadzące do węglowodorów aromatycznych
-rozerwanie pierścieni naftenów z wytworzeniem węglowodorów nienasyconych (olefinowych lub dienowych)
-polimeryzacja olefin wytworzonych w wymienionych reakcjach oraz ich kondensacja z dienami do węglowodorów aromatycznych
Katalizatorami reakcji krakingu katalitycznego sa glinokrzemiany, zeolity.
Katalityczny kraking przebiega wg. Mechanizmu jonowego. Istotnym elementem tego mechanizmu jest tzw. Rozklad β powstających pośrednio jonow karbonowych - analogiczny do rozkładu β wolnych rodnikow.
Schematy technologiczne przemysłowych instalacji krakingu katalitycznego musza oprocz reaktora obejmowac aparaty, w których przebiega regeneracja katalizatora. Regeneracja polega na wypaleniu koksu osadzonego na katalizatorze podczas procesu w reaktorze. Ściśle związany układ technologiczny dwóch podstawowych aparatów instalacji krakingu wraz z bezpośrednio współpracującymi z nimi innymi aparatami nazywa się zwykle blokiem reaktorowym.
Hydrokraking
W ogólnym rozumienie na reakcje hydrokrakingu składają się dwie następcze reakcje: krakowania węglowodorów i uwodornienia powstałych produktów krakingu. Katalityczne reakcje hydrokrakowania różnych rodzajów węglowodorów oraz związków zawierających azot i siarkę stanowią istotę przemysłowych procesów hydrokrakingu, które umożliwiają przeróbkę ciężkich surowców naftowych (destylatów próżniowych, pozostałości) na produkty o mniejszej masie molowej i niższej temperaturze wrzenia (gazy, benzynę, hydrokrakingowi, frakcje olejowe). Są to produkty analogiczne do produktów krakingu katalitycznego, z tym że nie zawierają one siarki i węglowodorów nienasyconych, gdyż hydrokraking prowadzony jest pod ciśnieniem wodoru. Gaz płynny z hydrokrakingu nie zawiera zatem ani propylenu ani butenów.
Hydrokraking destylatów olejowych można ukierunkować na produkcję: benzyn silnikowych, gazu płynnego, paliw odrzutowych, oleju napędowego lub olejów smarowych. W każdym z nich można przerabiać jako surowiec różne destylaty naftowe, jak: destylaty atmosferyczne i próżniowe z DRW, destylaty z koksowania, recyrkulaty z procesu krakingu katalitycznego oraz oleje pizolityczne. Ta duża elastyczność procesu jest jego bardzo istotną zaletą, umożliwiającą pełne wykorzystanie wszelkich strumieni destylatów olejowych, jakimi dysponują rafinerie, jako surowców do wytwarzania paliw silnikowych i gazu płynnego.
Hydrorafinacja
Przykładowa produkcja paliw w rafinerii
Biopaliwa (I i II-giej generacji), Technologiczne rozwiązania otrzymywania biopaliw z udziałem procesów wodorowych (Technologia NExBTL, wsad olejów roślinnych na instalację hydrotreatingu)
Biomasa jest biodegradowalną częścią produktów i odpadów rolniczych, w tym subsstacjami pochodzenia roślinnego lub zwierzęcego, z leśnictwa oraz związanych z nim przemysłów. Biomasa drewna składa się głównie z celulozy (ok. 45%), hemicelulozy (20-30%) oraz ligniny (20-30%). Pojęciem biomasy obejmuje się również biodegradowalne odpady przemysłowe i komunalne.
Według Dyrektywy 2003/30/EC do biopaliw zalicza się : bioetanol (otrzymywany z fermentacji cukrów, otrzymanych z przerobki trzciny lub buraków cukrowych, lub z hydrolizy skrobii), biom etanol, biodiesel, bio-DME (otrzymywany z biometanolu), bio-ETBE (otrzymywany z izobutylenu i bioetanolu), bio-MTBE.
Do głównych kierunków przeróbki drewna należą:
-otrzymywanie żywic i garbników
-sucha destylacja do węgla drzewnego i gazu oraz tzw. Octu drzewnego (kwas octowy, aldehyd octowy, metanol, aceton)
-hydroliza kwasowa do pentoz i heksoz przerabianych w procesie fermentacji na furfurol oraz etanol.
-roztwarzanie metodą siarczynową do celulozy, z której otrzymuje się papier oraz modyfikowane polimery
6. Procesy przemysłu rafineryjnego dla pozyskiwania surowców do syntez
a) Wytwarzanie węglowodorów aromatycznych (reforming i ekstrakcyjne wydzielanie węglowodorów aromatycznych, hydrodealkilowanie węglowodorów alkiloaromatycznych, piroliza frakcji oleju napędowego)
benzen - obecnie, obok ekstrakcji ze smoły pogazowej, stosowanych jest wiele innych metod otrzymywania benzenu. Wśród nich należy wymienić metody takie jak:
kraking parowy - piroliza lekkich frakcji ropy naftowej z parą wodną
reforming lekkich frakcji ropy naftowej - reformingowi poddaje się frakcje benzynowe wrzące w 80-180C otrzymane w wyniku rozdestylowania ropy naftowej. Proces prowadzi się w T=500C, p=1-4MPa, katalizator: Pt na tlenku glinu. Selektywność katalizy zwiększa się w przypadku zastosowania katalizatorów Bi- oraz polimetalicznych. Podczas reformingu zawarte w benzynach węglowodory ulegają różnym reakcjom chemicznym:
odwodornienie sześcioczłonowych naftenów do węglowodorów aromatycznych
dehydroizomeryzacja pięcioczłonowych naftenów (np. metylocyklopentan -> cykloheksan -> benzen)
dehydrocyklizacja (np. heksan -> cykloheksan -> benzen)
izomeryzacja
hydrogenoliza i hydrokrakowanie
dealkilacja toluenu - polega na przepuszczeniu mieszaniny toluenu i wodoru nad katalizatorem w temperaturze 500-600C p=3-5MPa. Reakcja: toluen + wodór -> benzen + metan. Katalizatorem jest CoO-Mo2O5 na tlenku glinu. Dodatek NaOH zwiększa selektywność reakcji. Czasami zamiast użycia katalizatora stosuje się jeszcze wyższą temperaturę - proces termiczny. Instalacje są prostsze, ale wymagają użycia stali stopowych i specjalne konstrukcji reaktorów. Oba powyższe procesy umożliwiają uzyskanie benzenu z wysoką wydajnością i o wysokiej czystości.
odwodornienie cykloheksanu - prowadzone w temperaturze 300C w obecności katalizatora (głownie platyny naniesionej na tlenek glinu).
polimeryzacja acetylenu w podwyższonej temperaturze w obecności węgla aktywnego. Schematyczne: 3*acetylen -> benzen
toluen - do niedawna toluen otrzymywany był w wyniku destylacji smoły węglowej. Obecnie uzyskuje się go w wyniku:
dehydrogenacji (odwodornienia) odpowiednich pochodnych alkilowych cykloheksanu
reakcji alkilowania benzenu halogenkami alkilowymi w obecności katalizującego reakcję kwasu Lewisa.
b) Procesy wytwarzania węglowodorów olefinowych (piroliza olefinowa), surowce warunki procesu, względny udział węglowodorów olefinowych.
Piroliza olefinowa - krakowanie termiczne surowca do alkenów by otrzymać niższe, nienasycone węglowodory alifatyczne, takie jak etylen, propylen, buten, butadien itd. Proces bezkatalityczny. Temperatura rozrywa wiązania C-C i H-H. Surowce do pirolizy: gaz płynny, benzyna, lekki olej napędowy, nafta. T=700-950C, p=0.1-0.3MPa. Piroliza zachodzi wg mechanizmu wolnorodnikowego.
Przykład: piroliza etanu
inicjacja: CH3-CH3 -> 2CH3*
propagacja: CH3-CH3 + CH3* -> CH4 + CH3-CH2*
CH3-CH2* -> CH2=CH2 + H*
H* + CH3-CH3 -> CH3-CH2* + H2
terminacja: rekombinacja, dysproporcjonowanie
Po reakcji pirolizy następuje rozdział produktów: wyodrębnienie frakcji składających się z węglowodorów o tej samej liczbie atomów węgla (etan-etylen; propan-propylen itd.) Następnie każda z tych frakcji rozdzielana jest na składniki parafinowe i olefinowe.
7. Procesy alkilowania
Alkilowanie i-butanu olefinami C3-C4, wykorzystanie produktu jako komponenta benzyn
Proces polega na wprowadzeniu rodnika alkilowego do cząsteczki związku organicznego. Izobutanem alkiluje się propylen, izobutylen, but-1-en oraz but-2-en. Produktem tej reakcji alkilowania są węglowodory rozgałęzione, wyżej wrzące, a także mające wyższą liczbę oktanową (nawet powyżej 100), co jest istotne przy wykorzystywaniu produktów reakcji jako komponentów benzyn. Proces można prowadzić bez katalizatora, wymaga on wtedy stosowania temperatury rzędu 500C i ciśnienia 20-40MPa. Przy zastosowaniu katalizatora (HF lub H2SO4) proces prowadzi się w T<50C i p=3MPa. Surowcem w procesie jest frakcja węglowodorów C3-C4 pochodząca z procesu pirolizy olefinowej, bądź krakingu katalitycznego. Surowce wymagają oczywiście odpowiedniego przygotowania jak usunięcie związków siarki, olefin, wody, etc.
Reakcje:
izobutan + propylen -> 2,3-dimetylopentan lub 2,4-dimetylopentan
izobutan + but-1-en -> 2,3- lub 2,4-dimetyloheksan
izobutan + but-2-en -> 2,2,4- lub 2,3,3- lub 2,3,4- lub 2,2,3-trimetylopentan
izobutan + izobutylen -> 2,2,4-trimetylopentan
Alkilowanie benzenu α-olefinami (alkilowanie benzenu propylenem) propylobenzen (kumen)
Kumenowa metoda produkcji fenolu; utlenienie kumenu → rozkład wodoronadlenku kumenu do fenolu i acetonu.
Alkilowanie węglowodorów aromatycznych i ich pochodnych prowadzi się najczęściej w fazie ciekłej z zastosowaniem olefin lub chloro parafin jako czynników alkilujących. Jako katalizatory stosuje się kwasy (siarkowy, fosforowy, fluorowodorowy), oraz katalizatory Friedel-Craftsa (kwasy Lewisa posiadające zdolność tworzenia połączeń koordynacyjnych ze związkami zawierającymi pi elektrony lub mającymi niesparowane elektrony).
W procesie alkilowania powstają kationy alkilowe, wysoce aktywne, atakujące elektrofilowo pierścień aromatyczny. Prowadzi to do podstawienia atomu wodoru w pierścieniu grupą alkilową.
Sumarycznie i skrótowo proces można przedstawić następująco:
CH3-CH=CH2 + H+ -> CH3-CH+-CH3
Bądź: CH3-CHCl-CH3 +AlCl3 -> CH3-CH+-CH3 + AlCl4-
Karbokation atakuje elektrofilowo pierścień, potem następuje odłączenie atomu wodoru z pierścienia i dostajemy kumen (izopropylobenzen). W przypadku katalizatora F-C powstaje dodatkowo HCl.
Alkilowanie benzenu prowadzi się w fazie ciekłej (ściśle jest to zawiesina kompleksu katalizatora w benzenie i polialkilobenzenach) z intensywnym mieszaniem barbotującym propylenem. T=95-1200C, p=0.5MPa. Dla samego alkilowania wystarcza temperatura do 500C, ale zwiększa się ją dla zwiększenia szybkości reakcji trans alkilowania polialkilobenzenów w celu zwiększenia ilości wytwarzanego kumenu. Mieszanina poreakcyjna zawiera ok. 40% mas. kumenu.
Zastosowanie kumenu:
Kumen + O2 -> 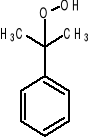
(wodoronadtlenek kumenu - WNTK) (reakcja rodnikowa - inicjator odłącza proton od kumenu, tworząc rodnik kumylowy, który następnie reagując z tlenem daje rodnik nadtlenkowy. Ten z kolei reaguje z kolejnym kumenem, dając wodoronadtlenek kumenu i kolejny rodnik kumylowy.)
Reakcja w fazie ciekłej, t=110-120C, p=0.5-1MPa. Utlenia się tlenem z powietrza. Mieszanina poreakcyjna ma ok. 25% WNTK. Zatęża się ją destylując próżniowo do 90%.
Dalej wodoronadtlenek kumenu pod wpływem stężonego kwasu siarkowego (0.1%mas w stosunku do WNTK) rozkłada się do fenolu i acetonu. Reakcja egzotermiczna, mieszaninę poreakcyjną chłodzi się i rozdziela w kolumnach rektyfikacyjnych na poszczególne składniki.
Alkilowanie benzenu α-olefinami (alkilowanie benzenu etylenem); etylobenzen
Wykorzystanie etylobenzenu (Odwodornienie etylobenzenu → styren )
Mechanizm alkilowania j/w.
Zastosowanie:
Etylobenzen -> styren + H2
Reakcja endotermiczna Tmax=630C (wyższa intensyfikuje reakcje uboczne) p=0.01MPa. Przereagowanie 70-80%.
Reakcje uboczne:
Etylobenzen -> acetylen + benzen
Etylobenzen + wodór -> toluen + metan
Reakcję prowadzi się wprowadzając parę wodną podgrzaną do 720C do mieszaniny etylobenzenu świeżego i obiegowego. Katalizatorem jest Fe2O3, ponadto Cr2O3 i K2CO3 jako aktywatory (charakteryzują się odpornością na działanie pary wodnej). Po ochłodzeniu mieszaniny poreakcyjnej rozdziela się ją w separatorze, do styrenu dodaje się hydrochinonu w celu inhibicji reakcji polimeryzacji. Ciekły produkt zawiera styren, etylobenzen i niepożądaną pozostałość (polimery styrenu, benzen, toluen).
O-alkilowanie izobutylenu metanolem (zastosowanie MTBE)
Pozyskiwanie surowca: piroliza olefinowa bądź FCC żeby uzyskać frakcję C4. Izomeryzacja n-butanu do izobutanu, następnie odwodornienie.
Reakcja:
1. utworzenie karbokationu tert-butylowego:
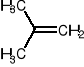
+ H+ -> 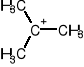
2. O-alkilowanie (egzotermiczne) t=90-100C, p=1.2MPa:
Katalizator - jonity sulfonowe (grupy SO3H)
MTBE jest stosowany przede wszystkim jako wysokooktanowy komponent benzyn bezołowiowych. Jest środkiem zastępującym tetraetyloołów, pozwala na zwiększenie liczby oktanowej (LO MTBE=110) dzięki zmniejszeniu zjawiska przedwczesnego zapłonu - jest środkiem przeciwstukowym.
8. Wykorzystanie etylenu do syntez
Otrzymywanie etanolu. Proces pośredniej i bezpośredniej hydratacji etylenu. Porównanie
procesów. (Inna metoda otrzymywania etanolu). Najważniejsze zastosowania etanolu.
Proces pośredniej (zwanej też kwasową) hydratacji etylenu
Jako surowca węglowodorowego używa się w tym procesie najczęściej nie czystego etylenu, ale frakcji etanowo-etylenowej zawierającej 56-60% etylenu. Etan wydzielony potem z mieszaniny poreakcyjnej (nie uczestniczy on w reakcjach) zwraca się do instalacji pirolizy.
Proces bezpośredniej hydratacji etylenu.
Bezpośrednią hydratację etylenu prowadzi się w temperaturze 265-300°C, pod ciśnieniem 7,5-8MPa i z molowym stosunkiem H2O:C2H6 w granicach 0,4-0,9. Jako katalizator stosuje się w zdecydowanej większości instalacji kwas fosforowy na nośniku mającym odpowiednią trwałość mechaniczną i odporność na działanie tego kwasu w warunkach prowadzenia hydratacji. Najczęściej tym nośnikiem jest krzemionka oczyszczona uprzednio z tlenków żelaza i glinu. W procesie bezpośredniej hydratacji etylenu uzyskuje się w jednym przejściu reaktora bardzo mały (4,5-5%) stopień przereagowania. Wynika stąd oczywiście konieczność stosowania dużej krotności cyrkulacji etylenu z czym jest związane znaczne zużycie energii na sprężanie recyrkulowanego strumienia.
Porównanie:
Zasadniczą wadą hydratacji pośredniej jest znaczne zużycie kwasu siarkowego i silna korozja aparatury, znacznie zwiększająca koszty eksploatacyjne. Ponadto w tym procesie uzyskuje się stosunkowo zanieczyszczony etanol, a obsługa instalacji jest skomplikowana i kłopotliwa. Hydratacja pośrednia ma również i zalety do których zalicza się:
- niskie temperatury i ciśnienia robocze
- duże przereagowanie etylenu w „jednym przejściu”, co umożliwia stosowanie surowców olefinowych bardziej rozcieńczonych, a więc tańszych np. frakcji etanowo-etylenowej, lub frakcji propanowo-propylenowej w przypadku produkcji alkoholu izopropylowego.
Obecnie praktycznie we wszystkich nowo uruchamianych instalacjach produkujących syntetyczny etanol jest stosowana bezpośrednia hydratacja etylenu.
Chlorek winylu. Trzy koncepcje syntezy chlorku winylu: oksychlorowanie etylenu, bezpośrednie chlorowanie etylenu, z uzyciem etylenu i acetylenu)
Zbilansowane chlorowanie mieszaniny etylenu i acetylenu:
CH2=CH2 + Cl2 -> CH2Cl-CH2Cl
CH2Cl-CH2Cl -> CH2=CHCl + HCl - kraking (t=500C, p=2MPa wydajność przy jednym przejściu przez reaktor rurowy 50-60%)
CH≡CH + HCl -> CH2=CHCl
Sumarycznie: CH≡CH + CH2=CH2 +Cl2 -> 2CH2=CHCl
Chlorowanie etylenu (f. ciekła - dichloroetan rozpuszcza etylen i chlor, kat. FeCl3. Nadmiar etylenu ogranicza reakcje uboczne. Dwa warianty procesu - wysokotemperaturowy 83-130C [powyżej temp. wrzenia dichloroetanu] oraz niskotemperaturowy - 30C):
CH2=CH2 + Cl2 -> CH2Cl-CH2Cl
CH2Cl-CH2Cl -> CH2=CHCl + HCl
Oksychlorowanie etylenu (w warstwie fluidalnej. kat CuCl2 na Al2O3. T=230-260C p=0.3MPa. Warstwę fluidalną tworzy katalizator unoszony przez etylen, techniczny tlen i chlorowodór)
2CH2=CH2 + 2HCl + 0.5O2 -> CH2Cl-CH2Cl + H2O
CH2Cl-CH2Cl -> CH2=CHCl + HCl
Często chlorowanie i oksychlorowanie prowadzi się w postaci zbilansowanej, aby wykorzystać powstający w reakcji chlorowania i oksychlorowanie HCl.
Metody syntezy z etylenu są częściej wykorzystywane, ze względu na niższy koszt i łatwiejszą dostępność etylenu.
Zastosowanie: w przeważającej większości chlorek winylu kieruje się do instalacji wytwarzania PVC.
Otrzymywanie kwasu octowego i bezwodnika octowego. Najważniejsze zastosowania
Otrzymywanie:
Karbonylowanie metanolu - najczęściej stosowane, najbardziej ekonomiczne. Reakcja: metanol + CO -> kwas octowy. T=200C, p=2MPa, kat. kompleksy rodu promotowane jodem. Proces przebiega w fazie ciekłej. Mieszanina jest ciągle mieszana za pomocą barbotującego CO. Reakcje uboczne: konwersja CO z parą wodną, w nieznacznym stopniu reakcja metanizacji, oraz reakcja, której produktem jest kwas propionowy. W procesie otrzymuje się lodowaty kwas octowy o b. dużej czystości (99.9%).
Katalityczne utlenianie aldehydu octowego. Reakcja czteroetapowa:
Utlenianie aldehydu do kwasu nadoctowego CH3-CHO + O2 -> CH3-COOOH (kat. octan kobaltu. Faza ciekła, reagent: tlen bądź powietrze wzbogacone w tlen)
Kwas nadoctowy wiąże następną cząsteczkę aldehydu, tworząc nadoctan 1-hydroksyetylu CH3-COOO-CH(OH)CH3. Te dwa pierwsze procesy powinny przebiegać z jednakową szybkością, aby unikać nagromadzenia się kwasu nadoctowego. Aby to zapewnić stosuje się nadmiar aldehydu w stosunku do tlenu i odpowiednie katalizatory.
Nadoctan ulega dehydratacji do bezwodnika octowego (kat. jony miedzi)
Bezwodnik ulega hydratacji do kwasu octowego
W reakcji otrzymuje się 65-70% bezwodnika octowego. Kwas octowy otrzymuje się ubocznie w ilości 15-20%.
Utlenianie n-butanu. Proces prowadzony w fazie ciekłej w roztworze lodowatego kwasu octowego. T=170-180C, p=6-7MPa. Proces autokatalityczny, bądź w obecności soli kobaltu lub manganu.
Utlenianie benzyny lekkiej C5-C6. Utlenianie w fazie ciekłej za pomocą powietrza. T=170-200C, p=5MPa, kat. nafteniany kobaltu. Benzyna C5-C6 jest tańsza od n-butanu. Musi być czysta (nie więcej niż 0.05% siarki, 0.5% węglowodorów C1-C3 i 5% C4). Otrzymuje się produkt zawierający 70-80% kwasu octowego, 15% kwasu mrówkowego, do 10% kwasu propionowego i kilka % kwasu bursztynowego.
Zastosowanie: Octan winylu, octan celulozy i inne estry. Kwas chlorooctowy. W przemyśle do produkcji barwników, leków, perfum, octu.
Otrzymywanie glikolu etylenowego. Integracja produkcji tlenku etylenu i glikolu etylenowego
Poniżej temperatury 200C katalizatory srebrowe są mało aktywne. Natomiast w zakresie 200-300C przyspieszają stosunkowo selektywnie reakcję:
CH2=CH2 + 0.5 O2 = CH2-O-CH2 (tlen: czysty lub z powietrza)
W celu uzyskania wysokiej selektywności proces prowadzi się w temperaturze 240-275 st. C ze stopniem przereagowania etylenu w granicach 40% (w wyższych temperaturach - niepożądane reakcje). Im niższa temperatura tym korzystniej jest stosować wyższe ciśnienie (do 2,5 MPa).
Selektywność katalizatora srebrowego możemy zwiększyć przez promotowanie go odpowiednimi dodatkami (nadtlenkiem baru, metalami alkalicznymi) oraz związkami chloroorganicznymi np. 1,2-dichloroetanem.
55-80% wytwarzanego tlenku etylenu przerabia się na glikol etylenowy.
Transport glikolu jest znacznie bezpieczniejszy.
Między innymi dlatego wytwarzanie tych dwóch produktów bardzo często realizuje się w zintegrowanym kompleksie instalacji.
Reakcja: (CH2)2O + H2O -> CH2OH-CH2OH
Reakcja hydratacji tlenku etylenu nie zatrzymuje się tylko na wytworzeniu glikolu monoetylenowego. Glikol ten może bowiem dalej reagować z tlenkiem etylenu i tworzyć glikol dietylenowy, a następnie trietylenowy itd.
Reakcje te mogą przebiegać bez katalizatora lub wobec katalizatorów kwasowych.
Przemysłowe procesy hydratacji tlenku etylenu są jednak w większości instalacji prowadzone bez katalizatora, z zastosowaniem dużego nadmiaru wody (molowo 15-20 krotnego), temperatury 180-200 st. C oraz ciśnienia 1,8-2MPa nieodzownego by utrzymać mieszaninę w fazie ciekłej.
9. Wykorzystanie propylenu do syntez
Chlorek allilu- epichlorohydryna - glicerol
chlorek allilu - otrzymuje się go na skale przemysłową na drodze chlorowania propenu w temp 450-500C. W niższych temperaturach następuje addycja chloru do wiązania podwójnego. Do chlorowania używa się 95% propenu, dobrze oczyszczonego od propanu i innych węglowodorów, które w podanej temperaturze mogą również ulegać chlorowaniu, zwiększając zużycie chloru i powodując zanieczyszczenie zasadniczego produktu. W praktyce stosuje się 5-6moli propenu na 1 mol chloru, przy czym chlorek allilu tworzy się z 80% wydajnością. Oprócz chlorku allilu powstają również inne produkty chlorowania propylenu: 2-chloropropen, 1-chloropropen, 1,3-dichloropropen, 3,3-dichloropropen.
Reakcja: CH3-CH=CH2 + Cl2 -> ClCH2-CH=CH2 + HCl
Epichlorohydryna - praktycznie jedyną drogą jej otrzymania jest metoda chlorohydrynowa polegająca na:
Chlorohydroksylowaniu chlorku allilu za pomocą wodnego r-ru chloru (t=25-50C).
CH2=CH-CH2Cl +Cl2 + H2O -> CH2OH-CHCl-CH2Cl + CH2Cl-CHOH-CH2Cl
Odchlorowodorowanie do epichlorohydryny przy użyciu Ca(OH)2. T=90C, p<atm.
CH2OH-CHCl-CH2Cl + CH2Cl-CHOH-CH2Cl +Ca(OH)2 -> epichlorohydryna + CaCl2 + 2H2O
glicerol - prowadzi się hydrolizę epichlorohydryny do glicerolu za pomocą wodnego roztworu węglanu sodowego. Proces ten zachodzi w temp 150C, p=1.4MPa w atmosferze CO2. Wydajność glicerolu w stosunku do epichlorohydryny wynosi 98%.
Reakcja (zalosowałem): epichlorohydryna +2H2O -> gliceryna + HCl
Produkcja gliceryny z propylenu tzw. metodą chlorową (przez chlorek allilu) jest kosztowna i kłopotliwa technologicznie. Stanowiło to inspirację do opracowania nowego procesu, w którym półproduktem jest akroleina otrzymywana metodą utleniania propylenu.
Utlenianie propylenu do akroleiny. Produkcja kwasu akrylowego
Katalityczne utlenienie propylenu do akroleiny
CH2=CH-CH3 + O2 -> CH2=CH-CHO + H2O
Katalizatorem jest Bi2O3-MoO3, T=370-400C
Dalej akroleina zostaje utleniona do kwasu akrylowego:
CH2=CH-CHO + 0.5O2 -> CH2=CH-COOH
T=250-300C, kat. tlenki Mo i Bi (j/w) z dodatkiem tlenków P, Co, Te.
Utlenianie propylenu, a potem akroleiny prowadzi się w osobnych reaktorach, aby można było dobrać parametry każdego procesu, dzięki czemu uzyskuje się wysokie wydajności akroleiny i kwasu akrylowego. Otrzymany produkt etapu 1 zawiera 99%wag akroleiny. Czysty kwas akrylowy otrzymuje się z wydajnością 80-85%.
Akroleina współcześnie stosowana jest głównie jako monomer do produkcji poliakroleiny.
Kwas akrylowy stosuje się również jako monomer do produkcji poliakrylanów, kwasu poliakrylowego oraz do produkcji żywic akrylowych.
10. Wykorzystanie węgla.
a) Energetyczne wykorzystanie węgla
Chemiczna przeróbka węgla; koksowanie (wykorzystanie produktów), upłynnianie, zgazowanie (rozwiązania technologiczne procesu zgazowania, kierunki wykorzystania gazu procesowego)
Koksowanie węgla to proces termicznego odgazowania węgla lub mieszanki węglowej w wysokiej
temperaturze (450-1000 C) bez dostępu powietrza. Głównym produktem tego procesu jest stała pozostałość (koks, karbonizat) o dużej wytrzymałości mechanicznej i dużej reaktywności chemicznej. Ponadto w procesie tym powstają produkty ciekłe (smoła węglowa) oraz gaz.
Zastosowanie koksu:
- koks wielkopiecowy-paliwo, reduktor, czynnik zapewniający gazoprzepuszczalność wsadu
- koks do produkcji żelazostopów (Fe+Si, Mn, Cr)
- koks odlewniczy (paliwo, czynnik zapewniający gazoprzepuszczalność wsadu)
- koks do wytopu metali nieżelaznych Zn, Pb, Cu (paliwo, reduktor, czynnik zapewniający
gazoprzepuszczalność wsadu)
- koks opałowy
- koks do produkcji karbidu
- koks do produkcji wapna palonego
Upłynnianie węgla
Proces upłynniania węgla jest trudny w realizacji, chociaż jego zasady wydają się dość proste. Węgiel wprowadzany jest do młyna, w którym zostaje rozdrobniony i wymieszany ze specjalną pastą. Tę mieszankę rozgrzewa się do temperatury 420C i wprowadza do 15-metrowego, wieżowego reaktora, do którego doprowadzany jest również wodór. Tutaj, w temperaturze 450C i pod ciśnieniem 20 MPa, następuje uwodornienie. Następnie w separatorze rozdzielone zostają poszczególne frakcje - oleje ciężkie, średnie i lekkie. Część średniego i ciężki wykorzystywane są m.in. do produkcji pasty, zaś cała frakcja lekka i część średniej stanowią ropę węglową. Poza tym otrzymuje się koks elektrodowy, włókno węglowe i czystą siarkę.
Ropa węglowa jest cieczą o kolorze mocnej herbaty. Można z niej otrzymywać dowolne produkty końcowe - różne rodzaje benzyn, olej napędowy, oleje silnikowe, paliwa lotnicze. Przetwórstwo ropy węglowej jest przy tym łatwiejsze od tradycyjnego przetwórstwa ropy naftowej. Ropa węglowa ma bowiem bardziej stabilne własności.
Procesy techniczne zgazowania
Zgazowanie prowadzi się w reaktorach zwanych dawniej generatorami obecnie zgazowywaczami. Skłąd gazu syntezowego zależy od składu chemicznego węgla.
Koppers-Totzek
Lurgi
Winkler (fluidalny)
Texaco
Przebieg procesu Koppers-Totzek
- Mielenie i suszenie węgla gazmi spalinowymi 90μm 2% wilgotność
- Transport pneumatyczny w strumieniu azotu
- Rozdział powietrza na tlen i azot. Zużycie tlenu 200 do 550 Nm3/tonę węgla (duży udział w kosztach zgazowania)
- Zgazowanie, ciśnienie atmosferyczne, temp. 1500-1900ºC
- Chlodzenie dostrzykiem wody (wypłukiwanie popiołu)
- Elektrofiltry - usuwanie popiołu ze strumienia gazu
Inny proces: LURGI, charakterystyka
* Reaktor - pionowa wieża
* Surowiec: węgiel ziarno 5-50mm, przesuwa się z góry na dół
* W przeciw prądzie od dołu płynie para wodna i tlen
* Temperatura do 1000ºC
* Skład gazu do 30% CO2, 20% CO, 38% H2, 11% CH4 (efekt niższej temperatury), większe wymagania jakościowe w stosunku do węgla niż w metodzie Koppers-Totzek
Zastosowanie gazu syntezowego (CO + H2):
Surowiec do syntez chemicznych
Gaz o małej wartości opałowej dla celów energetycznych
Gaz o właściwościach redukcyjnych, hutnictwo i przemysł maszynowy
Surowiec do wytwarzania syntetycznego gazu metanowego (metanizacja)
11. Gaz ziemny - procesy oczyszczania i wykorzystania
W zależności od złoża gazy zawierają więcej lub mniej składników niewęglowodorowych - azotu, siarkowodoru, dwutlenku węgla i pary wodnej. Różnią się także znacznie zawartością węglowodorów o masie molowej większej niż metanu.
Osuszanie. Gaz ziemny wydobywany ze złoża praktycznie zawsze zawiera parę wodną. Jej obecność jest przyczyną poważnych trudności eksploatacyjnych w instalacjach niskotemperaturowej przeróbki gazu oraz podczas jego transportu gazociągami. Trudności te są przyczyną skraplania się wody oraz tworzenia się lodowych korków i krystalicznych hydratów blokujących aparaturę lub rurociągi. Występuje także intensywna korozja (zwłaszcza gdy oprócz pary wodnej gaz zawiera siarkowodór i dwutlenek węgla).
Usuwanie H2S i CO2. Oczyszczanie gazu ziemnego z siarkowodoru i dwutlenku węgla jest jedną z najważniejszych czynności przygotowujących gaz do transportu lub przeróbki. Obecność większych ilości siarkowodoru w gazie ziemnym jest niedopuszczalna z następujących powodów:
*Toksyczne właściwości siarkowodoru uniemożliwiają skierowanie gazu do użytkowników komunalnych
*Siarkowodór jest związkiem silnie korodującym metale, dlatego jego obecność w gazie ziemnym powoduje szybkie niszczenie gazociągów, armatury oraz aparatury w instalacjach przerabiających ten gaz.
*Jeśli gaz ziemny ma stanowić surowiec do syntez chemicznych, to usunięcie siarkowodoru jest konieczne, bo dezaktywuje katalizatory. Poza tym pogarsza jakość produktów
*Obecność siarkowodoru w gazie przetłaczanym gazociągami intensyfikuje powstawanie hydratów. Siarkowodór jest najaktywniejszym czynnikiem „hydratotwórczym”, gdyż powoduje wytwarzanie się centrów krystalizacji, wokół których narastają kryształy hydratów węglowodorów.
Odgazolinowanie. Węglowodory o masie molowej większej niż etanu usuwa się z gazu przez tzw. Odgazolinowanie. Otrzymane gaz płynny i benzyna lekka są cennymi paliwami i surowcami do syntez chemicznych. Dlatego należy je dokładnie wydzielić z kondensatowego gazu ziemnego. Wydzielenie to można traktować jako konieczne oczyszczanie gazu suchego (metan, etan) przed jego gazociągowym transportem. Obecność węglowodorów o większej masie molowej intensyfikuje tworzenie się hydratów, a ponadto skraplanie się tych węglowodorów podczas transportu gazociągiem zmniejsza jego przepustowość.
Stabilizacja gazoliny i rozdzielanie mieszanin węglowodorów gazowych. Otrzymana w procesie odgazolinowania gazolina musi zostać poddana stabilizacji tj. rozdzieleniu na gazolinę stabilizowaną i gaz płynny. Często też gaz płynny rozdziela się jeszcze na poszczególne węglowodory: propan, n-butan, izobutan. Jest to koniecznie gdy gaz płynny ma być użyty jako surowiec do syntez.
Odazotowanie. Gazy zawierające duży procent azotu (tzw. Gazy zaazotowane) mają znacznie mniejszą wartość opałową niż gazy zawierające mało tego składnika niepalnego, a w szczególności gazy składające się prawie wyłącznie z węglowodorów. Transport gazu zaazotowanego wiąże się zatem ze stratą energii na przemieszczanie balastu azotowego.
Odgazolinowanie gazu ziemnego. Cel i schemat technologiczny
Proces odgazolinowania gazów prowadzi się w przemyśle trzema metodami: absorpcyjną, destylacji niskotemperaturowej pod zwiększonym ciśnieniem oraz adsorpcyjną. Niskotemperaturowe odgazolinowanie absorpcyjne (w temp. -10 do -40 °C) jest stosowane coraz częściej. Jest ono jednym z najbardziej ekonomicznych sposobów odgazolinowania gazu ziemnego zawierającego do 100g/m3 propanu i węglowodorów ciężkich. W niskiej temperaturze i pod niskim ciśnieniem absorbuje się bowiem mniej metanu i etanu niż w wyższej temperaturze i pod wyższym ciśnieniem, przy tym samym stopniu zaabsorbowania węglowodorów C3-C6. Ma to duże znaczenie technologiczne. W procesie odgazolinowania najlepszymi absorbentami są frakcje naftowe zawierające dużo węglowodorów parafinowych (benzyna, nafta, oleje).
Odgazolinowanie metodą niskotemperaturowej kondensacji kondensatowych gazów ziemnych polega na sprężeniu gazu w wymiennikach chłodzonych odparowującym ciekłym propanem lub ciekłym etanem.
Osuszanie gazu ziemnego, cel i główne metody oraz ich efekty
Do głównych metod osuszania gazu ziemnego należą :
*osuszanie gazów metodą absorpcji w roztworach glikoli etylenowych
*osuszanie w instalacjach kolumnowych za pomocą glikoli
*osuszanie adsorpcyjne (sita molekularne, tlenek glinu, żel krzemionkowy, węgiel aktywny)
Osuszanie gazu w instalacjach kolumnowych za pomocą glikoli
Jest to najbardziej rozpowszechniony sposób osuszania gazów ziemnych oraz gazów węglowodorowych w przemyśle rafineryjno-petrochemicznym Gaz do osuszania wprowadzany jest do dolnej części absorbera, w której oddzielają się niesione przez strumień gazu krople wody i ewentualnie krople ciężkich węglowodorów. Gaz przepływa następnie do górnej części tego absorbera, w której kontaktuje się przeciwprądowo ze spływającym po półkach wodnym roztworem TEG lub DEG. Osuszany gaz kieruje się ze szczytu absorbera do aparatu, w którym oddzielają się unoszone krople absorbentu.
Osuszanie gazu metodami adsorpcyjnymi
Osuszanie gazów węglowodorowych metodami adsorpcyjnymi stosuje się wtedy, gdy zachodzi potrzeba osiągnięcia bardzo niskiej temperatury punktu rosy. Dokonuje się tego najczęściej, stosując wstępne osuszanie absorpcyjne, a następnie doduszanie na adsorbentach stałych. Umożliwia to znaczne zmniejszenie obciążenia adsorberów i uczynienie całego procesu osuszania bardziej ekonomicznym. Szczególnie powszechnie stosuje się adsorpcję pary wodnej na sitach molekularnych w przypadkach osuszania gazu otrzymanego w procesie pirolizy olefinowej przed jego niskotemperaturowym rozdzielaniem w celu demetalizacji i wydzielenia frakcji wodorowej. Regeneracja sit jest przeprowadzana za pomocą przedmuchiwania gorącym metanem lub frakcją metanowo-wodorową. Czynnik regenerujący jest ogrzewany stopniowo w piecu i kierowany do adsorbera. Następuje desorpcja pary wodnej. Końcowa temperatura regeneracji sit wynosi 350-400 °C.
Metody odsiarczania gazu ziemnego (wymienić)
*Usuwanie H2S i CO2 przez absorpcje z reakcją chemiczną (wodne roztwory etanoloamin) np. proces Ekonamin - oczyszczanie diglikoloaminą, absorpcja w gorących roztworach soli alkalicznych
*Usuwanie przez absorpcję fizyczną. Proces Rectisol
*Proces Sulfinol
*Proces Clausa
*Procesy łączące absorpcję siarkowodoru z jego utlenianiem do siarki np. proces Holmes-Stredford
*Procesy chylatowe
f) Metody absorpcyjne odsiarczania gazu ziemnego (etanoloaminy)
Procesy absorpcji H2S i CO2 w wodnych roztworach etanoloamin są bardzo rozpowszechnione. Spośród etanoloamin najczęściej stosuje się monoetanoloaminę (MEA) i di etanoloaminę (DEA). Pomiędzy MEA i siarkowodorem lub dwutlenkiem węgla z gazu zachodzą następujące reakcje:
2HO-C2H4-NH2 +H2S = (OH-C2H4-NH3)2S
2HO-C2H4-NH2 + CO2 + H2O = (HO-C2H4-NH3)2CO3
W temperaturze ok. 50°C reakcje przebiegają na prawo tj. w kierunku tworzenia się siarczku i węglanu monoetanoloaminy. Zwiększenie temperatury do 110 °C prowadzi do uwolnienia siarkowodoru i dwutlenku węgla. Takiemu przebiegowi reakcje sprzyja także obniżenie ciśnienia.
Gaz zawierający siarkowodór wprowadza się do dolnej części absorbera. Płynie on następnie ku górze, spotykając się przeciwprądowo z wodnym roztworem etanoloaminy (15-30% MEA) spływającym z góry na dół. Odsiarczony gaz odprowadza się ze szczytu absorbera, a nasycony siarkowodorem roztwór MEA zbiera się z jego dolnej części. Roztwór ten tłoczy się następnie przez wymiennik do desorbera.
g) Wytwarzanie gazu syntezowego z gazu ziemnego
Konwersja metanu z parą wodną
CH4 + H2O CO + 3H2 ΔH = 206kJ
Reakcja silnie endotermiczna
Lub konwersja utleniająca węglowodorów z parą wodną + tlenem
(półspalanie w reakcji z tlenem)
CH4 + ½ O2 CO + 2H2 ΔH= -35,7kJ
CH4 + H2O CO + 3H2 ΔH= 206 kJ
C + H2O CO + H2
Wyszukiwarka