I.
Jakie procesy przeprowadzają w war tlenowych i beztlenowych bakterie kumulujące fosfor?
W strefie beztlenowej obecne w ściekach bakterie fosforowe (np. Acinetobacter), które są ścisłymi tlenowcami nie mogą oddychać w tych warunkach. Z drugiej strony obecność dużej ilości pokarmu (szczególnie lotnych kwasów tłuszczowych - głównie octowego), ich ulubionego pożywienia, jest tak atrakcyjna, ze zdobywają się na energię, aby je przyswoić i zakumulować (w postaci polihydroksymaślanów lub poliwalerianianów) na "gorsze czasy", gdy o pożywienie będzie trudniej. To "zdobycie się na energię" oznacza, że rozładowują one swoją baterię polifosforanową i wydzielają fosfor do otoczenia. To uwolnienie fosforu do ścieków w strefie beztlenowej jest z nawiązką rekompensowane w następnej strefie, którą jest strefa napowietrzania. W warunkach tlenowych "objedzone" bakterie fosforowe zużywają zakumulowany pokarm na procesy życiowe do których jest potrzebna energia. Gdy jest większa konkurencja o pokarm i pokarmu zaczyna brakować, bakterie fosforanowe zaczynają gromadzić fosfor w postaci polifosforanów (na wypadek, gdyby ponownie znalazły się w warunkach beztlenowych). Dzięki temu, że w strefie napowietrzonej bakterie te rozmnożyły się, zostaje wchłonięte dużo więcej fosforu niż uwolniło się go w strefie beztlenowej.
Wiązania wodorowe i znaczenie ich dla organizmów
Wiązanie takie może się wytworzyć między atomem elektroujemnym i atomem wodoru połączonym kowalencyjnie z tlenem lub azotem. Atomy biorące udział w wiązaniu wodorowym mogą się znajdować w dwóch częściach tej samej
cząsteczki lub należeć do różnych cząsteczek. Wiązania wodorowe decydują w znacznym stopniu o właściwościach wody. Wiązania te należą do oddziaływań słabych: łatwo się tworzą, łatwo również ulegają zerwaniu. Wiązania wodorowe są najsilniejsze wtedy, gdy 3 atomy ułożone są liniowo. Tworzenie się wiązania wodorowego wpływa znacznie na właściwości fizyczne i chemiczne substancji, np. na podwyższenie temp wrzenia np. białka i kwasy nukleinowe wiele ze swoich cech strukturalnych , a więc i właściwości zawdzięczają wiązaniu wodorowym. Wiązanie wodorowe między atomami wiązania peptydowego i łańcuchem bocznym aminokwasu ułatwiają stabilizację struktury przestrzennej białka.
Wydzielenie grupy α-aminowej od aminokwasu.
Głównym miejscem rozkładu aminokwasów u ssaków jest wątroba. Grupy α-aminowe z wielu różnych aminokwasów są przenoszone na α-ketoglutaran, co prowadzi do otrzymania glutaminianu. Ten z kolei ulega deaminacji oksydacyjnej, dając NH4+. Przeniesienie grup α-aminowych z α-aminowkasu na α-ketokwas katalizują aminotransferazy. Ogólnie te enzymy skierowują spływ grup aminowych z różnych aminokwasów na α-ketoglutaran w celu przemiany w NH4+. Najważniejsza z tej grupy enzymów aminotransferaza asparaginianowa katalizuje przeniesienie grupy aminowej z asparaginianu na α-ketoglutaran.
Kinetyka enzymów. Model Michaelisa-Mentena.
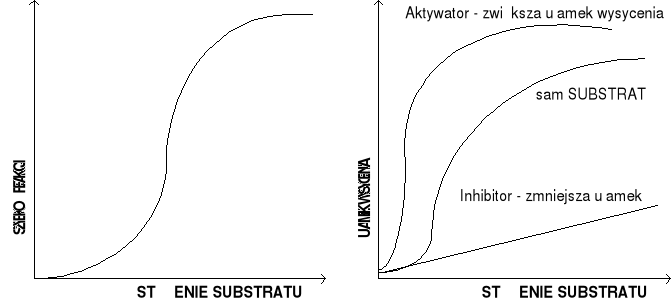
Kinetykę enzymów opisuje model M-M. Zasadniczym założeniem teorii MM jest konieczność wytworzenia odwracalnego kompleksu między enzymem, a substratem: E + S← k1k2→ ES →k3→ E+P. Kompleks ten podlega przemianie nieodwracalnej do produktu P z uwolnieniem enzymu. Stężenie enzymu jest wielokrotnie mniejsze w porównaniu ze stężeniem substratu i produktu, dlatego można przyjąć, że stężenie kompleksu ES jest stałe i zależy od stężenia enzymów w strukturze. Im większe jest stężenie kompleksu ES, tym więcej produktu powstanie.
←z wykresu wynika, że szybkość reakcji V początkowo jest wprost proporc. do stężenia substratu S i przebiega zgodnie z reakcją I rzędu. W miarę zwiększenia się stężenia substratu widać coraz mniejszy wpływ na szybkość reakcji. A po osiągnięci szybkości Vmax dalsze zwiększenie substratu nie odnosi żadnego skutku. Reakcja osiągnie szybkość max, gdy całkowita ilość enzymu E znajdzie się w postaci kompleksu ES, bo wtedy wszystkie cząsteczki enzymu są wysycone substratem. Km - stała Michaelisa - równa się takiemu stężeniu substratu, w którym szybkość reakcji enzymu równa się połowie szybkości Vmax, tzn. KM=K2+K3/K1 W warunkach stanu ustalonego szybkość tworzenia ES jest równa szybkości rozpadu ES. K1[E][S]=(K2+K3)[ES] stąd: [ES]=([E] [S])/KM
przekształcając: [ES]=[EC] * ([S]/[S]+ KM). [EC] - całkowite stężenie enzmu.
kiedy [S]/[S]+KM ≈ 1 → VMAX=K3 [EC]. stąd Równanie MM: V=VMAX * ([S]/[S]+KM)
Założenia graniczne:
• gdy [S] jest bardzo małe [S]<< KM to V=VMAX ([S]/KM) - reakcja I rzędu. Czyli szybkość reakcji jest wprost prop do [S].
• gdy [S]>> KM to V=VMAX - reakcja zerowego rzędu. Szybkość jest maksymalna i niezależna od stężenia substratu.
• gdy [S] = KM to V = VMAX/2 KM jest równe takiemu stężeniu substratu, dla którego szybkość reakcji osiąga połowę swej wartości maks.
Wykres Lineweavera-Burka.
1/V=1/VMAX+KM/VMAX*1/[S]
Stała KM dla większości enzymów waha się od 10-7 do 10-1 mol/dm3.
II.
Oksydacyjna deaminacja.
W procesie deaminacji aminokwasu wydziela się amoniak, w wyniku czego powstaje α-ketokwas. Jednym z typów tego procesu jest deaminacja oksydacyjna. Enzymy mogą współdziałać z NAD+ lub NADP+ względnie z FAD lub FMN. Najważniejszym przykładem enzymów enzymów deaminujących, współdziałających z NAD+ jest dehydrogenaza glutaminianowa. Enzym ten katalizuje przemianę kwasu glutaminowego do α-ketoglutarowego i amoniaku. Podstawowe znaczenie dehydrogenazy glutaminianowej, przy pełnej jej odwracalności, polega na wprowadzaniu amoniaku o związków organicznych oraz na powiązaniu przemiany aminokwasów z cyklem kwasów trójkarboksylowych, w którym α-ketoglutaran jest produktem pośrednim.
Właściwości kinetyczne enzymów allosterycznych.
Enzymy alloster mają sigmoidalną kinetykę. Zbudowane są z podjednostek. Mają symetrie cząsteczkową, znajdują się w miejscach rozgałęzień dróg metabolicznych. Enzymy alloster. mają specjalne miejsce receptorowe (centrum allosteryczne). W innym miejscu niż centrum aktywne. Większość enz alloste zwiera więcej niż jeden łańcuch polipeptydowy. Są bardziej skomplikowane. Enz allost mogą przyłączać w miejscu alloster efektory alloster (ligandy), które oddziałują na aktywność enzymu. Zmienia się aktywność enzymu, wywołana zmianą kształtu enzymu lub jego centrum aktywnego. Efektorami - mogą być same substraty reakcji. W ich nieobecności enzymy alloster przejawiają niewielką aktywność katalityczną. Podczas wiązania substratu następuje zmiana konformacyjna miejsca aktywnego i wzrost jego właściwości katalitycznych. Następne cząsteczki substratu wiązane są w miejscach aktywnych z większą szybkością i w większym tempie ulegają reakcjom chem. Współdziałanie miejsc aktywnych różnych podjednostek enzymu alloster jest odpowiedzialne za sigmoidalny charakter krzywej szybkości reakcji. Inhibitor wiązany w miejscu allosterycznym jednej z podjednostek obniża reaktywność miejsca katalitycznego tej podjednostki, jak również blokuje możliwość przejścia miejsc aktywnych w pozostałych podjednostkach enzymu alloster ze stanu niskiej aktywności do konformacyjnego stanu wysokoreaktywnego.
Model sekwencyjny, model jednoprzejściowy
Rola transaminacji.
Transmicja, w której może uczestniczyć wiele naturalnych aminokwasów, ma ogromne znaczenie w przemianie materii, gdyż pozwala organizmowi oszczędnie gospodarować azotem i wytwarzać aminokwasy z odpowiadających im szkieletów węglowych. Przemiana ta, katalizowana przez enzymy zwane aminotransferazami, polega na przeniesieniu grupy aminowej z aminokwasu na α-ketokwas. Reakcja transaminacji przez udział α-ketokwasów, a szczególnie szczawiooctanu, α-ketoglutaranu i pirogronianu, stanowi ważne powiązanie między przemianą azotową a przemianami cukrowców, a zwłaszcza z końcowym etapem ich rozkładu w cykl kwasów trójkarboksylowych. Szkielety węglowe takich aminokwasów, jak asparaginian, glutaminian czy alanina dzięki temu powiązaniu mogą być w miarę potrzeby spalone w tej przemianie do CO2 i H2O.
Tetrahydrofolian
Bardzo uniwersalny nośnik aktywowanych fragmentów jednowęglowych, odgrywa ważną rolę w metabolizmie aminokwasów i nukleotydów Koenzym ten przenosi fragmenty jednowęglowe o trzech wzajemnie zamiennych stanach utlenienia; najbardziej zredukowanym - grupę metylową; w stanie pośrednim - grupę metylenową; bardziej utlenionym - grupę formylową, formiminową i metenylową. Składa się z 3 grup: podstawowej pterydyny, p-aminobenzoesanu i glutaminianu. Ssaki mogą syntetyzować pierścień pterydyny, ale nie są zdolne połączyć go z dwiema pozostałymi jednostkami i dlatego pobierają tetrahydrofolian z pożywienia lub z mikroorganizmów żyjących w przewodzie pokarmowym. Pochodne tetrahydrofolianu służą jako donory fragmentów jednowęglowych w różnych biosyntezach. Służy jako akceptor fragmentów jednowęglowych w reakcjach rozpadu. Przenosi on aktywowane fragmenty jednowęglowe o kilku poziomach utlenienia.
III.
Podstawowe cechy i rola przemiany materii.
Podstawowe zadania przemiany materii: • dostarczenie substancji konstrukcyjnych; • wytworzenie odpowiedniej ilości energii; • stworzenie odpowiedniego potencjału redukcyjnego; • zabezpieczenie organizmu przed raptownymi zmianami warunków środowiska.
Cechy: • Kompartmentacja - podział na przedziały subkomórkowe przemian metabolicznych w komórce eukariotycznej; • Istnienie wyspecjalizowanych organów metabolicznych w wielokomórkowym organizmie eukariotycznym, (mózg, mięśnie szkieletowe, układ pokarmowy, nerki, tkanka tłuszczowa); • sieć reakcji biochemicznych w komórkach podlega złożonym mechanizmom kontrolnym. Proces metabolizmu musi być stale kontrolowany i regulowany. Warunek ten jest spełniany przez bardzo liczne mechanizmy regulacyjne, funkcjonujące na wszystkich poziomach organizacji biologicznej: regulacja hormonalna i nerwowa, czy też wytwarzanie enzymów pod kontrolą genetyczną. Regulacja aktywności enzymów: -interakcje allosteryczne, modyfikacje kowalencyjne (kaskada amplifikacyjna); utrzymanie odpowiedniego poziomu enzymu; przeciwstawne szlaki metaboliczne prawie zawsze różnią się od siebie częścią reakcji.
Metabolizm jest integralnie związany z żywym organizmem i ustaje dopiero w chwili śmierci. Dla podtrzymania metabolizmu każda komórka musi być układem otwartym, przez który przepływają w sposób ciągły materia i energia.
Rola, przebieg i regulacja cyklu pentozofosfora-nowego.
Szlak pentozofosforanowy wytwarza w cytozolu NADPH i rybozo-5-fosforan. NADPH jest zużywany w biosyntezach redukcyjnych, natomiast rybozo-5-fosforan jest konieczny do syntezy DNA i RNA i koenzymów nukleotydowych. Szlak pentozofosforanowy rozpoczyna się od dehydrogenacji glukozo-6-fosforanu w wyniku której powstaje lakton, hydrolizowany następnie do 6-fosfoglukonianu (oksydacyjna dekarboksylacja tego związku prowadzi do utworzenia rybulozo-5-fosforanu) - akceptorem elektronów jest NADP+. Ostatnim etapem szlaku jest izomeryzacja rybulozo-5-fosforanu (ketozy) katalizowana przez izomerazę pentozofosforanową do rybozo-5-fosforanu (aldozy).
Odwodorowanie glukozo-6-fosforanu, pierwsza reakcja w odgałęzieniu utleniającym szlaku pentozofosforanowego, jest zasadniczo nieodwracalna. W rzeczywistości ta reakcja ogranicza szybkość procesu w warunkach fizjologicznych i stanowi miejsce kontroli szlaku. Najważniejszym czynnikiem regulującym tą reakcję jest poziom NADP+ - akceptora elektronów podczas utleniania glukozo-6-fosforanu do 6-fosfoglukono-δ-laktonu. NADPH współzawodniczy z też z NADP+ w wiązaniu enzymu. Wyraźny wpływ poziomu NADP+ na przebieg utleniającego odgałęzienia szlaku pentozowego dowodzi, że tworzenie się NADPH jest ściśle powiązane z jego wykorzystaniem w redukcyjnych biosyntezach. Nieutleniające odgałęzienie szlaku pentozowego jest regulowane głównie przez dostępność substratów.
O odpływie glukozo-6-fosforanu decyduje zapotrzebowanie na NADPH, rybozo-5-fosforan i ATP.
Jeśli organizm potrzebuje znacznie więcej rybozo-5-fosforanu niż NADPH, czynne jest tylko nieutleniające odgałęzienie szlaku. W tych warunkach fruktozo-6-fosforan i aldehyd 3-fosfoglicerynowy, utworzone w procesie glikolizy, ulegają przekształceniu w rybozo-5-fosforan bez jednoczesnej produkcji NADPH. Natomiast rybozo-5-fosforan utworzony w utleniającym odgałęzieniu szlaku może być przekształcany do pirogronianu poprzez fruktozo-6-fosforan i aldehyd 3-fosfoglicerynowy. W tych warunkach tworzy się ATP i NADPH, a 5 z 6 węgli glukozo-6-fosforanu pojawia się w pirogronianie. Wzajemna oddziaływania szlaku glikolitycznego i pentozofosforanowego umożliwiają dostosowanie stężenia NADPH, ATP i związków budulcowych, takich jak rybozo-5-fosforan i pirogronian do stale zmieniających się potrzeb komórki. Reakcje szlaku pentozofosforanowego przebiegają znacznie mniej intensywnie w mięśniu szkieletowym, niż w tkance tłuszczowej.
Narysuj dowolny aminokwas. Przedstaw jego stałą dysocjacji i co z niej wynika?
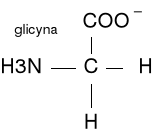
Dla glicyny pK grupy karboksylowej wynosi 2,3, a grupy aminowej 9,6. Punkt środkowy pierwszej jonizacji znajduje się przy pH=2,3, a drugiej, przy pH=9,6. Łańcuchem bocznym w glicynie jest tylko atom wodoru. Wartości pK zależą od temp, siły jonowej i mikrośrodowiska grup ulegających jonizacji.
Synteza „de novo” pierścienia purynowego i pirymidynowego.
Ważnym etapem w syntezie nukleotydów purynowych de novo jest powstanie 5-fosforybozyloaminy z PRPP i glutaminy. Grupa amidowa bocznego łańcucha glutaminy wchodzi na miejsce grupy pirofosforanowej, związanej z C-1 PRPP, z odwróceniem konfiguracji α do β, charakterystyczną dla normalnie występujących nukleotydów. Z fosforybozyloaminą łączy się glicyna, dając rybonukleotyd glicynoamidu. Wiązanie amidowe między grupą karboksylową glicyny i grupą aminową fosforybozyloaminy powstaje kosztem ATP. Pochodne folianu, jako aktywowane związki pośrednie, biorą udział w kilku etapach biosyntezy nukleotydów. Rybonukleotyd formyloglicynoamidyny ulega cyklizacji i powstaje rybonukleotyd 5-aminoimidazolu. Ten pośredni związek zawiera już kompletny pięcioczłonowy pierścień szkieletu purynowego.
W syntezie nukleotydów pirymidynowych najpierw powstaje pierścień pirymidyny, który następnie łączy się z rybozofosforanem tworząc nukleotyd pirymidynowy, odwrotnie niż w sekwencji syntezy de novo nukleotydów purynowych. PRPP ponownie jest donorem reszty rybozofosforanowej. Synteza pierścienia pirymidynowego zaczyna się od utworzenia karbamoiloaparaginianu z karbamoilofosforanu i asparaginianu w reakcji katalizowanej przez karbamoilotransferazę asparaginianową.
IV.
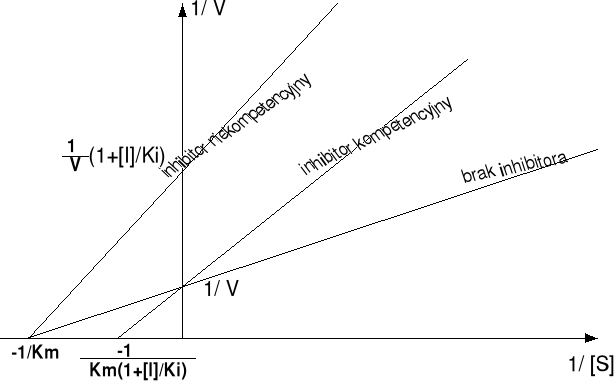
Porównać inhibitory kompetencyjne i niekompetencyjne. Jak zmienia się Vmax i Km?
Ihibicja - unieczynnienie enzymu przez inhibitor. Inhibicja odwracalna:
kompetencyjna: inhibitor jest cząsteczką bardzo zbliżoną strukturalnie do właściwego substratu, która pasuje do centrum aktywnego i wiąże się z enzymem. Nie jest na tyle podobny do substratu, by go w pełni zastąpić. Nie dochodzi więc do reakcji i do wytworzenia produktu. Enzym jest natomiast zablokowany dopóty, dopóki inhibitor pozostaje w miejscu aktywnym.
niekompetencyjna: inhibitor i substrat mogą się równocześnie wiązać z cząsteczką enzymu. Działanie inhibitora niekompetycyjnego polega na zmniejszeniu liczby obrotów enzymu, a nie na zmniejszeniu liczby cząstek enzymu. na skutek tego czas reakcji, w której następuje przemiana substratu, jak i szybkość wytworzenia produktu końcowego, znacznie się wydłuża.
Równanie Michaelisa Mentena w obecności inhibitora kompetycyjnego:
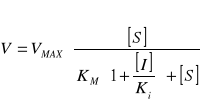
Ki - stała dysocjacji kompleksu I
Wykres Lineweavera-Burka:
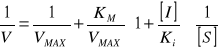
bez inhibitora: tgα=KM/VMAX
z inhibitorem
: 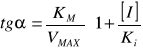
Równanie Michaelisa Mentena w obecności inhibitora niekompetycyjnego:
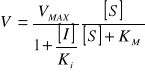
Wykres Lineweavera-Burka
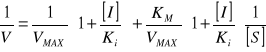
bez inhibitora: tgα=KM/VMAX
z inhibitorem: 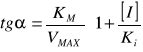
Wpływ inhibitorów na kinetykę reakcji enzymatycznej:
Porównać fotosystem I i fotosystem II.
Chociaż równanie fotosyntezy pozornie jest bardzo proste: 6CO2 + 6H2O—energia świetlna → C6H12O6 + 6O2 to w rzeczywistości proces ten jest niezwykle złożony i wymaga współdziałania 2 fotosystemów. Fotosystem I (fosforylacja cykliczna) wytwarza potencjał redukujący w postaci NADPH, a w pewnych warunkach wytwarza też ATP. Fotosystem II (fosforylacja niecykliczna) rozszczepia wodą, produkuje wolny tlen i dostarcza reduktora.
Centrum reakcji fotosystemu I jest cząsteczka chlorofilu A. Liczne cząsteczki chlorofilu absorbując światło ulegają wzbudzeniu a energię wywołującą ten stan przenoszą na centrum P700 (tzw. chlorofil a bo max absorbcji światła wynosi 700nm). P700 przenosi elektron na akceptor zwany w skrócie FRS, jego zredukowana forma przenosi elektron na ferredoksynę, która ulega redukcji i przekazuje elektron na NADP tworząc NADPH + H+. Ferredoksyna po przekazaniu elektronu na następny akceptor ulega utlenieniu. Elektrony pochodzące z centrum reakcji fotosystemu I mogą się przemieszczać innym torem → w wyniku tego tworzy się cząsteczka ATP. Wtedy elektron z FRS lub z ferredoksyny jest przeniesiony do cytochromu bf zamiast NADP+. Elektron ten powraca do formy utlenionej P700. Podczas tego powrotu wytwarzana jest energia, która zużyta jest na wytworzenie ATP. Warunkiem funkcjonowania fosforylacji cyklicznej jest mała ilość NADP+.
Fotosystem II funkcjonuje na zasadzie współdziałania chlorofilu a i chlorofilu b. Wybite elektrony z chlorofilu a są przenoszone na ferredoksynę, a następnie na NADP+. Związek ten reaguje z protonami, które powstają na skutek fotolizy wody i daje postać zredukowaną na NADPH+H+. Równocześnie chlorofil b pod wpływem światła ulega wzbudzeniu, a oderwane elektrony są przekazywane na chlorofil a i powodują jego powrót do stanu wyjściowego. Po przekazaniu elektronów na chlorofil z cytochromu na chlorofil a, następuje fosforylacja i powstaje ATP. Wzbudzony chlorofil b staje się silnym utleniaczem i przechwytuje elektrony od powstałych w wyniku fotolizy wody jonów OH- stanu wyjściowego. Wytworzone rodniki OH łączą się ze sobą dając cząsteczkę wody i tlen.
Model chemio-osmotyczny w oddychaniu tlenowym.
Zasadniczą rolę w oddychaniu tlenowym pełni łańcuch oddechowy z jego stopniami oksydoredukcyjnymi. Znaczenie łańcucha oddech. polega na tym, że na poszczególnych jego etapach część energii swobodnej zostaje zachowana pod postacią energii chemicznej zmagazynowanej w ATP (oksydacyjna fosforylacja). Mechanizm chemiczny sprzęgający reakcje oksydoredukcji w łańcuchu oddechowym, z powstaniem wysokoenzymatycznych wiązań jest opisany teorią chemioosmotyczną. Zgodnie z modelem chemio-osmotycznym łańcuch transportu elektronów (zlokalizowany w wewnętrznej błonie mitochondriów) działa jak pompa protonowa. W trzech miejscach łańcucha oddechowego energia uwolniona podczas transportu elektronów jest wykorzystywana do przenoszenia protonów z mitochondriów do przestrzeni międzybłonowej. Mogą one przenikać przez specjalne kanały w syntezie ATP.
Reakcje enzymatyczne w biosyntezie pirymidyn.
Biosynteza pirymidyny zaczyna się od utworzenia karbamoilofosforanu, który powstaje z udziałem różnych syntetaz karbamoilofosforanowych. Następny etap (utworzenie N-karbamoiloasparaginianu z asparaginianu i karbamoilofosforanu) katalizuje karbamoilotransferaza asparaginianowa - enzym regulujący. Orotan (powstał przez odwodorowanie dihydroortanu) reaguje z fosforybozylopirofosforanem (PRPP) i powstaje ortodylan (nukleoid pirymidynowy). Reakcję ta katalizuje fosforyozylotransferaza orotanowa. Udział w ostatnim z procesów ma też dekarboksylaza orotydylanowa. U organizmów wyższych biosynteza pirymidyn jest katalizowana przez enzymy wielofunkcyjne.
Odkryto, że syntetaza karbamoilofosforanowa, karbamoilotransferaza asparaginianowa i dihydroorotaza są kowalencyjnie połączone w pojedynczym łańcuchu polipeptydowym. Kowalencyjnie połączony kompleks wielofunkcyjny jest przypuszczalnie bardziej stabilny niż utworzony przez oddziaływania niekowalencyjne.
V.
Rola NAD+ i NADP+ w metabolizmie.
Wodory i elektrony odłączone od substancji przez odpowiednie dehydrogenazy w cyklu Krebsa są przekazywane na główne akceptory elektronów, do których należy dwunukleotyd nikotynamidoadeninowy NAD lub fosforan tego nukleotydu NADP. Większość dehydrogenaz przekazująca elektrony na te właśnie przenośniki jest ściśle swoista i współdziała z NAD lub NADP. Niektóre dehydrogenazy mogą przekazywać wodór i elektrony na oba nukleotydy, jednak szybsze działanie wykazują zawsze we współdziałaniu tylko z jednym z nich. Po przyjęciu H i elektronów od dehydrogenaz wymienione nukleotydy redukują się do NADH lub NADH+H według reakcji: NAD++H++2e-↔ NADH; NADP+←+2H -2H→ NADPH + H. NAD+ jest koenzymem oksydoreduktaz. Pełni rolę przenośnika elektronów. Rola NADP polega na podobnej reakcji odwracalnej, przenoszenia wodoru jak w przypadku NAD.
Wiązania chemiczne stabilizujące strukturę białek. Różnica między wiązaniami.
Grupa peptydowa jest sztywnym i płaskim elementem strukturalnym. Usztywnienie wiązania peptydowego umożliwia białkom tworzenie ściśle zdefiniowanych struktur przestrzennych.Wiązanie peptydowe Jest wiązaniem kowalencyjnym. Łączy poszczególne aminokwasy w cząsteczce białka. Wiązania peptydowe tworzą się w wyniku reakcji grupy karboksylowej jednego aminokwasu z grupą aminową drugiego. W budowie białek wyróżnia się cztery zasadnicze poziomy organizacji łańcuch polipeptydowego.
I-rzędowa. Kolejność aminokwasów w łańcuchu polipeptyd jest określana jako pierwszorzędowa struktura cząsteczki i jest utrwalona wyłącznie wiązaniami peptydowymi. Taki łańcuch może być w różnorodny sposób usytuowany w przestrzeni, co stanowi wtórną strukturę białka.
II-rzędowa. Sposób i stopień zwinięcia łańcucha polipeptyd w formie śruby prawej (α-heliks) lub lewej (β-heliks). Struktura jest ustabilizowana wiązaniami wodorowymi, które tworzą się w wyniku powinowactwa atomów H do takich elektronów ujemnych atomów jak azot, czy tlen, dzielących swe elektrony z wodorem. Stuktura α-heliks powstaje dzięki oddziaływaniom słabych wiązań (wodorowe, jonowe) między resztami aminokwasów, wchodzących w skład tego samego łańcuch polipeptydowego. Wiązanie wodorowe w spirali α powstaje między grupami CO i grupami NH, położonymi w sąsiednich pętlach spirali.
III-rzędowa. przedstawia wyższy poziom organizacji cząsteczek tych związków. Określa trójwymiarową pofałdowaną strukturę łańcucha polipeptydowego. Struktura ta jest zasadniczo stabilizowana dzięki oddziaływaniom między bocznymi łańcuchami aminokwasów. Ważną rolę wśród nich odgrywają mostki dwusiarczkowe o charakterze wiązań kowalencyjnych. Powstaję one między dwoma resztami cysteiny. Mostki wodorowe stabilizują również strukturą III rzędową. Tworzą się między grupami C=O i H=N. Trwałość struktury zapewniają też wiązania elektrostatyczne (jonowe).
IV-rzędowa. określa stopień asocjacji lub polimeryzacji poszczególnych cząsteczek białkowych lub łańcuchów polipeptydowych w większe agregaty. Ta struktura jest utrwalana przede wszystkim przez wiązania dwusiarczkowe, a także przez kleszczowe, tworzące się z udziałem reszt fenolowych, aminowych i karboksylowych i za pośrednictwem jonów metali oraz siłami Van der Waalsa.
Generalnie białka są stabilizowane przez liczne wzmacniające wiązania wodorowe, oddziaływania van der Waalsa, a także oddziaływania hydrofobowe
.
Synteza i rozkład glikogenu-przykład kaskady AMP
Metabolizm glikogenu w znacznym stopniu podlega wpływowi kilku hormonów. Insulina indukuje syntezę glikogenu. Glukagon i adrenalina wyzwalają rozkład glikogenu. Adrenalina silnie stymuluje rozpad glikogenu w mięśniach i w mniejszym stopniu w wątrobie.
Adrenalina i glukagon wiążą się do receptorów w błonie komórkowej komórek docelowych i wyzwalają aktywację białka pobudzającego, której podjednostka połączona z GTP aktywuje cyklazę adenylanową, enzym transbłonowy, który katalizuje przemianę ATP w cykliczny AMP. Wzrost poziomu cyklicznego AMP (cAMP) powoduje aktywację kinazy białkowej, która to z kolei katalizuje fosforylację zarówno kinazy fosforylazowej, jak i syntazy glikogenowej. Fosforylacja, prowadzona przez kinazę białek zależną od cAMP włącza fosforylazę i równocześnie wyłącza syntazę glikogenową. Syntaza glikogenowa jest także fosforylowana przez kinazę fosforylazową, czego dalszą konsekwencją jest brak syntezy glikogenu w czasie, gdy jest on rozkładany przez fosforylazę. W efekcie kinaza białkowa kontroluje proces syntezy i rozkładu glikogenu. Kaskada cyklicznego AMP silnie zwielokrotnia działanie hormonów.
Glikogen to polimer reszt glukozy połączonych wiązaniami α-1,4-gikozydowymi, od którego odchodzą odgałęzienia pojawiające się co 10 reszt.
ROZKŁAD GLIKOGENU (glikogeneza)
glikogen (n reszt) tPi ↔glikogen (n-1 reszt) + glukoza - 1 - fosforan
•fosforylaza glikozydowa - rozbija wiązania α-1,4-glikozydowe i usuwa kolejno reszty glukozy z nieredukującego końca (koniec z wolną grupą n-OH cząst. glikogenu)
•enzym usuwający rozgałęzienia - usuwa wiązanie α -1,6-glikozydowe w miejscach rozgałęzienia
glukozo-1-fosforan←fosfoglukomutaza→glukozo-6-fosforan - sam metabolizowany w szlaku glikolizy w mięśniach (energia)
+ H2O + glukozo-6-fosfataza→glukoza + Pi: w wątrobie →glukoza dyfunduje do krwi, utrzymuje się w możliwie stałym stężeniu.
Synteza glikogenu (GLIKOGENOGENAZA)
UTP+glukozo-1-fosforan ↔ UDP-glukoza + PPi
PPi + H2O→2Pi + energia.
UTP = urydyno-trifosforan.
Syntaza glikogenowa przenosi reszty glikozylowe z UDP-glukozy do gup C4OH przy końcu nieredukującym cząsteczki glikogenu tworząc wiązania α-1,4-glikozydowe.
Gdy pewna liczba cząstek glukozy zostanie już połączona w prosty łańcuch, enzym rozgałęziający rozcina jedno wiązania α-1,4-glikozydowe i przenosi odcinek (ok. 7 reszt) do bardziej wewnętrznego miejsca cząsteczki i go przyłącza przez wiązania α-1,6-glikozydowe
Równoczesna synteza i degradacja glikogenu powoduje netto hydrolizę UTP.
Rozkład puryn i pirymidyn.
U człowieka puryny są rozkładane do moczanu i wydalany z moczem.
VI.
Glukoneogeneza (rola, przebieg, własności). Czy jest to proces odwrotny do glikolizy?
Jest syntezą glukozy z prekursorów nie będących cukrowcami. Odgrywa on ważną rolę w mógu, dla którego glukoza stanowi podstawowy materiał energetyczny. Odgrywa również ważną rolę podczas intensywnego wysiłku organizmu. Podczas glukoneogenezy pirogronian przekształca się w glukozę. Niecukrowcowe prekursory wchodzą na szlak glukoneogenezy głównie jako pirogronian, szczawiooctan i fosforan dihydroksyacetonu. Najważniejszymi prekursorami niecukrowcowymi glukozy są mleczan, aminokwasy i glicerol. Głównym miejscem glukoneogenezy jest wątroba i nerki. Utrzymują one stały poziom we krwi, skąd cukier ten pobierają intensywnie metabolizujące go mięśnie i mózg.
W procesach glikolizy glukoza jest przekształcana w pirogronian, w procesach glukoneogenezy pirogronian przekształca się w glukozą. Niemniej glukoneogeneza nie jest odwróceniem glikolizy. Wymaga to kilku reakcji różnych od procesu glikolizy, ponieważ równowaga termodynamiczna glikolizy sprzyja tworzeniu się pirogronianiu. Ta odrębność dróg jest konieczna, ponieważ glikoliza jest procesem egzoergicznym. Nieodwracalne reakcje zachodzące podczas glikolizy sa w procesie glukoneogenezy omijane przez następujące przemiany: • fosfoenolopirogronian powstaje z pirogronianu przez produkt pośredni - szczawiooctan; • fruktozo-6-fosforan powstaje z fruktozo-1,6-bisfosforanu w wyniku hydrolitycznego odłączenia fosforanu estryfikującego; • glukoza powstaje z glukozo-6-fosforanu w wyniku reakcji hydrolizy wiązania fosfoestrowego
Oba procesy wykazują różnice enzymatyczne.
Rola NAD+ i cytochromów w procesie oddychania.
Cytochromy są białkami transportującymi elektrony w łańcuchu oddechowym, które zawierają hem jako grupę prostetyczną. Rola cytochromów w łańcuchu oddechowym polega na utlenieniu zredukowanych koenzymów flawionowych. Cytochromy są rozpowszechnione we wszystkich żywych tkankach. Pomimo, że katalizują proces przeniesienia elektronów, a więc działają jak oksydoreduktazy, są raczej uważane nie za enzymy, a jedynie za jednostki transportujące elektrony zgodnie z kierunkiem wzrastającego potencjału oksydoredukcyjnego. Heminy współdziałające z cytochromami nie są koenzymami, lecz grupami prostetycznymi.
NAD+ dinukleotyd nikotynoamidoadeninowy jest głównym akceptorem elektronów w reakcjach utleniania substratów oddechowych. Reaktywną część NAD+ stanowi jego pierścień nikotynoamidowy, pochodna pirydynowa. W reakcji utleniania substratu nikotynoamidowy pierścień NAD+ przyjmuje jon wodorowy i dwa elektrony, które są ekwiwalentem jonu hydroniowego. Zredukowana forma tego przenośnika - NADH. W formie utlenionej atom azotu tworzy cztery wiązania kowalencyjne i ma dodatni ładunek - NAD+.W formie zredukowanej NADH, atom azotu tworzy trzy wiązania kowalencyjne.
Cykl mocznikowy w metabolizmie i procesie syntezy aminokwasów.
Część grup NH4+ powstających przez rozkład aminokwasów jest używana w biosyntezie związków azotowych. U większości kręgowców nadmiar NH4+ jest przekształcany w mocznik i wydalany. Ptaki i gady przed wydaleniem NH4+ przekształcają go w kwas moczowy, a niektóre zwierzęta wydalają go bezpośrednio. Cykl mocznikowy jest tzw. cyklicznym torem metabolicznym. W moczniku 1 atom azotu pochodzi z NH4+ a drugi z asparaginianu. Atom węgla w moczniku pochodzi z CO2. Nośnikiem atomów węgla i azotu wchodzących w cykl mocznikowy jest ornityna - aminokwas, który nie stanowi jednostki budulcowej białek.
Bezpośrednim prekursorem mocznika jest arginina, która z udziałem arginazy ulega hydrolizie do mocznika i ornityny.
Pozostałe reakcje cyklu mocznikowego prowadzą do syntezy argininy z ornityny. Najpierw na ornitynę zostaje przeniesiona grupa karbamoilowa, co prowadzi do otrzymania cytruliny. Reakcję tą katalizuje karbamoilofosforan, który dzięki wiązaniu bezwodnikowemu charakteryzuje się dużym potencjałem przenoszenia. W następnym etapie syntetaza argininobursztynianowa katalizuje kondensację cytuliny z asparaginianem do argininobursztynianu. Reakcja ta przebiega kosztem energii ATP, który ulega przy tym hydrolizie do AMP i pirofosforanu, oraz dzięki następnej hydrolizie pirofosforanu.
W końcu liaza argininobursztynianowa rozszczepia argininobursztynian na argininę i fumuran. Należy zauważyć, że szkielet węglowy asparaginianu nie zostaje podczas tej reakcji naruszony, natomiast grupa aminowa zostaje przeniesiona na powstającą argininę. Karbamoilofosforan niezbędny do utworzenia cytruliny jest syntetyzowany z NH4+, CO2, ATP i H2O w skomplikowanej reakcji. Istotne znaczenie w cyklu mocznikowym ma synteza fumuranu, przez którą cykl ten zazębia się z cyklem kwasu cytrynowego.
Wiązania kowalencyjne i jonowe. Porównać.
Atomy mogą uzyskać stabilniejszy układ elektronów w zewnętrznej powłoce elektronowej przez oddziaływanie z innymi atomami. Gdy elektrony są przekazywane z jednego atomu na drugi, tworzy się wiązanie jonowe. Wiązanie kowalencyjne powstaje, jeśli elektrony są wspólnie użytkowane przez dwa atomy. Często tworzą się wiązania kowalencyjne, w których elektrony są częściowo przesunięte w kierunku jednego z atomów (nierówny udział w użytkowaniu elektronów), tworzy się wówczas wiązanie kowalencyjne spolaryzowane. Atomy połączone dwoma lub więcej wiązaniami kowalencyjnymi nie mogą się swobodnie obracać wokół osi wiązania.
VII.
W jaki sposób szlak C-4 wpływa na cykl Calvina pewnych roślin?
Szlak C4 jest dodatkowym mechanizmem wprowadzania CO2 do cyklu Calvina, który jest ostatnim etapem fotosyntezy zachodzącym bez udziału światła. Pobrany ze środowiska zewnętrznego CO2 podlega przekształceniu do poziomu heksozy (glukoza, fruktoza).
U roślin tropikalnych obserwuje się duże miejscowe stężenie CO2 w komórkach, gdzie zachodzi cykl Calvina. CO2 jest przenoszony w postaci związków czterowęglowych z komórek mezofilu, które kontaktują się z atmosferą zewnętrzną do komórek pochwy okołowiązkowej, gdzie są umiejscowione główne reakcje fotosyntezy. Dekarboksylacja związku C4 w komórkach pochwy okołowiązkowej pozwala na utrzymanie dużego stężenie CO2 w miejscu funkcjonalnym cyklu Calvina. Szlak C4 rozpoczyna się w komórkach mezofilu od kondensacji CO2 z fosfoenolopirogronianem z udziałem karboksylazy fosfoenolopirogronianowej. W wyniku czego powstaje szczawiooctan. Uwolniony CO2 kondensuje z rybulozo-1,5-bisfosforanem i ta reakcja rozpoczyna cykl Calvina.
Jeżeli CO2 jest doprowadzony do miejsca funkcjonowania cyklu Calvina z udziałem szlaku C4, to 30 cząsteczek ATP jest niezbędnych do wytworzenia jednej heksozy, natomiast tylko 18 cząsteczek potrzeba, jeżeli nie pośredniczy szlak C4. Dzięki szlakowi C4 w roślinach tropikalnych fotooddychanie jest ograniczone, gdyż duże stężenie CO2 w komórkach pochwy okołowiązkowej przyspiesza reakcję karboksylazową w stosunku do reakcji oksygenazowej. Zjawisko to szczególnie ważne w warunkach wysokiej temp.
Transport elektronów w warunkach beztlenowych.
Jakie produkty metaboliczne tłuszczy lipidów wpływają na metabolizm glikolizy?
Tłuszcze stanowią wyjątkowo bogatą w energię i mogącą ulec w miarę potrzeby szybkiemu uruchomieniu rezerwę odkładaną w tkankach żywych organizmów.
Bilans energetyczny i kontrola oddechowa glikolizy.
Reakcja sumaryczna przekształcania glukozy w pirogronian: glukoza + 2Pi + 2 ADP + 2 NAD+ → 2 cząsteczki pirogronianu + 2 ATP + 2 NADH + 2H+ + 2 H2O. W ten sposób podczas tego przekształcenia w dwie cząsteczki pirogronianu powstają dwie cząsteczki ATP. Porównując z wydajnością utleniania cukru w cyklu pentozofosforanów, glikoliza łącznie z dalszym utlenieniem pirogronianu do CO2 i H2O jest procesem bardziej wydajnym energetycznie.
Najważniejszym miejscem kontroli glikolizy jest fosfofruktokinaza, enzym katalizujący kluczowy etap tego procesu. Duże stężenie ATP hamuje działalność fosfofruktokinazy - enzymu, którego miejsca allosteryczne są różne od miejsc substratowych mających mniejsze powinowactwo do nukleotydu. Szybkość glikolizy zależy od zapotrzebowania na ATP, czego sygnałem jest stosunek ATP/ADP, a także od zapotrzebowania na materiał budulcowy, co sygnalizuje poziom cytrynianu.
VIII.
Różnice i podobieństwa w rozpadzie nasyconych i nienasyconych kwasów tłuszczowych.
Reakcje zachodzące podczas rozkładu kwasów tłuszczowych przebiegają w kolejności: utlenienie, uwodnienie, utlenienie, tioliza. W rezultacie każdego cyklu tych reakcji łańcuch węglowodorowy grupy acylowej jest krótszy o dwa atomy węgla, oraz powstaje FADH2, NADH i acetylo-CoA. W każdym obrocie cyklu degradacji kwasów tłuszczowych pierwszą reakcją jest utleniania acylo-CoA z udziałem dehydrogenazy acylo-CoA do enoilo-CoA. Odwodorowanie acylo-Co jest bardzo podobne do dehydrogenacji bursztynianu w cyklu kwasu cytrynowego. Akceptorem elektronów jest FAD, a nie NAD+. Elektrony z FADH2 - grupy prostetycznej zredukowanej formy dehydrogenazy acylo-CoA - są przekazywane na inną flawoproteinę, która je z kolei przekazuje na reduktazę ubichinonu, białko żelazo-siarkowe. Ubichinon ulega redukcji do ubichinolu, który przekazuje elektrony o wysokim potencjale przenoszenia na drugie miejsca pompujące protony w łańcuchu oddechowym.
Następnym etapem rozkładu kwasów tłuszczowych jest uwodnienie (hydratacja) podwójnego wiązania pomiędzy C-2 i C-3; reakcję katalizuje hydrataza enoilo-CoA. Uwodnienie enoilo-CoA jest wstępem do kolejnej reakcji utleniania, w rezultacie której grupa hydroksylwa przy węglu 3 ulega przekształceniu w grupę ketonową oraz powstaje cząsteczka NADH. Reakcję utleniania katalizuje dehydrogenaza L-3-hydroksyacylo-CoA. Trzy pierwsze reakcje degradacji kwasu tłuszczowego ściśle odpowiadają ostatnim etapom cyklu kwasu cytrynowego. Reakcje te doprowadziły do utlenienia grupy metylenowej przy C-3 do grupy ketonowej. Następny etap polega na rozszczepieniu 3-ketoacylo-CoA z udziałem grupy tiolowej drugiej cząsteczki CoA - na acetylo-CoA i acylo-CoA mający łańcuch o dwa atomy węgla krótszy od wyjściowego kwasu tłuszczowego. Tiolityczne rozszczepienie jest katalizowane przez β-ketotiolazę. Skrócony acylo-CoA wchodzi ponownie w cykl reakcji utleniania.
W procesie utleniania nienasyconych kwasów tłuszczowych wiele reakcji przebiega tak samo. Do rozkładu różnych nienasyconych kwasów tłuszczowych potrzebne są tylko dwa dodatkowe enzymy: izomeraza i reduktaza. Wiązania podwójne przy węglach nieparzystych w kwasach tłuszczowych przekształca izomeraza, a przy parzystych - reduktaza i izomeraza.
Kwasy tłuszczowe mające nieparzystą liczbę atomów węgla występują w przyrodzie rzadziej, niż z parzystą liczbą. Są one utleniane w ten sam sposób, jak parzystowęglowe, z tą różnicą, że w końcowej reakcji ostatniego cyklu tworzy się jedna cząsteczka acetylo-CoA i jedna propionylo-CoA, nie zaś dwie cząsteczki acetylo-CoA.
α-helisa i harmonijka β. Różnice i podobieństwa
Helisa α i harmonijka β to dwie regularne struktury łańcucha polipeptydowego.
Helisa α ma kształt cylindra. Ciasno skręcony łańcuch główny polipeptydu tworzy wewnętrzną część cylindra, a łańcuchy boczne aminokwasów występują na zewnątrz w ułożeniu helikalnym. Helisę α stabilizują wiązania wodorowe między grupami NH i CO głównego łańcucha. Grupa CO aminokwasu wiąże się wzajemnie wiązaniem wodorowym z grupą NH aminokwasu zajmującego w sekwencji liniowej pozycję wysuniętą do przodu o 4 reszty aminokwasowe. Helisa występująca w białkach jest prawoskrętna. Zawartość struktury α jest bardzo różna i waha się od prawie zerowej do niemal 100%.
Struktura harmonijki β różni się bardzo wyraźnie od cylindrycznej helisy α. W strukturze harmonijki β łańcuch polipeptydowy (nić β) jest prawie całkowicie rozciągnięty w odróżnieniu od ciasno upakowanego w helisie α. Odległość sąsiednich aminokwasów wzdłuż osi długiej cząsteczki wynosi 0,35 nm, podczas gdy w helisie α - 0,15nm. Różnica dotyczy także wiązań wodorowych stabilizujących strukturę przestrzenną. Harmonijkę β stabilizują wiązania wodorowe pomiędzy grupami CO i NH należącymi do odrębnych łańcuchów polipeptydowych, natomiast w helisie α wiążą się wodorowo grupy CO i NH aminokwasów należących do tego samego łańcucha polipeptydowego. Sąsiadujące ze sobą łańcuchy armonijki mogą być ułożone w jednym kierunku lub w przeciwnych.
Rola dUMP (metylacji deoksyurydylanu).
Uracyl nie jest składnikiem DNA. Natomiast DNA zawiera tyminę, metylowany analog uracylu. Deoksyurydylan (dUMP) jest metylowany do deoksytymidalanu (dTMP) w reakcji katalizowanej przez syntazę tymidylanową. Donorem grupy metylowej w tej reakcji jest pochodna tetrahydrofiolianu. Grupa metylowa wprowadzona jest do deoksyurydylanu jest bardziej zredukowana niż grupa metylenowa. W tej redukcji dwa elektrony w formie jonu wodorkowego pochodzą z samego tetrahydrofolianu. Wodór ten staje się częścią grupy metylowej w dTMP. Tetrahydrofolian zostaje utleniony do dihydrofolianu. Pochodne tetrahydrofolianu odgrywają kluczową rolę. Metabolizm nukleotydów oraz metabolizm aminokwasów są ściśle powiązane przez przenośniki fragmentów jednowęglowych.
IX.
W jaki sposób produkty rozkładu białek włączane są do szlaku energ. utleniania glukozy?
Rola FAD i FMN. Jakie ma znaczenie w metabolizmie komórki?
Są to nukleotydy flawinowe. Są pierwszymi akceptorami wodoru w łańcuchu oddechowym.
Dinukleotyd flawinoadeninowy FAD jest syntezowany z ryboflawiny i dwóch cząsteczek ATP. Ryboflawina ulega fosforylacji przez ATP do mononukleotydu flawinowego FMN. Reaktywną częścią FAD jest jego pierścień izoalaksazynowy. FAD, podobnie jak NAD+ może przyjmować dwa elektrony. Czyniąc to FAD, w przeciwieństwie do NAD+, wiąże proton tak samo, jak jon hydroniowy. FMN może przyjmować jeden elektron (albo FMNH2 może oddawać jeden elektron), tworząc pośrednią formę rodnika semichinonowego. Ryboflawina jest składnikiem FMN i FAD. Koenzymy flawinowe współdziałają z enzymami przenoszącymi elektrony i protony ze zredukowanego NAD+, czyli z reduktazami, lub w niektórych przypadkach bezpośrednio z substratu, czyli dehydrogenazami. Przyjęte przez układ flawinowy atomy wodoru są w dalszych reakcjach przekazywane na kolejne przenośniki tzw. łańcucha oddechowego i za ich pośrednictwem na tlen. Są też enzymy flawinowe, które odwodorowują substrat bezpośrednio, przekazując atomy wodoru na dalsze przenośniki, bądź bezpośrednio na tlen (oksydazy aminokwasowe, aminowe...). W przypadku bezpośredniego przeniesienia atomów wodoru z flawiny na tlen, produktem końcowym jest nadtlenek wodoru H2O2. Nukleotydy flawinowe są względnie mocno połączone z białkami enzymów i nie oddzielają się w czasie oczyszczania enzymów.
Synteza kwasów tłuszczowych.
Synteza rozpoczyna się od karboksylacji acetylo-CoA prowadzącej do malonylo-CoA. W reakcji tej katalizowanej przez karboksylazę acetylo-CoA zawierającą biotynę jest zużywany ATP. Intermediaty syntezy kwasów tłuszczowych są związane z białkowym nośnikiem grup acetylowych (ACP) przez kowalencyjne wiązanie z siarką jego fosfopantoteinowej grupy prostetycznej. Acetylo-ACP i malonylo-ACP kondensują tworząc acetoacetylo-ACP; reakcja ta jest napędzana przez uwalnianie CO2 z aktywowanej jednostki malonylowej. Po tym etapie następuje redukcja, dehydratacja i druga redukcja; reduktorem tutaj jest NADPH. Utworzony w ten sposób butyrylo-ACP wchodzi w następny cykl elongacji, rozpoczynający się od dołączenia jednostki dwuwęglowej, pochodzącej z cząsteczki malonylo-CoA. Siedem cykli elongacji prowadzi do palmitoilo-ACP; jego hydroliza daje palmitynian.
Biosynteza kwasów tłuszczowych nie jest odwróceniem kierunku reakcji prowadzących do rozkładu tych związków. Cechy biosyntezy kwasów tłuszczowych: • synteza ma miejsce w cytozolu, w przeciwieństwie do rozkładu kwasów tłuszczowych przebiegającego w matriks mitochondrialnej; • związki pośrednie biosyntezy kw. tłuszczowych są kowalencyjnie związane z grupą hydrosulfidową białkowego nośnika grup acetylowych, podczas, gdy produkty pośrednie rozkładu kw. tłuszczowych wiążą się z koenzymem A. • enzymy uczestniczące u wyższych organizmów w biosyntezie kwasów tłuszczowych są połączone w jeden łańcuch polipeptydowy, zwany syntazą kwasów tłuszczowych. • rosnący łańcuch kwasu tłuszczowego ulega elongacji przez kolejne dobudowywanie jednostek dwuwęglowych pochodzących z acetylo-CoA. Reakcja elongacji przebiega kosztem energii powstającej przez uwolnienie CO2. • w syntezie kw. tłuszcz związkiem redukującym jest NADPH; • elongacja katalizowana jest przez kompleks syntazy kwasów tłuszczowych zatrzymuje się po zsyntetyzowaniu łańcucha 16-o węglowego. Do dalszej elongacji i syntezy podwójnych wiązań potrzeba udziału innych układów enzymatycznych.
IMP.
Inozynian, produkt syntezy de novo jest prekursorem AMP i GMP. AMP (adenylan) powstaje z inozynianu przez wprowadzenie grupy aminowej przy C-6 zamiast tlenu karbonylowego. Donorem grupy aminowej jest asparaginian. Reakcja przebiega przez przyłączenie tego aminokwasu i następnie usunięcie fumuranu. Synteza adenylobursztynianu z inozynianu i asparaginianu zachodzi kosztem wysokoenergetycznego wiązania fosforanowego GTP.
GMP guanylan jest syntetyzowany przez utlenianie inozynianu i następnie wprowadzenie grupy aminowej przy C-2. W utlenianiu inozynianu do ksantylanu akceptorem wodoru jest NAD+. Grupa aminowa, wprowadzona do ksantyny, pochodzi z bocznego łańcucha glutaminy (z grupy amidowej). W przemianach inozynianu do adenylanu i guanylanu karbonylowy atom tlenu zostaje zamieniony na grupę aminową.
IMP jest inhibitorem (obok AMP i GMP) biosyntezy nukleotydów purynowych przez sprzężenie zwrotne. Inozynian jest miejscem rozgałęzienia w syntezie AMP i GMP. Reakcje prowadzące dalej z inozynianu są miejscami inhibicji przez sprzężenie zwrotne. AMP hamuje przemianę inozynianu w adenylobursztynian, swój bezpośredni prekursor. Podobnie GMP jest inhibitorem przejścia inozynianu w bezpośredni prekursor - ksantylan.
X.
Model chemioosmotyczny w fotosyntezie.
Synteza cysteiny.
Cysteina jest aminokwasem endogennym, który jest potrzebny człowiekowi do biosyntezy białka - powstaje w wyniku biosyntezy wewnątrzkomórkowej. Jest syntezowana z seryny, która jest syntezowana z kolei z metabolitu, który występuje w czasie glikolizy (3-fosfoglicerynian). Związkiem pośrednim w syntezie cysteiny jest homocysteina. Seryna podlega kondensacji z homocysteiną, dając cystationinę. Następnie cystationina ulega dezaminacji i rozpada się, przy udziale enzymu cystationazy, na cysteinę i α-ketomaślan.
homocysteina+seryna→H2O↑→cystationina→H2O↓→cysteina + α-ketomaślan
Synteza NAD, FAD, koenzymów kwasów nukleinowych.
Synteza NAD (dinukleotydu nikotynoamidoadeninowego) zaczyna się od utworzenia rybonukleotydu nikotynowego z nikotynianu i PRPP. Potem z ATP zostaje przeniesiona reszta ANP na rybonukleotyd nikotynianu i tworzy się deamido-NAD+. Końcowym etapem jest przeniesienie grupy amidowej glutaminy na grupę karboksylową nikotynianu i utworzenie NAD+.
FAD jest syntetyzowany z ryboflawiny i dwóch cząsteczek ATP. ryboflawina + ATP -> ryboflawino-5-fosforan + ADP, ryboflawino-5-fosforan + ATP ↔ dwunukleotyd flawinoadeninowy + PPi.
Synteza koenzymu A zaczyna się fosforylacją pantotenianu za 4-fosfopantotenian, który przyłącza grupę aminową z cysteiny i po odszczepieniu CO2 powstaje 4-fosfopantoteina. Potem zostaje przniesiona z ATP reszta ANP tworząc defosfokoenzym A. Następuje fosforylacja tego związku kosztem ATP, który przechodzi w ADP, co prowadzi do powstania koenzymu A.
Synteza DNA zwana jest replikacją (doprowadza do podwojenia liczby cząsteczek tego związku). Proces ten rozpoczyna się od enzymatycznego rozplatania obu nici (enzym helikaza) w czym biorą udział także białka. Pojedyncze łańcuchy DNA stają się wzorcem dla syntezy nowych łańcuchów. Kolejność zasad w nici wyznacza miejsce nukleotydu na zasadzie komplementarności (łaczenie się nukleotydów w pary). Reakcje łączenia nukleotydów w łańcuch polinukleotydowy katalizują enzymy zwane polimerazami DNA. Synteza DNA rozpoczyna się w miejscu incjacji replikacji (występuje tam specyficzna sekwencja zasad) przyłącza się tam krótki łańcuch tzw. starterowego DNA. Przy udziale polimerazy DNA nast. ępuje dobudowanie nukleotydów i wydłużenie (elongacja łańcucha polinukleotydowego). Po zakończonej replikacji obie nici macierzysta i nowa tworzą dwuniciową strukturę. W ten sposób każda powstająca cząsteczka DNA składa się z jednej nici otrzymanej z cząsteczki macierzystej oraz drugiej zsyntetyzowanej w czasie replikacji.
Synteza RNA zwana jest transkrypcją [bo informacja genetyczna zawarta w sekwencji nukleotydów w DNA jest przekazana na sekwencję nukletydów w RNA]. W tym procesie informacja zostaje powielona i przekazana z jednego kwasu, będącego materiałem genetycznym DNA na drugi kwas RNA. Część RNA, która w czasie transkrypcji przejmuje informacje o strukturze pierwszerzędowej białka zwana jest matrycowym (informacyjnym) m-RNA. Transkrypcja rozpoczyna się od rozpoznania przez polimerazę RNA specyficznej sekwencji zasad w DNA tzw. promotorem. Synteza RNA nie wymaga startera, Zapoczątkowanie transkrypcji przebiega na zasadzie komplmentarności zasad pomiędzy matrycą a nowo syntetyzowaną nicią RNA. mRNA ma jeszcze dodatkowe sekwencje (oprócz tych kodujących białko) np. liderową - są w niej miejsca rozpoznawcze dla rybosomu, które umożliwiają prawidłowe połączenie rybosomu z mRNA. Za sekwencją liderową jest sekwencja kodująca, a za nią miejsce terminacji, czyli zakończenia syntezy mRNA. Nowe cząsteczki mRNA opuszczają jądro i kierują się do rybosomów - miejsc syntezy białek.
Transportujący RNA (tRNA) dokonuje przemieszczenia wolnych aminokwasów do rybosomów. tRNA jest frakcją krótkołańcuchowych cząsteczek o charakterystycznym kształcie. środkowa pętla, w skład której wchodzą 3 zasady stanowi antykodon, którym tRNA rozpoznaje miejsce na mRNA zlokalizowanym na rybosomie.
Fermentacja mleczanowa i alkoholowe - porównanie.
Organizmami zdolnymi do fermentacji alkoholowej są w pierwszym rzędzie drożdże oraz niektóre pleśnie. Organizmy te zawierają komplet enzymów łańcucha glikolizy oraz zasadnicze dla fermentacji alkoholowej enzymy: dekarboksylazę pirogronianową i dehydrogenazę alkoholową. Enzymy te katalizują kolejno dekarboksylację pirogronianu do aldehydu octowego i przeniesienie na ten związek atomów wodoru z NADH, powstałego przez uwodorowanie NAD+ przy utlenianiu gliceraldehydo-3-fosforanu. Fermentacja alkoholowa może być modyfikowana w celu innych niż alkohol etylowy produktów końcowych.
Mleczanowa.
W mięśniach zwierząt wykonujących intensywną pracę tlenowy rozkład glikogenu, który dostarcza energii musi być często, z powodu niedostatku tlenu, zastąpiony beztlenowym procesem glikolizy - fermentacją mleczanową. W wyniku tego tworzy się pirogronian, który w warunkach beztlenowych jest akceptorem atomów wodoru z NADH i po ich przyłączeniu przechodzi w kwas mlekowy.
XI.
β-oksydacja palmitynianu. Obliczyć ilość ATP, jaka powstanie w wyniku jego utlenienia?
Oblicza się ilość energii uzyskiwanej w procesie utleniania kwasu tłuszczowego. W każdym cyklu reakcji łańcuch acylo-CoA ulega skróceniu o dwa atomy węgla i powstaje NADH, FADH2 oraz acetylo-CoA.
Cnacylo-CoA+FAD+NAD++H2O+CoA→Cn-2-acyloCoA+FADH2+NADH + acetylo-CoA + H+. Degradacja palmitoilo-CoA wymaga siedmiu reakcji. (C16-acylo-CoA). Podczas siódmego cyklu C4-ketoacylo-CoA ulega tiolizie do dwóch cząsteczek acetylo-CoA. pamitoilo-CoA + 7FAD + 7NAD+ + 7CoA + 7H2O → 8 acetylo-CoA + 7FADH2 + 2 NADH + 7H+. Podczas utleniania każdej cząsteczki NADH w łańcuchu oddechowym powstaje 2,5 cząsteczki ATP, a 1,5 cząsteczki ATP na skutek utleniania każdej cząsteczki FADH2, ponieważ jej elektrony wchodzą do łańcucha oddechowego na poziomie ubichinonu. Utlenienie acetylo-CoA w cyklu kwasu cytrynowego prowadzi do wytworzenia 10 cząsteczek ATP. Podczas całkowitego utlenienia pamitoilo-CoA powstaje zatem cząsteczek ATP: 10,5 z siedmiu FADH2, 17,5 z siedmiu NADH i 80 z ośmiu cząsteczek acetylo-CoA, co w sumie daje 108 cząsteczek ATP. W procesie aktywacji palmitynianu (wiązanie go z koenzymem A_ zużywane są dwa wysokoenergetyczne wiązania fosforanowe. Więc zupełne utlenienie pamitynianu dostarcza netto 106 cząsteczek ATP.
Losy węglowych szkieletów aminokwasów.
Większość intermediatów metabolicznych, które powstają podczas rozkładu aminokwasów, może może być przekształcona do glukozy lub utleniana w cyklu kwasów cytrynowych. W rezultacie część lub wszystkie atomy węgla rozkładanych 20 aminokwasów białkowych można znaleźć w zaledwie 7 rodzajach cząsteczek. Są nimi: pirogronian, acetylo-CoA, α-ketoglutaran, bursztynylo-CoA, fumaran i szczawiooctan. Aminokwasy, których rozkład prowadzi do acetylo-CoA lub acetoacetylo-CoA, określa się jako ketogenne, gdyż powstają z nich związki (ciała) ketonowe. Aminokwasy ulegające rozkładowi do pirogronianu, α-ketoglutaranu, bursztynylo-CoA, fumuranu lub szczawiooctanu nazywa się aminokwasami glukogennymi. Synteza glukozy z tych aminokwasów jest możliwa dlatego, że intermediaty cyklu kwasu cytrynowego i pirogronian mogą być przekształcone w fosfoeneolopirogronian, a ten z kolei w glukozę. Spośród 20 podstawowych aminokwasów białkowych wyłącznie ketogenne są leucyna i lizyna. Izoleucyna, fenyloalanina, tryptofan i tyrozyna są zarówno ketogenne, jak i glukogenne. Niektóre atomy węgla tych aminokwasów pojawiają się w acetylo-CoA lub acetoacetylo-CoA, inne występują w potencjalnych prekursorach glukozy. Pozostałe 14 aminokwasów - typowo glukogenne.
Rozpad i synteza hemu.
4 cząsteczki porfobilinogenu kondensują liniowo i w reakcji katalizowanej przez deaminazę porfobilinogenu powstaje łańcuchowy tetrapirol. Utworzenie każdego mostka metylowego wiąże się z uwolnieniem jonu amonowego. Łańcuchowy tetrapirol cyklizuje do uroporfirynogenu III o asymetrycznym układzie łańcuchów bocznych. Reakcje te wymagają obecności syntazy i kosyntazy. W obecności samej syntazy tworzy się uroporfirynogen I symetryczny izomer niefizjologiczny. Uroporfirynogen III jest również kluczowym związkiem pośrednim w syntezie witaminy B12 przez bakterie oraz chlorofilu przez bakterie i rośliny.
Teraz tworzy się szkielet porfiryny. Dalsze reakcje przekształcają łańcuchy boczne i zmieniają stopień nasycenia pierścienia porfirynowego. Koproporfirynogen III powstaje przez dekarboksylację bocznych łańcuchów octanowych. Utworzenie się układu sprzężonych wiązań podwójnych pierścienia porfirynowego i przejście dwóch bocznych łańcuchów propionowych w grupy winylowe prowadzi do powstania protoporfiryny IX. Chelatowe związanie żelaza powoduje w końcu utworzenie się hemu, grupy prostetycznej takich białek jak: mioglobina, hemoglobina, katalaza, peroksydaza i cytochrom c.
Rozpad. Pierwszy etap rozpadu grupy hemowej do bilirubiny to rozszczepienie jej mostka α-metinowego i utworzenie biliwerdyny, łańcuchowego tetrapirolu. Reakcję te katalizuje oksygenaza hemowa. Do reakcji rozszczepienia konieczne są O2 i NADPH (mostek metinowy uwalniany jest w postaci tlenku węgla). Centralny mostek metinowy biliwerdyny jest redukowany przez reduktazę biliwerdynową i powstaje bilirubina. Reduktorem jest tu NADPH.
ATP (uzyskiwanie energii) związek między egzo- i endogenicznymi reakcjami.
ATP - adenozynotrójfosforan - jest przenośnikiem energii swobodnej (pełni rolę w wymianie, transporcie, magazynowaniu energii). Jest on nukleotydem zbudowanym z adeniny, rybozy i trójfosforanu. Jest cząsteczką bogatą w energię, gdyż jego składowa trójfosforanowa zawiera 2 bezwodnikowe wiązania fosforanowe. Podczas hydrolizy ATP uwalniana jest duża ilość energii swobodnej i powstaje ADP - adenozynodwufosforan i ortofosforan. ATP + H2O → ADP + ortofosforan. W wyniki tej egzoergicznej reakcji uwalniane jest około 7,3 kcal energii z 1 mola ATP. Energia uwalniana podczas hydrolizy ATP jest sprzężona z procesami endoergicznymi w komórce, z udziałem pewnych enzymów. Są to reakcje syntezy składników komórkowych. Wraz z przemianami energetycznymi następuje też przepływ grup fosforanowych z ATP na inne związki - fosforylacja. Przemiany energetyczne przebiegają odwrotnie, gdy komórka przeprowadza np. utlenianie substratów energetycznych - zostaje wtedy uwolniona energia swobodna służąca do syntezy cząsteczek ATP z ADP lub AMP. Są to reakcje egzoergiczne, które zachodzą podczas dysymilacji, będącej częścią metabolizmu komórkowego. ATP jest więc ważnym związkiem wysokoenergetycznym łączącym reakcje egzoergiczne z endoergicznymi i niezbędnym składnikiem każdej żywej komórki. Cząsteczki ATP powstają podczas absorpcji energii słonecznej w procesie fotosyntezy oraz przy utlenieniu substratów organicznych podczas dysymilacji. Aby nie doszło do strat energii w komórce, musi być stale obecna rezerwa ADP i ortofosforanów. Dlatego energia z ATP po pokryciu zapotrzebowania na reakcje endoergiczne może być użyta do syntezy substancji zapasowych (takich jak polisacharydy, lipidy), to znaczy, że ATP są krótkotrwałym magazynem energii.
XII.
Co sprawia, że białka tracą swoją funkcje biologiczną.
Denaturacja. Przemiana związana ze zniszczeniem wtórnej struktury białka. Może wyst pod wpływem czynników fizycznych (podw temp, napromieniowanie lub zmiany pH) oraz chemicznych (stężony mocznik, kwasy aromatyczne, detergenty lub wyższe stężenie jonów metali ciężkich. Powoduje zmiany chemiczne i fizyczne: spadek rozpuszczalności w punkcie izoelektrycznym, utrata aktywności biologicznej, wzrost liczby grup jonizujących, wzrost kąta skręcania płaszczyzny światła spolaryzowanego w lewo, przyrost asymetrii cząstek, utrata zdolności krystalizacji); Różne białka są w różnym stopniu wrażliwe na poszczególne czynniki, a wrażliwość potęguje się przy działaniu dwóch lub więcej czynników.
Denaturacja jest procesem nieodwracalnym. Odwracalność stwierdzono tylko dla białek o prostej budowie - renaturacja.
Wbudowanie grupy α-aminowej do aminokwasów.
Wbudowanie NH4+ do aminokwasów. W procesie tym kluczową rolę odgrywają glutaminian i glutamina. Grupa α-aminowa w większościa aminokwasów pochodzi z grupy α-aminowej glutaminianu, przenoszonej do nich w reakcji transaminacji. Glutamina, drugi ważny donor azotu, dostarcza go z bocznego łańcucha do biosyntezy wielu ważnych związków. Glutaminian jest syntetyzowany z NH4+ i α-ketoglutaranu, związku pośredniego w cyklu kwasu cytrynowego z udziałem dehydrogenazy glutaminianowej. Jon amonowy zostaje wprowadzony do glutaminy podczas działania syntetazy glutaminowej na glutaminian. Amidacja jest zależna od hydrolizy ATP.
Aktywność oksygenowa i karboksylowa - różnice.
Przechodzenie glukozy w związki trójwęglowe (w jakich szlakach i gdzie one przebiegają, znaczenie).
Zasadniczym szlakiem degradacji glukozy jest glikoliza - szlak EMP, w czasie której jest ona przekształcana w duże trójwęglowe cząsteczki pirogronianu, cząsteczki ATP i NADH. Umożliwia ona zapoczątkowanie i dalszy przebieg reakcji składających się na cykl Krebsa. W ten sposób zdobywają energię bakterie tlenowe, mimo, że glikoliza przebiega bez udziału tlenu. Pirogronian jest podstawowym metabolitem, od którego rozdzielają się drogi oddychania tlenowego i fermentacji (oddychanie beztlenowe). Reakcje glikolizy zachodzą w cytoplazmie, natomiast dalsze przemiany pirogronianu - w matriks mitochondrium i dostarczają one energii namagazynowanej w ATP.
Innym sposobem przekształcenia glukozy jest szlak ED. Degradacja glukozy rozpoczyna się od powstania 6-fosofoglukonianu, koniec obejmuje przemianę aldehydu 3-fosfoglicerynowego do pirogronionanu. Szlak ten funkcjonuje u wielu bakterii z rodzaj Pseudomonas, Acetobacter i Gluconobacter. Dostarcza on komórce prekursorów do procesów biosyntetycznych. Kolejną reakcją rozkładu glukozy do związków trójwęglowego - pirogronianu, jest cykl pentozowy HMP - heksozomonofosforanowy. Pirogronian powstaje poprzez ciąg reakcji czynnych w szlaku EMP. Zgodna z glikolizą jest tylko jedna fermentacja mlekowa. Rozkładu glukozy (w war beztlenowych) przeprowadzają tu bakterie z rodzaju Loctobacillus i Streptococcus. Produktem końcowym tej reakcji jest tylko kwas mlekowy, dlatego jest to tzw. homofermentacja. C6H12O6 → 2CH3CHOH.COOH. Wydajność energetyczna fermentacji jest niższa, niż gdyby glukoza była rozkładana w warunkach tlenowych.
XIII.
Własności wody.
Dipolowy charakter cząsteczki wody stwarza możliwości przeprowadzenia do roztworu o polarnym i jonowym charakterze, np. zasad organ i nieorgan., ich soli oraz pochodnych, kwasów, sacharydów, itp.
Wiele niezwykłych właściwości wody, jak duże napięcie powierzchniowe, ciepło właściwe i ciepło parowania, wynika z kohezyjnego charakteru wody. Rola wody: • powszechny rozpuszczalnik związków ustrojowych, • jest czynnikiem umożliwiającym wymianę cieplną • związek uczestniczący w przebiegu większości reakcji metabolicznych; • środek transportu wewnątrzustrojowego; • utrzymuje odpowiednie wymiary i kształty komórek, warunkuje jędrność komórki (turgor). • uczestniczy w regulacji temp, ciśnienia osmotycznego, pH.
Stan uwodnienia ma zasadnicze znaczenie dla działalności enzymów. Białka rozpuszczalne (w tym enzymy) są utrzymywane w komórce w stanie cząsteczkowego rozproszenia dzięki odpowiedniej liczbie cząsteczek wody.
W przypadku niedoboru wody w komórce następują poważne zakłócenia w jej metabolizmie.
Wszystkie substancje dzielą się na hydrofilowe (rozpuszczalne w wodzie) i hydrofobowe (niezdolne do łączenia się z wodą).
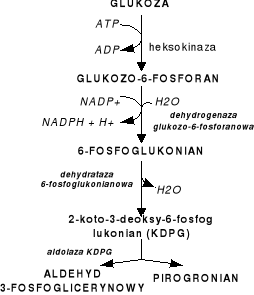
Szlak ED.
Szlak ten funkcjonuje u wielu bakterii (m.in.: Pseudomonas, Acetobacter, Gluconobacter). Rozpoczyna się od degradacji glukozy pod wpływem heksokinazy (ATP→ADP) i powstaje glukozo-6-fosforan utleniony; dehydrogenaza przekształca go w 6-fosfoglukonian (NADP+→NADPH+H+→syntezy komórkowe). Na skutek dehydrotazy powstaje 2-keto-3-deoksy-6-fosfoglukonian (KDPG). Wskutek rozszczepienia KDPG powstaje aldehyd 3-fosfoglicerynowy i pirogronian. Szlak ten dostarcza komórce prekursorów do procesów biosyntetycznych. Bilans: glukoza→2 pirogronian + 1NAD(P)H2 + 1 NADH2 + 1 ATP
Oddychanie tlenowe i beztlenowe w aspekcie pozyskiwania energii.
Dysymilacja (oddychanie wewnątrzkomórkowe) jest podstawowym szlakiem przemian katabolicznych prowadzonych w celu uzyskania energii. Jej źródłem są wiązania chemiczne związków organicznych (substratami: cukier, kwasy, związki aromatyczne i inne). Całość tych przemian tworzy skomplikowany szlak metaboliczny. Podstawowym warunkiem zdobywania energii z węglowodanów, białek i tłuszczów jest posiadanie przez komórkę odpowiednich enzymów dostosowanych do metabolizowania nie tylko substratu wyjściowego, ale i do wszystkich produktów pośrednich, jakie w trakcie procesu biokatalizy powstają. Koniecznym warunkiem jest obecność tlenu. Organizmy (w tym i człowiek) pozyskują w ten sposób energię. Oddychanie tlenowe (aerobowe) jest dwuetapowe: I faza - wymiana gazów O2 i CO2 w układzie oddechowym; II-faza - wewnątrz komórki, w mitochondriach - właściwy proces oddychania. Tu zostaje uwolniona energia magazynowana w ATP. Oddychanie wewnątrzkomórkowe ma na celu zdobycie energii swobodnej, użytecznej dla celów biologicznych. Najdostępniejsze źródło energii - glukoza (najpowszechniejszy sposób - dysymilacja glukozy).
Oddychanie beztlenowe (anaerobowe) odbywa się bez udziału tlenu. Końcowym akceptorem elektronów są wówczas pewne związki nieorganiczne, azotany (NO3-), azotyny (NO2-), tlenki azotu, siarczany i siarczyny, a nawet CO2. Donorami elektronów są związki organiczne, które mogą być utlenione do produktów końcowych, takich jak woda i CO2. Zdolność takiego oddychania mają niektóre bakterie beztlenowe. Przeprowadzają one: denitryfikację (redukcję azotanów); desulfurikację (redukcję siarczanów), czy też fermentacje: alkoholowa, mlekowa, masłowa, propionowa.
Synteza aminokwasów aromatycznych (rola fosfoenolopirogronianu).
Syntezę aminokwasów aromatycznych zapoczątkowuje reakcja kondensacji kwasu fosfoenolopirogronowego, który jest jednym z produktów pośrednich łańcucha glikolizy oraz erytrozo-4-fosforanu, który jest produktem pośrednim cyklu pentozofosforanów. Reakcja ta jest katalizowana przez transaldolazę, a jej produktem jest kwas 3-dezoksy-7-fosfo-heptulozonowy. Związek ten - przez kwas dehydrochinowy - przekształca się do kwasu szikimowego i następnie 5-fosf-szikimowego. Ten ostatni jest kluczowym produktem w syntezie aminokwasów aromatycznych, następuje rozwidlenie dróg w kierunku tryptofanu oraz fenyloalaniny i tyrozyny. Synteza aminokwasów aromatycznych jest ściśle powiązana z przemianami cukrowców.
1
Wyszukiwarka
Podobne podstrony:
II Rok OŚ Biochemia Ćwiczenia harmonogram, II rok, I semestr, biochemia
Faza ciemna fotosyntezy, II rok, I semestr, biochemia
MATEUSZ ROGACKI- opracowanie na egzamin z biochemii, analityka medyczna UMP II ROK 2015, BIOCHEMIA,
Biochemia egzamin czesc II opracowanie dowydruku, Biotechnologia POLSL, Semestr V, Biochemia, Egzami
Szymura, II ROK, SEMESTR II, psychologia różnic indywidualnych, opracowania
Nęcka r. 6, II ROK, SEMESTR II, psychologia różnic indywidualnych, opracowania
metodologia - zagadneinia na egzamin, UKSW - Pedagogika, II rok - I semestr, Metodologia Badań Pedag
JA, II ROK, SEMESTR I, psychologia społeczna I, opracowania
Skrypt(2), II ROK, SEMESTR II, psychologia różnic indywidualnych, opracowania
02.Psychologia Zdrowia opracowanie(1), psychologia UŚ, II rok, I semestr, Prop. psychologii zdrowia
zuckerman stu, II ROK, SEMESTR II, psychologia różnic indywidualnych, opracowania
zuckerman+klucz, II ROK, SEMESTR II, psychologia różnic indywidualnych, opracowania
Opracowanie Marii, Uczelnia, Filologia polska, II rok, semestr I, HLP - Romantyzm
egzaminy9pyt), analityka medyczna UMP II ROK 2015, BIOCHEMIA, EGZAMIN
material do pominiecia, II ROK, SEMESTR II, psychologia różnic indywidualnych, opracowania
opr bioch Piotrek, analityka medyczna UMP II ROK 2015, BIOCHEMIA, EGZAMIN
więcej podobnych podstron