1. Model atomu wg. Bohra.
Jądrowy model atomu Rutherforda w połączeniu z klasyczną mechaniką i elektrodynamiką nie jest w stanie wyjaśnić ani stabilności atomu, ani charakteru widma atomowego.
W 1913 r. duński fizyk Niels Bohr znalazł wyjście
z powstałego tu impasu, co prawda, kosztem wprowadzenia założeń sprzecznych z klasycznymi wyobrażeniami. Przyjęte przez Bohra założenia zawarte są w dwóch sformułowanych przezeń postulatach.
2. Postulaty Bohra.
Pierwszy postulat Bohra
Elektron nie może krążyć po dowolnej orbicie, lecz tylko po tych,
dla których moment pędu elektronu jest wielokrotnością h/2p .
h - stała nazwana stałą Plancka wynosząca 6,62*10-34J*s
Każdej orbicie odpowiada inny stan energetyczny atomu. Znajdując się na orbicie dozwolonej elektron nie promieniuje energii. Orbity dozwolone zostały nazwane stacjonarnymi.
Drugi postulat Bobra.
Atom absorbuje lub emituje promieniowanie w postaci kwantu
o energii hu przechodząc z jednego stanu energetycznego En do drugiego Ek (czyli przejściu elektronu z jednej orbity dozwolonej na inną).
Różnica energii tych stanów atomów równa się energii wypromieniowanego kwantu:
hn = En - Ek
We wzorze tym En oznacza energie atomu w stanie początkowym,
Ek - w stanie końcowym, v - jest częstotliwością emitowanego lub zaabsorbowanego promieniowania. Energia zostaje wypromieniowana, gdy En>Ek, pochłonięta zaś, jeżeli En< Ek.
Trzeci postulat Bohra
Prawa mechaniki opisują równowagę dynamiczną elektronów w stanach stacjonarnych, ale nie stosują się do przechodzenia elektronu pomiędzy dwoma stanami stacjonarnymi.
Zgodnie z pierwszym postulatem Bohra możliwe są tylko takie orbity, dla których moment pędu elektronu mevr spełnia warunek
mevr = nh (n = 1, 2, 3, ....)
Liczbę n nazywamy główną liczbą kwantową. Rozpatrzmy elektron poruszający się w polu jądra atomowego o ładunku Ze. Przy Z = 1 taki układ odpowiada atomowi wodoru, przy innych Z - jonowi wodoropodobnemu, tj. atomowi o liczbie atomowej Z, z którego usunięto wszystkie elektrony oprócz jednego.
Obecnie teoria Bohra ma głównie znaczenie historyczne.
Po pierwszych sukcesach tej teorii coraz bardziej widoczne stawały się jej niedociągnięcia. Szczególnie przygnębiające były niepowodzenia wszystkich prób skonstruowania teorii atomu helu - jednego z najprostszych atomów, następnego atomu bezpośrednio po atomie wodoru.
3. Dualizm korpuskularno-falowy - cecha wielu obiektów fizycznych (np: światła czy elektronów) polegająca na tym, że w pewnych sytuacjach, zachowują się one jakby były cząstkami (korpuskułami),
a w innych sytuacjach jakby były falami.
Wg mechaniki kwantowej właściwie całą materię charakteryzuje ten dualizm. Każdej cząstce, a nawet każdemu obiektowi makroskopowemu można przypisać charakterystyczną dla niego funkcję falową.
Z drugiej strony każde oddziaływanie falowe można opisać
w kategoriach cząstek.
Efekt fotoelektryczny
Emisja elektronów z katody zachodzi
tylko wówczas, gdy długość fali światła
padającego nie przekracza pewnej
wartości progowej „czerwonej linii
efektu fotoelektrycznego”. Ta graniczna
wartość l0 = c/n0 jest charakterystyczna
dla metalu katody.
Wynika z tego, że energia fotonu musi posiadać pewną minimalną
wartość, poniżej której nie jest możliwe wybicie elektronu z katody.
Nadmiar energii h(n - n0) jest zamieniany na energię kinetyczną
elektronu.
Dualizm korpuskularno-falowy
Efekt Comptona
Doświadczenie polegało na rozproszeniu promieniowania
Röntgena na parafinie. Compton stwierdził, że długość fali promieniowania rozproszonego
jest większa niż pierwotnie. W przeciwieństwie do zjawisk interferencji czy dyfrakcji promieniowania elektromagnetycznego efektu fotoelektrycznego oraz efektu Comptona
nie da się wyjaśnić na drodze teorii falowej.
Dyfrakcja - odchylenie się fal od prostoliniowego kierunku
rozchodzenia się, np. po przejściu przez szczelinę.
Interferencja - nakładanie się fal prowadzące do zwiększenia
lub zmniejszenia amplitudy fali wypadkowej.
4. Hipoteza de Broglie'a.
Dualizm korpuskularno-falowy jest cechą nie tylko światła
(fotonów), lecz i wszystkich cząstek materialnych o masie
spoczynkowej różnej od zera, np. elektronów
Przykład: dla kuli o masie 1g, wystrzelonej z prędkością
300 m/s, l jest rzędu 10-31 cm (h =6,624 · 10-34 J · s).
5. Liczby kwantowe n, l, m i s
n - główna liczba kwantowa, określa przede wszystkim energię
elektronu w atomie. Główna liczba kwantowa może przybierać
tylko wartości równe liczbom naturalnym: n = 1, 2, 3, .....
n określa przynależność elektronu do poziomów: K, L, M, N, O, P, Q
l - poboczna liczba kwantowa, kwantuje moment pędu związany
z ruchem elektronu w polu jądra. Dla danego n wartości l wynoszą:
0, 1, 2, ..., (n-1). Liczba l określa przynależność do orbitali:
s, p, d, f, g...
m - magnetyczna liczba kwantowa, decyduje o możliwych wartościach
składowej momentu pędu w wyróżnionym kierunku (z). Dla danego l
możliwe wartości m wynoszą: 0, Ⴑ1, Ⴑ 2, ..., Ⴑ l.
s - spinowa liczba kwantowa, kręt elektronu, może przyjmować
wartości ±1/2. Ma ona duże znaczenie przy tworzeniu wiązań
chemicznych.
Zakaz Pauliego
Możliwe są tylko takie stany elektronowe w atomie, w których
żaden elektron nie ma identycznych wszystkich czterech
liczb kwantowych.
6. Kolejnośc zapełniania poziomów energetycznych .
7. Wiązania chemiczne.
Tworzenie się związków chemicznych i powstawanie odpowiednich wiązań chemicznych tłumaczy się charakterystycznym kwantowo-mechanicznym oddziaływaniem pomiędzy elektronami i jądrami łączących się atomów.
Aby utworzona molekuła była trwała, musi być uboższa energetycznie niż wchodzące w jej skład oddzielne atomy. Oznacza to, że proces tworzenia się molekuł powinien być energetycznie korzystny, a więc powinien prowadzić do osiągnięcia przez układ minimum energii.
Podstawowymi wielkościami charakteryzującymi wiązanie jest:
energia wiązania (energia dysocjacji),
odległość pomiędzy atomami,
kąt pomiędzy kierunkami wiązań.
Wiązanie chemiczne jest siłą przyciągającą do siebie atomy
i utrzymującą je razem.
Molekuła jest to zgrupowanie atomów powstające w wyniku tych oddziaływań.
Długość wiązania jest związana z jego siłą: im wiązanie silniejsze,
tym bliżej siebie znajdują się atomy. Dwa atomy wodoru połączone wiązaniem kowalencyjnym znajdują się w odległości 0,74 angstremów, podczas gdy w przypadku oddziaływań van der Waalsa odległość ta wynosi 1,2 angstrema.
Elektronowa teoria wiązania chemicznego opiera się na trwałości konfiguracji oktetowej i w sposób jednolity na podstawie reguły oktetu wyjaśnia różne typy i liczby wiązań w związkach chemicznych.
Rodzaje wiązań:
wiązania jonowe,
wiązania kowalencyjne,
wiązania kowalencyjne spolaryzowane,
wiązania koordynacyjne,
wiązania metaliczne,
wiązania wodorowe,
oddziaływania van der Waalsa,
oddziaływania hydrofobowe
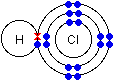
Wiązania jonowe występują w układach złożonych z atomów skrajnie różniących się elektroujemnością.
W czasie powstawania wiązania jonowego atom pierwiastka elektrododatniego oddaje, a atom pierwiastka elektroujemnego przyłącza elektrony. Tworzą się dwa jony o różnoimiennych ładunkach, przyciągające się dzięki działaniu sił elektrostatycznych
Na Cl
Wiązania atomowe (kowalencyjne) powstają również, gdy łączą się ze sobą atomy pierwiastków elektroujemnych o takich samych wartościach elektroujemności.
Podobnie jak w wiązaniu jonowym, wiążące się atomy dążą do osiągnięcia struktury oktetowej najbliższego gazu szlachetnego.
Wiązania tego typu występują w cząsteczkach H2, Cl2, O2, N2 itp.
Przykładem jest wodór dla którego pojedynczy atom ma jeden elektron.
Gdy dwa atomy wodoru tworzą cząsteczkę, ich elektrony rozmieszczają się symetrycznie wokół obydwu jąder, tworząc parę elektronową.
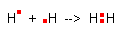
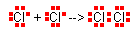
W wiązaniu atomowym wiążąca para elektronowa znajduje się
w jednakowej odległości od jąder atomów tworzących wiązanie.
Wiązanie atomowe spolaryzowane jest wiązaniem pośrednim między jonowym a atomowym; powstaje wówczas, gdy łączą się ze sobą atomy pierwiastków różniących się elektroujemnością, lecz nie tak znacznie jak w przypadku tworzenia wiązania jonowego.
Cechą charakterystyczną tego wiązania jest przesunięcie pary elektronowej wiążącej atomy w kierunku atomu pierwiastka bardziej elektroujemnego.
Cząsteczki z wiązaniami kowalencyjnymi spolaryzowanymi z powodu nierównomiernego, niesymetrycznego w stosunku do środka cząsteczki, rozmieszczenie ładunków wykazują biegunowość. W cząsteczkach tych wyróżnić można biegun dodatni i ujemny.
Cząsteczki o budowie polarnej nazywamy dipolami, tzn. cząsteczkami dwubiegunowymi.
Cząsteczki dwubiegunowe mają tzw. moment dipolowy u.
Wiązanie koordynacyjne tym różni się od wiązania atomowego lub atomowego spolaryzowanego, że para elektronowa tworzących wiązanie oddawana jest przez jeden z dwóch łączących się atomów. Najprostszym przykładem powstawania wiązania koordynacyjnego jest tworzenie się jonu amonowego
Wiązania wodorowe- Jest to słabe oddziaływanie elektrostatyczne pomiędzy elektroujemnym atomem (akceptorem), a atomem wodoru, który jest kowalencyjnie połączony z innym atomem elektroujemnym (donorem). W wiązaniu tym wodór pełni rolę mostka łączącego dwa elektroujemne atomy.
Siły van der Wasala
Siły van der Waalsa są bardzo słabymi oddziaływaniami zachodzącymi pomiędzy wszystkimi typami atomów (zarówno polarnymi jak i niepolarnymi). W chmurze elektronowej otaczającej jądro pojawiać się mogą chwilowe fluktuacje rozmieszczenia ładunków ujemnych - wynik normalnego ruchu elektronów. Takie nieregularności prowadzą do powstawania i zanikania dipoli, które wpływają na rozmieszczenie elektronów w sąsiednich atomach.
Gdy atomy znajdujące się w bezpośrednim sąsiedztwie mają spolaryzowane chmury elektronowe, pojawiać się może słabe przyciąganie. Zanika ono bardzo szybko wraz ze wzrostem odległości między atomami, natomiast gdy atomy znajdą się zbyt blisko siebie zostają odepchnięte przez jednoimienne ładunki chmur elektronowych.
Oddziaływania Van der Wasala
Oddziaływania van der Waalsa odpowiadają
na przykład za przyciąganie się cząsteczek niepolarnych cieczy oraz stanowią siły łączące składniki ścian komórkowych (poprzez przyciąganie się niepolarnych łańcuchów fosfolipidów).
8. Prawo działania mas Guldberga i Waagego.
Stała równowagi
- prawo działania mas Guldberga i Waagego.
Substancje reagują tak długo, dopóki stosunek iloczynu stężeń
produktów do iloczynu stężeń substratów nie osiągnie pewnej
stałej wartości, charakterystycznej dla danej reakcji i dla temperatury
Prawo Daltona
p = p1 + p2 + ........
Ciśnienie wywierane przez mieszaninę gazów nie reagujących
ze sobą jest równe sumie ciśnień jakie wywierałby każdy z gazów
wchodzących w skład mieszaniny, gdyby sam został umieszczony
w całej objętości zajmowanej przez mieszaninę.
Równanie stanu gazu doskonałego:
9. Kinetyczne uzasadnienie prawa działania mas.
W stanie równowagi dynamicznej: nHJ = nH2 +J2
;
9. Stała i stopień dysocjacji.
Dysocjacja elektrolityczna- Samorzutny rozpad cząsteczek na jony pod wpływem
rozpuszczalnika (woda, ciekły amoniak)
Stopień dysocjacji:
a = 1 - elektrolity mocne
Stała dysocjacji:
Położenie równowagi chemicznej zależy nie tylko od rodzaju
reakcji chemicznej, lecz także od stężeń składników
reagujących.
10. Iloczyn rozpuszczalności.
Iloczyn rozpuszczalności jest to wartość iloczynu jonowego
całkowicie zdysocjowanego elektrolitu będącego w równowadze
z formą niezdysocjowaną.
11. Wpływ HCl na stopień dysocjacji kwasu octowego.
Położenie równowagi chemicznej zależy nie tylko od rodzaju
reakcji chemicznej, lecz także od stężeń składników
reagujących.
Przykład:
Mieszanina kwasu solnego i kwasu octowego o stężeniach, kolejno
0.05 i 0.1 mol/dm3
W formie uproszczonej
Dla porównania liczba moli zdysocjowanego samego kwasu octowego
wynosi:
W formie uproszczonej
Przykład zależności stałej dysocjacji od stopnia dysocjacji:
12. Wpływ HCl na rozpuszczalnośc chlorku srebra.
Przykład
Jak wpłynie 0,1 molowy roztwór kwasu solnego na liczbę jonów
srebra obecnych w roztworze w stanie równowagi.
13. Iloczyn jonowy wody, pH.
14. Szybkośc i rząd reakcji.
Szybkość reakcji chemicznej wyraża szybkość przybywania lub ubywania składnika w wyniku przebiegu reakcji chemicznej.
szybkość jest wyrażona tak :
Jeżeli w równaniu występują współczynniki stechiometryczne, to będą występowały także w mianowniku wyrażenia na szybkość - choć nie zawsze muszą im ściśle odpowiadać.
Z drugiej strony eksperymentalnie dowiedziono, że szybkość reakcji można wyrazić tak:
v = k[A]n[B]m
przy czym wykładniki potęgowe nie wynikają z równania sumarycznego, a jedynie z mechanizmu reakcji, dokładnie z jej rzędowości. Dla prostych reakcji wykładniki te przyjmują zwykle wartości całkowite, zaś dla reakcji złożonych mają wartości ułamkowe. W tym wyrażeniu mogą występować produkty, lecz tylko w szczególnych sytuacjach. Łącząc te dwa zapisy dochodzi się do równania kinetycznego.
Jednostka szybkości reakcji chemicznej zależy od jej równania kinetycznego. Może to być:
1/czas (reakcja zerowego rzędu)
masa/(objętość*czas) (reakcja rzędu pierwszego)
lub bardziej złożone układy jednostek (dla reakcji wyższych rzędów lub rzędów ułamkowych).
Rząd reakcji chemicznej
Rząd reakcji jest to suma wykładników potęg występujących przy stężeniach związków chemicznych, w równaniach kinetycznych danej reakcji chemicznej.
Istnieją różne rodzaje rzędów reakcji:
Rzędem zewnętrznym reakcji nazywa się sumę wszystkich wykładników, występujących w ostatecznym, sumarycznym równaniu kinetycznym danej reakcji.
Rzędem względnym (względem danego substratu) - nazywa się wartość pojedynczego wykładnika, występującego przy stężeniu danego substratu. Rozróżnia się tu jeszcze dodatkowo rząd względny:
zewnętrzny - czyli występujący w ostatecznym równaniu kinetycznym
Wewnętrzne rzędy względne wynikają bezpośrednio z liczby cząsteczek danego substratu uczestniczących w pojedynczym akcie elementarnym i dlatego przyjmują one zawsze wartości całkowite - zwykle 0, 1 lub 2.
Czasami zdarza się, że względny rząd wewnętrzny równa się zewnętrznemu rzędowi względnemu - co świadczy o tym, że dany substrat uczestniczy w ramach danej reakcji tylko w jednym akcie elementarnym. Często jednak w końcowych równaniach kinetycznych występują rzędy ułamkowe, co wynika z faktu dużej złożoności mechanizmu takiej reakcji.
Ustalenie rzędu zewnętrznego reakcji jest zazwyczaj bardzo proste i wymaga tylko wykreślenia zależności stężenia wszystkich substratów od czasu reakcji, a następnie dobrania do tak otrzymanych krzywych odpowiednich równań, zwykłymi metodami statystycznymi. Ustalenie rzędów wewnętrznych jest znacznie trudniejsze i wymaga stosowania wielu złożonych technik (np: znakowania izotopami) ale jest często konieczne do pełnego zbadania mechanizmu danej reakcji.
Np: w poniższym równaniu :
zewnętrzny rząd reakcji wynosi (n+m+p).
Gdy można opisać zmianę stężenia substratu A równaniem:
to względny rząd zewnętrzny dla substratu A wynosi 0;
gdy stężenie to zmienia się zgodnie z równaniem:
to względny rząd zewnętrzny dla substratu A wynosi 1;
gdy poprawne jest równanie:
to względny rząd zewnętrzny dla substratu A wynosi 2;
I wreszcie, gdy zmiany stężenia substratów A i B są zgodne z równianiem:
to wówczas względne rzędy zewnętrzne dla obu substratów są równe 1, zaś rząd zewnętrzny całej reakcji wynosi 2.
15. Równanie Arrheniusa.
Równanie Arrheniusa, podane przez szwedzkiego chemika Svantego Arrheniusa, wiąże ze sobą stałą szybkości reakcji z energią aktywacji i temperaturą, w której reakcja zachodzi:
gdzie:
A - stała dla danej reakcji, zwana też czynnikiem przedwykładniczym
Zaletą równania jest łatwa do wykorzystania w praktyce zależność liniowa między lnk i 1/T. Równanie Arrheniusa w postaci logarytmicznej przedstawia linię prostą:
Znając doświadczalne wartości stałych szybkości reakcji w kilku temperaturach, można łatwo wyznaczyć zarówno stałą A, jak i energię aktywacji dla danej reakcji.
16. Absorpcja i adsorpcja.
Absorpcja (fizyka, chemia) (łac. absorbere, wchłaniać) to proces polegający na wnikaniu cząsteczek, atomów lub jonów do wnętrza innej substancji tworzącej dowolną fazę ciągłą - (gazu, cieczy, ciała stałego itp.) Absorpcji nie należy mylić z adsorpcją, która jest zjawiskiem powierzchniowym. Absorpcja, adsorpcja i wymiana jonowa są wspólnie nazywane procesami sorpcji.
Mechanizm absorpcji polega na podziale absorbowanego składnika pomiędzy dwie fazy (ośrodki) objętościowe. Zjawisko to opisuje prawo podziału Nernsta, a w szczególnym przypadku równowagi gaz/ciecz prawo Henry'ego.
Zjawiska absorpcji są powszechne w naturze, np. oddychanie jest procesem absorpcji tlenu do krwi. Absorpcja jest też stosowana na masową skalę w procesach technologicznych. Stanowi np. podstawowy mechanizm umożliwiający oczyszczanie związków chemicznych przez ekstrakcję. Jest to proces dyfuzyjny zachodzący podczas bezprzeponowego zetknięcia cieczy z gazem zawierającym składnik, który chcemy z niego usunąć. Składnik gazowy pochłaniany przez ciecz nazywa się absorbatem, a ciecz używana do pochłaniania określonego składnika nazywa się absorbentem. W chemii mamy najczęściej do czynienia z absorpcją jednej substancji (absorbat) i przez (absorbent) znajdująca się w jednej fazie oraz z absorpcją promieniowania (elektromagnetycznego, korpuskularnego, fal akustycznych) przez różne substancje.
Absorpcja jest pochłanianiem całą objętością absorbentu, którym najczęściej jest ciecz (rzadko ciało stałe np. gazowy wodór pochłaniany przez pallad) absorbująca gazy lub inne ciecze. Metodą absorpcji można np. oczyszczać gazy selektywne, absorbując niepożądane składniki (np. tlenki siarki pochłaniane w roztworze wodorotlenku wapnia). Absorpcja jest związana z reakcjami chemicznymi pomiędzy absorbatem i absorbentem.
Adsorpcja — to proces wiązania się cząsteczek, atomów lub jonów na powierzchni lub granicy faz fizycznych, powodujący lokalne zmiany stężenia. Adsorpcji nie należy mylić z absorpcją, która jest procesem wnikania do wnętrza fazy. Adsorpcję, absorpcję i wymianę jonową przyjęło się wspólnie nazywać procesami sorpcji.
17. Adsorpcja fizyczna i chemiczna.
Adsorpcja fizyczna lub mniej precyzyjnie fizysorpcja to zjawisko lub proces adsorpcji na skutek działania sił oddziaływania międzycząsteczkowego - sił van der Waalsa (z wyłączeniem wiązań chemicznych - wówczas mówimy o adsorpcji chemicznej, czyli chemisorpcji) pomiędzy cząsteczką adsorbatu a powierzchnią adsorbentu. Energia związana z procesem adsorpcji fizycznej (ciepło adsorpcji) jest z reguły rzędu kilku kJ/mol (rzadko kilkunastu kJ/mol), poczas gdy w przypadku adsorpcji chemicznej są to wielkości o rząd większe (ciepło reakcji chemicznej). Wielkość adsorpcji fizycznej silnie zależy od temperatury oraz ciśnienia lub stężenia adsorbatu, a obniżenie ciśnienia lub podwyższenie temperatury prowadzi do łatwej i szybkiej desorpcji.
Oprócz oddziaływań adsorbat-adsorbent w układzie takim istnieją zawsze zwiększające wielkość adsorpcji oddziaływania adsorbat-adsorbat. Jako tzw. oddziaływania boczne zwiększają wartość adsorpcji w obrębie monowarstwy adsorpcyjnej, prowadzą również do pojawienia się kolejnych warstw adsorbatu na już zapełnionej monowarstwie - tzw. zjawisko formowania wielowarstwy odpowiadające skraplaniu pary cieczy przy ciśnieniu obniżonym na skutek przyciagających sił adsorpcyjnych.
Siły van der Waalsa zależą od rodzaju powierzchni adsorbentu i adsorbatu, jednak mają charakter w dużym stopniu niespecyficzny, gdyż wynikają przed wszystkim z powszechnego istnienia sił dyspersyjnych (Londona) związanych z budową materii (dodatnio naładowane jądro i krążące wokół jądra ujemnie naładowane elektrony). Poza przypadkiem trwałych dipoli oraz wiązań wodorowych są również bezkierunkowe.
Adsorpcja fizyczna jest jednym ze zjawisk tworzących w sumie zjawisko adsorpcji, a to ostatnie jednym ze zjawisk określanych wspólną nazwą sorpcji (nazwa niezbyt precyzyjna nazwa obejmuje zjawiska adsorpcji, absorpcji gdzie mechanizmem jest podział objętościowy, zjawiska tzw. sorpcji wymiennej oraz inne w wyniku których określone substancje przemieszczają się z jednej fazy do drugiej z wytwozeniem pewnego stanu równowagi).
Chemisorpcja - adsorpcja chemiczna, polegająca na tworzeniu się silnych wiązań chemicznych między adsorbentem i adsorbatem.
Aby usunąć chemisorbowaną cząsteczkę nie wystarczy silnie obniżyć ciśnienie lub stężenie adsorbatu, wystarczające w przypadku adsorpcji fizycznej. Należy jeszcze silnie podnieść temperaturę. W niektórych przypadkach usunięcie adsorbatu może być niemożliwe bez destrukcji adsorbentu. Przykładem jest tlen chemisorbowany na węglu aktywnym po ogrzaniu wydziela się ale w postaci tlenków węgla.
Chemisorpcja jest adsorpcją jednowarstwową, co oznacza, że na powierzchni adsorbentu może się zaadsorbować jedynie jedna warstwa (monowarstwa) adsorbatu. Jednak adsorpcji chemicznej zawsze towarzyszy adsorpcja fizyczna w obrębie monowarstwy oraz jako adsorpcja wielowarstwowa - na istniejącej chemisorbowanej monowarstwie. Adsorpcja fizyczna stanowi również ważny etap pośredni pomiędzy adsorbatem gazowym a chemisorbowanym.
Oddziaływania związane z chemisorpcją są oddziaływaniami specyficznymi. Związane są z efektem orientacyjnym (efekt Keesoma) i efektem indukcyjnym (efekt Debye'a), a w przypadku adsorpcji jonów także z oddziaływaniami elektrostatycznymi.
18. Izotermy Langmuira.
Izoterma Langmuira to podstawowa izoterma adsorpcji wprowadzona w 1916 r. przez Irvinga Langmuira, laureata nagrody Nobla w 1932 r.
Ta teoria kinetyczna zakłada, że adsorbat może tworzyć na powierzchni adsorbentu tzw. monowarstwę czasteczek oddziaływujących z miejscami adsorpcyjnymi (oddziaływanie "pionowe") a nie oddziaływujacymi (albo słabo oddziaływującymi) ze sobą (oddziaływania "poziome"). Cząsteczki adsorbatu obecne w fazie gazowej uderzają w powierzchnię - prawdopodobieństwo zaadsorbowania rośnie wraz z dostępną wolną powierzchnią. Zaadsorbowane cząsteczki charakteryzuje pewne prawdopodobieństwo desorpcji (proces przeciwny do adsorpcji). Oba prawdopodobieństwa zależą od temperatury i wielkości energii adsorpcji. Wraz z ciśnieniem rośnie częstość uderzeń cząsteczek w powierzchnię, a wraz z ilością zaadsorbowanych cząsteczek maleje dostępna powierzchnia.
W założeniach równania jest: brak możliwości tworzenia wielowarstwy, stałość energii adsorpcji (powierzchnia energetycznie jednorodna, czyli homogeniczna), zaniedbywalność oddziaływań bocznych.
równanie izotermy Langmuira
gdzie: a - adsorpcja rzeczywista, am - wielkość adsorpcji odpowiadająca zapełnieniu monowarstwy, K - stała równowagi adsorpcji, p - ciśnienie adsorbatu
stała równowagi adsorpcji
gdzie Ko to tzw. czynnik przedeksponencjalny albo czynnik entropowy, ΔS - entropia adsorpcji, E - energia adsorpcji (ciepło adsorpcji) - w konwencji: dodatnia wartość oznacza wydzielenie się energii, w termodynamice przyjęte jest zwykle odwrotnie.
W badaniach nad adsorpcją często używa się pojęcia względnego pokrycie powierzchni oznaczanego θ, które ma prosty sens geometryczny dla adsorpcji monowarstwowej:
względne pokrycie powierzchni
Inne postacie izotermy Langmuira
Jeżeli znamy wielkość pojemności monowarstwy (np. z innych badań), możemy używać inną postać izotermy Langmuira:
lub
Jeżeli znana jest wartość pojemności monowarstwy (np. z innych badań) liniowa forma logarytmiczna jest przydatna zwłaszcza do sprawdzania zgodności danych doświadczalnych z modelem oraz wyznaczania stałej równowagi K:
Inne postacie liniowe izotermy Langmuira są powszechnie stosowane do wyznaczania parametrów równania bezpośrednio z danych doświadczalnych:
postać liniowa (1/a) = f(1/p) o podobnych właściwościach jak poprzednia zależność:
postać liniowa a = f(a/p), pozwalająca łatwo dostrzec wszelkie odchylenia systematyczne od modelu teoretycznego (ale dość wrażliwa na rozrzut eksperymentalny):
Izoterma Freundlicha to równanie o charakterze eksperymentalnym opisujące dobrze adsorpcję na powierzchniach heterogenicznych (energetycznie niejednorodnych) oraz na adsorbentach mikroporowatych.
Różne formy równania Freundlicha dla adsorpcji z fazy gazowej:
a = kp1 / n
a = am(Kp)m
a = am(p / ps)m
gdzie: a - adsorpcja rzeczywista, am - wielkość adsorpcji odpowiadająca zapełnieniu warstwy adsorpcyjnej lub zapełnieniu mikroporów, k - stała, K - stała równowagi adsorpcji, p - ciśnienie adsorbatu, x = p/ps - tzw. ciśnienie względne (ps - ciśnienie pary nasyconej), n,m - empirycznie określone tzw. parametry heterogeniczności (m = 1/n ≤ 1 - im wartość m jest mniejsza tym większa jest niejednorodność energetyczna układu adsorpcyjnego).
Równanie to stosuje się szczególnie do adsorpcji na mikroporowatych węglach aktywnych z rozcieńczonych roztworów wodnych związków organicznych - w powyższych równaniach należy zastąpić ciśnienie stężeniem:
a = kc1 / n
a = am(Kc)m
a = am(c / cs)m
Należy zwrócić uwagę na fakt, że ograniczonej zgodności danych adsorpcji z równaniem izotermy Freundlicha można oczekiwać dla praktycznie dowolnych układów eksperymentalnych (przybliżenie fragmentu krzywej odcinkiem prostej). Niektóre izotermy teoretyczne również redukują się do izotermy Freundlicha (np. izoterma GF dla niskich stężeń). W ramach teorii adsorpcji zlokalizowanej na niejednorodnych energetycznie ciałach stałych izotermie Freundlicha odpowiada eksponencjalnie malejąca funkcja rozkładu energii, f(E) = A exp(-mE), jednak w przeciwieństwie do równań opartych o monowarstwowe równanie Langmuira, izoterma Freundlicha nie zawiera ograniczenia wielkości adsorpcji monowarstwą.
19. Równanie Nernsta.
E0 - normalny potencjał elektrody, tj. potencjał elektrody
w roztworze o aktywności jonów metalu aMn+ = 1
R - stała gazowa,
T - temperatura [K],
F - stała Faradaya,
N - liczba elektronów biorących udział w reakcji.
aM0 = const. = 1
20. E0 - potencjał standardowy, potencjał normalny.
Potencjał standardowy można obliczyć z danych termodynamicznych - standardowych potencjałów chemicznych substancji biorących udział w reakcji elektrochemicznej posługując się równaniem: 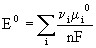
Przykład
Oblicz potencjał elektrody żelaznej zakładając, że na jej powierzchni ustala się równowaga:
wiedząc że:

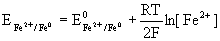
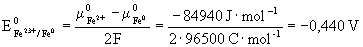
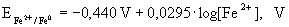
Potencjał elektrody żelaznej zależy od stężenia kationów Fe2+
w roztworze.
21. Siła elektromotoryczna ogniwa (SEM)
SEM = E1 - E2
Potencjał pojedynczej elektrody możemy mierzyć przy pomocy ogniwa w którym jedną z elektrod jest elektroda wzorcowa, elektroda o znanym, niezmiennym w czasie potencjale, zwanym potencjałem odniesienia.
22. Elektroda szklana.
Elektrody stałomembranowe- Elektrody szklane do oznaczania stężenia jonów wodorowych
Elektrody szklane do oznaczania stężenia kationów jednowartościowych
Selektywność elektrod jest zależna od składu chemicznego szkła,
z którego została wykonana membrana.
Skład elektrody szklanej do oznaczania pH:
Na2O - 14%, SiO2 - 86% (% molowy), wsp. czułości Ag/Na - 1400
Skład elektrody szklanej do oznaczania jonów Ag+ i Na+:
Na2O - 11%, Al2O3 - 18%, SiO2 - 86% (% molowy)
Skład elektrody szklanej do oznaczania jonów K+ i NH4+:
Na2O - 27%, Al2O3 - 4%, SiO2 - 69% (% molowy) wsp. czułości K/NH4 - 1400
Elektrody krystaliczne homogeniczne
Membranę stanowi monokryształ lub układ polikrystaliczny trudno
rozpuszczalnych związków.
Jedną z pierwszych elektrod krystalicznych była elektroda fluorkowa
opisana w 1966 r. przez Franta i Rossa. Membranę stanowił w niej
monokryształ fluorku lantanu lub fluorku innych pierwiastków ziem
rzadkich. Fluorki te wykazują elektryczne przewodnictwo wynikające
z ruchliwości jonów fluoru w sieci krystalicznej.
Schematyczny mechanizm przenoszenia ładunków:
LaF3 + dziura cząsteczkowa LaF2+ + F-
23. Prawa Absorpcji: Bouguera-Lamberta, Beera.
Prawo Lamberta-Beera (prawo Beera-Lamberta-Bouguera) - opisuje pochłaniane promieniowania elektromagnetycznego przy przechodzeniu przez częściowo absorbujący i rozpraszający ośrodek.
Prawo to głosi, że stopień atenuacji (uwzględniającej absorpcję oraz rozpraszanie) światła jest proporcjonalny do grubości warstwy i jej własności optycznych , np. w przypadku roztworów należy uwzględnić stężenie molowe czynnika powodującego pochłanianie. Ogólnie mówiąc, prawo to jest spełnione dla wiązki światła: a) monochromatycznej, b) skolimowanej, chociaż jest często używane także dla sytuacji wąskich przedziałów pasmowych, zwłaszcza jeżeli zależność spektralna atenuacji nie jest silna w tym paśmie. Rejestrowane natężenie I0 jest natężeniem również monochromatycznym i skolimowanym. Wartość końcowa natężenia promieniowania I1 jest mniejsza od I0 o wartość natężenia promieniowania pochłoniętego (zaabsorbowanego). Jest kilka metod w jakie to prawo może być matematycznie sformułowane:
{kind=link}
Absorpcja promienia światła przechodzącego przez kuwetę o na odcinku o długości l.
Gdzie:
I1 - natężenie światła po przejściu przez ciało
l - droga jaką pokonuje światło w ciele.
c - stężenie molowe substancji absorbującej w roztworze
α - współczynnik absorpcji zwany poprawnie absorbancją molową
λ - długość fali pochłanianego światła
24. Ekstrakcja jest operacją służącą do rozdzielenia mieszanin ciał stałych i ciekłych. Rozdział następuje przez rozpuszczenie niektórych składników mieszaniny w cieczach, zwanych rozpuszczalnikami. Proces ekstrakcji może zachodzić zatem w układach dwufazowych: ciało stałe - ciecz lub ciecz - ciecz.
Najprostszy układ ekstrakcyjny składa się z dwóch nie mieszających się cieczy "A" i "C" oraz ciała "B" rozpuszczającego się w obu cieczach. "A" nazywamy rafinatem (rozpuszczalnikiem pierwotnym), a "C" ekstrahentem (rozpuszczalnikiem wtórnym). Ciało rozpuszczone "B" zwane ekstraktem, może być cieczą lub ciałem stałym. W przypadku gdy ekstrahent jest mieszaniną dwóch lub więcej cieczy, proces ten nosi nazwę ekstrakcji frakcyjnej.
W ekstrakcji siłą napędową procesu jest różnica stężeń ekstrahowanego składnika w rozpuszczalniku pierwotnym i rozpuszczalniku wtórnym, zatem proces ten jest procesem dyfuzyjnym. Zjawisko przebiega tak długo, aż układ osiągnie stan równowagi fizykochemiczne.
25. Konduktometria- polega na pomiarze przewodnictwa elektrycznego lub oporu roztworu znajdującego się między dwiema elektrodami obojętnymi w warunkach stosowania zmiennego napięcia, o częstotliwości nie przekraczającej
105 Hz.
Konduktancja (przewodność elektryczna) jest odwrotnością rezystancji. Jest więc miarą podatności elementu na przepływ prądu elektrycznego.
Zwyczajowo konduktancję oznacza się symbolem G (wielka litera G).
Jednostką konduktancji w układzie SI jest simens (1 S).
Miarą podatności materiału na przepływ prądu elektrycznego jest konduktywność. Dla znanych wymiarów geometrycznych przewodnika i konduktywność materiału, z jakiego został wykonany, jego konduktancję określa wzór:
,
gdzie: l - długość przewodnika, S - pole przekroju poprzecznego elementu, σ - konduktywność właściwa materiału.
Konduktancja dotyczy obwodów prądu stałego, a w obwodach prądu zmiennego tylko elementów rezystancyjnych (rezystor). Uogólnieniem i rozwinięciem pojęcia konduktancji na elementy pojemnościowe (kondensator) i indukcyjne (cewka) jest admitancja.
Podział technik konduktometrycznych:
konduktometria klasyczna,
konduktometria bezelektrodowa małej częstotliwości,
konduktometria wielkiej częstotliwości.
Konduktometria klasyczna, polega na pomiarze przewodnictwa słupa cieczy zawartego między dwiema elektrodami platynowymi, do których przykłada się napięcie prądu zmiennego o częstotliwości ok. 105 Hz
(na ogół od 1 do 10 kHz).
Ładunek elektryczny w roztworach elektrolitów przepływa w wyniku uporządkowanego ruchu jonów w polu elektrycznym. Zgodnie z prawem Ohma rezystancja (opór) wyraża się wzorem:
Konduktometria bezelektrodowa
wielkiej częstotliwości (oscylometria
Oscylometria polega na pośrednim pomiarze admitancji
(przewodności pozornej) lub impedancji (oporu pozornego) naczyńka pomiarowego zawierającego badany roztwór, podczas przyłożenia napięcia przemiennego wielkiej częstotliwości. Zmiany własności elektrycznych są związane ze zmianami stężenia, można więc z pomiaru parametrów obwodu wnioskować o stężeniu roztworu.
Konduktywność (przewodność elektryczna właściwa) to miara podatności materiału na przepływ prądu elektrycznego.
Konduktywność materiału wyznaczyć można znając wymiary geometryczne i konduktancję jednorodnego bloku danego materiału:
,
W ogólności konduktywność metali spada przy wzroście temperatury, a konduktywność półprzewodników wzrasta wraz z temperaturą.
26. Zależnośc konduktywności od stężenia elektrolitu.
Konduktancja elektrolityczna i przewodność właściwa elektrolitów zależą od:
rodzaju elektrolitu,
stężenia,
b. mocno od temperatury - zamienia się zarówno
koncentracja nośników jak i
ruchliwość.
W danej temperaturze przewodnictwo właściwe elektrolitu jest funkcją stężenia. W roztworach o małym stężeniu konduktywność elektrolitu zwiększa się niemal liniowo ze wzrostem stężenia. Ten wzrost przewodnictwa właściwego w obszarze o małych stężeniach jest wynikiem zwiększania się koncentracji jonów. W roztworach elektrolitów o dużym stężeniu konduktywność początkowo rośnie wraz ze wzrostem stężenia, a następnie maleje.
27. Miareczkowanie konduktometryczne - w chemii analitycznej chemiczna technika miareczkowa polegająca na pomiarze zmian przewodnictwa elektrycznego analizowanego roztworu w trakcie stopniowego dodawania do niego odczynnika miareczkującego.
Miareczkowanie konduktometryczne przeprowadzane jest zwykle w układzie kwas-zasada. O przewodnictwie układu kwas-zasada decydują głównie bardzo ruchliwe jony hydroniowe, a zatem jest ono funkcją pH układu.
Przy miareczkowaniu słabych kwasów, nie zauważa się początkowego spadku przewodnictwa, gdyż od razu powstaje mocny elektrolit. Pomimo tego po osiągnięciu punktu równoważnikowego, stężenie jonów hydroksylowych i ciągłe zwiększanie się sumy stężeń wszystkich jonów, powoduje powstanie wyraźnego załamania w punkcie zobojętnienia na krzywej miareczkowania. Przy miareczkowaniu mieszaniny mocny - słaby kwas, najpierw zostaje zneutralizowany mocny kwas.
28. Podział elektrod jonoselektywnych.
29. Elektrody stałomembranowe
Selektywność elektrod jest zależna od składu chemicznego szkła,
z którego została wykonana membrana.
Skład elektrody szklanej do oznaczania pH:
Na2O - 14%, SiO2 - 86% (% molowy), wsp. czułości Ag/Na - 1400
Skład elektrody szklanej do oznaczania jonów Ag+ i Na+:
Na2O - 11%, Al2O3 - 18%, SiO2 - 86% (% molowy)
Skład elektrody szklanej do oznaczania jonów K+ i NH4+:
Na2O - 27%, Al2O3 - 4%, SiO2 - 69% (% molowy) wsp. czułości K/NH4 - 1400
ektrody szklane do oznaczania stężenia kationów jednowartościowych
Elektrody krystaliczne homogeniczne
Membranę stanowi monokryształ lub układ polikrystaliczny trudno
rozpuszczalnych związków.
Jedną z pierwszych elektrod krystalicznych była elektroda fluorkowa
opisana w 1966 r. przez Franta i Rossa. Membranę stanowił w niej
monokryształ fluorku lantanu lub fluorku innych pierwiastków ziem
rzadkich. Fluorki te wykazują elektryczne przewodnictwo wynikające
z ruchliwości jonów fluoru w sieci krystalicznej.
Schematyczny mechanizm przenoszenia ładunków:
LaF3 + dziura cząsteczkowa LaF2+ + F-
Elektrody krystaliczne heterogeniczne- Elektrody heterogeniczne składają się z substancji aktywnej
zdyspergowanej na obojętnym nośniku. Zadaniem nośnika jest
polepszenie właściwości mechanicznych membrany.
Jako substancje aktywne stosuje się trudno rozpuszczalne sole
lub wymieniacze jonowe, natomiast jako nosniki stosuje się
parafinę, polistyren, PCV lub gumę silikonową. Nośnik powinien być substancją hydrofobową.
30. Elektrody ciekłomembranowe
Mała ruchliwość dwuwartościowych kationów w stałych membranach zainspirowała opracowanie membran ciekłych. Jedną z pierwszych była opracowana przez Rossa w 1967 r. elektroda z ciekłą membraną do oznaczania jonów wapnia. Membrana z hydrofobowego materiału (octan celulozy, spiekane szkło), nasycona roztworem wymieniacza organicznego. Oddziela ona zewnętrzny roztwór badany od wewnętrznego roztworu membrany o stałym stężeniu jonów, na które czuła jest elektroda.
Elektrody zawierające kationity.
W składzie roztworu wymieniacza organicznego są stosowane ciekłe kationity. Najczęściej stosuje się sole diestrów kwasu fosforowego lub sole kwasów karboksylowych zawierające grupę tioeterową. Służą m. in. do oznaczania: Zn2+, Cu2+, Ni2+, Ca2+.
Elektrody zawierające anionity.
W skład roztworu wymieniacza organicznego wchodzą najczęściej czwartorzędowe sole amonowe. Służą m. in. do oznaczania: NO3-, ClO4-, BF4-, I-.
Elektrody zawierające obojętne związki makrocykliczne, np. roztwory makrocyklicznych antybiotyków. Umożliwiają one np. pomiar stężenia jonów potasu w obecności jonów sodu.
31. Elektrody uczulane składają się z właściwej elektrody jonoselektywnej i układu przetwarzającego lub wyodrębniającego oznaczany składnik z próbki analitycznej. Do elektrod jonoselektywnych uczulanych należą elektrody enzymatyczne i gazowe
Elektrody uczulane, enzymatyczne
Klasycznym przykładem elektrody enzymatycznej jest elektroda służąca do oznaczania mocznika. Cząsteczki mocznika dyfundujące z objętości roztworu przez pierwszą błonę półprzepuszczalną do powierzchni elektrody ulegają w warstwie żelu zawierającego enzym ureazę rozkładowi enzymatycznemu zgodnie z reakcją enzymatyczną:
Powstałe w wyniku reakcji jony amonowe dyfundują przez drugą półprzepuszczalną błonę do powierzchni elektrody szklanej powodując zmianę jej potencjału.
Elektrody uczulane, gazowe- Wykorzystuje się je m. in. do pomiarów stężenia CO2, SO2 i NH3. Gaz dyfunduje przez membranę
i rozpuszcza się w roztworze wewnętrznym (roztwór wodny), na skutek czego w roztworze zmienia się stężenie jonów wodorowych:
CO2 + H2O HCO3- + H+
SO2 + H2O HSO3- + H+
NH3 + H2O NH4+ + OH-
32. Pomiary konduktometryczne
Podział technik konduktometrycznych:
konduktometria klasyczna,
konduktometria bezelektrodowa małej częstotliwości,
konduktometria wielkiej częstotliwości.
Konduktometria klasyczna, polega na pomiarze przewodnictwa słupa cieczy zawartego między dwiema elektrodami platynowymi, do których przykłada się napięcie prądu zmiennego o częstotliwości ok. 105 Hz
(na ogół od 1 do 10 kHz). Ładunek elektryczny w roztworach elektrolitów przepływa w wyniku uporządkowanego ruchu jonów w polu elektrycznym. Zgodnie z prawem Ohma rezystancja (opór) wyraża się wzorem:
Zakładając, że roztwór jest dostatecznie rozcieńczony (siły oddziaływania międzyjonowego są pomijalnie małe) i jony są kulami o średnicy niezależnej od natury rozpuszczalnika (pomijamy solwatację) wówczas ruchliwość jonu można określić następującą zależnością:
Zastosowanie:
Oprócz ogólnie znanych przykładów wykorzystania metod konduktometrii klasycznej (pomiar stopnia zasolenia oraz analiza miareczkowa), dzięki dużej czułości (na poziomie 10-7 - 10-6 mol/dm3), są one stosowane do badania wielu procesów chemicznych i biologicznych, m. in.:
do wyznaczania stopni i stałych dysocjacji elektrolitów słabych,
do wyznaczania iloczynów rozpuszczalności soli trudno rozpuszczalnych,
- do wyznaczania stałych trwałości kompleksów,
do badania kinetyki procesów dyfuzyjnych,
do badania rozwoju kultur bakteryjnych,
w medycynie i inżynierii materiałowej,
w inżynierii rolniczej.
Konduktometria bezkontaktowa (bezelektrodowa)
Stosowana w przypadkach, gdy nie jest możliwe lub wskazane zanurzanie elektrod w analizowanym roztworze.
Badany roztwór umieszczany jest w zamkniętym, niemetalowym (np. szklanym,
z tworzywa sztucznego) naczyniu, umieszczonym pomiędzy uzwojeniami dwóch tran Konduktometria bezelektrodowa wielkiej częstotliwości (oscylometria
Oscylometria polega na pośrednim pomiarze admitancji
(przewodności pozornej) lub impedancji (oporu pozornego) naczyńka pomiarowego zawierającego badany roztwór, podczas przyłożenia napięcia przemiennego wielkiej częstotliwości. Zmiany własności elektrycznych są związane ze zmianami stężenia, można więc z pomiaru parametrów obwodu wnioskować o stężeniu roztworu.
33. Prawa gazowe.
Prawo Boyle'a - Mariotte'a (1672)
p1v1 = p2v2 = pv = const.
Prawa Charlesa i Gay-Lussaca (1802)
Prawo to podaje zależność objętości gazu od temperatury,
a więc obowiązuje przy stałym ciśnieniu (p = const.).
Jest to równanie dla prze vt = v0x (1 + a x t)
mian izobarycznych: gdzie: v0 - objętość gazu w temp. 0 0C,
vt - objętość gazu w temp. t,
a - współczynnik rozszerzalności gazów, jednakowy dla wszystkich
gazów równy 1/273,15 = 0,0036
Prawa Charlesa i Gay-Lussaca (1802)
II prawo Prawo to podaje zależność ciśnienia gazu od temperatury, w pewnej stałej objętości gazu, czyli v = const.
Jest to równanie dla przemian izochorycznych:
pt = p0x (1 + a x t)
gdzie: p0 - ciśnienie danej objętość gazu w temp. 0 0C,
pt - ciśnienie gazu w temp. t,
a - współczynnik rozszerzalności gazów, jednakowy dla wszystkich
gazów równy 1/273,15 = 0,0036
Prawo objętościowe Gay-Lussaca (1809)
Objętości gazów wchodzących w połączenia chemiczne pozostają do siebie w prostych stosunkach liczbowych. Objętość powstającego reakcji chemicznej produktu gazowego pozostaje również w prostym stosunku liczbowym do objętości gazów wyjściowych.
wodór + chlor = chlorowodór
1 objętość + 1 objętość = 2 objętości
wodór + tlen = woda
2 objętości + 1 objętość = 2 objętości
Hipoteza Avogadra (1811
Objętości 1 mola gazów w warunkach normalnych wynosi 22,214 dm3.
Liczba Avogadra:
liczba cząstek N, zawarta w 1 molu gazu, czyli w objętości 22,214 dm3.
N = 6,024 x 1023
34. Równanie Clapeyrona (stanu gazu doskonałego)
Równanie to podaje zależność pomiędzy ciśnieniem, objętością, temperaturą a liczbą moli gazu i można je wyprowadzić z wcześniej przedstawionych praw gazowych.
35. Teoria kinetyczna gazów
W temperaturach powyżej zera absolutnego (tj. powyżej -273,15 oC) atomy
i cząsteczki substancji znajdują się w nieustannym ruchu. W ciałach stałych ruch ten nie jest wielki i odnosi się głównie do oscylacji atomów i cząsteczek względem węzłów sieci krystalicznej.
W miarę ogrzewania rośnie energia kinetyczna ruchów a w temperaturze zwanej temperaturą topnienia cząsteczki lub pojedyncze atomy tracą zdolność powrotu do swych pierwotnych położeń i substancja stała przechodzi w ciecz.
Wzajemne oddziaływanie między atomami i cząsteczkami jest jednak ciągle tak duże, że objętość cieczy w danej temperaturze pozostaje stała.
Dalsze ogrzewanie cieczy prowadzi wreszcie do stanu, w którym cząsteczki uzyskują energię kinetyczną wystarczającą do pokonania sił wzajemnego przyciągania i ciecz zamienia się w gaz, zdolny do rozprzestrzeniania się w całej dostępnej przestrzeni.
W danej temperaturze średnia wartość energii kinetycznej układu jest stała.
Nie oznacza to jednak, ze każda indywidualna cząsteczka znajdująca się
w tym układzie ma tę właśnie średnią wartość energii.
Ek,śr = mvśr2 / 2 = 3/2 x R/N x T
gdzie: m - masa molekuły, v - szybkość poruszania się molekuł,
R - stała gazowa, N - liczba Avogadra, T - temperatura [K]
Średnia energia kinetyczna molekuł jest jednakowa
dla wszystkich gazów w tych samych warunkach ciśnienia
i temperatury.
36. Gaz rzeczywisty.
Gazy rzeczywiste wykazują pewne odstępstwa od praw stanu gazu doskonałego.Odstępstwa te są tym większe, im gaz rzeczywisty znajduje się pod większym ciśnieniem, a jego temperatura jest bliska temperatury skroplenia. Van der Waals wprowadził poprawki do gazowego równania stanu gazowego uwzględniające wzajemne oddziaływanie cząsteczek i objętość własną cząsteczek. Dla 1 mola gazu rzeczywistego równanie ma postać:
(p + a/v2)(v-b) = RT
gdzie: v - objętość gazu rzeczywistego, p - ciśnienie, R - stała gazowa, T - temperatura,
a i b - stałe charakterystyczne dla danego gazu rzeczywistego.
Wartość a jest stałą wynikającą z istnienia sił przyciągania międzycząsteczkowego, natomiast b jest po Poprawka a/v2 nosi nazwę ciśnienia wewnętrznego gazu. Dodaje się ją do ciśnienia zewnętrznego p dlatego, że ciśnienia te mają zgodny kierunek działania.
Poprawka b zależy od wielkości i kształtu cząsteczki gazu rzeczywistego, oznacza tzw. sferę działania cząsteczek i jest równa w przybliżeniu poczwórnej objętości własnej cząsteczek. Poprawkę b wynikającą z istnienia objętości własnej cząsteczek odejmuje się od całkowitej objętości v zajmowanej przez gaz. Zachowanie się gazu rzeczywistego poddanego sprężaniu w różnych temperaturach można przedstawić graficznie. Na rysunku przedstawiono szereg izoterm otrzymanych doświadczalnie dla CO2 w różnych temperaturach.
38. Wykres fazowy wody
.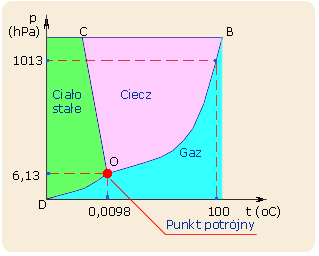
39. Reguła faz W. Gibbsa
Dla układu dwu- lub więcej składnikowego (n = 2, 3, ...), reguła faz Gibbsa przyjmuje postać:
n +2 = f + s
n - minimalna liczba składników niezależnych, tj. takich, przy pomocy których zbudować można wszystkie fazy występujące w danym układzie
40. Mieszanina azeotropowa (azeotrop) - ciekła mieszanina (roztwór) dwóch lub więcej związków chemicznych, która jest w równowadze termodynamicznej z parą nasyconą powstającą z tej mieszaniny. Gdy mówimy o azeotropie w kontekście wykresu fazowego, parametry takie określamy nazwą punktu azeotropowego.
Proporcje molowe (stężenie) związków chemicznych obecnych w parze nasyconej, powstającej w trakcie parowania mieszaniny azeotropowej, są dokładnie takie, jak w samej cieczy. Oznacza to, że po skropleniu pary znad mieszaniny azeotropowej uzyskuje się ciecz o dokładnie takim samym składzie chemicznym, jak wyjściowa mieszanina, co uniemożliwia jej rozdzielenie przez destylację, a nawet rektyfikację.
Mieszaniny azeotropowe rozdziela się zazwyczaj przez ekstrakcję, sorpcję lub poprzez dodanie do układu jeszcze jednego związku chemicznego, który tworzy azeotrop z jednym ze składników wcześniejszej mieszaniny i umożliwia dzięki temu wydestylowanie potrzebnego składnika.
Klasycznym przykładem azeotropu jest spirytus rektyfikowany, czyli mieszanina wody z etanolem o stężeniu alkoholu (zależnie od temperatury i ciśnienia) od 95,5% do 97,5%. Aby uzyskać prawie czysty etanol, do azeotropu można dodać benzen i kontynuować rektyfikację. Pozwala to uzyskać roztwór etanolu zawierający około 99,8% alkoholu i niewielkie ślady benzenu. Obecnie, ze względu na dużą toksyczność benzenu (nawet rzędu 1 ppm w powietrzu), tej metody już się nie stosuje.
.
S
-
+
G
K
A
h n/c
h n'/c
m v
Rozproszony foton
Odrzucony elektron
![]()
+
2p
2s
1s
H
H
H
H
N
+ Cl-
+
H
H
H
H
N
H
H
H
N
+ H+ Cl-
![]()
nA +mB = qC + rD
![]()
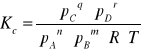
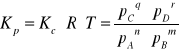
![]()
![]()
![]()
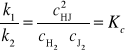
![]()
![]()
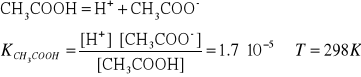

![]()
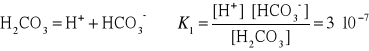
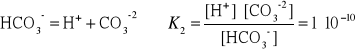

![]()
![]()
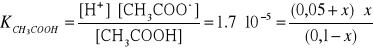
![]()
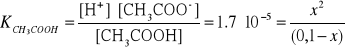
![]()
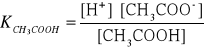
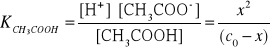
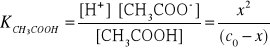
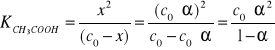
![]()
![]()
![]()
![]()
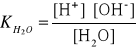
![]()
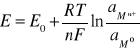
![]()
![]()
![]()
krystaliczne
homogeniczne
heterogeniczne
szklane
do oznaczania stężenia
jonów wodorowych
do oznaczania stężenia
kationów jednowartościowych
zawierające
kationity
zawierające anionity
zawierające związki makrocykliczne
enzymatyczne
gazowe
Elektrody jonoselektywne
stałomembranowe ciekłomembranowe uczulane
![]()
![]()
![]()
Przemiana izotermiczna
p0,v1, t = 1
p1,v0, t = 1
Przemiana izobaryczna
p0,v1, t = 1
p0,v0, t = 0
pv = p1v0 = povo(1+at) = povo x T/273,15
Wyszukiwarka