CHOROBY GENETYCZNE
Trzy procent dzieci rodzi się z wadami wrodzonymi,
z tego 85% defektów jest uwarunkowanych genetycznie.
Powstawanie wad jaja płodowego wiąże się ściśle z uszkodzeniem aparatu genowego chromosomów. Może dojść do tego podczas zapładniania komórki jajowej przez plemniki, gdy przekazane zostaną np. nieprawidłowe chromosomy. Może też mieć to miejsce w okresie gametogenezy (wytwarzania gamet). Do powstawania wad płodu może przyczynić się także uszkodzenie genu - lub kilku genów - w wyniku zadziałania niekorzystnego czynnika fizycznego lub chemicznego podczas różnicowania się listków zarodkowych.
Choroby genetyczne są chorobami przekazywanymi z pokolenia na pokolenie. Są to też choroby powstające de novo na skutek zmian i zaburzeń w mechanizmach przekazywania cech dziedzicznych. Te dopiero powstałe nieprawidłowości mogą być przekazywane potomstwu jako choroby dziedziczne. Jednym słowem, choroba genetyczna może mieć swój początek - na skutek sprzężenia się różnych czynników - w każdej chwili u każdego z nas.
ZESPÓŁ DOWNA - TRISOMIA autosomów 21 pary
Zespół Downa występuje u jednego na 600-800 żywourodzonych noworodków. Ryzyko urodzenia dziecka z zespołem Downa wzrasta wraz z wiekiem matki- jest znacznie większe wśród dzieci urodzonych z kobiet, które ukończyły 37 rok życia, niż u młodych matek.
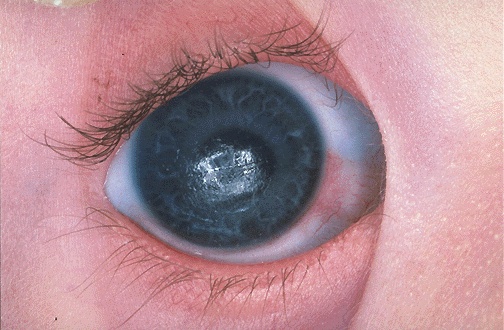
Zespołowi Downa towarzyszy wiele różnych objawów. Nie ma takiej cechy, która występowałaby u wszystkich chorych.
Większość dzieci dotkniętych tym schorzeniem ma charakterystyczny wyraz twarzy- skośne ustawienie szpar powiekowych (stąd skojarzenie z rasą Mongolską), opadające ku dołowi kąciki ust, otwarte usta, stosunkowo duży język, spłaszczoną nasadę nosa, czy bruzdę poprzeczną dłoni. Dodatkowo stwierdza się tzw. plamki Brushfielda na tęczówce, zmniejszenie nosa, małżowin usznych (nisko osadzonych i często zniekształconych) oraz żuchwy. Często stwierdzanym objawem jest także duży odstęp pomiędzy pierwszym i drugim paluchem stopy.
Dodatkowo noworodki z zespołem Downa są zwykle bardziej wiotkie niż ich rówieśnicy, często sprawiają problemy przy karmieniu- te dolegliwości są przejściowe i stosunkowo niegroźne, natomiast niektóre - takie jak wady serca (ok 40% chorych), mogą być groźne dla życia. Wady serca związane są głównie z zaburzeniem rozwoju poduszeczek wsierdziowych. Zazwyczaj są to wady dotyczące przegród międzyprzedsionkowej lub międzykomorowej, wady zastawek przedsionkowo-komorowych. Wady te stanowią najczęstszą przyczynę zgonów w wieku noworodkowym i niemowlęcym. Dzieci z zespołem Downa nieco częściej niż zdrowe chorują na białaczki. Dlatego bardzo ważna jest opieka lekarska przez cały okres życia. Inne częste problemy to przepukliny i brak jąder w worku mosznowym u chłopców (jądra niezstąpione). Zwykle w dalszej perspektywie obserwuje się opóźnione dojrzewanie płciowe i niski wzrost (u obu płci). Mężczyźni przeważnie są bezpłodni. Dziewczynki mogą miesiączkować, a niektóre zajść w ciążę.
Najczęstszym objawem jest upośledzenie umysłowe -zwykle znacznego stopnia (ok. 80% chorych wykazuje IQ w granicach 25-50).

Obecnie niemal 80% chorych z zespołem Downa przeżywa więcej niż 30 lat. Niemal wszyscy z trisomią 21 po czterdziestym roku życia zapadają na chorobę Alzheimera.
Dzieci z zespołem Downa z reguły są pogodne, ciepłe, bardzo przywiązane do rodziców. Niemniej są indywidualistami- każde z nich jest nieco inne, każde potrzebuje od nas czegoś innego, ale równocześnie czym innym może nas obdarować.
ZESPÓŁ TURNERA - całkowita utrata jednego chromosomu X (bądź Y) , oznaczana zapisem kariotypu 45,X lub 45,XO
Występuje on u 2-3/1000 żywo urodzonych dzieci płci żeńskiej. Częstotliwość tej choroby nie jest zależna od wieku matki i najprawdopodobniej polega na pomejotycznej utracie X lub Y w trakcie spermatogenezy.
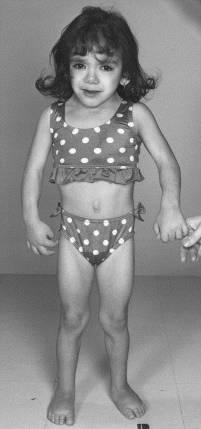
Dziecko z tym zespołem charakteryzuje się niskim wzrostem. ZT manifestuje się również opóźnionym dojrzewaniem, brakiem pierwszej miesiączki i owulacyjnych cykli miesięcznych, pierwotną niepłodnością, brakiem rozwoju gruczołów sutkowych, obniżonym stężeniem estrogenów i podwyższonym gonadotropin. U 20 - 30 % osób z ZT czynność jajników jest częściowo zachowana, występuje pierwsza miesiączka, jednak cykle menstruacyjne zanikają po kilku miesiącach (rzadziej latach). Tylko w sporadycznych przypadkach cykle są owulacyjne, zachowana jest płodność i prawidłowo rozwijają się piersi.
Wśród cech fenotypowych charakterystyczne są : zniekształcenia twarzy, płetwistość szyi, obrzęki rąk i nóg, niska linia włosów na karku, koślawość łokci, skrócenie IV kości śródręcza, skrócenie paznokci, niskie osadzenie i (lub) odstawanie małżowin usznych, szeroki rozstaw brodawek sutkowych, puklerzowata klatka piersiowa, opadanie powieki. Bardzo ważnym zaburzeniem jest obniżenie mineralizacji kośćca, co prowadzi do osteoporozy.
W ZT nie obserwuje się upośledzenia umysłowego. Iloraz inteligencji (IQ) jest normalny.
Leczenie polega na poprawie wzrastania - hormonem wzrostu oraz na wywołaniu feminizacji - estrogenami.
ZESPÓŁ KLINEFELTERA
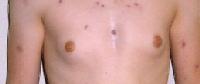
Choroba genetyczna spowodowana obecnością dodatkowego chromosomu X (lub większej ilości dodatkowych chromosomów X) w komórkach organizmu mężczyzny (np. genotyp 47,XXY lub 48, XXXY).
Osoby cierpiące na zespół Klinefeltera są najczęściej bezpłodne w wyniku nieprawidłowego rozwoju jąder. Sylwetka chorych może być zbliżona do kobiecej; może też występować ginekomastia, czyli rozrost gruczołów piersiowych. Czasami współistnieje lekkie upośledzenie umysłowe.
Zespół ten występuje od urodzenia i rozpoznaje się go tylko u mężczyzn. Jest chorobą genetyczną, związaną z posiadaniem dodatkowego jednego, bądź kilku, chromosomu X np. genotyp 47,XXY lub 48, XXXY. Jest rozpoznawany nieco częściej niż Zespół Downa i wiąże się z charakterystycznymi dla niego objawami fizykalnymi jak i psychicznymi.
{kind=link}
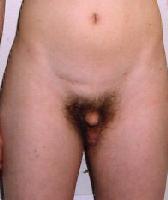
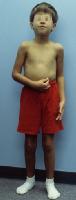
W dzieciństwie nie zauważa się dalekich różnic między chłopcami z Zespołem Klinefeltera a ich rówieśnikami, jednak już w okresie dorastania wyróżniają się znaczącym wzrostem (nieproporcjonalnie dłuższe kończyny dolne) oraz prawdopodobieństwem występowania zrośnięcia kości przedramienia. Mężczyźni z Zespołem Klinefeltera maja małe, twarde jądra, które wytwarzają mniej testosteronu niż w normie, dlatego też prącie jest niewielkie, owłosienie ciała skąpe i występuje bezpłodność. Obserwuje się także powiększone piersi. Mężczyźni z Zespołem Klinefeltera są przeważnie otyli, niektórzy chorują na cukrzyce. Mogą także pojawić się zaburzenia funkcjonowania tarczycy.
Osoby z Zespołem Klinefeltera cierpią na liczne problemy psychiczne: depresję, psychozę, niską aktywność płciową, parafilię (podpalenia oraz inne akty przemocy). Chorobie może towarzyszyć lekki niedorozwój umysłowy. Podawanie testosteronu może złagodzić skutki Zespołu Klinefeltera, zmniejszyć liczbę kryminogennych zachowań.
{kind=link}
Leczenie tej choroby polega na podawaniu testosteronu. Uzupełnianie tego hormonu umożliwia „korektę” sylwetki ciała i innych cech psychofizycznych, których stan zależy od prawidłowego poziomu testosteronu w organizmie.
ZESPÓŁ EDWARDSA - trisomia chromosomu 18
Częstotliwość występowania tej wady wynosi 0,3/1000 żywo urodzonych niemowląt, z tym że u dziewczynek występuje on 3 razy częściej. Wiek matki wpływa na częstotliwość występowania tego zespołu.
Większość dzieci przeżywa 3-4 miesięcy, wyjątkowo do 2 lat. W przypadku wczesnego zdiagnozowania wiek ten może się znacznie wydłużyć.
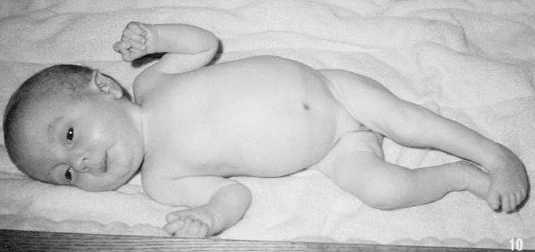
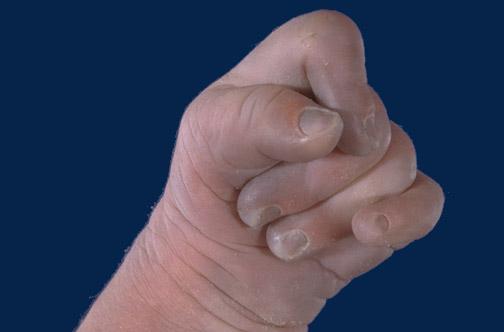
Dzieci z tym zespołem charakteryzują się m.in. wzmożonym napięciem mięśniowym, przykurczem kończyn, nisko osadzonymi uszami, pojawiają się wady stóp (tzw. suszkowate stopy), niemowlęta zaciskają charakterystycznie piąstkę. Często zespołowi Edwardsa towarzyszą wady serca bądź ośrodkowego układu nerwowego.
Jak w większości chorób genetycznych zespół objawów może znacznie różnić się od najczęściej wymienianych. W przypadku zespołu Edwardsa znane są sytuacje kiedy chorobie tej towarzyszyło zmniejszone napięcie mięśniowe i padaczka.
ZESPÓŁ PATAU - trisomia chromosomu 13
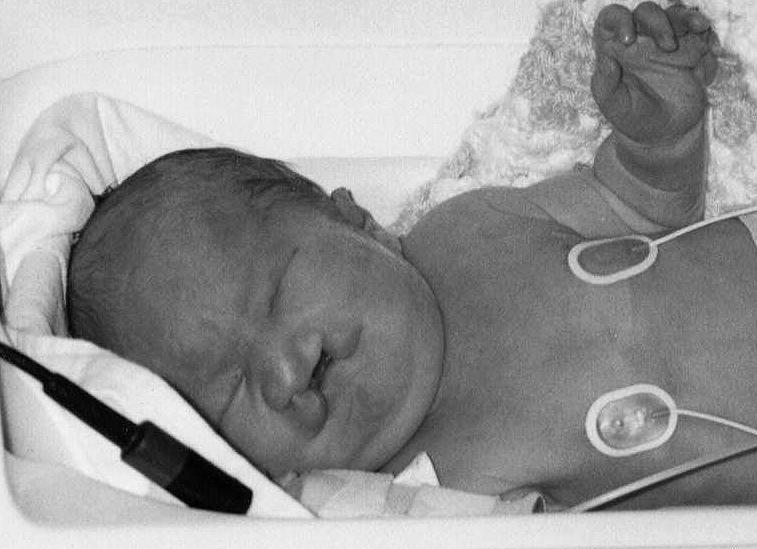
Częstotliwość występowania tej wady wynosi 0,2/1000 żywo urodzonych niemowląt. Wiek matki, choć w niewielkim zakresie, wpływa na częstotliwość występowania tego zespołu.
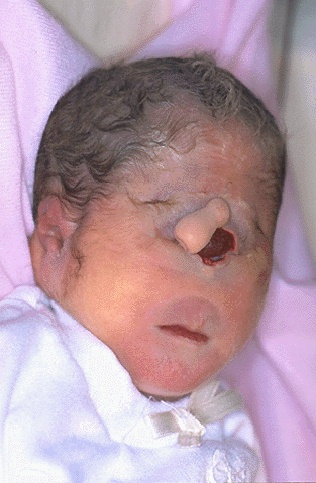
Dzieci z zespołem Patau umierają zazwyczaj w kilka godzin lub dni po urodzeniu. Tylko 1 dziecko z 20 przeżywa dłużej niż 6 miesięcy. Jednakże niektóre dzieci przeżywają okres dojrzewania. Osoby dorosłe z tym zespołem są rzadkością.
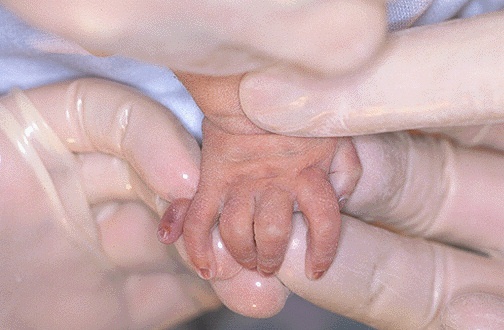
Chorzy z Zespołem Patau charakteryzuje się ubytkami owłosienia na głowie, oczy są osadzone blisko siebie (może dojść nawet do ich "połączenia").
Zespołowi temu towarzyszy często rozczep wargi i podniebienia, polidaktylia czyli dodatkowy palec, najczęściej od strony palca małego, zdarzają się wytrzewienia.
Z chorób towarzyszących liczne są wady serca, występują one w 80% przypadków. Zazwyczaj obejmują one takie zaburzenia jak : przetrwały przewód tętniczy, defekt przegrody międzyprzedsionkowej i międzykomorowej, dextrocardia (prawostronne położenie serca).
Kliniczne fenotypy zespołu Patau i zespołu Edwardsa mogą być podobne i zdarza się że lekarze mylą te choroby.
ZESPÓŁ RUSSEL-SILVER (RSS)
Jednym z interesujących a jednocześnie sprawiającym wiele trudności aspektów tej choroby jest bardzo szeroki i zróżnicowany zespoły objawów.

Pomimo różnorodnych objawów można dostrzec pewne prawidłowości. Prawie u każdego chorego pojawiają się : mała waga urodzeniowa, zmniejszenie długości noworodka, zbliżony do trójkątnego kształt twarzy, normalna wielkość głowy, choć z powodu zmniejszenia długości ciała i wagi może się ona wydawać większa, pourodzeniowe opóźnienie wzrostu, słaby apetyt w pierwszych latach, klinodaktylia (zakrzywienie) piątego palca.

Do często towarzyszących temu zespołowi objawów należą: hipoglikemia (w okresie niemowlęcym i wczesnym dzieciństwie (2-3 roku)), asymetria długości kończyn, mały podbródek, nieprawidłowo osadzone uszy (zazwyczaj niżej niż normalnie o mniejszym kształcie i często odstające), wnętrostwo (nie zstąpienie jąder), zmniejszone napięcie mięśniowe.
Rozpoznanie tego zespołu nie oznacza zmniejszenia jakości życia. Osoby z RSS prowadzą normalne życie. Jednakże niski wzrost i pewne trudności w wieku dziecięcym takie jak brak apetytu, zmniejszenie napięcia mięśniowego, asymetria i inne z wcześniej wymienionych objawów, mogą powodować pewne trudności. Zazwyczaj jednak włożony wysiłek, dodatkowa troska i opieka przynoszą rezultaty. U wielu pacjentów znika "trójkątny" kształt twarzy, wzmaga się napięcie mięśniowe, poprawia się apetyt, mowa staje się bardziej wyraźna, rozwija się koordynacja ruchów, zdolność nauki poprawia się. Dzieci z zespołem Russel-Silvera zazwyczaj stają się zdrowymi i szczęśliwymi dorosłymi.
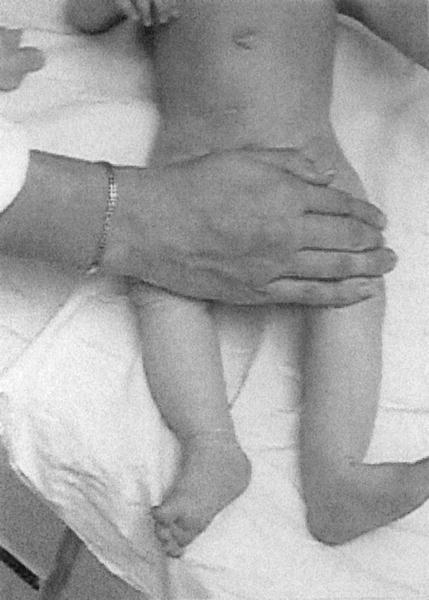
Etiologia tej choroby jest nadal nieznana. Niektóre przypadki RSS występowały wśród kilku członków rodziny, jednak zazwyczaj choroba ta pojawia się sporadycznie, bez obciążenia genetycznego, a także bez określenia konkretnych mutagenów. Prace nad dokładną lokalizacją genów odpowiedzialnych za rozwój tej choroby nadal trwają. W wielu przypadkach pojawiały się dwa chromosomy 7 pochodzące od matki (normalnie matka przekazuje tylko jeden chromosom swojemu dziecku).
ZESPÓŁ ANGELMANA

Przyczyną tego zespołu jest mikrodelecja odcinka 15 chromosomu pochodzącego od ojca (gdy dojdzie do mikrodelecji odc. 15 chromosomu pochodzącego od matki mamy wtedy do czynienia z zespołem Prader-Williego). Brakujący odcinek chromosomu odpowiedzialny jest m.in za regulację białka zwanego ubikwityną.
Zespół Angelmana nie jest zazwyczaj rozpoznawany w okresie noworodkowym i niemowlęcym, dopiero gdy pojawiają się problemy rozwojowe, gdy ujawnia się charakterystyczne zachowanie, rodzice zaczynają podejrzewać istnienie choroby (następuje to zwykle miedzy 3 a 6 rokiem życia).

Do cech charakterystycznych tego zespołu należy: opóźnienie w rozwoju, upośledzenie mowy, zaburzenia ruchu i równowagi (ataksja, drżące ruchy kończyn), częsty śmiech bez powodu, osoba chora często się rozkojarza, łatwo się ekscytuję, odznaczą się nadmierną pobudliwością.
Do równie częstych objawów (ok. 80%) należą: nieproporcjonalny rozwój obwodu głowy (zazwyczaj prowadzący do małogłowia), napady padaczki (zwykle pojawiają się przed 3 rokiem życia), nieprawidłowy zapis EEG.
Wśród osób z zespołem Anglemana pojawiają się także : zez, bruzda tętnicy potylicznej, grzybica języka, problemy z karmieniem (głównie w okresie niemowlęcym), wysunięcie żuchwy do przodu (prognathia), zmniejszona ilość barwnika w skórze (również we włosach i w oczacz, stąd zwykle osoby z ZA mają jasne włosy i niebieski kolor oczu), zaburzenia snu, zwiększone zainteresowanie wodą (mogące nawet przechodzić w fascynację).
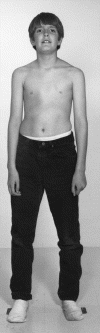
ZESPÓŁ MARFANA - jest chorobą tkanki łącznej, o zmiennej ekspresji, objawy mogą być ostre lub bardzo łagodne, mogą pojawić się tuż po urodzeniu, w dzieciństwie bądź dopiero w dorosłym życiu.
W 70- 85% przypadków choroba jest dziedziczona autosomalnie dominująco; u pozostałej części chorych (ok. 1/4) nie stwierdza się u rodziców żadnych zaburzeń. W tych przypadkach choroba jest wynikiem nowo powstałych, spontanicznych mutacji.
Molekularnym podłożem tej choroby jest mutacja genu strukturalnego fibryliny zlokalizowanego na ramieniu długim chromosomu 15. (loci genu: 15q2) Białko to jest jednym z głównych składników włókien tworzących macierz zewnatrzkomórkową. Stanowi ona swego rodzaju rusztowanie, do którego przyłączają się podjednostki tropoelastyny; wszystkie te elementy wchodzą w skład włókien elastynowych. Szczególnie duże ilości tego rodzaju włókien znajdują się w ścianie aorty, ścięgnach oraz w obwódce rzęskowej soczewki i te narządy są miejscem głównych zaburzeń charakterystycznych dla zespołu Marfana.
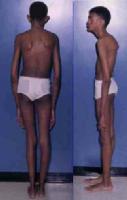
U chorych z zespołem Marfana mogą pojawić się dolegliwości ze strony:
- układu szkieletowego; rozluźnienie połączeń stawowych, wysoka i szczupła sylwetka ciała, nieproporcjonalnie długie ręce i nogi w stosunku do tułowia, arachnodaktylia (pająkowate palce); zdarza się, że połówki ciała chorego są asymetryczne np. jedna ręka jest grubsza i dłuższa od drugiej. Ta asymetria ciała może być przyczyną bocznego skrzywienia kręgosłupa, który jest słaby i wiotki. Czasami pojawia się wydrążona (szewska) klatka piersiowa (pectus excavatum). Stopy są zwykle albo nadmiernie wysklepione, albo płaskie. Ścięgna rąk i stóp są wiotkie, umożliwiając wykonywanie ruchów niemożliwych do wykonania w prawidłowo zbudowanych stawach, np. charakterystyczny jest nadmierny przeprost kciuka. Często również czaszka jest wydłużona (dolichocephalia), ze szczególnie uwypuklonymi wyniosłościami czołowymi i silnie zarysowanymi wałami nadoczodołowymi. Podniebienie twarde jest wysoko wysklepione.

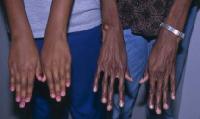
- narządu wzroku: przemieszczenie soczewki (jednostronne lub obustronne), najczęściej do przedniej komory oka i ku górze. Ze względu na nieprawidłowe napięcie włókien obwódkowych soczewka przyjmuje kształt bardziej kulisty niż prawidłowy, a kształt gałki ocznej w osi optycznej jest wydłużony, co powoduje krótkowzroczność (myopia). U niektórych chorych dochodzi również do odklejania siatkówki, co z czasem może prowadzić do jej rozdarć i zwyrodnienia. Pojawia się też czasami jaskra czy też zaćma.
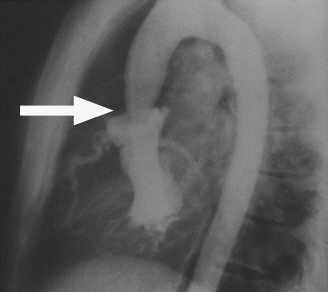
- układu krążenia: nieprawidłowa budowa ścian naczyń krwionośnych z powodu złej struktury tkanki łącznej. Nieprawidłowości dostrzegane są głównie w obrębie aorty, która jest osłabiona (zazwyczaj ulega rozszerzeniu w wyniku czego dochodzić może do jej rozwarstwienia a nawet pęknięcia). Często występującym i łatwym do wykrycia przez lekarza objawem jest słyszalny, charakterystyczny szmer w sercu, pojawiający się w następstwie defektu zastawek serca
- układu nerwowego: w miarę upływu lat, u osób starszych z zespołem Marfana, dochodzić może do osłabienia i rozciągnięcia opon otaczających struktury ośrodkowego układu nerwowego (dura ectasia), głównie w odcinku lędźwiowo-krzyżowym.
Poziom umysłowy dziecka nie odbiega zwykle od normy.
Z powodu słabości mięśni klatki piersiowej , a także skrzywienia bocznego kręgosłupa - zwłaszcza jeżeli jest duże - upośledzone jest oddychanie dziecka. Dlatego dzieci z zespołem Marfana często chorują na zapalenie oskrzeli, płuc, a wielu dorosłych cierpi na astmę.
U schyłku XIX wieku i do lat 60-tych XX wieku chorzy z zespołem Marfana umierali w młodości (do 30 roku życia). Najczęstszą przyczyną było pęknięcie tętniaka aorty (np. u młodego mężczyzny, który chorował na zespół Marfana nie wiedząc o tym, pęknięcie aorty nastąpiło w czasie wysiłku fizycznego, młode kobiety pod koniec ciąży umierały z tego samego powodu). Obecnie istnieją znacznie większe możliwości pomocy chorym i dlatego mogą oni dożyć późnej starości.
MUKOWISCYDOZA - zwłóknienie torbielowate (cystic fibrosis,CF)
Mukowiscydoza jest najczęstszym schorzeniem genetycznym białej rasy, występującym raz na 3000 urodzeń. Przyczyną choroby jest mutacja genu CFTR (ang. cystic fibrosis transmembrane regulator). W 80% przypadków mutacja polega na delecji trzech nukleotydów kodujących fenyloalaninę. Gen tej choroby umiejscowiony jest na długim ramieniu chromosomu VII.
CFTR jest transbłonowym białkiem związanym z błonowym kanałem dla jonów chlorkowych. Brak CFTR powoduje zaburzenia wydzielania elektrolitów i wody w nabłonkach. Wskutek tego dochodzi do zalegania gęstej śluzowej wydzieliny i zmian z tym zwiazanych, przede wszystkim w płucach i trzustce.
Organizm chorego produkuje nadmiernie lepki śluz, który powoduje zaburzenia we wszystkich narządach posiadających gruczoły śluzowe (m.in. w płucach, układzie pokarmowym).
Mukowiscydoza jest chorobą ogólnoustrojową, objawiającą się przede wszystkim przewlekłą chorobą oskrzelowo-płucną oraz niewydolnością enzymatyczną trzustki z następowymi zaburzeniami trawienia i wchłaniania. Gruczoły potowe wydalają pot o podwyższonym stężeniu chloru i sodu (tzw. "słony pot").
ZESPÓŁ WILLIAMSA
Za przyczynę zespołu Williamsa uważa się delecje genu elastyny i enzymu nazywanego LIM kinazą na chromosomie 7. W normalnych komórkach elastyna jest kluczowym skłądnikiem tkanki łącznej odpowiadającym za jej elastyczność. Mutacja lub delecja genu elastyny prowadzi głównie do zmian w naczyniach krwionośnych. Z drugiej strony LIM kinaza występuje głównie w mózgu i przypuszcza się że ma ona związek ze zdolnością do oceny relacji przestrzennych. Mogłoby to wyjaśnić, dlaczego ludzie z zespołem Williamsa mają trudności z narysowaniem z pamięci znanych prostych przedmiotów.
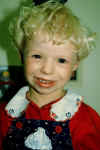

Ludzie z zespołem Williamsa przeważnie cierpią na różnego stopnia - od niewielkiego do znacznego - nadzastawkowe zwężenie tętnicy głównej (SVAS - supravalvular aortic stenosis). Potem lekarze dostrzegli jeszcze inne cechy. Niektóre z nich ujawniają się w bardzo wczesnym okresie życia. Już w niemowlęctwie mogą wystąpić trudności z przyjmowaniem pokarmu, bóle brzucha i przepuklina. Dzieci źle sypiają, bywają drażliwe i miewają kolkę - co niekiedy wynika z często obserwowanego u nich podwyższonego poziomu wapnia we krwi. Od samego początku mówią ochrypłym głosem i są opóźnione w rozwoju fizycznym oraz umysłowym. Zaczynają chodzić na ogół w wieku 21 miesięcy, zazwyczaj niezgrabnie, często stawiając stopę "z palców", co zostaje im na całe życie. Mają również upośledzoną kontrolę ruchów precyzyjnych. Co więcej, ludzie z zespołem Williamsa są niezmiernie wrażliwi na hałas, niżsi niż rówieśnicy i przedwcześnie się starzeją (np. siwieją i robią im się zmarszczki w stosunkowo młodym wieku). Zespołowi temu też towarzyszą nieprawidłowości w budowie nerek.
Twarz chorego z Zespołem Williamsa nazywana jest "twarzą elfa". Charakteryzuję się ona: krótkim zadartym nosem, płaskim grzbietem nosa, długą rynienką podnosową (philtrum), płaską okolicą jarzmową, szerokimi ustami, nieprawidłowym zgryzem i małą żuchwą (micrognathia).
ZESPÓŁ CRI DU CHAT
Przyczyną choroby jest częściowa delecja krótkiego ramienia 5 chromosomu.
Charakterystyczny płacz jest podobny do miauczenia kota. Jest on spowodowany nieprawidłową strukturą krtani. Zazwyczaj nieprawidłowości dotyczą asymetrycznej budowy strun głosowych, opadającej nagłośni, małej krtani. Jednakże przyczyna charakterystycznego płaczu nie może być wyłącznie kojarzona z nieprawidłowościami w obrębie krtani. Obszar mózgu odpowiedzialny za kierowanie prawidłową czynnością krtani jest też prawdopodobnie zdeformowany. Charakterystyczny płacz zazwyczaj ustępuje z czasem, u około 1/3 dzieci dzieje się to przed ukończenie drugiego roku życia.
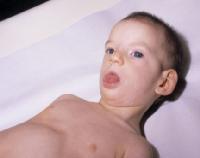
Osoby z zespołem cri-du-chat mogą odznaczać się hiperaktywnością, agresją, nagłym wybuchem złości, zdolnością do samookaleczeń, powtarzającymi się ruchami, nadwrażliwością na dźwięk, niezgrabnością.
Do komplikacji występujących u noworodków zalicza się: słabe ssanie, potrzebę opieki w inkubatorze, zespół zaburzeń oddechowych, żółtaczkę, zapalenie płuc i odwodnienie. Dodatkowo pojawia się niska waga urodzeniowa, obniżone napięcie mięśniowe, małogłowie, opóźnienie wzrostu, okrągła twarz z "pełnymi" policzkami, hiperteloryzm, nisko osadzone uszy, krótkie palce, micrognathia (nieprawidłowo mała żuchwa), zmarszczka nakątna (skóra nasady nosa rozciąga się na wewnętrzny kąt oka). Pojawiają się też zaburzenia ze strony układy krążenia: nieprawidłowość w budowie przegrody międzyprzedsionkowej i międzykomorowej , przetrwały przewód tętniczy czy też tetralogia Fallota.

W okresie dzieciństwa nieprawidłowości obejmują: niedorozwój umysłowy, małogłowie, wzmożone napięcie mięśniowe, przedwczesne siwienie włosów, mała, pociągła, często asymetryczna twarz, opuszczona żuchwa, otwarte usta- jako następstwo zwiotczenia twarzy, krótka rynienka podnosowa, nieprawidłowy zgryz, krótkie kości śródręcza (skrócenie obejmuje zazwyczaj od 3 do 5 kości śródręcza). W tym okresie zdarzają się nawracające choroby takie jak: infekcje górnych dróg oddechowych, zapalenie ucha środkowego, ciężkie zaparcia.
W okresie późnego dzieciństwa i w okresie dojrzewania można wyróżnić takie problemy jak opóźnienie w rozwoju, małogłowie, głęboko osadzone oczy, nieprawidłowy zgryz i skoliozę.
Wymienione wyżej objawy mogą wystąpić z różnym nasileniem (jak w większości chorób genetycznych)
ZESPÓŁ WERNERA / (PROGERIA Hutchinsona-Gilforda) / HGPS / Przedwczesny starczy wygląd - nazwa tej choroby wywodzi się z języka greckiego i oznacza "przedwczesną starość". Zespół ten charakteryzuję się przyspieszonym procesem starzenia. Dziedziczy się jako cecha autosomalna dominująca.

Progeria u osób dorosłych nazywana jest zespołem Wernera.
U dzieci z progerią później formują się zęby, mają starszy wygląd skóry (m.in. liczne zmarszczki), są zwykle łyse i karłowate. Z tychże powodów wyglądają one o wiele starzej. Nos jest "ściągnięty", policzki zapadnięte (z powodu utraty tkanki tłuszczowej).
Utrata tkanki tłuszczowej może być również przyczyną owrzodzeń na przedniej powierzchni goleni i na stopach.
Mogą też pojawiać się problemy ze sztywnością stawów, problemy z przemieszczaniem się stawu biodrowego. Zdarzają się krwawienia z nosa, bóle głowy, stwardnienie tętnic i problemy sercowo-naczyniowe. Zanotowano także przypadki gdy dochodziło do zaniku mięśni. Charakterystyczny jest także wysoki ton głosu.
Czaszka jest w porównaniu z resztą ciał duża, z nieproporcjonalną dolną częścią twarzy.
Aktualny wiek ciała chorych zmienia się o 8-10 lat na każdy przeżyty przez chorego rok. Jednak rozwój umysłowy nie jest zaburzony. Osoby z tym zespołem wykazują iloraz inteligencji powyżej przeciętnej. Chorzy wykazują też zwiększone ryzyko wystąpienia nowotworów.
Pierwsze objawy progerii można dostrzec u dzieci powyżej 2 roku życia. Należą do nich: napięcie skóry, nieprawidłowy przyrost wagi, utrata włosów na głowie. Na tym etapie zazwyczaj rodzice zaczynają przypuszczać istnienie jakieś choroby.
Podsumowując, do charakterystycznych cech w pełnoobjawowej progerii należą:
- niski wzrost
- niska masa ciała
- niedojrzałość płciowa
- duża głowa w stosunku do twarzy
- niedorozwój żuchwy
- przepełnienie żył skóry głowy
- uogólnione łysienie (brak rzęs i brwi)
- wyłupiaste oczy
- opóźniony rozwój uzębienia
- gruszkowata klatka piersiowa
- krótkie obojczyki
- koślawe biodra
- chód na szerokiej podstawie
- cienkie kończyny
- wystające cienki stawy
- cienka, sucha skóra z brązowymi plamami i zmarszczkami
- „rzeźbiony”, ptasi koniec nosa
- dystroficzne paznokcie
- wysoko brzmiący głos
Czasami wygląd opisywany jest jako: „oskubanego ptaka”.
Na dzień dzisiejszy nie wynaleziono lekarstwa na tę chorobę ani nie określono ścisłych reguł w terapii. Przeciętna długość życia osób z progerią wynosi pomiędzy 7 a 27 lat (średnio około 13 lat). Zgon następuje najczęściej z powodu postępującej miażdżycy naczyń wieńcowych i mózgowych.
Stosuje się jedynie leczenie fizykoterapeutyczne w celu zapobiegania przykurczom mięśni.
ACHONDROPLAZJA - jest genetycznie uwarunkowaną chorobą spowodowaną mutacją pojedynczego genu (FGFR-3), kodującego receptor 3 czynnika wzrostu fibroblastów. (loci genu: 4p16.3)
Osoby z tym schorzeniem odznaczają się głównie niskim wzrostem (karłowatością) i zaburzeniami proporcji ciała. Częstość występowania choroby wynosi 1:25000 urodzeń i dotyczy w równym stopniu obu płci i wszystkich ras. Średnia wzrostu osiąganego przez chorych z achondroplazją wynosi u mężczyzn 131 cm, a u kobiet 124 cm.
W większości przypadków zaburzenie jest wynikiem mutacji genetycznej (żadne z rodziców nie przekazuje dziecku choroby), ale czasami dzieci dziedziczą chorobę po rodzicu. Ryzyko zachorowania jest większe, jeżeli w rodzinie wcześniej wystąpił przypadek choroby
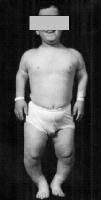
Do występujących nieprawidłowości należą:
- skrócenie długości kończyn (głównie części bliższych kończy tzn. ud i przedramion)
- duża głowa z zapadniętą nasadą nosa
- brachydaktylia (krótkopalczastość)
- genu varum (kolano szpotawe)
- przykurcze w stawach
- krótka szyja
- kifoza kręgosłupa w odcinku lędźwiowym
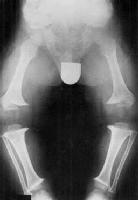
- krótkie nasady łuków kręgów, które mogą powodować zwężenie kręgosłupa, a także związane z patologią kanału kręgowego różnego rodzaju objawy neurologiczne
- dysplastyczna kość biodrowa
- zaburzenia proporcji ciała (jako wynik skrócenia długości kończyn w stosunku do długości tułowia)
Rozwój intelektualny chorych jest z reguły prawidłowy.
Choroba ta znana była w starożytności. Egipskiego boga Ptaha wyobrażano w plastyce jako achondroplastycznego karła. Achondroplastyków zatrudniano jako błaznów nadwornych, później jako wróżbitów czy też klaunów cyrkowych. Znanym achondroplastykiem był malarz francuski Henri Toulouse-Lautrec.
Klasyczna FENYLOKETONURIA (phenyloketonuria - PKU) - jest rzadko występującą chorobą metaboliczną. Jej przyczyną jest zwykle niedobór enzymu : hydroksylazy fenyloalaninowej (PAH) - enzymu wytwarzanego w wątrobie. Defekt ten prowadzi do zwiększenia stężenia fenyloalaniny (Phe) we krwi i w innych tkankach. Nieleczona choroba charakteryzuje się występowaniem niedorozwoju umysłowego, małogłowia, opóźnienia rozwoju mowy, drgawek, wyprysku, zaburzeń zachowania oraz innych objawów.
Blokada normalnej drogi przekształcenia fenyloalaniny do Tyrozyny powoduje wzrost koncentracji Phe we krwi. U zdrowego dziecka poziom ten wynosi do 2mg/100 ml krwi, zaś u nie leczonego chorego na fenyloketonurię jest zazwyczaj dziesięciokrotnie wyższy. Wysoki poziom fenyloalaniny we krwi powoduje zaburzenia rozwoju układu nerwowego prowadząc do ciężkiego uszkodzenia mózgu. Jest to na niebezpieczne szczególnie w pierwszych latach życia dziecka, gdyż mózg rozwija się wtedy najintensywniej. Im wcześniej rozpoczyna się leczenie fenyloketonurii, tym jest ono skuteczniejsze, dlatego też każdy noworodek poddany jest badaniu przesiewowemu w kierunku fenyloketonurii.
Organizm osoby chorej na fenyloketonurię nie jest zdolny do prawidłowego metabolizmu jednego ze składników diety - fenyloalaniny. Aminokwas fenyloalanina znajduje się w pokarmach zawierających dużo białka, takich jak: mięso, jaja, ryby, mleko, ser oraz (w mniejszych ilościach) w produktach zbożowych, warzywach i owocach.
Aktualnie leczenie fenyloketonurii polega na utrzymywaniu ścisłej kontroli metabolicznej za pomocą diety o małej zawartości fenyloalaniny, co wymaga stosowania specjalnych leczniczych produktów żywnościowych. Elementami, które należy w znacznym stopniu wyeliminować z jadłospisu, są produkty mięsne, ryby, jaja, ser, mleko i jego przetwory oraz chleb. Zamiast nich używa się sztucznie przyrządzone preparaty, takie jak Lofenalac, oraz inne produkty.
Skuteczność programów badań przesiewowych noworodków w kierunku PKU jest duża - niemowlęta, u których chorobę rozpoznano we wczesnym okresie życia i natychmiast rozpoczynano leczenie, uzyskując dobrą metaboliczną kontrolę choroby, pozostawały zdrowe i prawidłowo się rozwijały, a ich oczekiwana długość życia prawdopodobnie będzie taka sama, jak w populacji ogólnej.
CHOROBA HUNTINGTONA - jest dziedziczną chorobą mózgu
"Jej nazwa pochodzi od nazwiska lekarza, Georga Huntingtona, który jako pierwszy opisał ją w 1872 roku. Choroba ta nazywana była początkowo pląsawicą Huntingtona.
Choroba Huntingtona powoduje obumieranie komórek (neuronów) w niektórych częściach mózgu: jądrze ogoniastym i skorupie oraz w miarę rozwoju choroby w korze mózgowej. Jądro ogoniaste i skorupa powiązane są z wieloma innymi rejonami mózgu i pomagają w kontrolowaniu ruchów ciała, emocji, myślenia oraz w postrzeganiu świata.
W miarę obumierania komórek mózgu, osoby z chorobą Huntingtona tracą możliwość kontrolowania ruchów i emocji, przypominania sobie ostatnich zdarzeń oraz podejmowania decyzji. Choroba prowadzi do niepełnosprawności i śmierci.[..]"
"Choroba Huntingtona jest genetycznie uwarunkowana, spowodowana obecnością "wadliwego" (zmutowanego) genu na chromosomie 4. Gen choroby jest dominujący, co oznacza, że dzieci chorego rodzica (każde) mają 50% ryzyko odziedziczenia tej choroby i określa się je jako "osoby z ryzykiem zachorowania". Ryzyko odziedziczenia choroby jest takie samo dla mężczyzn i kobiet.
Choroba Huntingtona występuje u przedstawicieli wszystkich ras, ale najczęściej spotykana jest u osób pochodzenia europejskiego.
Choroba Huntingtona dotyczy przede wszystkim osób dorosłych. Objawy pojawiają się zazwyczaj pomiędzy 30 i 45 rokiem życia, ale choroba może ujawniać się nawet u 5-cio letnich dzieci, jak również u dorosłych po 70 roku życia. [...]"
CHOROBA ATAXIA FREDREICHA
Ataxia Fredreicha (FA) jest chorobą genetyczną. Powoduje ją uszkodzony gen (fragment chromosomu) zlokalizowany na chromosomie 9. Jak każdy inny gen, gen odpowiedzialny za FA zawiera informację o produkcji białek. Ten gen zawiera informację o 216 aminokwasowym białku frataksynie. Istotną rzeczą jest, żeby wiedzieć, że chromosomy to maleńkie struktury zlokalizowane w jądrze komórek i że to one właśnie przekazują informację genetyczną. Chemicznie chromosomy są uformowane z molekuł (cząsteczek) DNA.DNA zawiera całą informację genetyczną i dzięki niej wszystkie cechy mogą być przekazywane z pokolenia na pokolenie. DNA jest zbudowany z 4 podstawowych aminokwasów: adeniny, guaniny, cytozyny i tymidyny. Genetyczna informacja jest utworzona z setek tych aminokwasów ułożonych w trójki w różnych kombinacjach.U większości pacjentów z FA występuje uszkodzenie kolejności aminokwasów w genie odpowiedzialnym za budowę frataksyny. W DNA odpowiedzialnym za to ważna jest ilość nieprawidłowych „powtórzeń trójek aminokwasowych”. U zdrowego pacjenta takie powtórzenia występują 7-22 razy. U pacjenta z FA zdarza się to 150-1000 razy. To powoduje niedobór prawidłowo zbudowanej i działającej frataksyny.
Rola frataksyny nie jest jeszcze do końca poznana ale na pewno wiemy, że:
Frataksyna jest ważnym białkiem dla funkcji ważnych narządów
Powinna być obecna w mitochondriach (są to organy komórkowe znajdujące się w cytoplaźmie wewnątrzkomórkowej). Wszystkie komórki zawierają mitochondria. Jest to miejsce produkcji energii; mitochondria to małe agregaty produkujące energię w komórce. Produkują one 90% energii potrzebnej tkankom, organom i całemu organizmowi żeby funkcjonował. Dzięki nim możemy myśleć, biegać, stać itp. Mitochondria zawierają enzymy które przetwarzają żywność w energię i pozwalają komórkom zużywać tlen, którego potrzebują. Można powiedzieć, że mitochondria to płuca komórki
Frataksyna odgrywa ważną rolę w określaniu jak dużo żelaza wchodzi do mitochondrium i jak ono będzie zużyte. Jest więc regulatorem wejścia żelaza do mitochondrium. Kiedy poziom frataksyny jest obniżony (a jest znacznie u pacjentów z FA) wejście żelaza nie jest kontrolowane i zbyt dużo jego dostaje się do mitochondriów. To żelazo reaguje z tlenem i formuje toksyczne cząstki zwane wolnymi rodnikami. Wolne rodniki blokują funkcję mitochondriów i powstrzymują je od produkcji potrzebnej komórce energii
Oczywiście organizm posiada naturalne sposoby eliminacji wolnych rodników jak enzymy i naturalne antyoksydanty jak dysmutaza nadtlenkowa, koenzym Q, glutation i katalaza. Gdyby pacjenci z FA mogli eliminować wolne rodniki nie dochodziłoby do uszkodzeń neuronów. Czym pacjent jest starszy tym system eliminacji jest gorszy a to powoduje gorsze uszkodzenie nerwów. Poprzez wykonywanie biopsji u pacjentów z FA określiliśmy deficyt białek żelazowo-siarkowych w mitochondriach. Jest on obecny tylko w nerwach, i komórkach serca. Komórki mięśni, limfocyty i fibroblasty są „normalne”. Ponieważ akumulacja wolnych rodników u pacjentów z FA uszkadza białka żelazowo siarkowe, lekarze wpadli na pomysł podawania pacjentom antyoksydantów (wymiataczy wolnych rodników).
Wiele antyoksydantów zostało przebadanych ale wybrano jeden (MNESIS czyli IDEBENONE) ponieważ:
Łatwo przechodzi przez błony komórkowe i ścianę mitochondriów
Atakuje i niszczy wolne rodniki i jony tlenu
Usuwa je na zewnątrz poprzez drogi oddechowe przy pomocy czynnika III zanim mogą zaatakować białka żelazowo siarkowe
Nie daje efektów ubocznych
HEMOFILIA
Hemofilia jest chorobą krwi, w której jedno z białek osocza odpowiedzialne za krzepliwość krwi występuje w organizmie chorego w niewielkich lub niemal zerowych ilościach. Istnieje kilka postaci hemofilii. Najpowszechniej występuje w najcięższej postaci tzw. hemofilii A, charakteryzującej się niedoborem czynnika VIII. Rzadszym przypadkiem jest Hemofilia B, w której występuje niedobór czynnika IX. Najrzadziej spotykaną jest tzw, "choroba von Willebrandta", zaliczana przez niektórych do specyficznych odmian hemofilii.
Gdy człowiek chory na hemofilię skaleczy się lub zrani, zostaje narażony na krwawienie, które nie jest ani mocniejsze, ani bardziej intensywne, niż u innych ludzi, jednakże z reguły trwa znacznie dłużej z uwagi na to, że jego krew nie jest w stanie utworzyć odpowiednio solidnego zakrzepu. Niewielkie "zacięcia" lub ranki nie stanowią problemu, jednak wszelkie głębsze krwawienia mogą trwać nawet wiele dni. Niektóre krwotoki występują jako skutek zranień lub uszkodzeń skóry. Hemofilik jest jednakże narażony również na krwotoki wewnętrzne, niewidoczne z zewnątrz, powstające często samoistnie lub na skutek niewielkich nawet obrażeń fizycznych, trudne do opanowania i kontrolowania.
Specjaliści szacują, że średnio co dziesięciotysięczny mieszkaniec Ziemi cierpi na jedną z postaci Hemofilii. W USA np. zarejestrowanych jest około 20.000 hemofilików. W Polsce, niestety, wiedza o Hemofilii w społeczeństwie jest jeszcze dość niska i nie wszyscy hemofilicy są oficjalnie zarejestrowani w głównych ośrodkach medycznych, zajmujących się jej leczeniem. Ocenia się jednak, że ok. 4 - 5 tysięcy obywateli naszego kraju to hemofilicy. Występowanie hemofilii nie jest uzależnione od rasy, narodowości, czy przynależności do określonej grupy społeczno-ekonomicznej.
Hemofilia jest chorobą dziedziczną, przekazywaną z pokolenia na pokolenie za pośrednictwem chromosomu X w kodzie genetycznym, gdzie nosicielami hemofilii są zawsze kobiety, a chorują - niemal wyłącznie mężczyźni. Hemofilią nie można się zarazić, tak jak grypą. Jest to choroba, z którą człowiek się po prostu rodzi.
Znanych jest jednak w świecie kilka przypadków tzw. hemofilii nabytej. Mamy w tych przypadkach zjawisko spontanicznego tworzenia się w organizmie chorego przeciwciał, zwalczających własny czynnik VIII. Takie "uszkodzenie" systemu immunologicznego sprawia, że chory może mieć objawy niemal identyczne z tymi, jakie spotykane są u hemofilików
"z urodzenia".
Wśród genów i chromosomów (przekaźników dziedziczności), jakie każdy człowiek przejmuje od swoich rodziców, występują dwa chromosomy płci oznaczone literami X i Y. Kobieta przejmuje po swoich rodzicach dwa chromosomy X: jeden od matki i jeden od ojca. To czyni ją kobietą. Mężczyzna przejmuje jeden chromosom X od swojej matki oraz jeden chromosom Y od ojca, co czyni go mężczyzną. Jeżeli wszystkie chromosomy X danej osoby zawierają gen hemofilii, to osoba ta będzie miała hemofilię. Warunek ten spełniony jest znacznie rzadziej u kobiet, niż u mężczyzn.
W licznych przypadkach hemofilia może być "utajona" przez wiele pokoleń, jeżeli na świat nie są wydawane dzieci rodzaju męskiego z ujawnioną chorobą. Wówczas gen zawierający hemofilię jest "przenoszony" przez całe pokolenia kobiet, które, z uwagi na to, że drugi chromosom X jest normalny, same nie chorują na hemofilię. W niektórych przypadkach, nie udokumentowanych w historiach rodowych odnotowywano zmianę w chromosomie X, zwaną niekiedy mutacją genu. Z reguły jednak matki dzieci z hemofilią miały ojców, dziadków, braci lub innych "męskich" krewnych ze strony "żeńskiej", którzy urodzili się z hemofilią. Do bardzo rzadkich przypadków należą kobiety z wrodzoną hemofilią, gdzie na ogół matka była "nosicielką" choroby, a ojciec był hemofilikiem.
http://kobieta.gazeta.pl/edziecko.html
http://samisobie.clan.pl/pshem.htm
Choroby dziedziczne człowieka -c.d.
Spośród kilku tysięcy znanych i opisanych chorób człowieka wiele jest wynikiem zmian w naszych genach, i to zmian związanych z jednym tylko, konkretnym genem. W zasadzie choroby takie klasyfikuje się na różne sposoby. Ciekawy wydaje się tu następujący problem: Czy aby zaistniała choroba, defekt musi wystąpić w jednym allelu, czy w obu obecnych kopiach genu? Jeśli konieczne są dwie uszkodzone kopie genu, mutacja jest recesywna, jeśli wystarcza jedna - mutacja jest dominująca. Ponadto mutacja może być autosomalna lub sprzężona z płcią. Istnieje też wiele chorób ludzi spowodowanych zmianą liczby chromosomów. Pamiętajmy też, że w chwili obecnej leczenie chorób dziedzicznych (jeśli w ogóle jest kuracja) ma charakter objawowy.
Niektóre choroby dziedziczne człowieka wywołane mutacjami genowymi
Opisana wcześniej hemofilia jest dziedzicznym zaburzeniem krzepliwości krwi. Obecnie możliwe jest skuteczne, choć bardzo kosztowne, leczenie tej choroby przez podawanie (w zastrzykach) brakującego białka - czynnika krzepliwości krwi uzyskanego technikami inżynierii genetycznej. Innym zaburzeniem jest daltonizm polegający na nieprawidłowym rozróżnianiu barw. Ma on różne warianty, ale tak jak hemofilia zawsze jest związany z mutacją genu położonego w chromosomie X. Nie ma innych objawów, a daltonistami są niemal wyłącznie mężczyźni.
Większość chorób powodowana jest przez mutacje w genach położonych w autosomach (są ich 22 pary, a chromosomów płci - tylko jedna). Zwykle choroby te są powodowane przez mutacje autosomalne recesywne. Można to też ująć inaczej -jeden działający allel daje takie stężenie jego produktu, które wystarcza do pełnego realizowania funkcji w organizmie.
Wiele chorób jest związanych z zaburzeniami metabolizmu. Mutacje w genach kodujących enzymy określonych szlaków metabolicznych są przyczyną tak zwanych bloków metabolicznych. Na przykład nierozkładanie aminokwasów aromatycznych prowadzi do fenyloketonurii, alkaptonurii lub albinizmu, zaś nierozkładanie cukru - galaktozy - do galaktozemii (tab. 6.2).
Innym przykładem choroby powodującej defekty w ważnych funkcjonalnie białkach jest anemia sierpowata. U osób dotkniętych tą chorobą nieprawidłowa hemoglobina słabo wiąże tlen, a same krwinki czerwone mają charakterystyczny sierpowaty kształt. Na anemię sierpowata, rozpowszechnioną na znacznych obszarach Afryki, nie chorują osoby będące heterozygotami. Nie zapadają one także na malarię, ponieważ nieco zmienione właściwości krwinek skutecznie hamują wzrost pasożyta powodującego malarię (por. opis cyklu rozwojowego zarodźca malarii, Biologia 1. Zakres rozszerzony, rozdz. 4).
Najczęstszą chorobą powodowaną przez mutację autosomalna recesywna u rasy białej jest mukowiscydoza. Gen, w którym zachodzą mutacje, koduje białko odpowiadające za transport jonów chlorkowych przez błonę komórkową. Zaburzenia funkcjonowania tego białka przynoszą bardzo różne efekty. Między innymi zachodzą zmiany w śluzie w płucach, który staje się bardzo dobrą pożywką dla zakażających bakterii. Nawracające infekcje płuc są charakterystycznym objawem choroby, oprócz tego u niektórych zakażonych osób występują objawy związane z zaburzeniami wydzielania enzymów trzustkowych.
Mężczyźni dotknięci tą chorobą są na ogół bezpłodni. W Europie co 20-25 osoba jest nosicielem jednego zmutowanego genu.
Nazwa choroby |
Defekt/objawy |
Sposób dziedziczenia |
Częstość występowania (przybliżona) |
Uwagi (przeżycie lat) |
Hemofilia |
mutacja w genie jednego z czynników krzepnięcia krwi - krwawienia, wylewy wewnętrzne |
recesywna sprzężona z płcią |
1/5000 chłopców |
leczenie - podaje się czynnik krzepnięcia krwi uzyskany technikami inżynierii genetycznej (70) |
Daltonizm Fenyloketonuria |
zaburzenie w rozróżnianiu barw (najczęściej nieodróżnialnie barwy czerwonej) brak enzymu rozkładającego fenyloalaninę, upośledzenie umysłowe |
recesywna sprzężona z płcią autosomalna |
8/100 chłopców 1/10 000 |
nie wpływa na długość życia, nie jest też leczona dieta z bardzo matą ilością fenyloalaniny zapobiega objawom choroby u niemowląt i dzieci (70) |
Alkaptonuria |
defekt w rozkładaniu tyrozyny/odkładanie ciemnego barwnika w chrząstkach i stawach; w wieku powyżej 20 lat stany zapalne i zwyrodnieniowe stawów |
recesywna autosomalna |
1/100 000 |
|
Albinizm |
zaburzenie w wytwarzaniu melaniny barwnika skóry, włosów, oczu itd. - skóra biała, bardzo wrażliwa na stonce |
recesywna autosomalna |
1/110 000 |
|
Galaktozemia |
defekt w enzymie metabolizmu galaktozy; jeśli formą leczenia nie jest dieta, to gromadzące się produkty powodują toksyczne efekty m.in w układzie nerwowym, zaćmę |
recesywna autosomalna |
1/50 000 |
dieta bez mleka i produktów mlecznych przez całe życie; jeśli diagnoza-ostaje postawiona przed pierwszym miesiącem życia. to wprowadzenie diety zapobiega upośledzeniu umysłowemu |
Anemia sierpowata |
mutacja w genie kodującym jeden z łańcuchów hemoglobiny |
recesywna autosomalna |
1/625 u rasy czarnej |
transfuzje krwi heterozygoty nie chorują (mutacja rzadka u rasy białej) |
Mukowiscydoza |
mutacja w genie, którego produkt związany jest z transportem jonów chlorkowych, w efekcie zmiany w śluzie w płucach, częste zakażenia; bywają zaburzenia w działaniu trzustki |
recesywna autosomalna |
1/2500 |
średnio 25 lat, zależy od postaci (są mniej i bardziej ostre), leczenie - podawanie antybiotyków/preparatów z enzymami trzustkowymi (gdy są potrzebne), związków upłynniających śluz w płucach |
Choroba Huntingtona |
mutacja w genie białka występującego w mózgu, zaburzenia ruchu i otępienie |
dominująca autosomalna |
1/24 000 |
występuje na ogół u osób dorosłych (35-45 lat), od momentu rozpoznania średni czas przeżycia 16 lat; nie ma leczenia |
Badania nad alkaptonurią, mimo że choroba ta występuje nieczęsto, miały istotne znaczenie dla rozwoju genetyki. Pod koniec XIX wieku lekarz angielski Archibald Garrod stwierdził, że w moczu osób chorych na alkaptonurię występuje kwas homogentyzynowy i w kilka lat później, już w XX wieku, opisał tę chorobę w pracy naukowej, używając pojęcia „wrodzona wada metaboliczna”. Pojęcia tego używa się nadal przy określaniu wielu chorób genetycznych.
Ważna rolę w powstawaniu molekularnych podstaw chorób odegrało też badanie anemii sierpowatej. Wybitny chemik Linus Pauling jeszcze w roku 1949, przed odkryciem struktury DNA postawił hipotezę, że przyczyną tej choroby jest zmieniona budowa hemoglobiny, a w 1956 roku Vernon M. Ingram wykazał, że anemię sierpowatą powoduje zmiana tylko jednego aminokwasu w jednym z łańcuchów polipeptydowych hemoglobiny.
Choroby, wywołane mutacją autosomalną dominującą, są wynikiem zmiany tylko w jednym z pary alleli. Na ogół produkt jednego z alleli jest wadliwy i coś niszczy - istnieje bardzo wiele zaburzeń układów nerwowego i mięśniowego wynikających z nagromadzenia się różnych (w zależności od konkretnej choroby) wadliwych produktów białkowych. Jednym z najczęstszych zaburzeń tego rodzaju jest choroba Huntingtona (dawniej, ze względu na mimowolne ruchy chorych, zwana pląsawicą Huntingtona; tab.6.2).
Chorobami autosomalnymi dominującymi są też niektóre nowotwory (w sumie stanowią 5-10% przypadków wszystkich chorób nowotworowych). Osoba z takiej rodziny może odziedziczyć formę zmutowaną jednego z genów kontrolujących prawidłowe podziały komórki, co bardzo zwiększa ryzyko na zachorowanie na dany typ nowotworu (różny w zależności od odziedziczonego zmutowanego genu). Niektóre mutacje powodują występowanie tylko jednego typu nowotworu, inne wielu różnych.
Zdarzają się też choroby dziedziczone tylko po matce (jest to tzw. dziedziczenie pozajądrowe). Przykładem są niektóre choroby mięśni u człowieka (zwykle są skutkiem delecji lub innych mutacji punktowych w DNA mitochondrialnym). Objawiają się zmianami biochemicznymi w mitochondriach, osłabieniem mięśni szkieletowych i obecnością nieco poszarpanych włókien nerwowych. Innym przykładem może być zespół Lebera - niewielka mutacja w mitochondrialnym DNA prowadzi do dziedzicznego zaniku nerwu wzrokowego i utraty widzenia.
Czy istnieją choroby, które dziedziczy się tylko po ojcu? W starszych podręcznikach genetyki podawano przykłady cech determinowanych przez geny zlokalizowane w chromosomie Y, jednak nie należy wierzyć w ich prawdziwość. Chromosom Y ma pewne geny, a szczególnie jeden, bardzo ważny, którego obecność warunkuje płeć męską człowieka czy innego ssaka (SRY). W badaniach nie wykazano jednakże dziedziczenia jakichś cech przenoszonych z ojca na syna, choć oczywiście syn w całości dziedziczy chromosom Y.
Niektóre choroby dziedziczne człowieka wywołane mutacjami chromosowymi
Zmieniona liczba chromosomów prowadzi do poważnych zaburzeń rozwojowych. Stosunkowo łagodne objawy występują przy zespole Downa i zespole Turnera. Zespół Turnera wynika z obecności tylko jednego chromosomu X i występuje u 1 na 5000 nowo narodzonych dziewczynek. Oprócz bezpłodności występują u nich różne inne objawy, jednak są to wady strukturalne, nie występuje jednak upośledzenie umysłowe. Często charakterystyczny jest niski wzrost i na przykład u 20% chorych - wrodzone wady serca. Choroba nie wpływa również na długość życia. Ta jedyna znana monosomia wynika z faktu, że w komórkach kobiet czynny jest w zasadzie tylko jeden chromosom X, drugi zaś w znacznej swojej części jest unieczynniony. Tak więc „na co dzień” wystarcza jeden chromosom X, ale obecność drugiego jest potrzebna w czasie rozwoju, co jest przyczyna zaburzeń w zespole Turnera.
Zespół Downa - potrójny chromosom 21 (trisomia chromosomu 21) pojawia się raz na 700 noworodków, ale częstość zależy od wieku matki (od 1 na 1350 u kobiet 25-letnich do 1 na 28 u kobiet 45-letnich). Zatem dodatkowy chromosom pochodzi na ogół z komórki jajowej. Zespół Downa powoduje upośledzenie umysłowe (średni iloraz inteligencji wynosi 50, średni dla osób zdrowych wynosi 100). Dość często występują wrodzone wady serca, które u części dzieci mogą doprowadzić do przedwczesnej śmierci. Choroba ta wpływa więc pośrednio na długość życia (np. często wcześnie rozwija się u nich choroba Alzheimera).
Wydaje się, że ważna - przynajmniej dla niektórych genów - jest ich właściwa liczba - muszą to być dwa geny, nie zaś trzy lub jeden. Chromosom 21 jest najmniejszym chromosomem ludzkim z najmniejszą liczbą genów, jednak efekty istnienia dodatkowego chromosomu są znaczne. Rodzą się też dzieci z trisomią chromosomu 13 (zespół Pataua) albo 18 (zespół Edwardsa), są jednakże ciężko chore i nie przeżywają 1 roku.
Przykładem choroby wywołanej przez mutację chromosomową strukturalną jest przewlekła białaczka szpikowa (aberracja somatyczna pojawiająca się tylko szpiku kostnym). Na skutek translokacji fragmentu chromosomu 9 na chromosom 22 dochodzi do połączenia pewnych genów. Powstaje tzw. gen fuzyjny kodujący białko o odmiennych właściwościach w stosunku do białka kodującego.
Choroby wielogenowe
Przyczyną chorób trapiących ludzkość nie jest 1 gen, lecz interakcje kilku genów i środowiska. Choroby takie nazywamy wieloczynnikowymi (czasem niezbyt ściśle - wielogenowymi). Większość przypadków cukrzycy spotykanej u dorosłych osób jest wynikiem mutacji w kilku różnych genach, znaczenie ma tez tryb życia i odżywiania. Niestety, geny te nie są jeszcze do końca poznane. Podobnie jest z chorobami układu krążenia, podatnością na nowotwory (z wyjątkiem wspomnianych wcześniej 5 - 10%, za które odpowiedzialne są pojedyncze mutacje). Poznanie podłoża chorób wielogenowych jest więc jednym z wielkich wyzwań genetyki XXI wieku.
(Ewa Bartnik, Waldemar Lewiński, Biologia 3. Choroby dziedziczne człowieka, Gdynia 2004, 127-131)
Wyszukiwarka