KOMPUTEROWE MODELOWANIE PEPTYDÓW I BIAŁEK.
ZAGADNIENIA DO PRZYGOTOWANIA
1. Aminokwasy (budowa, grupy funkcyjne).
2. Wiązanie peptydowe, podstawowe struktury drugorzędowe łańcuchów polipeptydowych, wiązania stabilizujące struktury trzecio- i czwartorzędowe.
3. Modyfikacje potranslacyjne.
4. Centrum aktywne enzymów, koenzymy.

WPROWADZENIE
Rysunek 1 przedstawia model kanału wodnego białka AQP1 zbudowanego z czterech homomonomerów. Po lewej stronie widoczna jest struktura łańcuchów polipeptydowych. W środku - białko wraz z cząsteczkami wody przepływającymi przez cztery kanały wodne. Z prawej - układ cząsteczek wody w czasie przepływu przez błonę komórkową. AQP1jest białkiem błony erytrocytów; inne komórki posiadają analogiczne kanały wodne.
W 2001 roku jednocześnie w dwóch prestiżowych czasopismach naukowych, Science i Nature, dwie współzawodniczące grupy badaczy (jedna skupiona w Celera Genomics, druga w Human Genome Project) opublikowały pierwsze wyniki analizy sekwencji nukleotydów ludzkiego genomu.
Obecnie szacuje się, że liczba wszystkich genów człowieka nie przekracza 60 000, z czego 30 000 - 40 000 to geny kodujące białka. Do tej pory rozpoznanych zostało około 12 000 genów, wśród nich ok. 6 000 o znanej lub przewidywanej funkcji. Alternatywny splicing i modyfikacje potranskrypcyjne sprawiają, że w komórkach człowieka występuje najprawdopodobniej 85 000 różnych mRNA. W procesie pierwotnej obróbki potranslacyjnej wytwarzanych jest około 100 000 polipeptydów, które w wyniku dalszych modyfikacji mogą zostać przekształcone w ponad 500 000 różnych białek. Możliwości modyfikacji potranslacyjnych są tak duże, że kilkadziesiąt tysięcy ludzkich genów może kodować od 50 000 000 do 100 000 000 różnych funkcjonalnie cząsteczek białek.
Ze względu na olbrzymią liczbę możliwych konformacji łańcuchów polipeptydowych w białkach, określenie ich trzecio- i czwartorzędowej struktury jest bardzo trudne. W przypadku krótkich peptydów, zawierających kilkanaście aminokwasów, za pomocą odpowiednich algorytmów komputerowych można przewidzieć ich konformację. Większe cząsteczki wymagają już analizy empirycznej. Najczęściej stosowaną metodą jest badanie dyfrakcji promieniowania rentgenowskiego w kryształach białek (ang. X-ray crystallography); w ten sposób poznano ponad 80% znanych struktur. Drugą metodą jest magnetyczny rezonans jądrowy (NMR); około 16% rozpoznanych struktur. W dużym uproszczeniu, dzięki umieszczeniu badanej próbki w silnym polu magnetycznym, NMR obrazuje zmiany odległości pomiędzy sąsiadującymi atomami spowodowane oddziaływaniami fizyko-chemicznymi.
Ponieważ poznano już dość dużo konformacji różnych białek, możliwe stało się przewidywanie struktury na podstawie porównania podobieństwa sekwencji aminokwasów badanego polipeptydu z białkami o określonej empirycznie konformacji (jak dotąd tylko 2% znanych struktur opracowano tą metodą).
Prolina jest jedynym aminokwasem, którego azot wchodzący w skład wiązania peptydowego znajduje się w pierścieniu. Ta specyficzna budowa proliny powoduje, że łańcuch peptydowy załamuje się pod kątem 30 stopni i zostaje zaburzona zdolność do tworzenia wiązań wodorowych pomiędzy wiązaniami peptydowymi. Prolina jest często spotykana w białkach zawierających struktury β. Również specyficzna konformacja kolagenu jest zdeterminowana między innymi przez dużą zawartość proliny i glicyny, które stanowią 50% wszystkich aminokwasów tego białka.
W wielu białkach prolina znajduje się na powierzchni cząsteczki i wchodzi w skład strukturalnych domen odpowiedzialnych za interakcje pomiędzy białkami. W ostatnim czasie zidentyfikowano wiele białek, w których znajdują się fragmenty bogate w reszty prolilowe mające ważne znaczenie funkcjonalne. Fragmenty takie rozpoznawane są przez odpowiednie domeny (SH3 i WW) innych białek. Takie interakcje pomiędzy białkami należą do istotnych mechanizmów regulacyjnych metabolizmu komórek i całego organizmu.
Dystroglikan jest silnie glikozylowanym białkiem błony komórkowej i pełni rolę receptora dla różnych białek struktur pozakomórkowych łącząc je, poprzez dystrofinę, z łańcuchami aktyny szkieletu komórkowego. Właściwe funkcjonowanie tego połączenia jest warunkiem prawidłowego rozwoju i działania mięśni. W sekwencji reszt aminokwasowych β-dystroglikanu znajduje się motyw sekwencyjny zawierający trzy reszty prolilowe (-Ser-Pro-Pro-Pro-Tyr-), który łączy się z domenami WW znajdującymi się w dystrofinie. Domeny WW mają 35-45 reszt aminokwasowych i charakterystyczną budowę:
a) reszty tryptofanylowe przedzielone są 20-22 aminokwasami,
b) pomiędzy tryptofanami znajdują się dwa lub trzy aminokwasy aromatyczne,
c) w odległości trzech reszt za drugim tryptofanem w kierunku końca karboksylowego białka znajduje się prolina.
Wszelkie mutacje powodujące zmianę aminokwasów zarówno w bogatym w prolinę fragmencie dystroglikanu bądź w domenie WW dystrofiny powodują błędy w usieciowaniu szkieletu komórkowego. Mutacje te są przyczyną różnego rodzaju dystrofii mięśniowych, w tym spotykanej stosunkowo często dystrofii typu Duchenne (DMD). Przebieg tej choroby jest gwałtowny i ciężki. Pierwsze objawy występują przed trzecim rokiem życia. Uszkodzenie mięśnia sercowego diagnozuje się około 6 roku życia. W wieku 12 lat dziecko całkowicie przestaje chodzić, a około 15 roku życia występuje już poważna dysfunkcja mięśni gładkich. Pacjent umiera zazwyczaj przed 20 rokiem życia (mediana 17 lat). Choroba wywołana jest przez mutacje w genie dystrofiny znajdującym się na chromosomie X. W związku z taką lokalizacją choroba dotyka tylko chłopców, chociaż niekiedy u kobiet nosicielek mutacji mogą wystąpić znacznie łagodniejsze objawy dystrofii mięśniowej. Mutacje są stosunkowo częste; w zależności od populacji od 5 do 10% kobiet jest nosicielkami. Choroba dotyka jednego na 4 000 chłopców. Łagodniejszy przebieg ma dystrofia mięśniowa typu Beckera. W tej chorobie również obserwuje się mutacje genu dystrofiny, ale nie zaburzają one całkowicie funkcji białka.
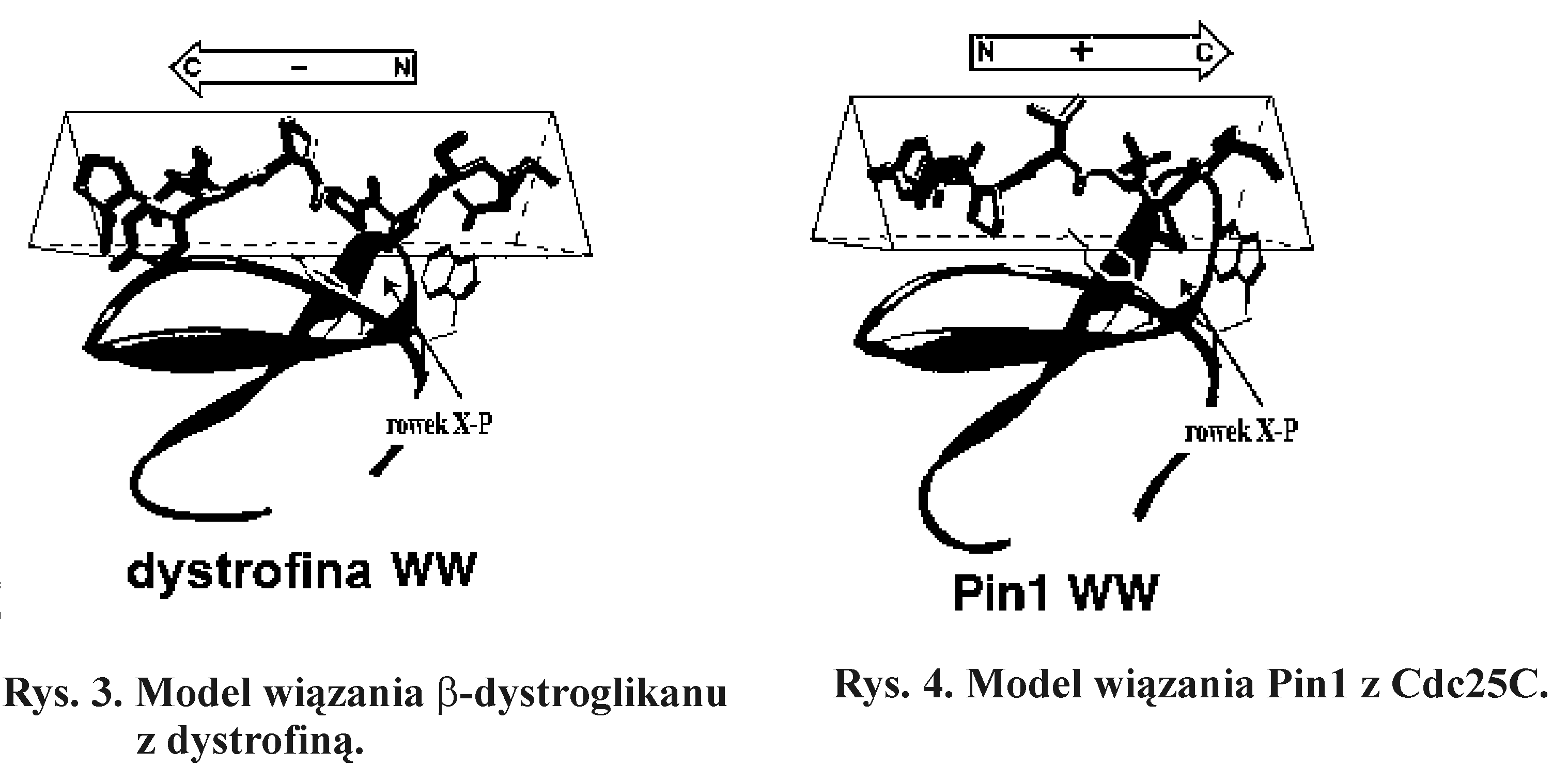
Na rysunku 4 przedstawiono schemat wiązania β-dystroglikanu z dystrofiną. Uwagę zwraca podobieństwo konformacji fragmentu łańcucha dystroglikanu do konformacji kolagenu (Rys. 2).
Kolejnym przykładem współdziałania białek zawierających domeny WW i prolinowe jest udział Pin1 i Cdc25C (Rys. 3) w regulacji cyklu komórkowego. Białko Cdc25C jest cyklinozależną kinazą tyrozynową indukującą mitozę, a dokładniej przejście z fazy G2 do fazy M cyklu komórkowego. W strukturze białka Cdc25C znajduje się sekwencja -Ser-Pro-, w której reszta serylowa może być ufosforylowana. Taka fosforylacja aktywuje kinazę Cdc25C i uruchamia mitozę. Z kolei ufosforylowana seryna (-fosfoSer-Pro-) jest rozpoznawana przez domenę WW białka Pin1. Drugą domeną funkcyjną Pin1 jest izomeraza peptydylo-prolilowa, która zmienia wiązanie peptydowe tworzone przez resztę prolilową z pozycji cis do trans. W konformacji trans fosfoseryna jest defosforylowana przez fosfatazę PP2A, co inaktywuje aktywność kinazy białka Cdc25C.
Inną funkcją białka Pin1 jest izomeryzacja i w konsekwencji defosforylacja białka tau. W chorobie Alzheimera obserwuje się nagromadzenie w neuronach ufosforylowanej formy tau i przypuszczalnie niższa aktywność Pin1 może być molekularną przyczyną tej choroby.
Zespół Liddle'a jest chorobą, której najważniejszym objawem jest nadciśnienie tętnicze. Przyczyną choroby jest zwiększona ilość kanałów sodowych (ENaC, Epithelial Sodium Chanel) w błonach komórek nabłonkowych. ENaC zbudowany jest z co najmniej trzech podjednostek: α, β i γ. Mutacje genów kodujących podjednostki beta lub gamma są odpowiedzialne za nadciśnienie tętnicze w zespole Liddle'a. Nadciśnienie to związane jest z hipokalemią, niską aktywnością reninową osocza i niskim poziomem aldosteronu w osoczu. Mutacje w podjednostce beta powodują wymianę lub delecję grupy aminokwasów (-Pro-Pro-Pro-Asn-Tyr-); podobna grupa aminokwasów ulega mutacji w podjednostce γ. Zanika wtedy specyficzna struktura łańcucha związana z obecnością kilku reszt prolilowych. Utrata lub zmiany w budowie bogatego w prolinę fragmentu uniemożliwiają ubikwitynację ENaC przez ligazy ubikwitynowe. Ligazy ubikwitynowe rozpoznają substraty poprzez swoje domeny WW, a następnie przenoszą z cząsteczki ubikwityny na rozpoznane białko krótki fragment polipeptydowy. Fragment ten jest sygnałem dla inaktywacji i degradacji zaznaczonego białka. Ligaza ubikwitynowa rozpoznająca łańcuch β ENaC to NEDD4. W efekcie mutacji błędy w budowie ENaC powodują zablokowanie internalizacji i inaktywacji kanału. W konsekwencji w błonie komórkowej znajduje się nadmierna liczba kanałów ENaC, co powoduje wzrost absorpcji jonów sodu w kanaliku dystalnym nerki i utratę jonów potasu, co powoduje nadciśnienie.
Zaburzenia interakcji białek zawierających motywy prolinowe z białkami partnerskimi posiadającymi domeny WW są też molekularną przyczyną choroby Huntingtona i odgrywają istotną rolę w kancerogenezie i progresji nowotworów.
CZĘŚĆ DOŚWIADCZALNA
UWAGA
W trakcie zajęć każda z grup studenckich wykonuje zadanie na zaliczenie, które polega na identyfikacji reszt aminokwasowych istotnych dla funkcji analizowanych białek.
Ćwiczenia polegają na analizie wybranych białek, odnalezieniu w nich grup funkcyjnych i identyfikacji reszt aminokwasowych istotnych dla funkcji i konformacji białka. Do analizy wykorzystuje się dane położenia poszczególnych atomów w białkach, uzyskane przy pomocy badań krystalograficznych (rentgenografii lub NMR). Dane takie można pobrać z udostępnionej w internecie bazy danych białek zdeponowanej w RCSB Protein Data Bank (http://www.rcsb.org/pdb/index.html). Do analizy wykorzystuje się program DeepView/Swiss-Pdb Viewer (http://www.expasy.org/spdbv/).
Panel kontrolny DeepView zbudowany jest z kilku okien. Najważniejsze z nich to:
1. Okno paska zadań Toolbar
służące do otwierania i zapisywania plików, pomiaru długości wiązań i kątów pomiędzy nimi, wprowadzania mutacji itp.
2. Okno Control Panel
które ułatwia podświetlanie, ukrywanie, obróbkę pojedynczych reszt aminokwasowych i ich łańcuchów bocznych.
3. Okno sekwencji Alignment
umożliwiające liniowe porównanie dopasowania sekwencji aminokwasów różnych polipeptydów.
4. Okno (Layers Infos)
umożliwiające przełączanie pomiędzy poszczególnymi białkami/peptydami otwartymi w projekcie.
W programie używane są następujące kolory atomów:
C --> biały O --> czerwony N --> niebieski
S --> żółty P --> pomarańczowy H --> błękitny
inne --> szary
1. Modelowanie cząsteczki dystrofiny i zapoznanie się z podstawowymi funkcjami programu DeepView
Najpierw należy otworzyć program SVPDB/DeepView z ikony znajdującej się na pulpicie
.
1. Z rozwijanego menu wybrać File -> Open PDB File... W otwartym oknie odszukać folder plików PDB ćwiczenia, otworzyć go i wybrać do otwarcia plik dystrofina.pdb. W pliku tym zapisane są dane koordynacyjne rozmieszczenia atomów w cząsteczce dystrofiny związanej z krótkim fragmentem dystroglikanu. Model białka otwiera się w oknie graficznym. Jest to tzw. model „druciany”, w którym poszczególne atomy przedstawione są łącznie z wiązaniami jako kolorowe krótkie odcinki. W celu poprawienia wyglądu z rozwijanego menu wybrać Display -> Use OpenGL Rendering i następnie Display -> Render in solid 3D.
2. Na pasku zadań można skorzystać z następujących narzędzi:
po kliknięciu na to narzędzie modelowana cząsteczka umieszczana jest w centrum okna.
tym narzędziem można przesuwać cząsteczkę w jednej płaszczyźnie. Kolejne
służy do zmiany wielkości cząsteczki.
to narzędzie służy do obracania cząsteczki (jeśli jednocześnie przytrzyma się klawisz F5 cząsteczka będzie przesuwana wzdłuż osi X, klawisz F6 umożliwia przesunięcie wzdłuż osi Y, a klawisz F7 wzdłuż osi Z).
Zestaw narzędzi
służy do (od lewej) ● pomiaru odległości pomiędzy wybranymi atomami; ● pomiaru kąta pomiędzy wiązaniami tworzonymi przez trzy atomy; ● mierzy kąty omega, psi i fi dla wybranego aminokwasu (użyte razem z klawiszem Ctrl umożliwia wybór 4 atomów i pomiar kąta skręcenia wiązania); ● narzędzie identyfikujące wybrany atom (jego współrzędne, przynależność do cząsteczki, aminokwas i jego numer kolejny); ● wyświetla bądź ukrywa grupy będące w zadanej odległości od wybranego atomu; ● przesuwa cząsteczkę ustawiając wybrany atom w centrum okna; ● dopasowuje dwie cząsteczki do siebie (działa tylko wtedy, kiedy cząsteczki wyodrębnione są w oknie Layers Infos); ● narzędzie wprowadzania mutacji, zamienia wybrany aminokwas na inny (pozwala też na wybór rotameru wybranego aminokwasu oraz oblicza dopasowanie energetyczne; najniższa wartość score oznacza najbardziej uprzywilejowaną konformację); ● okno umożliwiające zmianę kąta skrętu wybranego aminokwasu lub całej grupy (oblicza też dopasowanie energetyczne).
3. Z rozwijanego menu wybrać kolejno:
Window -> Control Panel;
Window -> Alignment;
Window -> Layers Infos.
Okno Control Panel podzielone jest na kolumny:
group - zawiera podkolumny (od lewej): ● litery oznaczające łańcuchy, wersalikami i małymi literami oznacza się odpowiednio łańcuchy polipeptydowe należące do różnych cząsteczek białek lub innych makromolekuł. (W Control Panel dla dystrofiny mamy dwa oznaczenia: litera A - oznacza łańcuch polipeptydowy dystrofiny; litera p - łańcuch peptydoglikanu); ● kolejna podkolumna określa tworzenie przez aminokwasy regularnych struktur (h - α helisa i s - struktura β); ● dalej umieszczone są trzyliterowe skróty aminokwasów i ich kolejność w całej badanej strukturze (można wybrać kursorem i klikając lewym klawiszem myszy dowolny aminokwas lub ich grupę, obowiązują takie same zasady jak w Wordzie tj. zaznaczanie przez przeciąganie, użycie klawiszy Shift (zaznaczanie grupy ciągłej) i Ctrl (zaznaczanie grupy z przerwami).
show - w tej kolumnie zaznacza się aminokwasy, które mają być widoczne w oknie graficznym.
side - tutaj zaznacza się czy mają być wyświetlone łańcuchy boczne aminokwasów, czy tylko rdzeń tworzący wiązania peptydowe (zaznaczenie można wybrać dla całej cząsteczki, pojedynczego aminokwasu lub grupy).
labl - wyświetla oznaczenie literowe i numer kolejny aminokwasu w oknie graficznym.
::v - wyświetla chmury elektronowe atomów należących do wybranej cząsteczki (posiada rozwijane menu z opcjami).
ribn - wyświetla wybrane aminokwasy w postaci wstęgi (ta opcja doskonale wizualizuje struktury α i β).
col - pozwala na wybranie koloru wyświetlanej cząsteczki (posiada rozwijane menu z opcjami wyboru struktur do kolorowania).
4. Wykorzystując funkcje Control Panel wybrać do wyświetlenia w postaci łańcucha aminokwasów tylko fragment dystroglikanu (użycie narzędzia show). Przyjrzeć się dokładnie łańcuchowi dystroglikanu. Obracając cząsteczkę zlokalizowć bogaty w prolinę fragment, który jest wychwytywany przez dystrofinę.
5. Wybrać wizualizację łańcucha dystrofiny w postaci wstęgi (użycie narzędzia ribn; najwygodniej wskazać kursorem na kolumnę ribn, „kliknąć” prawym przyciskiem myszy i następnie przytrzymując lewy przycisk skasować selekcję łańcucha dystroglikanu). Przesuwając i obracając cząsteczkę przyjrzeć się wzajemnemu układowi dystrofiny i dystroglikanu.
6. Korzystając z rozwijanego menu wybrać Edit -> Search for PROSITE Pattern. Funkcja ta wyszuka, korzystając z lokalnej bazy danych, znane domeny funkcyjne znajdujące się w cząsteczce dystrofiny. W pojawiającym się oknie tekstowym widać nazwy poszczególnych domen zidentyfikowanych w dystrofinie. Jeśli wybierze się kursorem i „kliknie” lewym przyciskiem myszy na nazwie zidentyfikowanej domeny to zostanie ona zaznaczona w łańcuchu polipeptydowym. W ten sposób można kolejno prześledzić lokalizację wszystkich odszukanych domen w cząsteczce białka. Jeśli wybierze się i „kliknie” na zaznaczony na czerwono numer domeny wyświetli się okno tekstowe z opisem domeny (tylko jeśli jest aktywne połączenie internetowe).
Wybrać domenę WW. W oknie Control Panel i w oknie Alignment zaznaczone zostaną aminokwasy charakterystyczne dla domen WW. Są to dwie reszty tryptofanylowe, na środku pomiędzy nimi dwie reszty tyrozylowe, w sąsiedztwie drugiego Trp reszta treonylowa i cysteilowa, a trzy aminokwasy dalej reszta prolilowa.
Przywrócić utracone kolorowanie dystroglikanu. W tym celu wskazać w Control Panel kolumnę col i przytrzymując lewy przycisk myszy zaznaczyć pozycje należące do dystroglikanu. Po otwarciu okna dialogowego kolorowania „kliknąć” OK. Następnie przystąpić do uwidocznienia domeny WW. Również w oknie Control Panel w kolumnie col w rozwijanym menu zaznaczyć ribbon, a następnie wybrać wszystkie aminokwasy należące do domeny WW zaczynając zaznaczenie na pierwszej reszcie Trp, a kończąc na Pro, które już są „podświetlone” w kolumnie. W otwartym oknie dialogowym wybierać kolor zaznaczenia i potwierdzić OK.
Uwidocznić jeszcze istotne dla funkcjonowania domeny WW reszty aminokwasowe. W tym celu w kolumnie show zaznaczyć odpowiednie reszty Trp, Tyr, Thr, Cys i Pro. Przyjrzeć się w jaki sposób odpowiednie łańcuchy boczne domeny WW są ułożone względem dystroglikanu (wykorzystać tu narzędzie obracania cząsteczki).
7. Spróbować w oknie graficznym pokazać tylko fragment dystroglikanu i domenę WW dystrofiny. Wskazać kursorem i „kliknąć” prawym klawiszem myszy na kolumnie ribn w Control Panel pozostawić w oknie tylko dystroglikan i funkcyjne reszty aminokwasowe domeny WW, można dokładniej przyjrzeć się wzajemnemu ułożeniu cząsteczek. Następnie zaznaczyć w kolumnie ribn tylko aminokwasy należące do domeny WW. Porównać otrzymany obraz z rysunkiem 3.
Zamknąć program wybierając File -> Exit.
2. Porównanie domen WW dwóch różnych białek
Wszystkie domeny WW mają podobną strukturę złożoną z trzech antyrównoległych łańcuchów β i podobny układ grup funkcyjnych aminokwasów. Drobne różnice w konformacji i charakter pozostałej reszty aminokwasów decydują o specyficzności wiązania z odpowiednim białkiem zawierającym bogate w prolinę fragmenty.
W ćwiczeniu tym wykonuje się zadanie na zaliczenie, które polega na identyfikacji funkcyjnych reszt aminokwasowych w domenach WW badanych białek. Należy podać nazwy aminokwasów i ich numery porządkowe z kolumny group w Control Panel.
Oprócz omówionych powyżej dystrofiny i Pin1 wykorzystuje się jeszcze dwa inne białka zawierające domeny WW.
NEDD4 - jest ligazą ubikwitynową znakującą w komórce cząsteczki białek przeznaczone do degradacji. NEDD4 bywa często nazywane białkiem śmierci, gdyż szczególnie dużą jego zawartość obserwujemy w apoptozie - programowanej śmierci komórki. W pliku znajdują się dane przedstawiające cząsteczkę NEDD4 związaną z fragmentem łańcucha β kanału sodowego ENaC.
FBP28 - białko o nieznanej funkcji, tworzące agregaty o strukturze amyloidu.
Wykonanie ćwiczenia.
1. Otworzyć program DeepView a następnie pierwszą cząsteczkę (NEDD4). Korzystając z odpowiednich funkcji przekształcić ją tak, aby widoczna była tylko domena WW (Control Panel => show. Zaznaczyć po dwa aminokwasy przed pierwszą resztą tryptofanylową i ostatnią resztą prolilową w domenie WW. Wybrany fragment cząsteczki zaznaczyć jednym kolorem. Dla uproszczenia widoku zlikwidować zaznaczenie atomów wodoru (Display -> Show Hydrogens) (to samo można wykonać w oknie Layers Infos w kolumnie H) i łańcuchów bocznych (Control Panel => side) (pamiętać o zapisaniu odpowiednich aminokwasów i ich liczb porządkowych).
2. Otworzyć drugą cząsteczkę (FBP28). W oknie Layers Infos w kolumnie vis zlikwidować zaznaczenie pierwszej cząsteczki i podświetlić na czerwono nazwę drugiej cząsteczki. Zmiana cząsteczki w oknie Control Panel następuje automatycznie. Podobnie jak w punkcie 1 zidentyfikować domenę WW (zapisać odpowiednie aminokwasy i ich liczby porządkowe). Tak jak w pkt. 1 zlikwidować zaznaczenie atomów wodorów i łańcuchów bocznych.
3. W oknie Layers Infos w kolumnie vis wybrać wizualizację obu fragmentów cząsteczek. W tym samym oknie w kolumnie mov można wybrać przemieszczanie jednej lub obu cząsteczek. Przesuwając ręcznie cząsteczki można porównać ich konformację.
4. Spróbować automatycznego dopasowania cząsteczek. Rozwijanego menu z Toolbar wybrać Fit -> Interative Magic Fit. W oknie dialogowym zaznaczyć All atoms i potwierdzić OK. Teraz obracając cząsteczki przyjrzeć się podobieństwom i różnicom w konformacji struktury β i położeniu grup bocznych funkcyjnych reszt aminokwasowych. W oknie Alignment można sprawdzić homologię aminokwasów w domenach WW.
5. Wyłączyć program File -> Exit.
3. Sprawdzenie w jaki sposób ligaza ubikwitynowa NEDD4 rozpoznaje łańcuch β kanału sodowego ENaC przeznaczonego do degradacji
W tym ćwiczeniu śledzisię, w jaki sposób kinaza ubikwitynowa NEDD4 rozpoznaje łańcuch β kanału sodowego ENaC przeznaczonego do degradacji.
1. Uruchomić program i wybrać do wyświetlenia plik NEDD4.pdb. Zidentyfikować i zaznaczyć domenę WW. Przedstawić ją w postaci wstęgi z łańcuchami bocznymi aminokwasów funkcyjnych i odpowiednio pokolorować. Pozostałe reszty aminokwasowe przedstawić tylko w postaci szkieletu wiązań peptydowych.
2. Odszukać fragment łańcucha ENaC ulegający mutacjom w zespole Liddle'a. W rozwijanym menu Toolbar wybrać Edit -> Find Sequence. W oknie dialogowym wpisać odpowiednią sekwencję aminokwasów (PPPNY). Zaznaczyć 0 w allowed mismatches (zapewnia to 100% zgodność odnalezionych sekwencji z sekwencją zadaną). Zaznaczyć highlight residues in structure (zaznacza odnalezioną sekwencję w cząsteczce). Potwierdzić OK.
3. W oknie Control Panel w kolumnie group znaleźć zaznaczone na czerwono odnalezione reszty aminokwasowe. Wyświetlić ich łańcuchy boczne i zaznaczyć odpowiednim kolorem.
4. Dla uproszczenia pozostawić widoczną tylko domenę WW NEDD4 z łańcuchami bocznymi aminokwasów funkcyjnych i fragment PPPNY łańcucha β ENaC.
5. Porównać otrzymaną strukturę z przedstawionymi modelami (Rys. 1, 2, 3).
6. Zamknąć program File -> Exit.
Piśmiennictwo
H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne: The Protein Data Bank. Nucleic Acids Research, 28, 235-242 (2000)
Guex, N. and Peitsch, M.C.: SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling.
Electrophoresis 18, 2714-2723 (1997).
Aminokwasy alifatyczne (hydrofobowe)
aminokwas |
kod trzyliterowy |
kod jednoliterowy |
glicyna |
Gly |
G |
alanina |
Ala |
A |
walina |
Val |
V |
leucyna |
Leu |
L |
izoleucyna |
Ile |
I |
metionina |
Met |
M |
fenyloalanina |
Phe |
F |
tryptofan |
Trp |
W |
prolina |
Pro |
P |
Aminokwasy polarne (hydrofilowe)
seryna |
Ser |
S |
treonina |
Thr |
T |
cysteina |
Cys |
C |
tyrozyna |
Tyr |
Y |
asparagina |
Asn |
N |
glutamina |
Gln |
Q |
Aminokwasy kwaśne (hydrofilowe)
Kwas asparaginowy |
Asp |
D |
Kwas glutaminowy |
Glu |
E |
Aminokwasy zasadowe (hydrofilowe)
lizyna |
Lys |
K |
arginina |
Arg |
R |
histydyna |
His |
H |
Wyszukiwarka
Podobne podstrony:
2012 I termin, medycyna, II rok, biochemia, giełdy
Pytania 2012!!!, medycyna, II rok, fizjologia, giełdy
koło 3, medycyna, II rok, biochemia, giełdy
2011 I termin odp, medycyna, II rok, biochemia, giełdy
koło 2, medycyna, II rok, biochemia, giełdy
1. termin EGZAMINU Z BIOCHEMII 2014!!, medycyna, II rok, biochemia, giełdy, GIEŁDY BIOCHEMIA 2014!!
2010 I termin odp, medycyna, II rok, biochemia, giełdy
GIEŁDY BIOCHEMIA-seminaria 2014!!, medycyna, II rok, biochemia, giełdy, GIEŁDY BIOCHEMIA 2014!!
2013 I termin, medycyna, II rok, biochemia, giełdy
2013 0 termin, medycyna, II rok, biochemia, giełdy
koło 1, medycyna, II rok, biochemia, giełdy
2010 II termin, medycyna, II rok, biochemia, giełdy
Notatki Ania Ciepiela, medycyna, II rok, patofizjologia, ćwiczenia
biochemia egz (1), Studia (2012-2017) SGGW - WNoŻ - Technologia Żywności i Żywienie Człowieka, II ro
wykład nr 9 2010 - węglowodany, MEDYCYNA - ŚUM Katowice, II ROK, Biochemia, 5. WYKŁADY
więcej podobnych podstron