
134
POLYMER BRUSHES
Vol. 11
POLYMER-SUPPORTED
REAGENTS
Introduction
Since the original reports by Merrifield, published some 40 years ago (1), solid
supports have been widely used as a synthetic device to avoid expensive and long
winded purification processes, principally due to the simplicity of isolating the
support and its bound material by direct filtration. There are a number of other
advantages when using solid supports; most notably reactions can be driven to
completion using high concentrations and mass action of reagents and synthesis
can be easily automated (2).
With the introduction of a range of high throughput synthesis methods
(3–5), solid supports have become even more indispensable (6), for example, with
the use of polymer-supported reagents and catalysts (7), which maintains the
target molecules in solution but immobilizes the other component, and renders
possible the parallel monitoring of supported reactions but in a solution sense.
The most utilized polymer support in solid-phase chemistry is still a copolymer of
styrene and divinyl benzene [DVB (2%)], known as a gel-type resin, because of its
“gelatinous” and nonmacroporous behavior, originally used by Merrifield for the
synthesis of the first tetrapeptide made by solid-phase synthesis (see P
OLYPEPTIDE
S
YNTHESIS
, S
OLID
-P
HASE
M
ETHOD
).
Nevertheless, there are some problems correlated with the use of this sup-
port, such as its voluminous nature when swollen, its lack of physical stability un-
der harsh handling conditions, and its incompatibility with a number of solvents
(8). For this reason, several groups of researchers have investigated a number
of alternative solid supports, for solid-phase chemistry, in order to improve resin
properties, such as physical robustness, ease of handling, enhancement in loading,
Encyclopedia of Polymer Science and Technology. Copyright John Wiley & Sons, Inc. All rights reserved.

Vol. 11
POLYMER-SUPPORTED REAGENTS
135
improvement of reaction kinetics, and so forth. This article presents an overview
of the materials commonly used in solid-supported chemistry and their evolution.
Styrene–DVB-Based Resins
Polystyrene resins (25–300
µm) are the most commonly used supports in solid-
phase chemistry and, in a variety of formats, are extensively used in a wide range
of industrial processes (see S
TYRENE
P
OLYMERS
). These basic materials can be eas-
ily obtained by radical polymerization of a suspension of an organic phase (the
styrene and divinylbenzene monomers in varying proportions) in water, in the
presence of a stabilizer (in order to reduce the surface tension of the droplets
and to prevent their aggregation) (9). A minimum level (usually at least 0.5%) of
DVB is required in order to ensure that the polymer obtained is insoluble in or-
ganic solvents. The percentage of DVB (cross-linker), influences the physical and
chemical properties of the support. High levels of cross-linking produce more rigid
supports; therefore, drastic conditions of reaction such as mechanical stirring and
high temperatures are permissible, but on the other hand slower reactions are of-
ten observed because of the fact that the reagents cannot easily diffuse within the
polymer network (10). On the other hand, low levels of cross-linking produce highly
swellable but more fragile beads, which, however, exhibit better reaction kinetics.
In most chemical reactions, the solvent plays a fundamental role and this
effect is enhanced in solid-supported chemistry. When in contact with so-called
“good” or “poor” solvents, the beads can swell or shrink (this effect depends prin-
cipally on the level of cross-linking and can be considered akin to the polymer
tending to dissolve in the solvent). This phenomenon can increase the ability of
reagents to diffuse into the beads, and therefore dramatically influences reaction
rates. Reactivity within beads is also highly dependent on resin loading. Loading
variations can for example vary, allowing concentrations within beads to range
between 50
µM and 0.5 M. As in solution chemistry, such concentration effects
can cause huge effects on reaction rates, depending upon the reaction under inves-
tigation as a result of electrostatics and/or steric effects of the reagents. To avoid
some of these type of issues, spacers are often placed between the polymer and
the reactive functionality of the resin (11), but the high concentration of material
on a bead is often ignored in solid-phase synthesis.
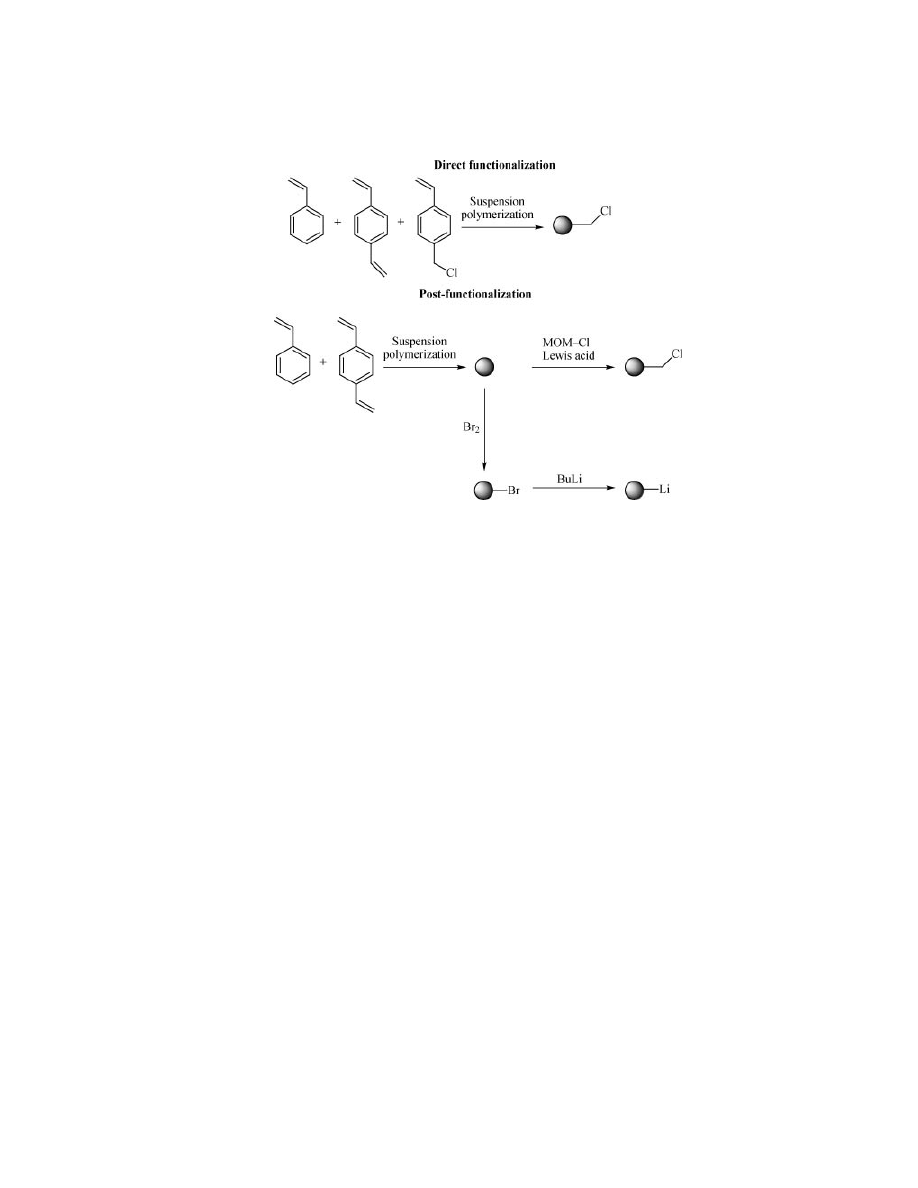
The reactive functionality (the site of chemistry) on a resin can be added
during or after polymerization as shown in Figure 1 (12). Direct functionalization
can be performed by including a third monomer into the original monomer mix-
ture, which already bears the desired functional group. Chloromethylstyrene is
often used in this regard because it can be easily transformed into a wide range of
alternatives (Fig. 2) and is a monomer that incorporates well into styrene/divinyl
benzene mixtures (Fig. 1).
Post-functionalization is usually carried out by electrophilic aromatic sub-
stitution in the presence of a Lewis acid (27) or by metallation of a brominated
resin (Fig. 1) (28).
Good quality polymers are obtained by direct functionalization with the re-
sulting functionality uniformly distributed throughout the bead. However, there
are other factors that influence the quality of the polymer. As mentioned by
136
POLYMER-SUPPORTED REAGENTS
Vol. 11
Fig. 1.
Functionalization of polystyrene resins.
Czarnik (29), resins from different batches often show different chemical be-
haviour, as the process of suspension polymerization depends upon many factors
such as reactor geometry, agitator design, and agitation rate (30). Uniform, spher-
ical beads are preferred for reproducible chemistry, irregular particles being much
more sensitive to mechanical destruction and often falling apart during the re-
action. Both 6- and 8-multiparallel polymerization systems have been designed,
and these systems show very good reproducibility when used for the synthesis
of polymer libraries (Fig. 3) (31), attesting to the parallel advantages of synthe-
sis and maintaining a consistent reaction geometry. A second-generation system
allowed 8-multiple polymerizations with the possibility of changing the stirring
rate of each reaction vessel.
The level of functional groups or “loading,” expressed in mmol/g, can cover a
wide range of values (from 0 to virtually 7 mmol/g), but this level varies greatly
depending on the application of the support. Generally, the loading and application
of polystyrene resins can be grouped as follows:
(1)
<0.1 mmol/g, suitable for oligonucleotide synthesis and the synthesis of
large peptides/proteins
(2) 0.1–0.7 mmol/g, suitable for supported catalysts (which are added in small
amounts)
(3) 0.5–1.5 mmol/g, suitable for solid-phase synthesis (small molecules and pep-
tides) avoiding problems of steric hindrance
(4)
>2 mmol/g, preferred for scavenger and reagents based resins, as these
types of reagents are added in excess
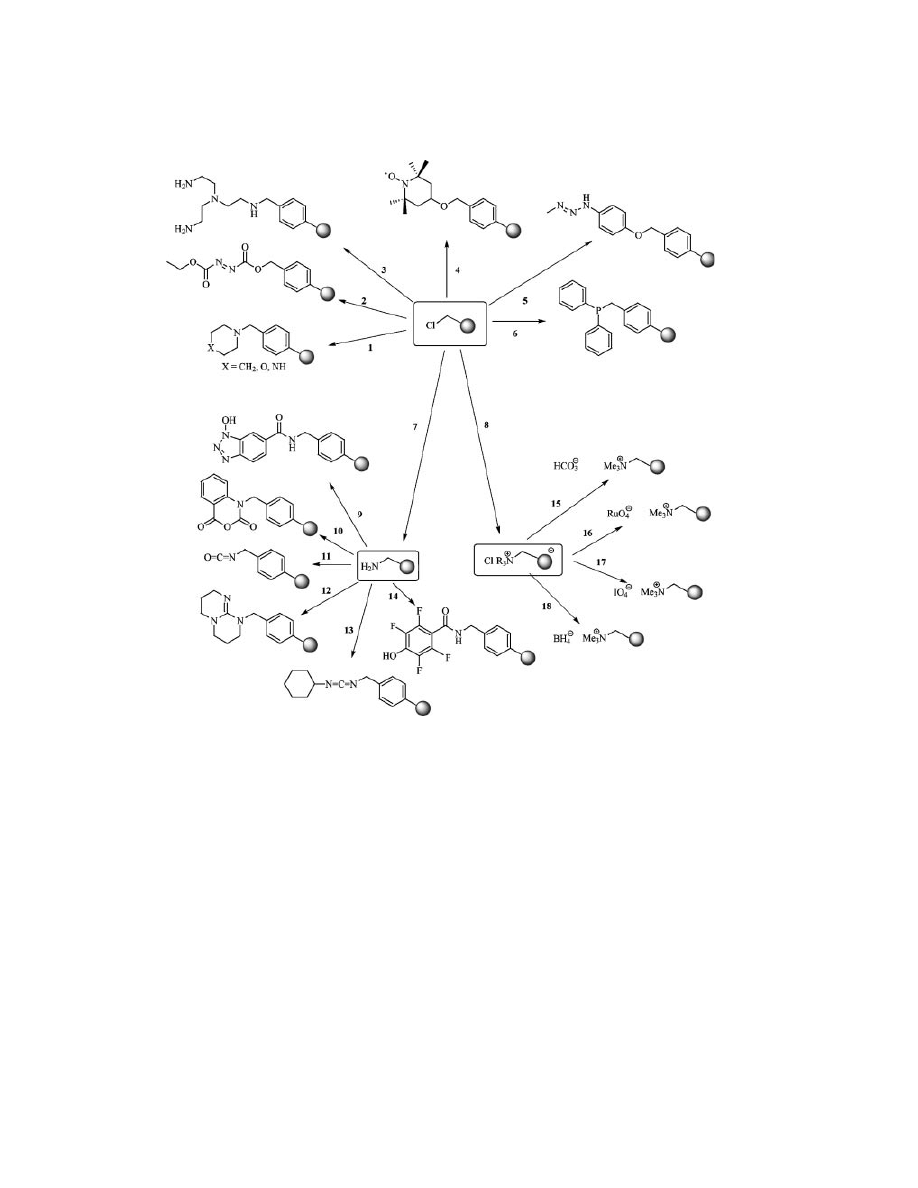
Vol. 11
POLYMER-SUPPORTED REAGENTS
137
Fig. 2.
Preparation of solid-supported reagents and catalysts by derivatization of
chloromethyl polystyrene. (1: Ref. 13, 2: Ref. 14, 3: Ref. 13, 4: Ref. 15, 5: Ref. 16, 6: Ref. 17,
7: Ref. 18, 8:NR
3
–DMF 110
◦
C, 9: Ref. 19, 10: Ref. 20, 11: Ref. 13, 12: Ref. 21, 13: Ref. 22,
14: Ref. 23, 15: Ref. 24, 16: Ref. 25, 17: Ref. 12, 18: Ref. 26).
Resin Types
Gel-Type Polystyrenes.
Gel-type polystyrenes can be synthesized by
radical suspension polymerization of styrene and DVB as reported in 1946 by
Hohenstein and Mark (9,32). When suspended in a solvent with good swelling
properties, these “white soft beads” assume a gelatinous state; their volume may
increase by 6–8 times and they become optically transparent. This process of
swelling, as well as the shrinking, occurs from the outside to the inside of the
polymer network and the increment of volume is in correlation with its solvation.
This plays an important role in chemistry, because it is through such processes
that the reactants diffuse into the polymer network where 99% of the functional
sites reside (33).

138
POLYMER-SUPPORTED REAGENTS
Vol. 11
Fig. 3.
(a) Multiparallel suspension polymerization system; (b) close-up of reaction vessel.
As a consequence, the solvent has to be chosen to allow swelling of the
resin (34). However, swelling is not the only “solvent dependency”, as the type
of functionality also affects solvent compatibility. Thus, while Merrifield resins
(chloromethyl polystyrene) shrink in highly polar solvents such as water, ion ex-
change resins show the opposite behavior and do not swell in “good swelling sol-
vents” such as dimethylformamide (DMF). Merrifield observed that the volume
of the beads and their behavior with the solvent changed continuously as the
supported peptide was grown, attesting to the importance of the resin and the
attached molecules, a factor which obviously is influenced by resin loading (35).
There are thus a number of other factors which need to be considered when han-
dling and evaluating gel-based supports. If the beads are fully swollen in a “good
solvent” and then shrunk rapidly in a bad solvent (eg washing step), “mechanical
shock” can take place and the beads may disintegrate, especially with large beads
(osmotic shock) (36). To avoid this issue, beads should be washed gradually, start-
ing from a good solvent to a poor solvent, while in the fully swollen state beads
should not be agitated with rigid materials, or they may become damaged.
Although a number of issues are correlated with the use of this material,
gel-type polystyrene resins are the most used supports in solid-phase chemistry,
because of their acceptable loading capacity and relative good kinetics when fully
swollen in good solvents.
Macroporous-Type Resins.
Macroporous resins were prepared as long
ago as 1962 by Millar (37). Macroporous resins have been used for many years

Vol. 11
POLYMER-SUPPORTED REAGENTS
139
Fig. 4.
Synthesis of macroporous polystyrene chloromethyl resin.
as ion-exchange materials, polymeric adsorbents (38), and preparative reverse
phases for purification of biomolecules (39), but there are also several examples
of this type of support in organic synthesis as carriers for reagents and catalysts
(40,41).
Even although the IUPAC definition for macroporous refers to materials
that have pores with a diameter greater or equal to 50 nm, the term has been
generalized in the resin area to include resins that present a porous structure in
the dry state (36).
The formation of the pores occurs when an inert solvent, or porogen, is
added to a polymerization mixture containing relatively high levels of cross-linker
(Fig. 4). After polymerization, removal of the solvent from the beads leaves intact
pores that do not collapse because of the high level of cross-linker used (
>20%).
The morphology of the pores (surface area, total pore volume, and average
pore diameters) can be adjusted and controlled by an appropriate choice of the
type and amount of porogen and the level of cross-linker used (42).
Several porogens (also called diluents) have been used to prepare macrop-
orous resins (43), and they can be divided in three main classes:
(1) Solvating (SOL), such as toluene or dichloroethane (small pore)
(2) Nonsolvating (NONSOL), such as n-heptane or alcohols (large pores)
(3) Polymeric (POLY), such as linear polystyrene (very large pores) (44)
Pore formation and their different morphologies have been explained in a
recent review (Fig. 5) (36).
When polymerization begins, droplets are formed by the suspension process.
The monomer droplets start forming small spherical, so-called microgel, particles.
Fig. 5.
Pore formation in macroporous resins. (1) Highly solvated swollen microgels, (2)
aggregation of swollen microgels, (3) phase separation, (4) solvent removal.

140
POLYMER-SUPPORTED REAGENTS
Vol. 11
Fig. 6.
SEM micrograph of macroporous resins synthesized using (a) toluene (solvating)
and (b) heptane (nonsolvating) porogen.
If these contain a good solvent for the monomers, such as toluene, these particles
are fully solvated until there is a high conversion to polymer. Thus, when phase
separation takes place (ie, the polymer is no longer soluble in the solvent), the envi-
ronment within the particle contains only a small amount of unreacted monomer.
These particles, therefore, retain their identity and the network of small pores
already generated between them is essentially retained. When a nonsolvating
porogen, such as heptane, is used, the microgel particles are poorly swollen and
phase separation takes place early (low conversion to polymer). In this case the
droplet contains a high level of unreacted monomers and these have the effect
not only of fusing the microgel particles together, but also of causing significant
filling of small pores between the microgel particles. The results of two extreme
cases are shown in the Figure 6. Smooth beads polymerized with 200% vol/vol
of toluene showed a rough surface only when magnified, whereas beads obtained
from heptane (150% vol/vol) gave an irregular morphology.
To improve the benefits of solid-supported reagents and scavengers, macrop-
orous beads need to have a large surface area and large pores. While a high surface
area provides more sites for functionalization, large pores provide efficient trans-
port of reagents through the beads. However, as suggested by Sherrington, these
two parameters are obviously related (12). If a resin bead has a large surface area
and a large number of pores, then each pore is likely to have a small diameter
and vice versa. Experimentally, these parameters are measured by nitrogen ad-
sorption/desorption (BET) (45), mercury intrusion porosimetry (46), and electron
microscope techniques.
Another consequence of the fixed pores and the rigid structure (high DVB
levels) is that macroporous resins have limited swelling. Thus a wide range of
solvents commonly used in organic synthesis can be used in solid-phase “macrop-
orous resin assisted chemistry” (even water), without modification of the reaction
conditions as required for the use of gel-type resins. Moreover, the rigid structure
makes these supports very resistant to mechanical agitation and easy to handle
(macroporous resins do not stick like gel type resins).

Vol. 11
POLYMER-SUPPORTED REAGENTS
141
A comparison between macroporous and 2% gel-based Merrifield resins was
performed by Janda (47) who demonstrated the efficiency of washing a dye from
pretreated resins was better with a macroporous resin than a gel-type resin. Com-
parison of a palladium catalyst, for Wacker olefin oxidation, supported on gel-type
and macroporous resins gave better results on the macroporous support. Con-
tributions in this area have also been made by Labadie who demonstrated good
results using tailored macroporous resins, commonly known as ArgoPore (these
resins had surface area, pore volume, and loading well defined for optimal perfor-
mance in solid-phase chemistry), for example, reactions in water such as the TiCl
3
-
mediated reduction of nitro compounds, periodate-mediated olefin oxidations (48),
Suzuki reactions on polymer-supported iodides and bromides (49), and alkylation
of supported acids via enolate formation at low temperature (50). Nowadays, it
is possible to find a wide range of macroporous polymer-supported reagents and
catalysts commercially available. In conclusion, well-designed macroporous resins
exhibit better efficiency and kinetics than gel-type resins in highly polar solvents
and have become a valid alternative for solid-phase chemistry, without significant
swelling and washing issues, although maximum available loadings cannot be
as high as the gel-type counterpart (commercially available macroporous resins
for solid-phase chemistry have a loading that depends on the manufacturer (ie,
Argonaut Technologies: 0.2–1.8 mmol/g; Polymer Labs: 3.0 mmol/g).
Microgel Polystyrenes.
Microgels are soluble polymers with a certain
degree of cross-linking, but can be precipitated using an appropriate solvent (51).
Beads with a diameter of 5–25
µm can be prepared by radical dispersion polymer-
ization in the presence of a surfactant.
It is imperative to use low levels of monomers during preparation to avoid
macrogelation due to the aggregation of the microgels to form large-size particles.
One of the main characteristics reported for microgels are their ease of handling
when solubilized. A consequence of their solubility is that reactions can be easily
monitored by classical solution-phase techniques (eg NMR) but that materials can
then be readily recovered in a solid-phase manner.
Janda reported a detailed investigation of polymerization parameters such
as the type of solvent, percentage of monomer, and level and different type of
cross-linker and showed that polymers with good characteristics could be obtained
using 5 wt% monomer solution in tetrahydrofuran (THF) (52). Good polymers were
achieved using 10% of DVB or 5% of alternative cross-linkers (Fig. 7). Precipitation
of the soluble polymers was achieved by addition of cold methanol and filtration
through a glass frit.
Becuse of their recent deployment there are only a few examples in litera-
ture on the use of this material in solid-phase chemistry. Wulff (53) used microgel
Fig. 7.
Cross-linker used in the preparation of microgels.
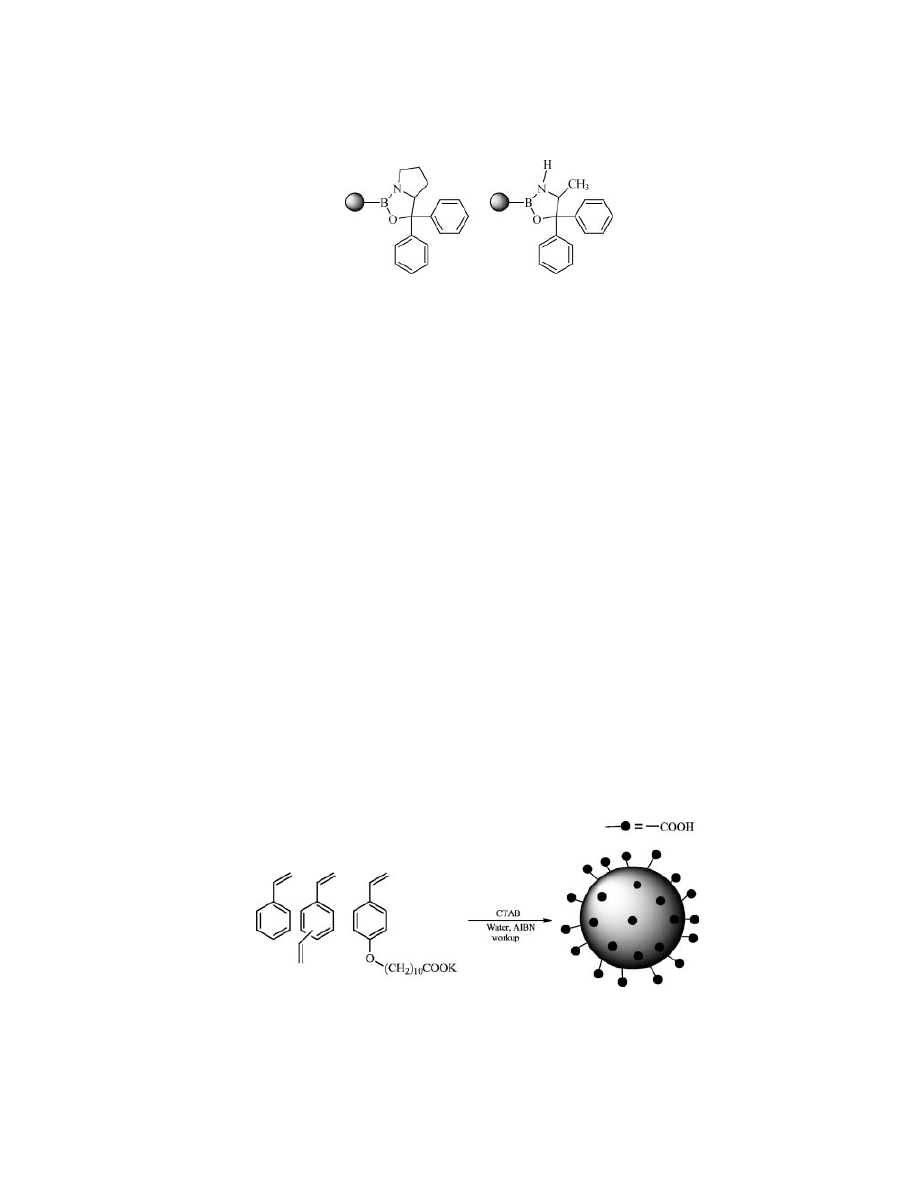
142
POLYMER-SUPPORTED REAGENTS
Vol. 11
Fig. 8.
Microgel-supported oxazaborolidines catalysts.
polymers for the preparation of supported oxazaborolidines as catalysts for asym-
metric reduction of ketones (Fig. 8).
This type of catalyst, supported on polystyrene gel type resins, gave results
comparable to the homogeneous counterpart. However, because of the poor me-
chanical properties of the resin under the reaction conditions, a more suitable poly-
mer support was needed. Unfortunately, highly cross-linked resins, with their poor
kinetics, decreased the overall selectivity of the catalyst. Although linear polymers
were used (54), microgels offered advantages because of their ease of handling.
Janda used these supports to perform the solid-phase synthesis of oxazoles (55)
and later produced a combinatorial library of phthalimides in good purities and
yields using these supports. In addition, microgels were used for the preparation
of supported tris(2-aminomethyl)amine and borohydrides to remove isocyanates
and to reduce aldehydes respectively.
Polystyrene Nanoparticles.
Polymer-based nanoparticles are used in
several areas of life science, for example drug delivery (56), and one such polymer
matrix commonly used is poly(
L
-lactic acid) (57). However, nanoparticles with a
polystyrene–DVB matrix have been also synthetized (58,59) and used as novel
solid supports for organic synthesis (60). The method commonly used to generate
these monodisperse particles is Microemulsion Polymerization (qv) (58). How-
ever, nanoparticles have also been prepared by precipitation polymerization (61).
A microemulsion of styrene and DVB, with an amphiphilic comonomer, in water
(three component oil-in-water) (62) allows the preparation of nanobeads with a
hard core of polystyrene and the amphiphilic comonomer dispersed on the surface
with diameters around 50 and 300 nm that can be precipitated by the addition of
methanol. Functionalization of the nanoparticles surface, can be easily achieved
using the functionalized amphiphilic comonomer (Fig. 9) (60).
Fig. 9.
Synthesis of functionalized polystyrene nanobeads.

Vol. 11
POLYMER-SUPPORTED REAGENTS
143
These types of beads were used by Cammidge (60) to perform a solid-phase
synthesis of porphyrin derivatives following the method described by Leznoff and
co-workers (63). Although the final yield of the product was not brilliant (4 and
8% for both the isomers), they were higher than that achieved using “conventional
polystyrene resin.”
PEG-Grafted Resins
Although gel-type resins are the most widely used supports in solid-phase chem-
istry, it has been found that such supports are not always the most suitable for pep-
tide synthesis. This is due to two main factors: polystyrene chains are hydrophobic
and are not completely compatible with peptides and, furthermore, bulky peptides
can adopt unfavorable conformations within the polystyrene cross-linked matrix.
These issues can be avoided by performing the synthesis on linear polymers
such as poly(ethylene glycol) (PEG) (64,65), as PEG is soluble in a wide range
of organic solvents and, when linked to polypeptides, enhances solubilization.
Furthermore, Bayer demonstrated that during coupling reactions, PEG-linked
amino acids had the same kinetic properties as amino acid esters (66). However,
the main problem in liquid-phase peptide synthesis is the separation of linear
PEG from the final polypeptide after cleavage.
Because of its compatibility with peptide chemistry, several authors began
to solve the issue of PEG isolation by grafting it onto polystyrene–DVB resin.
As reported by Bayer, polymer grafting by simple reaction with oligomeric
PEG and Merrifield resin did not give good results (68), because of the fact that
the hydroxylic PEG functionalities could react with two adjacent chloromethyl PS
groups, giving an additional degree of cross-linking and reducing the final loading
of the resin. However, Barany demonstrated that the grafting of PEG could be
achieved by the use of a bifunctional amino-PEG acid (Table 1, entry 2) (69,70).
Unfortunately, this support has not found wide use, probably because of its amide
linkage.
Bayer described grafting of PEG onto PS resin, by direct anionic polymer-
ization of ethylene oxide onto poly(styryl-methyltetraethylene glycol ether) (68).
The “tentacle polymer” so obtained is commercialized by Rapp Polymere un-
der the name of TentaGel and it is the second most used support for solid-phase
organic synthesis after polystyrene resin (Table 1, entry 1) (67). The main char-
acteristic of these resins is their greater swelling in polar solvents such as water,
methanol, ethanol, etc, compared to PS resins. As shown in Table 2, entry 2, the
swelling of TentaGel is uniform in high to medium polarity solvents. This behavior
allows the packing of resins into columns for continuous flow peptide syntheses
(68).
Moreover, because of the fact that molecules bound to the highly solvated
PEG chains are in a “solution-like” environment and the interactions with the
hydrophobic polystyrene backbone are reduced, reaction rates in solid-phase pep-
tide synthesis on TentaGel are greater than on the equivalent Merrifield resins
(74,75). Because of the high mobility of PEG chains, gel-phase and MAS NMR
result in narrow lines (76). Kinetic comparisons between TentaGel and other sup-
ports have been carried out (77,78), and there are several examples of “nonpeptide”
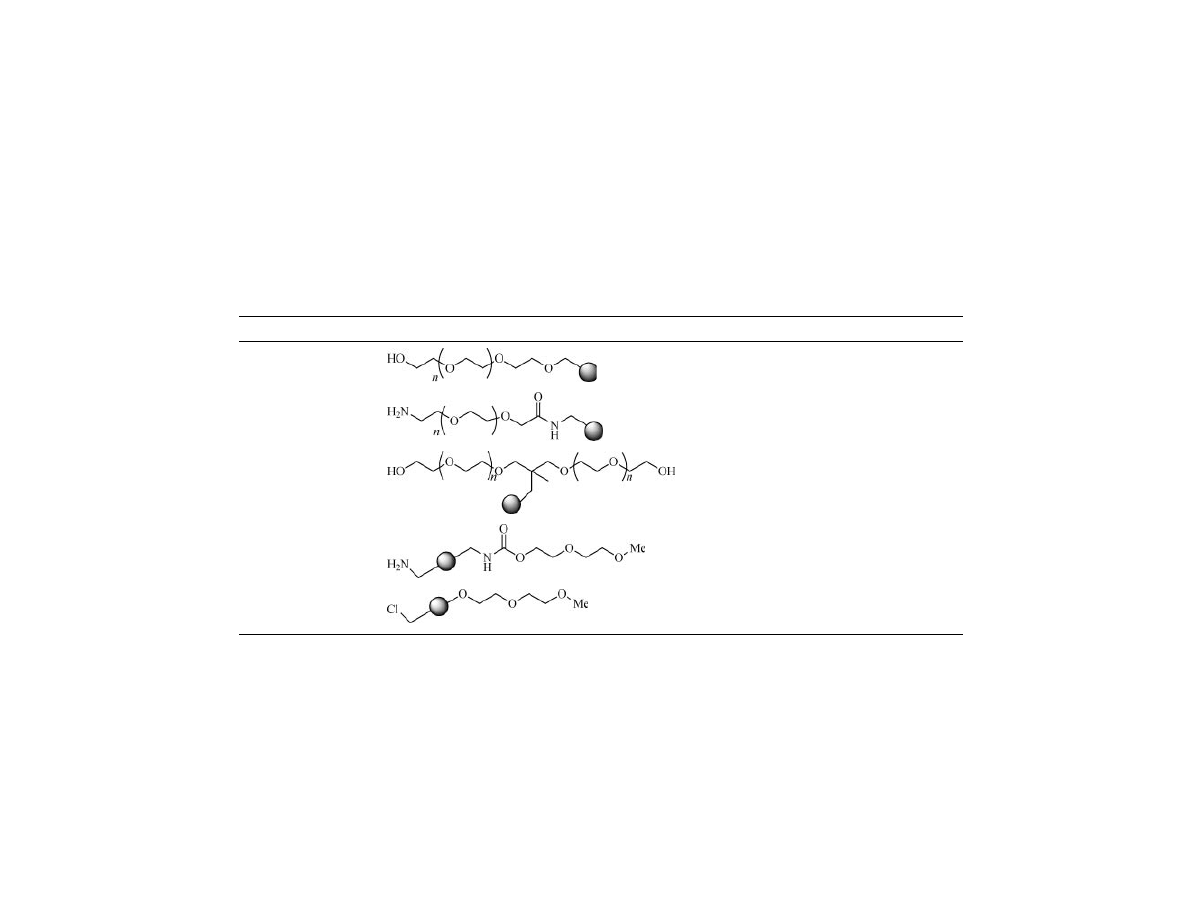
Table 1. PEG Branched Resins
Entry
Resin
Structure
Linkage
Author
References
1
TentaGel
Benzylether
a
Bayer Rapp
67,68
2
PS–PEG
Amide
Barany
69,70
3
ArgoGel
Diol
Gooding
71
4
NovaGel
Uretane
Hudson
72
5
PS–MPEG
Phenylether
Bradley
73
a
Original version of TentaGel.
144
Vol. 11
POLYMER-SUPPORTED REAGENTS
145
Table 2. Resin Swelling (mL/g of Resin)
Entry
Resin
H
2
O
CH
3
OH
CH
3
CN
DMF
CH
2
Cl
2
THF
1
TentaGel
a
3.6
3.6
4.2
4.7
6.3
5.0
2
PS–PEG
b
2.8
4.2
3.7
5.1
5.6
3.9
3
ArgoGel
c
3.9
4.7
—
6.0
7.5
5.8
4
NovaGel
d
—
4.0
5.0
7.0
10.0
7.5
5
PS–MPEG
e
2.4
2.8
—
5.8
8.2
8.4
6
Gel-type
f
—
1.6
3.2
5.6
8.3
8.8
a
Obtained from Rapp Polymere.
b
PS–PEG low loading.
c
Obtained from Argonaut Technologies.
d
Obtained from Novabiochem.
e
7% PEG derivative included.
f
PS-DVB (1%).
chemistry where the rates are faster on TentaGel than PS resins (ie, Suzuki reac-
tions using supported boronic acids (79), or Sm(II) mediated radical cyclizations
(80), or Sharpless asymmetric dihydroxylations) (81). Yan, however, demonstrated
that this rule was not always true (82), and found that some reactions, for exam-
ple terminal modification of aspartic acid or the synthesis of dansyl hydrazones,
were faster on PS resins. There are several drawbacks that limit the use of Tent-
aGel resin as a universal support for solid-phase chemistry. The substitutions of
TentaGel resins are lower than PS resins although loading can be increased by
decreasing the grafting level. However, Lee has demonstrated recently that the
swelling of these types of resins depends on the level of grafted PEG (83). When
the solvent used was water, there was a direct relationship between PEG level
and swelling, but in other solvent systems such as THF or DMF there was a
discontinuous trend until the level of PEG was greater than 72% by weight.
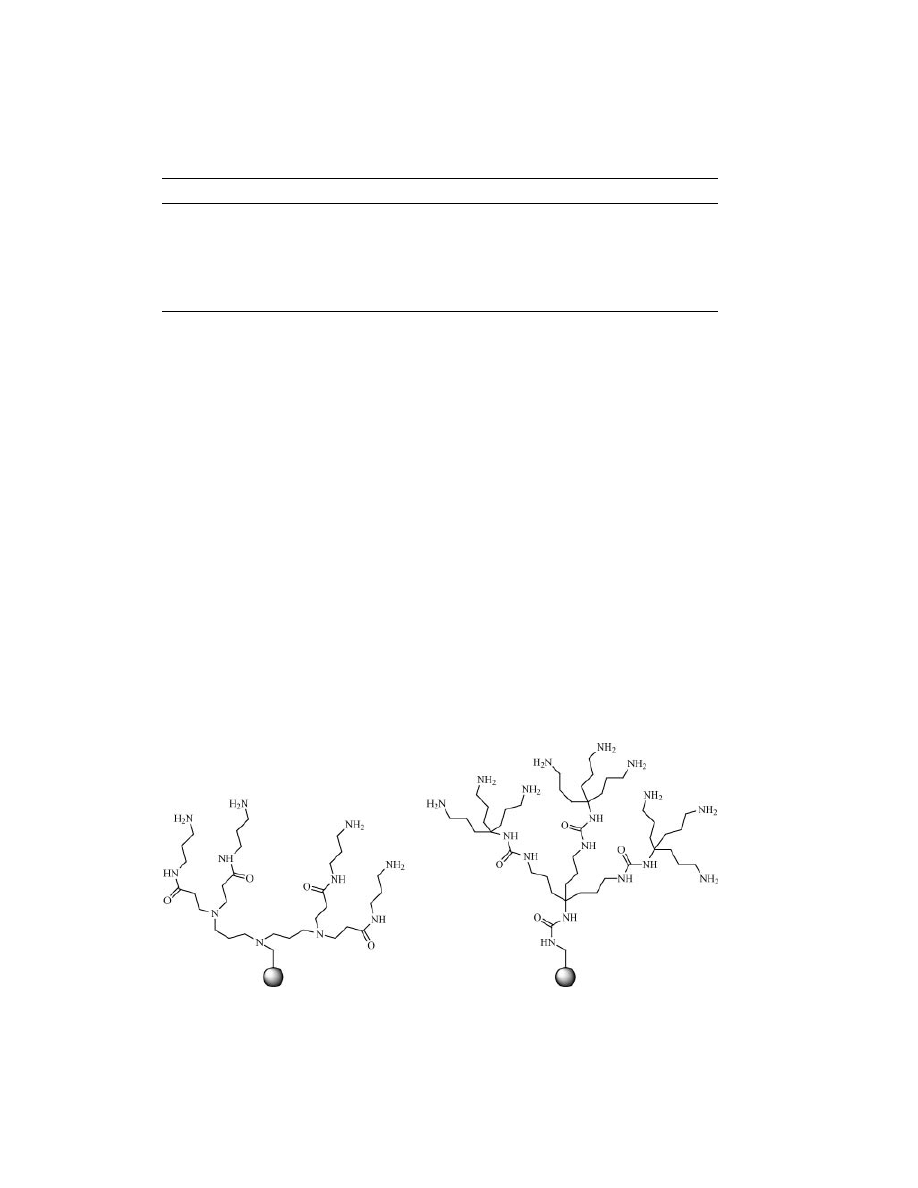
A method to increase loading was demonstrated by Bradley by dendrimer-
ization. In this way, TentaGel bead loading was amplified about 10 times (Fig. 10)
(84,85).
Fig. 10.
Dendrimeris for loading improvement.

146
POLYMER-SUPPORTED REAGENTS
Vol. 11
Fig. 11.
Preparation of ruthenium-supported catalysts.
There is at least one other main issue that has to be cited. It has been ob-
served that traces of linear PEG are found in final products after cleavage because
of the weak benzyl ether bond that links the PEG to the polystyrene backbone.
For this reason, Gooding prepared a PEG-grafted polystyrene resin using a
polystyrene diol as a scaffold for the PEG grafting (Table 1, entry 3) (71) [Rapp
also used hydroxyethyl PS in the new generation of TentaGel resins (TentaGel S)].
In this way, the chemical stability as well as the loading (0.4–0.5 mmol/g) of such
resins was improved. The higher stability of this support was proved by repeated
amide cleavages under acidic conditions, where no impurities of linear PEG were
found even after long treatments (86). This resin is commercially available from
Argonaut Technologies under the name of ArgoGel, and it has been used as a
support in the preparation of substituted imidazoles, N-alkyl sulfonamides, acy-
lamines, benzofurans, etc (71). De Miguel demonstrated the use of such flexible
supports for the immobilization of a highly selective ruthenium cluster catalyst
for hydrogenation of olefins and ketones (Fig. 11) (87). Recently, the same group
reported the use of such supported catalysts in applications such as gas sensors
for H
2
S, CO, and SO
2
by color changing of the resin (88).
During his research to improve PEG-grafted resins, Hudson, following the
example of Barany, by linking oligomeric PEG chains to aminomethylated resins
by a urethane linkage (Table 1, entry 4) (72). These “NovaGel” resins have been
commercialized by Novabiochem. The main difference between these supports and
the others previously described is that the reactive functionality is still bound to
the polystyrene backbone and not to the PEG chains. The effect of the PEG grafting
in this case improves swelling as shown in Table 2, entry 4.
However, even if the urethane linkage is more stable than the benzyl ether
linkage, leaching of linear PEG can contaminate the final product. Recently,
Bradley reported the preparation of novel supports using a principle similar to
NovaGel resin, where short PEG chains were linked in the polymer by a phenyl
ether and the functionality copolymerized into the beads backbone (Table 1, entry
5). Polystyrene–MPEG nongrafted resins were prepared by adding a new PEG
derivative comonomer during polymerization as shown in Table 1, entry 5 (73).
Such resins exhibit higher loading capacities (0.63–0.77 mmol/g) compared to
PEG-grafted resins and at the same time showed broad solvent compatibility (Ta-
ble 2, entry 5) and improved resistance to acidic and basic conditions. In addition,
during their synthetic evaluations, such supports showed similar, if not better,
results compared to grafted polymers without detection of PEG impurities in the
final products.

Vol. 11
POLYMER-SUPPORTED REAGENTS
147
Alternative Cross-Linkers for Polystyrene-Based Resins
To overcome the incompatibility of the hydrophobic polystryrene–DVB matrix
with highly polar solvents (aqueous), alternative types of cross-linkers have been
utilized over the last few years. Polyethers have been extensively used for this
purpose and properties such as swelling and mechanical stability have been im-
proved compared to gel-type resins. The cross-linking agents described herein are
(1) PEG and (2) (poly)tetrahydrofuran based.
Copoly(styrene–PEG).
As
explained
above,
PEG
grafted
onto
polystyrene improves its swelling properties in polar solvents (89). One of
the major drawbacks of this type of supports is their inherent low loading
(0.2–0.6 mmol/g) because a large part of the polymer mass is PEG (
∼70%)
(48). An alternative to this method is to include PEG in the polymer matrix
during polymerization as a cross-linker. Vinyl PEG derivatives have been used to
replace DVB as a cross-linker in the synthesis of a polymer matrix (TTEDA–PS:
tetraethyleneglycol diacrylate cross-linker–polystyrene) (90). Although this resin
showed comparable mechanical properties to gel-type resins, it showed much
more effective swelling and solvatation than PS–DVB in polar and nonpolar
solvents. The synthesis of an 18-residue peptide on such a chloromethylated resin
afforded better yield and purity than PS–DVB. Later bis-styrene–PEG monomers
were synthesized, copolymerized with styrene, and used in supporting catalyst in
Diels–Alder reactions (91). Oligo(ethylene glycol) styrene derivatives have also
been synthesized and copolymerized with styrene by Meldal to prepare polymers
used for peptide synthesis (92). Kurth reported an interesting evaluation of
the swelling and diffusion properties for these polymers compared to 2% DVB
polystyrene (93). In this study several PEG-based cross-linkers, linking two
styrene monomers (with different polyether chain lengths, n
= 1, 2, 4, 6; see
Fig. 12), were prepared and used to prepare a library of PEG-based resins using
different proportions of PEG cross-linking (2%, 10%, 20%).
As expected, the swelling of these beads in all solvents was much better
than their 2% PS–DVB counterpart. At low levels of PEG cross-linking (2%) the
swelling was independent of the PEG tether length, but at high levels (20%) better
swelling was observed when n
= 6. In conclusion, the swelling and at the same time
the physical stability of the beads were improved. The diffusion of the reagents
within these resins was measured by the inclusion of a vinyl dye derivative (dansyl
derivative) in the monomer mixture, and the fluorescence quenching of the result-
ing polymer bound dye was measured by adding triethyloxonium tetrafluoroborate
Fig. 12.
Synthesis of PEG cross-linked PS resin.
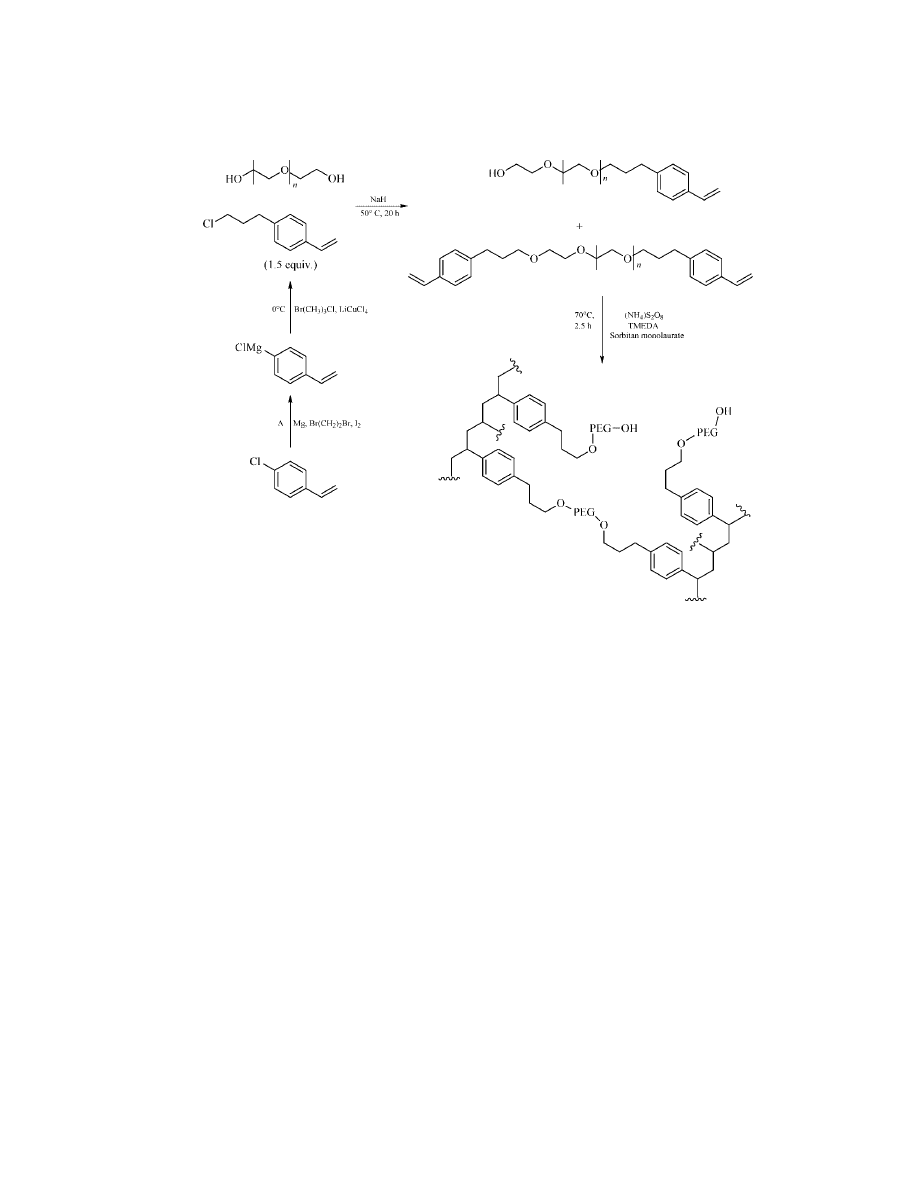
148
POLYMER-SUPPORTED REAGENTS
Vol. 11
Fig. 13.
Synthesis of POEPS-3.
in accordance with the method of Shea (94). These studies showed only a slight dif-
ference between conventional PS resins and that modified with PEG cross-linker
when the reactions were performed in a good swelling solvent (toluene); however,
in poor solvents the PEG-based cross-linked resins showed the best kinetics.
However, one of the drawbacks of using this type of cross-linker is that PEG
is connected to the polystyrene backbone by a benzyl ether bond that could be
cleaved by the use of Lewis or protic acids. Meldal reported the synthesis and the
use of an alternative cross-linker containing a three-carbon spacer between the
PEG chain and the polystyrene backbone (85) (Fig. 13).
Properties such as swelling, loading, diffusion, and ability to furnish high
quality MAS-NMR have been investigated and compared to commercial coun-
terparts such as TentaGel, ArgoGel, 2% PS-DVB, and macroporous PS (96). The
results of this study showed that POEPS-3 was comparable to TentaGel and Argo-
Pore, in terms of swelling, NMR quality, and diffusion. Moreover, these supports
showed better swelling in the range of solvents tested.
Copoly(styrene-butylene glycol).
Because of the favorable properties
shown by the PEG-based cross-linkers, Janda investigated other types of cross-
linkers (97). Because of the tendency of PEG to complex organometallic com-
pounds and its high hydrophilicity, new types of polystyrene resin containing
“polytetrahydrofuran”–styrene derivatives as cross-linkers were prepared to allow

Vol. 11
POLYMER-SUPPORTED REAGENTS
149
Fig. 14.
Cross-linker used in the synthesis of copoly(styrene-tetrahydrofuran) resins.
better solvation of the resin and its functionality that might show reactivity close
to homogeneous-phase conditions.
Resins with 1–2% of cross-linker (Fig. 14, A) degraded in the presence of
strong Lewis acids (TMSOTf) or strong bases (BuLi). However, increasing the
cross-linking level (5–10%) gave resins that were unaffected by these conditions.
Resins with 1–2% of cross-linker (Fig. 14, B) showed good stability under the
conditions reported above. The resin synthesized with 2% of cross-linker (Fig. 14,
B) is commercialized under the name of JandaJel.
The main characteristic of these resins was their incredible swelling in or-
ganic solvents, almost doubled compared to conventional gel-type 1% polystyrene
resin with 1% DVB (Table 3).
JandaJel was used to prepare a small library of phthalides where the key
step of the synthesis was the ortho-lithiation of the supported benzamide (98).
The same research group investigated several applications of these resins: Jacob-
son asymmetric epoxidation with a supported catalyst (99); synthesis of tertiary
amines using the REM linker (100); solid-phase synthesis of oligoesters (101);
solid-phase peptide synthesis using a routine Boc Protocol (102). Other benefits
were reported by Shibasaki during the development of new supported catalysts
for Reisser- and Strecker-type reactions (103,104). In a recent publication (105),
atom transfer radical polymerization ligands on JandaJel were used to remove a
copper catalyst, because of its better solvent compatibility compared to PS–DVB.
However, although JandaJel showed less efficiency compared to low molecoular
polyethylene capped with appropriate ligands previously developed (106), the re-
action times were shorter. In conclusion, JandaJel is more “organic solvent-like”
than PS–DVB. However, although its excessive swelling might improve reaction
kinetics and yields, it can be a drawback because of the difficulty of handling the
support during library synthesis.
Table 3. Swelling Comparison Between Gel-Type and JandaJel Resins
a
Volume of swollen resins, mL/g
Resins (cross-linking)
Dioxane
THF
DMF
Benzene
DCM
Merrifield (1)
6.0
6.4
4.8
6.6
6.0
Merrifield (2)
5.4
5.4
4.2
6.6
5.8
JandaJel (1)
14.8
14.0
10.4
14.6
15.0
JandaJel (2)
7.8
7.4
6.0
8.2
7.4
a
Ref. 97.

150
POLYMER-SUPPORTED REAGENTS
Vol. 11
Table 4. Scavenging Efficiencies Using Different Amines
Amine
Solvent
Amount scavenged %
Benzylamine
THF
75
Butylamine
THF
77
Phenylethylamine
DCM
78
Diethylamine
DCM
90
3,5-dimethylamine
THF
48
Alternative Formats for Polystyrene Supports
The synthesis of combinatorial libraries by the split and mix method allows thou-
sands of compounds to be obtained in just a few synthetic steps. In accordance
with the original concept, each resin bead bears one single compound, and iden-
tification of biologically active compounds can be achieved directly on the beads.
The drawback of this process is that the amount of compound that each bead can
carry is limited (a few hundreds of picomoles), which is not enough for chemical
identifications. To resolve this issue several approaches have been developed. The
original method by Bradley was the enhancement of loading by “dendrimeriza-
tion” (107). Bigger beads can be used, but such supports suffer from poor reaction
kinetics and a propensity to disintegrate. Several authors have proposed different
formats for solid-supported chemistry for split and mix as well as parallel synthe-
sis applications to improve the issues described above and to improve the ease of
handling.
Monoliths and Discs.
Fr´echet and co-workers introduced the monolith
format in the 1990s for applications in chromatography (108) and solid-supported
catalysts (109). In 1999, Sherrington described the preparation of polymer discs,
cut from monoliths, for synthetic purposes (110). Among the cross-linkers used,
PEG
1000
diacrylates showed better resistance to mechanical stirring and os-
motic shock than PS–DVB. After chloromethylations, the discs were coupled with
5-bromosalicylic acid and treated with phenylboronic acid in the presence of
Pd(PPh
3
)
4
. Unfortunately, the Suzuki products were not obtained in good yields. In
the same year, Fr´echet demonstrated the use of their porous-grafted macroporous
polystyrene–DVB monolith-disc in so-called “reactive filtration” with good results
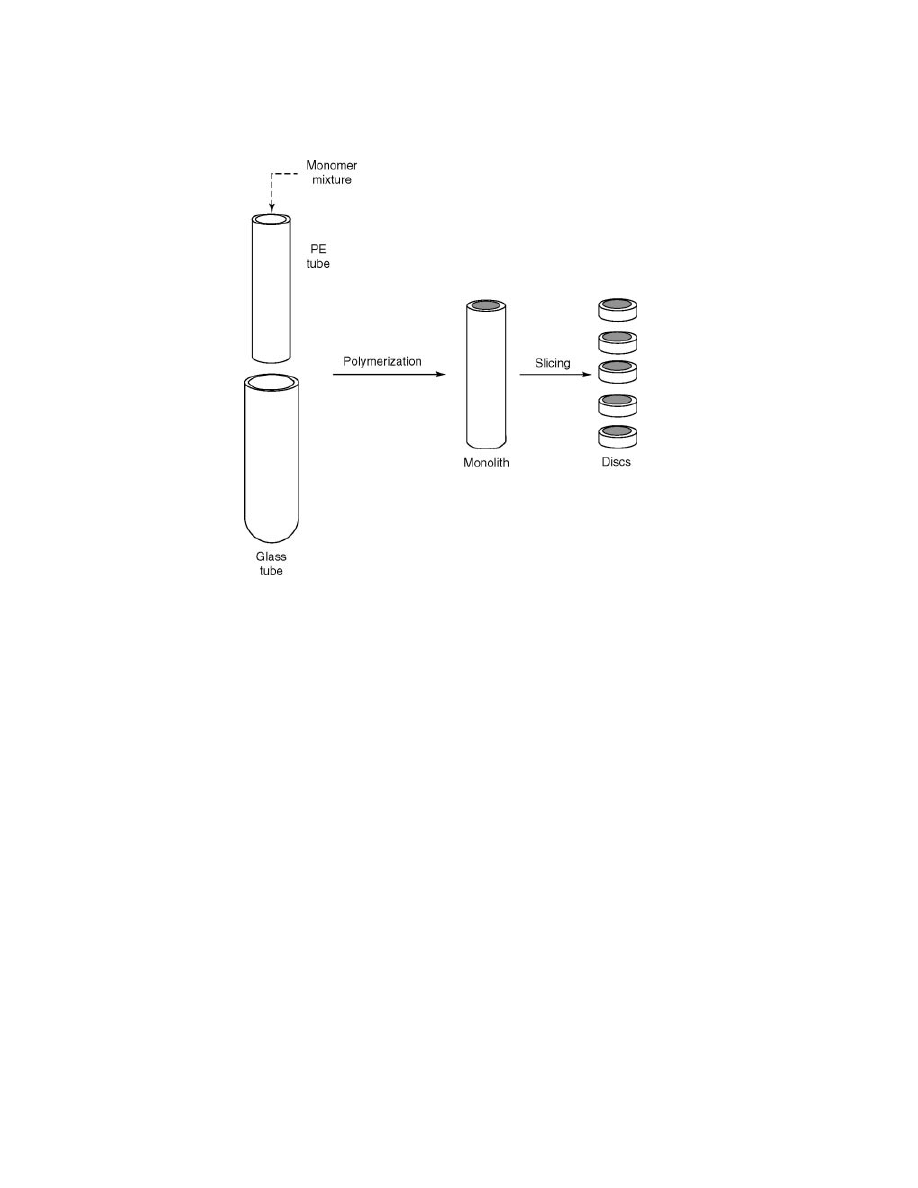
(111). For the preparation of the monoliths, the monomer mixtures and the poro-
gen were placed into a shrinkable polyethylene tube that acted as a mould inside a
glass tube. After polymerization, the glass tube was broken, leaving the monolith
tightly trapped in the polyethylene tube which could be cut into 5-mm disc shapes
(Fig. 15) (112). The resulting PE-encircled discs showed good mechanical stability
and also ease of handling.
To increase the accessibility of the inner reactive groups and also the num-
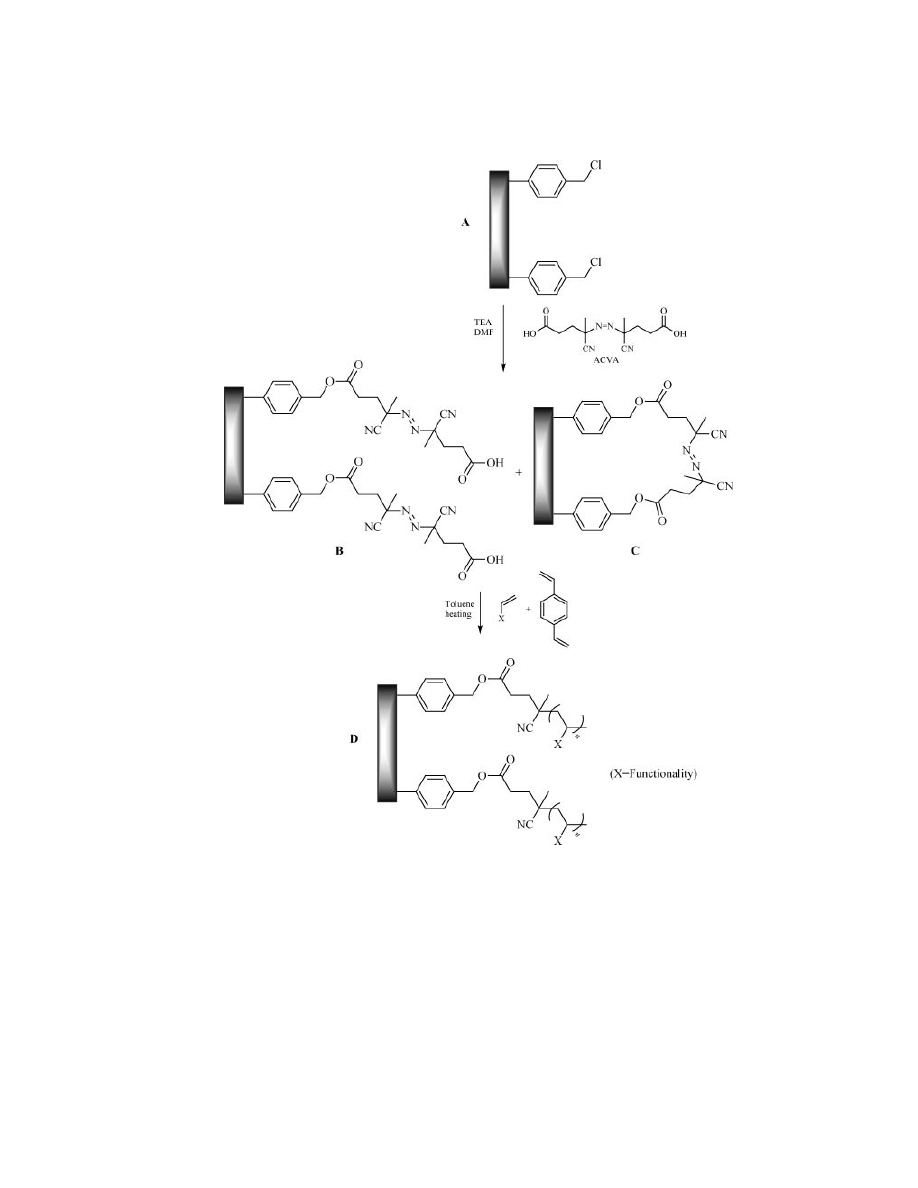
ber of functionalities, the monolith surface was grafted as showed in Figure 16.
The chloromethyl polystyrene discs (Fig. 16, A) reacted with azobis(4-cyanovaleric
acid) (ACVA), a symmetrical azo initiator, to give the polymer-supported acid (Fig.
16, B and C). The pores were then filled with an appropriate monomer and poly-
merized to afford the final products (Fig. 16, D). The initiator could also react
with two chloromethyl groups (Fig. 16, C) and this was indeed preferred to obtain
Vol. 11
POLYMER-SUPPORTED REAGENTS
151
Fig. 15.
Preparation of monolithic discs using the method of Fr´echet (112).
better grafting as propagation could occur from both the sides of the initiator and,
therefore, maximize grafting efficiencies.
Supported disc-reagents were prepared for acylation of several amines in a
flow system (113), affording very good conversions into the corresponding amides
(114).
The major advantage of this format is that the polymer could be fitted into a
cartridge, allowing easy interchangeability during automatic synthesis. Moreover,
they could be used to perform synthesis and purification in a flow system (114).
One other application of such monoliths, for the synthesis of a split and mix
library, was presented by Janda (115). The preparation of “euclidean-shape” mono-
liths was performed using a monomer mixture of styrene, chlroromethylstyrene,
and 1,4- bis(vinylphenoxy)butane as cross-linker. Dodecanol was used as inert
diluent and benzoyl peroxide as initiator.
Films.
PS-based films were introduced by Sherrington to be used as an easy
device for FTIR in situ monitoring (116). Thin films (76mm
× 14mm × 60–120µm)
were obtained by polymerization of monomers in presence of porogen (for macrop-
orous films), and a UV-sensitive initiatior, inserted between two microsocpe slides
(Fig. 17). The microscope slides were filled by capillarity, and polymerization ini-
tiated by UV irradiation.
Gel-type films were produced using different cross-linkers coupled with
styrene and chloromethylstyrene (CMS). The best films were obtained using a
152
POLYMER-SUPPORTED REAGENTS
Vol. 11
Fig. 16.
Grafting of polymer discs.
PEG
1000
/1500
-DVB cross-linker, but the PS–DVB films resulting were fragile. The
chemistry of these gel-type films was evaluated by nucleophilic displacement of
the chlorine with 5-bromosalicylic ester. Macroporous films were also produced
using styrene, DVB, and 4-vinylpyridine. Flexible and resistant films were ob-
tained using long-chain alcohols as porogens. To evaluate diffusion through the
macroporous films, the pyridine functionality was methylated with methyliodide

Vol. 11
POLYMER-SUPPORTED REAGENTS
153
Fig. 17.
Preparation of polymeric films (116).
in acetone and the yellow color of the film examined due to the pyridinium iodide
charge transfer absorption band. Treatment of the supported pyridinium salt with
KMnO
4
allowed the film to change color to purple because of the exchange of the io-
dide with the permanganate. Treating the supported pyridinium salt with CuSO
4
caused the film to turn blue because of coordination of the pyridine with Cu(II).
The supported-pyridinium salts were also used for the preparation of immobilized
rhodium complexes and for mechanistic studies on the carbonylation of olefins by
FTIR (117).
Plugs and Tablets.
In 2001, Bradley introduced resin plugs as a method
to handle polymer supports (118). Plugs were achieved by sintering resin beads
with an inert polymer matrix (ultrahigh molecular weight PE). The plugs were
obtained in cylindrical forms (9.9-mm length
× 7.5-mm diameter) and had loadings
between 50 and 150
µmol). Although both PS- and PEG- grafted resins were used
for the preparation of the plugs, they are now prepared using StratoSphere resins
and commercialized by Polymer Laboratories with the name of StratoSphere Plugs
(Fig. 18).
During their evaluations, plugs were used as supports for solid-phase syn-
thesis of small libraries of biaryl compounds, sulfonamides, tertiary amines, ureas,
and amides. Although yields and purities were similar to the loose beads counter-
part, the reaction times were slightly increased. Because of their mechanical sta-
bility and limited swelling, plugs are much easier to handle than the corresponding
Fig. 18.
(a) Cross-section of a resin plug (100
× magnification); (b) single bead within
the matrix of the resin plugs (270
× magnification); (c) illustration of the dimension of the
resins plugs.

154
POLYMER-SUPPORTED REAGENTS
Vol. 11
Fig. 19.
Preparation and use of palladium plug-supported catalyst.
loose beads. In solution-phase synthesis, plugs have been employed as supports
for immobilized palladium catalysts to prepare libraries of biaryl compounds (Fig.
19) with yields comparable to Pd(PPh
3
)
4
(119).
In conclusion, plug-supported palladium maintained comparable catalytic
activities, but were much easier to handle than the homogeneous analogue
Pd(PPh
3
)
4
.
An alternative to improve resin handling was introduced by Ruhland, who
described the preparation of tablets obtained from the compression of a blend of
resin with solid reagents or catalysts (120). The preparation of such tablets in-
volved three steps: swelling of the beads (and the solid reagent or catalyst) in a good
solvent; sieving; and compression. The first step was essential for the preparation
of mechanically stable tablets. The effect of swelling and drying resulted in ag-
glomerations caused by bead–bead interactions as observed by SEM. Observation
of the tablet structure showed that the beads were distorted after compression but
not destroyed. The disintegration time of the tablets, measured by gentle shaking,
was very solvent-depending and implicated that the swelling of the beads caused
disintegration. As a consequence, good solvents such as THF or DCM gave shorter
times of disruption (7–16 min) than DMSO or ethanol (
>30 min). Functionalized
tablets were employed to carry out Mitsunobu reactions and amine acylations,
giving results comparable to the corresponding loose resins. During syntheses,
tablets were loaded in 48-well plates in only 3 min, whereas the same procedure
applied to resin beads took much longer. Moreover, preparation of tablets allowed
the protection of sensitive functional groups from moisture or oxygen, and also a
drastic reduction in the time of exposure to toxic hazardous and dusty materials
in the working environment.
Nonpolystyrene Polymer Matrices
All the polymer supports described so far for solid-phase chemistry have a com-
mon factor: the main presence of a polystyrene backbone. Although this polymer
matrix has good characteristics, such as chemical and mechanical stability (de-
pending on the type and amount of cross-linker) and favorable loadings in solvent,
its compatibility with polar solvents and large biomolecules, such as enzymes and
receptors, is quite poor (121). In order to resolve these problems, several authors
have replaced hydrophobic polystyrene with a more amphiphilic backbone. In the
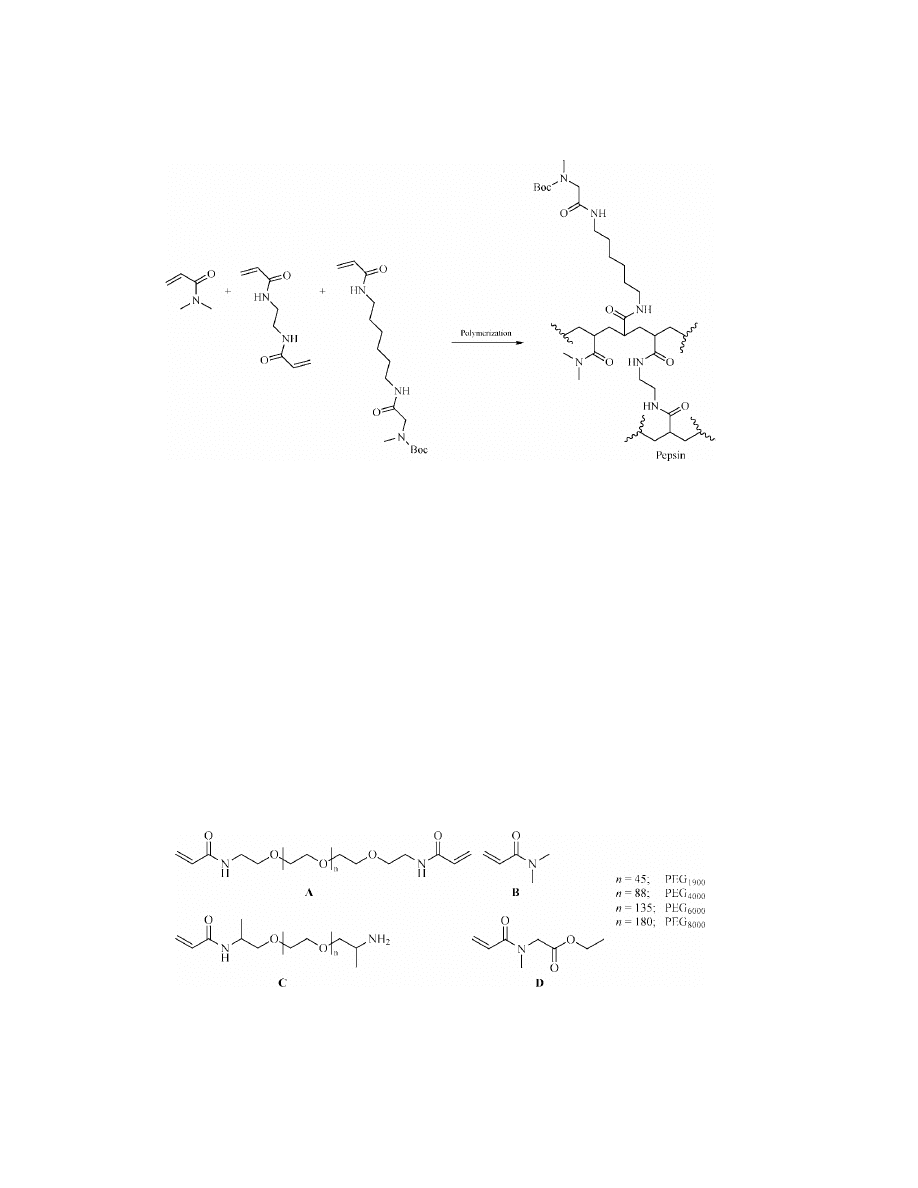
Vol. 11
POLYMER-SUPPORTED REAGENTS
155
Fig. 20.
Preparation of polyamide resins (Pepsin)
early 1970s, Sheppard introduced polyacrylamide supports, prepared by emul-
sion polymerization (Fig. 20), as an alternative to polystyrene gel-type resin for
polypeptide synthesis (122). Such resins showed good swelling in highly polar
solvents (see A
CRYLAMIDE
P
OLYMERS
). Moreover, polypeptides were obtained in
higher yields and purities and their syntheses were faster than with traditional
polystyrene gel-type resin. These commercialized resins are still in use for large-
scale preparation (33).
However, such supports, being very soft and fragile, are difficult to work with
(123). Pepsin K was prepared by polymerization of pepsin monomers within rigid
macroporous particles to help solve this problem (124).
In the 1990s, Meldal, following the work of Sheppard, developed an improved
support which was named PEGA (PEG–polyacrylamide) (125), obtained by in-
verse suspension polymerization (126) of N,N-dimethylacrylamide cross-linked
with different bis-acrylic-PEG derivatives (Fig. 21, A).
Functionalization was introduced by inclusion of acryloyl-PEG-NH
2
monomers (Fig. 21, C; loading
= 0.08–0.13 mmol/g) or acryloyl-sarcosine
Fig. 21.
Monomers used for the preparation of PEGA.
156
POLYMER-SUPPORTED REAGENTS
Vol. 11
Fig. 22.
PEGA supported ruthenium catalyst.
methylester monomer (Fig. 21, D; loading
= 0.22–1.0 mmol/g) (127). The first
version of such resins (PEG
1900
) allowed the diffusion of biomolecules with a
mass of
∼32 kDa into the beads. Although monomer A (Fig. 21), prepared us-
ing PEG
4000
,6000,8000
, improved the limit (
>120 kDa), handling and loadings were
poor. Recently, Gardossi observed that the introduction of a positive charge by tri-
alkylammonium species in PEGA
1900
allowed better swelling than neutral PEGA
and consequent inclusion of large proteins (128) although favorable charge–charge
interactions are perhaps a more reasonable explanation (but also see Section 3.1).
Although the main area of use of PEGA remains for biological based chemistry,
because of size accetability (129–131), it has also been used with water-insoluble
ruthenium catalysts for metathesis reactions in aqueous and protic solvents (in
such conditions the nonsupported catalyst does not work) (Fig. 22) (132).
This catalyst not only showed good activity in metathesis reactions but also
did not need to be used in degassed solution, as required by conventional Grubbs
catalysts (133). However, the main problem of PEGA is its difficulty of handling:
having to be handled in a solvated state as it may disintegrate when dry. A more
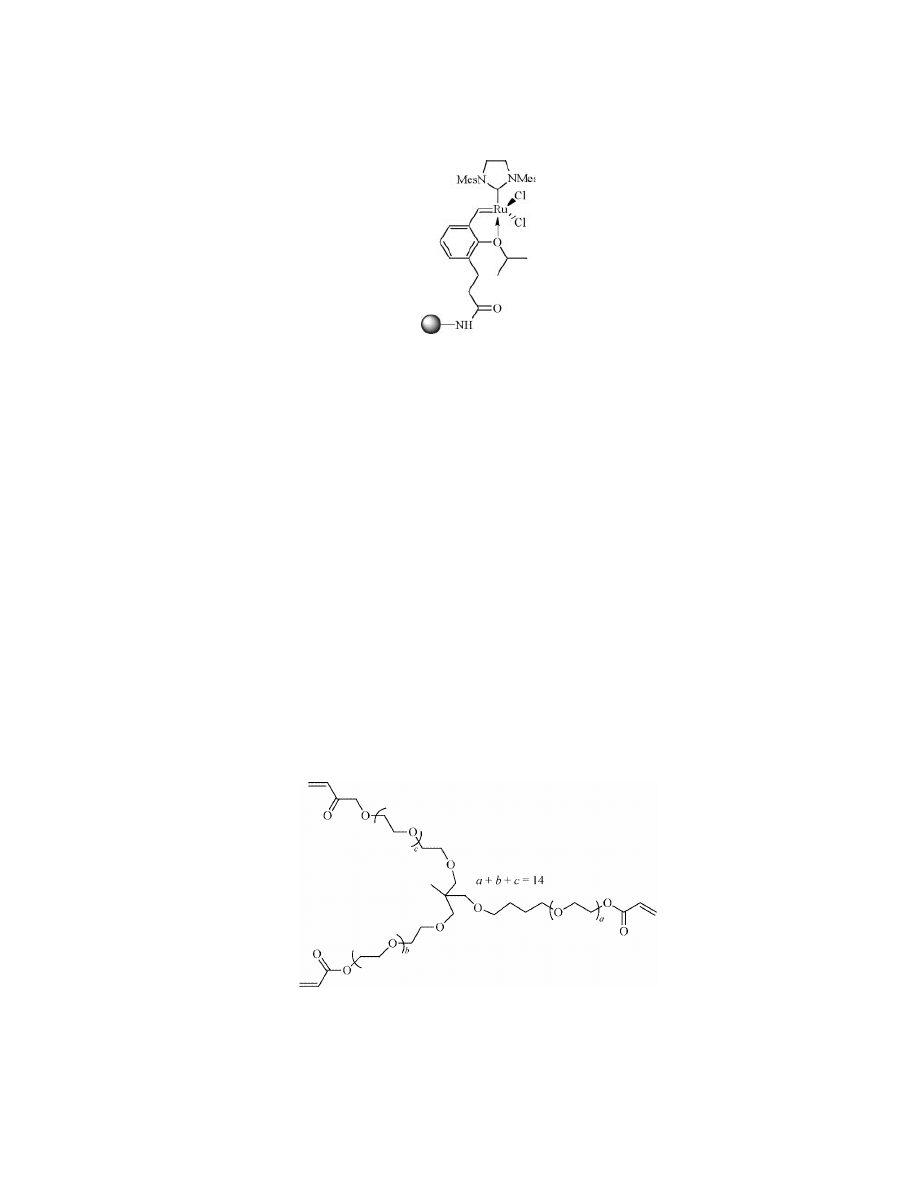
robust cross-linked ethoxylate acrylate resin (CLEAR) was presented in 1996 by
Kempe and Barrany (134). The main component of such supports is a branched
PEG-acrylate derivative used in
∼50 mol% (Fig. 23), which gave good swelling in
a range of solvents.
Fig. 23.
Trimethylopropane ethoxylate (14/3 EO/OH) triacrylate.
Vol. 11
POLYMER-SUPPORTED REAGENTS
157
Fig. 24.
Preparation of PEG-MA.
Unfortunately, CLEAR is vulnerable to strong bases (such as NaOH or NH
3
),
although complete polymer dissolution occurs only after long reaction times.
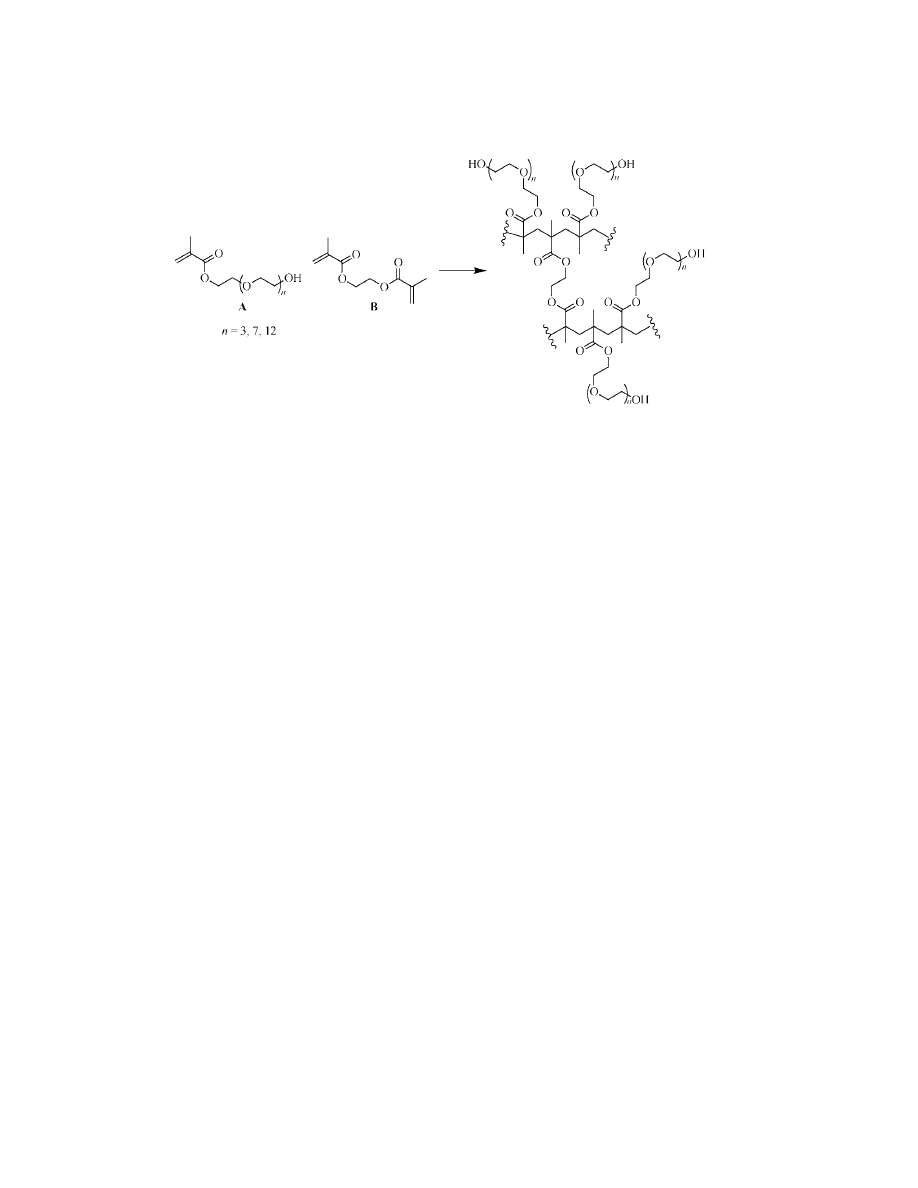
In 2001, Fr´echet introduced PEG-MA (PEG methacrylate) (135). The
monomers are PEG methacrylate derivatives (Fig. 24, A) and ethylene
dimethacrylate is used at 2 mol% as a cross-linker (Fig. 24, B). Although such
monomers are soluble in aqueous media, the preparation of PEG-MA was con-
ducted by a “modified suspension polymerization.”
As expected for resins that contained poly(ethylene glycol), PEG-MA was
compatible with a broad range of polar and nonpolar solvents. Swelling depended
on the chain length of the PEG incorporated in the monomer (Fig. 24, A), longer
chains giving better swelling. Although loadings calculated were much lower
(51–67%) than the ones expected, such values were still much higher than com-
mercial PEG-grafted resins (1.2–1.8 mmol/g). The PEG-MAs were used for the
preparation of a small library of hydantoins. The advantages of using PEG clearly
reside in the improved property of the tailored polymer matrix. Loading, solvent
compatibility (and so, swelling), and mechanical resistance depend on the level of
this polymeric monomer. In 2002, Meldal prepared a new class of polymers named
SPOCC (superpermeable organic combinatorial chemistry) resins (136). The main
characteristic of these polymers was that the backbones and cross-linker both con-
tained PEG. The second generation of such polymers (SPOCC
194
) was prepared
by cationic polymerization of the two monomers shown in Figure 25.
Novel SPOCCs exhibit great chemical resistance under various reaction con-
ditions, because there are no benzyl ether, ester, and amide linkages. The use of
shorter PEG chains in SPOCC
194
allowed higher loading compared to the previ-
ous generation resins (longer PEG chains), although swelling decreased. Although
mainly used in peptide chemistry, SPOCC resins have seen successfully tested in
a variety of different organic transformations (136,137). Direct comparison of this
support, PS resin, and TentaGel showed comparable if not better kinetics in pep-
tide synthesis.
Cellulose: From Paper to Beads.
The first use of cellulose (qv) as a solid
support for peptide chemistry can be attributed to Merrifield. Unfortunately, at

158
POLYMER-SUPPORTED REAGENTS
Vol. 11
Fig. 25.
Preparation of SPOCC
194
.
that time, only a dipeptide was obtained in unsatisfactory yield. Moreover, cellu-
lose was not chemically stable to the conditions used in the synthesis. Merrifield
decided to abandon this support and to search for others with better properties
(138). Peptides and oligonucleotides have been synthesized on paper and cotton
(139,140), and Frank demonstrated, in 1992, the flexibility of cellulose for the
preparations of large arrays of peptides on membranes by Spot synthesis (141).
At the end of the synthesis, biological assays could be performed directly on each
spot on the filter disk, as cellulose is compatible with many binding, enzymatic,
and cellular essays (142). However, paper membranes are not suitable for scav-
enger or supported reagents, because of difficulties in their handling and their
limited loading. In 1977, Stamberg found a method to prepare spherical cellu-
lose particles with a certain degree of porosity by heating a viscose suspension
(an aqueous solution of sodium cellulose xanthate) in a water-immiscible solvent
under stirring (143). Such beads became commercially available under the name
of Perloza. Because of the availability of three hydroxyl groups for each glucopy-
ranose monomer, such supports provided very high loadings. Functionalized cel-
lulose beads (0.37–0.65 mmol/g) were used for the synthesis of peptides (5–34
amino acids). The analysis of crude peptides (20 amino acids length) showed bet-
ter purity when assembled on Perloza than polystyrene (144). The application of
cellulose beads in solution-phase chemistry was demonstrated by Chesney and
Steel (145). In their procedure, the pendant hydroxyl groups first reacted with
allyl bromide; addition of bromine in water was followed by treatment with ex-
cess of tris(2-aminoethyl)amine and led to supported branched amino resins (2.2
mmol/g, 4 mmol/g). Such amino resins exhibited good and uniform swelling prop-
erties in polar and nonpolar solvents (between 8 and 10 mL/g in all solvents).
Moreover, they allowed the synthesis of small libraries of amides and ureas in
quite good purities and yields by the scavenging of excess acid chloride and iso-
cyanate derivatives (Fig. 26, B). Treatment of Perloza with the enzyme cellulase
allowed the complete biodegradation of the beads to glucose that, after treatment
with brewers’ yeast, led to ethanol, CO
2
, and H
2
O.
The supported allyl beads were also reactive enough to permit their ap-
plication as a scavenger of bromine (Fig. 26, A) (146). Recently, Perloza aniline
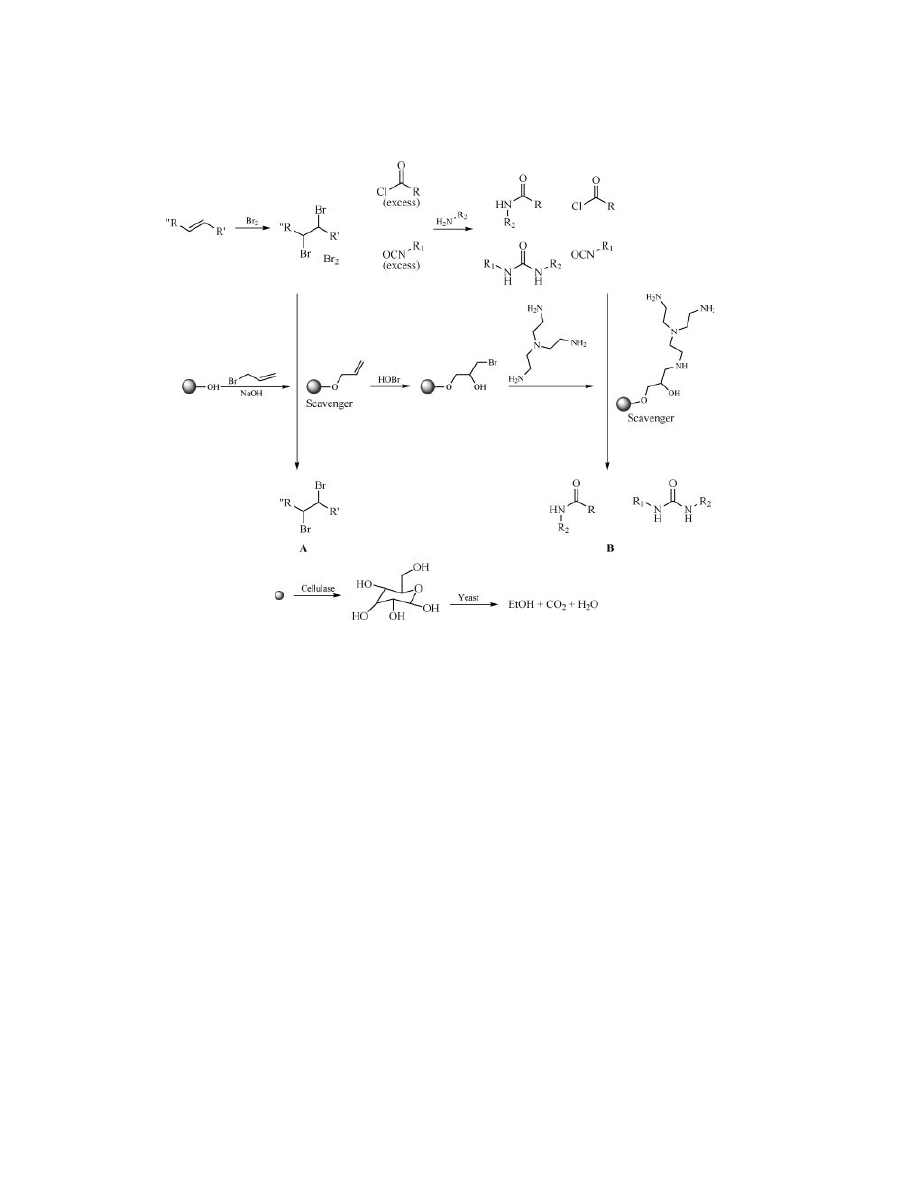
Vol. 11
POLYMER-SUPPORTED REAGENTS
159
Fig. 26.
Scavenger uses of biodegradable cellulose beads.
functionalized beads were used for a synthesis of a library of pyrazoles. Once the
reaction conditions were established, the “catch and release” method was adopted
and allowed the continuous recycling of the beads without decreasing yields and
purities (147).
In conclusion, cellulose beads have interesting properties, such as relatively
high loading, cheapness, chemical and mechanical stability, and recycling and easy
disposal, factors that could be of use in large-scale industrial processes.
As can be seen, over the years there has been huge development in supports
for synthetic applications, with variation of improvement in loading, morphologies,
and polymer type.
Gel-type polystyrene resins are still the most widespread supports in solid-
phase organic chemistry, exhibiting good characteristics of loading, swelling, and
handling. However, they are not universal. Although an ideal support should be
inert to reactions conditions, it does not mean that it does not have to interact
with the reagents. “Resin beads are like solvents,” suggested Czarnic (29), and
this concept could be expanded by saying that “different polymer supports are like
different solvents.” As the choice of a particular solvent can change the course of a
reaction, a polymer support has to be considered in the same way. For this reason,
research groups continue the early work of Merrifield (139) in order to screen

160
POLYMER-SUPPORTED REAGENTS
Vol. 11
and synthesize new and enhanced polymer supports for a range of chemical and
biological applications.
BIBLIOGRAPHY
“Polymers as Chemical Reagents” in EPST 1st ed., Suppl. Vol. 1, pp. 468–492, by A.
Patchornik and M. A. Kraus, The Weizman Institute of Science; in EPSE 2nd ed., Vol.
12, pp. 618–658, by Philip Hodge, University of Lancaster.
1. R. B. Merrifield, J. Am. Chem. Soc. 85, 2149–2154 (1963).
2. J. A. Porco, T. Deegan, W. Devonport, O. W. Gooding, K. Heisler, J. W. Labadie, B.
Newcomb, C. Nguyen, P. van Eikeren, J. Wong, and P. Wright, Mol. Diversity 2, 197–
206 (1996).
3. M. Lebl, J. Comb. Chem. 1, 3–24 (1999).
4. J. A. Ellman, Acc. Chem. Res. 29, 132–143 (1996).
5. L. A. Thompson and J. A. Ellman, Chem. Rev. 96, 555–600 (1996).
6. R. C. D. Brown, J. Chem. Soc., Perkin Trans. 1 1, 3293–3320 (1998).
7. S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leach, D. A. Longbottom,
M. Nesi, J. S. Scott, and R. I. T. S. J. Storer, J. Chem. Soc., Perkin Trans. 1 3815–4195
(2000).
8. A. Akelah and D. C. Sherrington, Chem. Rev. 81, 557–587 (1981).
9. W. P. Hohenstein, and H. Mark, J. Polym. Sci. A 1, 127–145 (1946).
10. S. Rana, P. White, and M. Bradley, J. Comb. Chem. 3, 9–15 (2001).
11. F. Guillere, D. Orain, and M. Bradley, Chem. Rev. 100, 2091–2157 (2000).
12. P. Hodge and D. C. Sherrington, eds., Polymer-Supported Reactions in Organic Syn-
thesis, John Wiley & Sons, Inc., New York, (1980).
13. R. J. Booth and J. C. Hodges, J. Am. Chem. Soc. 119, 4882 (1997).
14. L. D. Arnold, H. I. Assil, and J. C. Vederas, J. Am. Chem. Soc. 111, 3973–3976 (1989).
15. S. Weik, G. Nicholson, G. Jung, and J. Rademann, Angew. Chem., Int. Ed. 40, 1436–
1439 (2000).
16. J. Rademann, J. Smerdka, G. Jung, P., G., and D. Schmid, Angew. Chem., Int. Ed. 40,
381–385 (2001).
17. I. Fenger and C. Le Drian, Tetrahedron Lett. 39, 4287–4290 (1998).
18. M. S. Gibson and R. W. Bradshaw, Angew. Chem., Int. Ed. Engl. 7, 919 (1968).
19. E. P. Iuliana, P. D. Benolt, and A. L. Tartar, J. Org. Chem. 62, 2594–2603 (1997).
20. G. M. Coppola, Tetrahedron Lett. 39, 8233–8236 (1998).
21. K. IIjima, W. Fukuda, and M. Tomoi, J. Macromol. Sci., Pure Appl. Chem. 29, 249–261
(1992).
22. S. Crossigniani, P. D. White, and B. Linclau, Org. Lett. 5, 853–856 (2003).
23. J. M. Salvino, N. V. Kumar, E. Orton, J. Airey, T. Kiesow, K. Crawford, R. Mathew, P.
Krolikowski, M. Drew, D. Engers, D. Krolikowski, T. Herpin, M. Gardyan, G. McGee-
han, and R. Labaudiniere, J. Comb. Chem. 2, 691–697 (2000).
24. S. R. Stauffer and J. A. Katzenellenbogen, J. Comb. Chem. 2, 318–329 (2000).
25. B. Heinzen and S. V. Ley, J. Chem. Soc., Perkin Trans. 1 1907 (1997).
26. H. W. Gibson and J. J. Bailey, Chem. Commun. 815 (1977).
27. B. A. Bunin, in E. M. Gordon and J. F. Kerwin eds., The Combinatorial Index, Academic
Press, San Diego, 1998, pp. 11–15.
28. M. J. Farral and J. M. J. Fr´echet, J. Org. Chem. 41, 3877–3882 (1976).
29. A. W. Czarnik, Biotechnol. Bioeng. 61, 77–79 (1998).
30. E. Erbay, T. Bilgic, I. M. Karal, and O. T. Savasci, Polym. Plast. Technol. Eng. 31,
589–605 (1992).

Vol. 11
POLYMER-SUPPORTED REAGENTS
161
31. S. Alesso, Z. Yu, D. Pears, A. P. Worthington, R. W. A. Luke, and M. Bradley, J. Comb.
Chem. 3, 631–633 (2001).
32. R. Arshady, Colloid. Polym. Sci. 270, 717–732 (1992).
33. D. Hudson, J. Comb. Chem. 1, 333–360 (1999).
34. R. Santini and M. C. Griffit, Tetrahedron Lett. 39, 8951–8954 (1998).
35. V. K. Sarin, S. B. H. Kent, and R. B. Merrifield, J. Am. Chem. Soc. 102, 5463–5470
(1980).
36. D. C. Sherrington, Chem. Commun. 2275–2286 (1998).
37. J. R. Millar, D. G. Smith, and T. R. E. Kressmann, J. Chem. Soc. 304–31 (1965).
38. F. M. B. Couthino, M. A. F. S. Neves, and M. L. Dias, J. Appl. Polym. Sci. 65, 1257–1262
(1997).
39. J. M. J. Fr´echet and W. A. Rolls, J. Chromatogr. A 97–112 (1990).
40. M. Bernard and W. T. Ford, J. Org. Chem. 48, 326–332 (1983).
41. H. G. Tang and D. C. Sherrington, J. Mol. Catal. 94, 7–17 (1994).
42. K. L. Lewandowski, F. Svec, and J. M. J. Fr´echet, J. App. Polym. Sci. 67, 597–607
(1998).
43. O. Okay, Prog. Polym. Sci. 25, 711–719 (2000).
44. I. C. Ponescu and C.-D. Vlad, Eur. Polym. J. 33, 1515–1521 (1997).
45. H. Deleuzel, X. Shultze, and D. C. Sherrington, Polym. Bull. 44, 179–186 (2000).
46. P. A. Webb, C. Orr, Analytical Methods in Fine Particle Technology, Micrometrics In-
strument Corp., 1997.
47. M. Hori, J., G. D., P. Wentworth, and K. Janda, Bioorg. Med. Chem. Lett. 8, 2363–2368
(1998).
48. J. W. Labadie, Curr. Opin. Chem. Biol. 2, 346–352 (1998).
49. R. Frenette and R. W. Friesen, Tetrahedron Lett. 35, 9177–9180 (1994).
50. B. J. Backes and J. A. Ellman, J. Am. Chem. Soc. 116, 11171–11172 (1994).
51. W. Funke, O. Okay, and B. Joos-M ¨
uller, Adv. Polym. Sci. 136, 139–234 (1998).
52. O. Shinomura, B. Clapham, C. Spanka, S. Mahajan, and K. Janda, J. Comb. Chem. 4,
436–441 (2002).
53. C. Schunicht, A. Biffis, and G. Wulff, Tetrahedron 56, 1693–1699 (2000).
54. G. Giffels, J. Beliezey, M. Felder, and U. Kragl, Tetrahedron: Asymmetry 9, 691–696
(1998).
55. C. Spanka, B. Clapham, and K. Janda, J. Org. Chem. 67, 3045–3050 (2002).
56. B. Jeong, Y. H. Bac, D. S. Lee, and S. W. Kim, Nature 388, 860–862 (1997).
57. A. G. A. Combers, S. Tasker, M. Lindblad, J. Holmgrem, K. Hoste, V. Toncheva, E.
Schacht, M. C. Davies, L. Illum, and S. S. Davis, Biomaterials 18, 1153–1161 (1997).
58. M. Pyrasch and B. Tieke, Colloid Polym. Sci. 278, 375–379 (2000).
59. W.-H. Li, K. Li, and H. St¨over, J. Polym. Sci. A 37, 2295–2303 (1999).
60. A. N. Cammidge, S. Downing, and Z. Ngaini, Tetrahedron Lett. 44, 6633–6634 (2003).
61. K. Li and H. St¨over, J. Polym. Sci. A 31, 3257–3263 (1993).
62. M. Dreja, W. Pyckhout-Hintzen, and B. Tieke, Macromolecules 31, 272–280 (1998).
63. C. Leznoff and P. I. Svirskaya, Angew. Chem., Int. Ed. Engl. 17, 947 (1978).
64. E. Bayer and M. Mutter, Nature 237, 512–513 (1972).
65. M. Mutter, H. Hagenmaier, and E. Bayer, Angew. Chem., Int. Ed. Engl. 10, 811–812
(1971).
66. E. Bayer, M. Mutter, R. Uhmann, J. Polster, and H. Mauser, J. Am. Chem. Soc. 96,
7333–7336 (1974).
67. U.S. Pat. 4,908,405 (Mar. 3, 1990), E. Bayer and W. Rapp.
68. E. Bayer, Angew. Chem., Int. Ed. Engl. 30, 113–216 (1991).
69. S. Zalipsky, J. L. Chang, F. Albericio, and G. Barany, React. Polym. 22, 243–258 (1994).
70. S. A. Kates, B. F. McGuinnes, C. Blackburn, G. W. Griffin, N. A. Sol´e, G. Barany, and
F. Albericio Biopolymers 47, 365–380 (1998).

162
POLYMER-SUPPORTED REAGENTS
Vol. 11
71. O. W. Gooding, S. Baudart, T. L. Deegan, K. Heisler, J. W. Labadie, W. S. Newcomb, J.
A. Porco, and P. van Eikeren, J. Comb. Chem. 1, 113–122 (1999).
72. J. H. Adams, R. M. Cook, D. Hudson, V. Jammalamadaka, M. H. Lyttle, and M. F.
Songster, J. Org. Chem. 63, 3706–3716 (1998).
73. S. Alesso, Z. Yu, D. Pears, A. P. Worthington, R. W. A. Luke, and M. Bradley, Tetrahe-
dron 59, 7163–7169 (2003).
74. E. Bayer and W. Rapp, in M. E. Harris, ed., Poly(Ethylene Glycol) Chemistry: Biotech-
nical and Biomedica Applications, Plenum Press, New York, 1992, p. 325.
75. S. A. Kates, N. A. Sol´e, M. Beyermann, G. Barany, and F. Albericio, Peptide Res. 9,
106–113 (1996).
76. E. Bayer, A. Klaus, H. Willisch, W. Rapp, and B. Haemmasi, Macromolecules 23,
1937–1940 (1990).
77. D. P. Walsh, C. Pang, P. B. Parikh, Y.-S. Kim, and Y.-T Chang, J. Comb. Chem. 4,
204–208 (2002).
78. D. Walsh, D. Wu, and Y.-T. Chang, Curr. Opin. Chem. Biol. 7, 353–361 (2003).
79. B. Ruhland, A. Bombrun, and M. A. Gallop, J. Org. Chem. 62, 7820–7826 (1997).
80. X. Du and R. W. Armstrong, J. Org. Chem. 62, 5678–5679 (1997).
81. H. Han and K. Janda, Angew. Chem., Int. Ed. Engl. 36, 1731–1733 (1997).
82. W. Li and B. Yan, J. Org. Chem. 63, 4092–4097 (1998).
83. Y.-S. Lee and B.-D. Park, React. Fun. Polym. 44, 41–46 (2000).
84. V. Swali, N. K. Wells, J. Langley, and M. Bradley, J. Org. Chem. 62, 4902–4903 (1997).
85. S. Lebreton, B. Newcomb, and M. Bradley, Tetrahedron Lett. 43, 2475–2478 (2002).
86. E. Swayze, Tetrahedron Lett. 38, 8465–8468 (1997).
87. C. M. G. Judkins, K. A. Knights, B. F. G. Johnson, Y. R. de Miguel, R. Raja, and J. M.
Thomas, Chem. Commun. 2624–2625 (2001).
88. C. M. G. Judkins, K. A. Knights, B. F. G. Johnson, and Y. R. de Miguel, Polyhedron 22,
3–7 (2003).
89. Y. Kohno, N. Ogawa, K. B. Chung, and W. Fakuda, Makromol. Chem. 193, 3009–3021
(1992).
90. M. Renil, R. Nagaraj, and V. N. Rajasekharan, Tetrahedron 50, 6681 (1994).
91. K. Kamahori, K. Ito, and S. Itsuno, J. Org. Chem. 61, 8321 (1996).
92. M. Renil and M. Meldal, Tetrahedron Lett. 37, 6185 (1996).
93. M. E. Wilson, K. Paech, W.-J. Zhou, and J. Kurth, J. Org. Chem. 63, 5094–5099 (1998).
94. K. J. Shea, Y. Okahata, and T. K. Dougherty, Macromolecules 17, 296–300 (1984).
95. J. Buchardt and M. Meldal, Tetrahedron Lett. 39, 8695–8698 (1998).
96. M. Grøtli, C. H. Gotfredsen, J. Rademann, J. Buchardt, A. J. Clark, J. Ø. Duus, and
M. Meldal, J. Comb. Chem. 2, 108–119 (2000).
97. P. H. Toy and K. Janda, Tetrahedron Lett. 40, 6329–6332 (1999).
98. P. H. Toy, T. S. Reger, P. Garibay, J. C. Garno, J. A. Malikayil, G. Liu, and K. Janda,
J. Comb. Chem. 3, 117–124 (2001).
99. T. S. Reger and K. Janda, J. Am. Chem. Soc. 122, 6929–6934 (2000).
100. P. H. Toy, T. S. Reger, and K. Janda, Org. Lett. 2, 2205–2207 (2000).
101. O. Br ¨
ummer, B. Clapham, and K. Janda, Tetrahedron lett. 42, 2257–2259 (2001).
102. J. A. Moss, T. J. Dickerson, and K. D. Janda, Tetrahedron Lett. 43, 37–40 (2002).
103. H. Nogami, S. Matsanaga, M. Kanai, and M. Shibasaki, Tetrahedron Lett. 42, 279–283
(2001).
104. M. Takamura, K. Funabashi, M. Kanai, and M. Shibasaki, J. Am. Chem. Soc. 123,
6801–6808 (2001).
105. M. E. Honigfort and W. J. Brittain, Macromolecules 36, 3111–3114 (2003).
106. S. Liou, J. T. Rademarcher, D. Malaba, M. E. Pallack, and W. J. Brittain, Macro-
molecules 33, 4295–4296 (2000).
107. C. Fromont and M. Bradley, Chem. Commun. 283–284 (2000).

Vol. 11
POLYMER-SUPPORTED REAGENTS
163
108. F. Svec and J. M. J. Fr´echet, J. Anal. Chem. 64, 820–822 (1992).
109. F. Svec and J. M. J. Fr´echet, J. Science 273, 205–211 (1996).
110. N. Hird, I. Huges, D. Hunter, M. G. J. T. Morrison, D. C. Sherrington, and L. Stevenson,
Tetrahedron 55, 9575–9584 (1999).
111. J. A. Tripp, J. A. Stein, F. Svec, and J. M. J. Fr´echet, Org. Lett. 2, 195–198 (1999).
112. J. A. Tripp, F. Svec, and J. M. J. Fr´echet, J. Comb. Chem. 3, 216–223 (2001).
113. P. Hodge, Curr. Opin. Chem. Biol. 7, 362–373 (2003).
114. J. A. Tripp, F. Svec, and J. M. J. Fr´echet, J. Comb. Chem. 3, 604–611 (2001).
115. A. R. Vaino, and K. Janda, Proc. Natl. Acad. Sci. U.S.A. 97, 7692–7696 (2000).
116. P. H. Findlay, S.-M. Leinonen, T., M. G. J., E. E. A. Shepherd, and D. C. Sherrington,
J. Mater. Chem. 10, 2031–2034 (2000).
117. BP Chemicals Ltd, Purolite International, Johnson Matthey plc, University of
Sheffield, and University of Straclyde, ACCP News Applied Catalysis and Catalytic
Processes, No. 3, 2000, pp. 6–7.
118. B. Atrash, M. Bradley, R. Kobylecki, D. Cowell, and J. Reader, Angew. Chem., Int. Ed.
Engl. 40, 938–941 (2001).
119. B. Atrash and M. Bradley, Tetrahedron Lett. 44, 4779–4782 (2003).
120. T. Ruhland, P. Holm, and K. Andersen, J. Comb. Chem. 5, 842–850 (2003).
121. J. Vagner, G. Barany, K. S. Lam, V. Krchn ´ak, N. F. Sepetov, J. A. Ostrem, P. Strop,
and M. Lebl, Proc. Natl. Acad. Sci. U.S.A. 93, 8194–8199 (1996).
122. E. Atherton, D. L. J. Clive, and R. C. Sheppard, J. Am. Chem. Soc. 97, 6584–6585
(1975).
123. E. Atherton, E. Brown, R. C. Sheppard, and A. A. Rosevear, Chem. Commun. 1151
(1981).
124. R. Arshady, E. Atherton, D. L. J. Clive, and R. C. Sheppard, J. Chem. Soc., Perkin
Trans. 1 529–537. (1981).
125. M. Meldal, Tetrahedron Lett. 33, 3077–3080 (1992).
126. R. Arshady, Colloid. Polym. Sci. 268, 948–958 (1990).
127. M. Renil, M. Ferreras, J. M. Delaisse, N. T. Foged, and M. Meldal, J. Pept. Sci. 4,
195–210 (1998).
128. A. Basso, L. De Martin, L. Gardossi, G. Margetts, I. Brazendale, A. Y. Bosma, R. Ulijn,
and S. Flitsch, Chem. Commun. 1296–1297. (2003).
129. S. Leon, R. Quarrel, and G. Lowe, Bioorg. Med. Chem. Lett. 8, 2997–3002 (1998).
130. H. K. Smith and M. Bradley, J. Comb. Chem. 1, 326–332 (1999).
131. M. Conza and H. Wennemers, Chem. Commun. 866–867. (2003).
132. S. J. Connon and S. Blechert, Bioorg. Med. Chem. Lett. 12, 1873–1876 (2002).
133. D. M. Lynn, B. Mohr, L. M. Henling, M. W. Day, and R. H. Grubbs, J. Am. Chem. Soc.
122, 6601 (2000).
134. M. Kempe and G. Barrany, J. Am. Chem. Soc. 118, 7083–7093 (1996).
135. R. Kita, F. Svec, and J. M. J. Fr´echet, J. Comb. Chem. 3, 564–571 (2001).
136. J. Rademann, M. Grøtli, M. Meldal, and K. Bock, J. Am. Chem. Soc. 121, 5459–5466
(1999).
137. L. P. Miranda, W. D. Lubell, K. M. Halkes, T. Groth, M. Grøtli, J. Rademann, C. H.
Gotfredsen, and M. Meldal, J. Comb. Chem. 4, 523–529 (2002).
138. R. B. Merrifield, in J. I. Seeman, ed., Profiles, Pathways and Dreams: Autobiographies
of Eminents Chemists, American Chemical Society, Washington, D.C., 1993.
139. R. Frank, W. Heikens, G. Heistberg-Moutsis, and H. Bl¨ocker, Nucleic Acids Res. 11,
4365–4377 (1983).
140. R. Frank and R. Doring, Tetrahedron 44, 6031–6040 (1983).
141. R. Frank, Tetrahedron 48, 9217–9232 (1992).
142. U. Reineke, R. Volkmer-Engert, and J. S. Mergener, Curr. Opin. Biotechnol. 12, 59–64
(2001).

164
POLYMER-SUPPORTED REAGENTS
Vol. 11
143. U.S. Pat. 4,055,510 (Oct. 25, 1977), J. Peska, J. Stamberg, and Z. Blace. (Ceskosloven-
ska Akademie Ved).
144. D. R. Englebretsen and D. R. K. Harding, Int. J. Peptide Protein Res. 43, 546–554
(1994).
145. A. Chesney, P. Barnwell, D. F. Stonehouse, and P. G. Steel, Green Chem. 2, 57–62
(2000).
146. A. Chesney, P. G. Steel, and D. F. Stonehouse, J. Comb. Chem. 2, 434–437 (2000).
147. L. De Luca, G. Giacomelli, A. Porcheddu, M. Salaris, and M. Taddei, J. Comb. Chem.
5, 465–471 (2003).
GENERAL REFERENCES
For nice, historical review of solid-supported reagents catalysts, and sorbents, see
D. C.
Sherrington, J. Polym. Sci. A 39, 2364–2377 (2001), and also D. Hudson, J. Comb. Chem.
1, 336–360 (1999); J. Comb Chem. 1, 403–457 (1999).
M
ARK
B
RADLEY
N
ICOLA
G
ALAFFU
University of Southampton
POLYMETHACRYLATES.
See M
ETHACRYLIC
E
STER
P
OLYMERS
.
POLY(METHACRYLIC ACID).
See A
CRYLIC
(
AND
M
ETHACRYLIC
) A
CID
P
OLYMERS
.
POLYNUCLEOTIDES.
See Volume 3.
POLYOXYMETHYLENE.
See A
CETAL
R
ESINS
.
Wyszukiwarka
Podobne podstrony:
Polymer supported catalysis in synthetic organic chemistry
polymer supported sharpless catalysts
solid supported reagents
Degradable Polymers and Plastics in Landfill Sites
alcatel support document for cable system in cuba
Development of Carbon Nanotubes and Polymer Composites Therefrom
Polymer Processing With Supercritical Fluids V Goodship, E Ogur (Rapra, 2004) Ww
Inorganic Polymers
cg100 prog iii airbag restore devices support list
Propylene Polymers
Fundamentals of Polymer Chemist Nieznany
Intelligence Support Activity
MCWP 4 11 1 Health Service Support Operations
Guidelines for Persons and Organizations Providing Support for Victims of Forced Migration
Electrochemical properties for Journal of Polymer Science
więcej podobnych podstron