
STANY SKUPIENIA
GAZOWY, CIEKŁY I STAŁY
STAN GAZOWY
Gaz
– ciało, które nie ma określonego kształtu i objętości, słabe
oddziaływania międzycząsteczkowe.
Gaz
składa się z oddalonych od siebie cząstek (atomów, cząsteczek)
poruszających się szybko i chaotycznie.
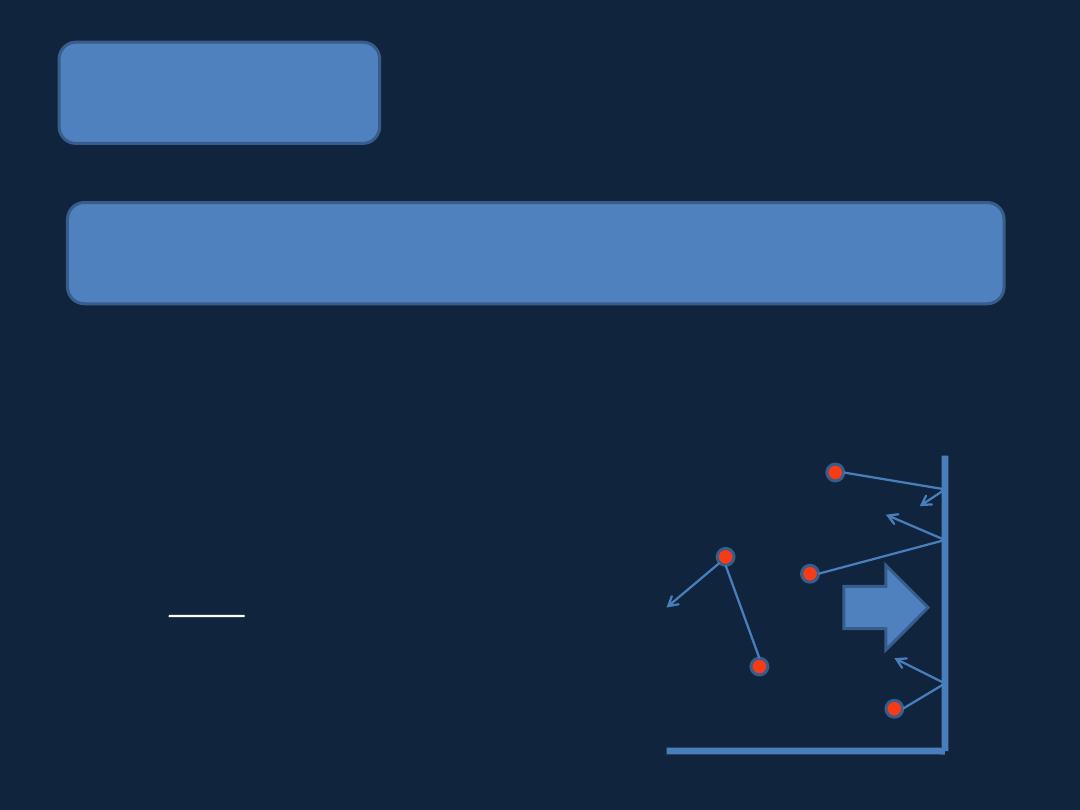
Jedną z wielkości charakteryzujących
stan gazu jest ciśnienie (p):
F
p =
A
F
– siła wywierana przez gaz
A
– pole powierzchni na która działa gaz
ścianki naczynia
PRAWA GAZOWE
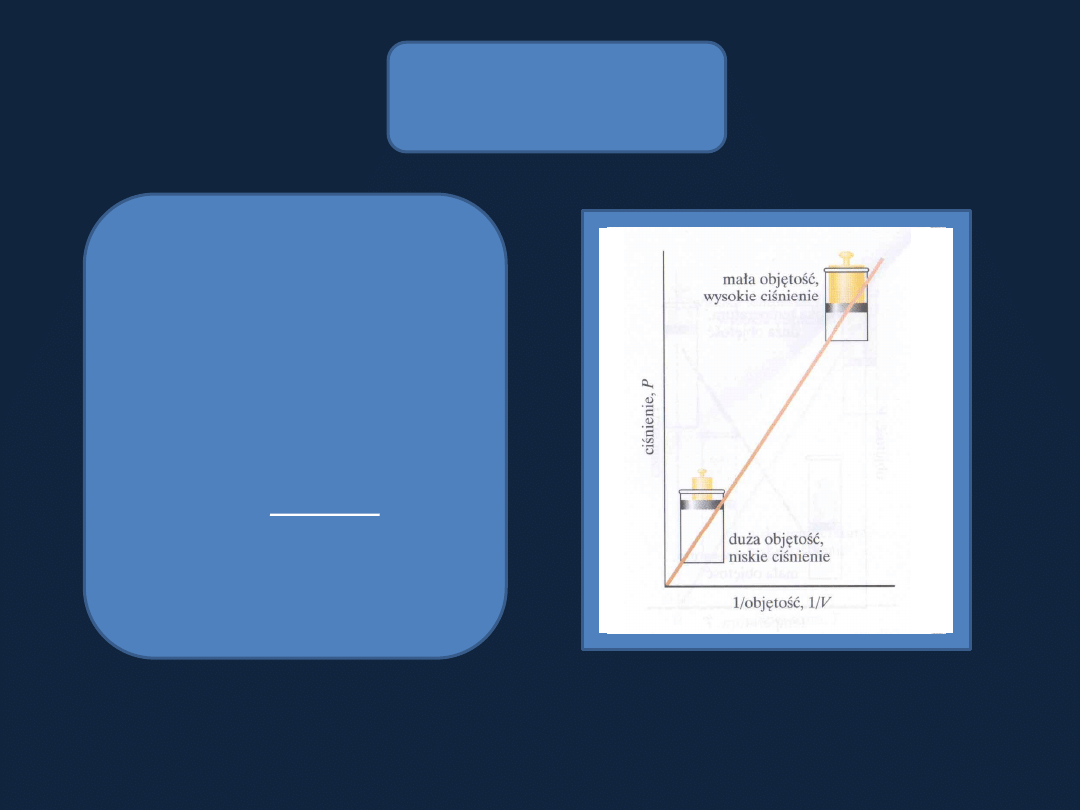
Prawo
Boyle’a
Dla stałej ilości gazu w stałej
temperaturze ciśnienie gazu
jest odwrotnie proporcjonalne
do jego objętości.
const
p =
v
p ∙ v = const
Wykorzystanie
– obliczenie zmiany ciśnienia lub objętości po zmianie warunków
w
układzie – p
1
v
1
= p
2
v
2
(gdzie 1 i 2
odpowiadają odpowiednio warunkom początkowym
i
końcowym).
Rys. 5.8 str
189 Atkins

Prawo Charlesa i Gay-Lussaca
Objętość stałej ilości gazu pod stałym ciśnieniem jest proporcjonalna
do temperatury
bezwzględnej
oraz
ciśnienie stałej ilości gazu w stałej objętości jest proporcjonalne
do temperatury
bezwzględnej
v = T
∙ const oraz p = T ∙ const
Rys. 5.11 i
5.12 str 191
Atkins

Prawo Avogadra
Objętość próbki gazu w stałej temperaturze i pod stałym ciśnieniem jest
proporcjonalna do liczby moli
cząsteczek tego gazu
v = n
∙ const
n
– liczba moli
Gaz
Objętość molowa (dm
3
/mol)
Gaz doskonały
22,41
Azot
22,40
Tlen
22,40
Wodór
22,43
Objętości molowe różnych gazów w temp. 273 K i pod ciśń. 1 atm

Gaz
doskonały
Założenia:
-
cząstki (atomy, cząsteczki) znajdują się w nieustannym i bezładnym
ruchu,
-
cząstki nie oddziaływają ze sobą w inny sposób niż poprzez zderzenia
sprężyste,
-
objętość cząstek gazu jest zaniedbywalnie mała.
Gazy rzeczywiste
wykazują oddziaływania siłami van der Waalsa
i
posiadają określoną objętość.
Dlatego
gaz
może być traktowany jako gaz doskonały tylko w wysokiej
temperaturze
(energia kinetyczna
większa od energii oddziaływań
elektromagnetycznych) i
pod niskim
ciśnieniem
(rozmiary
cząstek o wiele
mniejsze
niż odległości pomiędzy nimi).
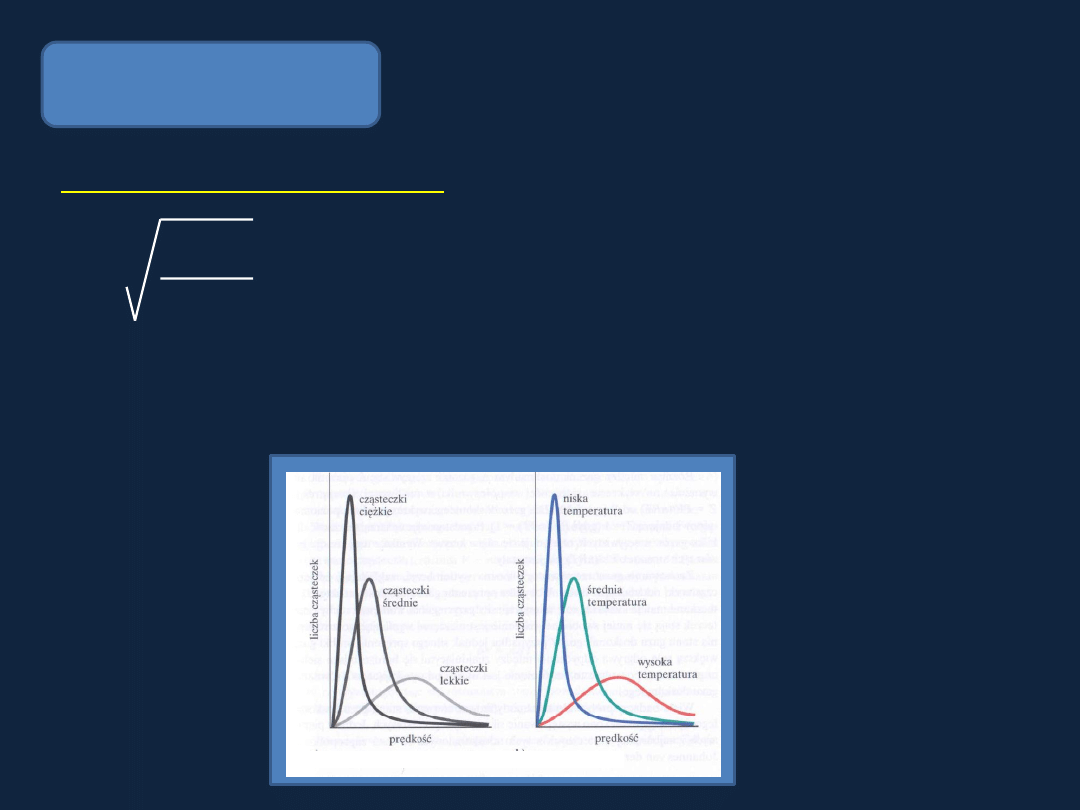
Prędkość cząstek gazu
Średnia prędkość cząstek gazu:
3RT
v =
gdzie: M
– masa molowa, R – stała gazowa, T - temperatura
M
Np. O
2
– prędkość (20°C, 1atm.) – ok. 500 m/s, liczba zderzeń ok. 7∙10
9
1/s,
przeciętna droga swobodna cząsteczki ok. 5 ∙10
-5
cm.
Rys 5.28 str 215
Atkins

Równanie stanu gazu doskonałego
Z prawa Avogadro wynika, że v jest proporcjonalna do n
Z prawa Charlesa i Gay Lussaca wynika, że v jest proporcjonalna do T
Z prawa
Boyle’a wynika, że v jest odwrotnie proporcjonalna do p
stąd:
pv = nRT
gdzie:
R
– stała proporcjonalności,
p
–ciśnienie,
v
– objętość,
T
– temperatura
n
– liczba moli
R = 0,0820578 atm
/(K ∙ mol)
R = 8,31451 kPa
/(K ∙ mol)
R = 8,31451 J/(K ∙ mol)

Gazy rzeczywiste - poprawka van der Waalsa
(p + an
2
/v
2
) (v
– nb) = nRT
Poprawka uwzględnia efekt oddziaływania między cząstkami gazu.
Stała a - uwzględnia efekt przyciągania między cząstkami
Stała b - uwzględnia efekt odpychania między cząstkami
n
– liczba moli gazu
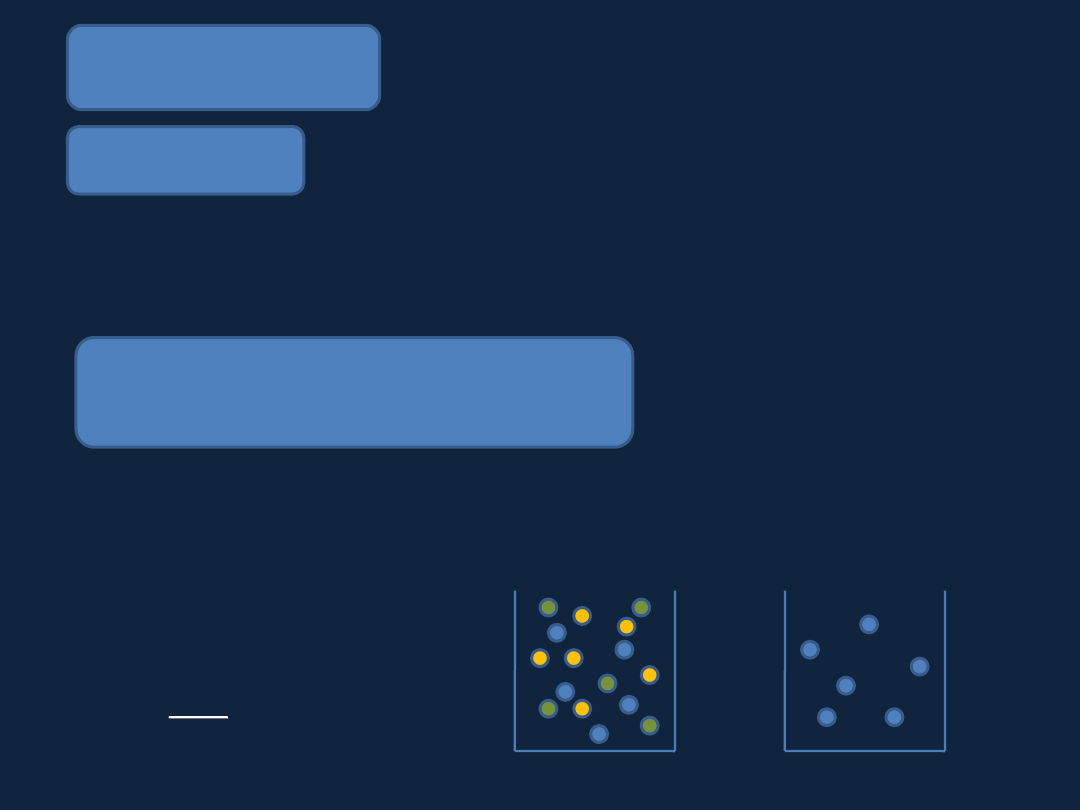
Mieszanina gazów
Prawo Daltona
Ciśnienie mieszaniny gazów jest równe sumie ciśnień cząstkowych
składników mieszaniny.
n
p = p
A
+ p
B
+ p
C
+… + p
i
= ∑ p
i
i=1
Cząstkowe ciśnienie gazu, to ciśnienie, które wywierałby ten gaz, gdyby
sam wypełniał całe naczynie.
n
i
/n = x
i
– ułamek molowy
n
i
p
i
= ∙ p = x
i
∙ p
n

STAN CIEKŁY
Ciecz
– ciało posiadające zdefiniowaną objętość, ale nie ma określonego
kształtu (przyjmuje kształt zbiornika)
Charakteryzuje
się niewielką ściśliwością, mniejszą ruchliwością cząstek
niż w przypadku gazu – silniejsze oddziaływania międzycząsteczkowe.
Cząsteczki w cieczy mogą się przemieszczać w różnych kierunkach
i
zderzać ze sobą.
W
cieczy
mogą tworzyć się obszary
o
uporządkowanej strukturze – jednak ich
istnienie ma charakter dynamiczny , tzn.
że
tworzą się i szybko zanikają, odtwarzając
w innym miejscu.

Właściwości cieczy
Lepkość cieczy
Jest to opór (tarcie pomiędzy warstwami cieczy) przeciwdziałający jej
płynięciu.
Rys. str 449 dół
Atkins
Przyczyną lepkości jest
występowanie sił
międzycząsteczkowych, które
wiążą ze sobą cząsteczki i
utrudniają ich przemieszczanie
się względem innych cząsteczek.
Ciecze zawierające wiązania
wodorowe mają z reguły dużą
lepkość.
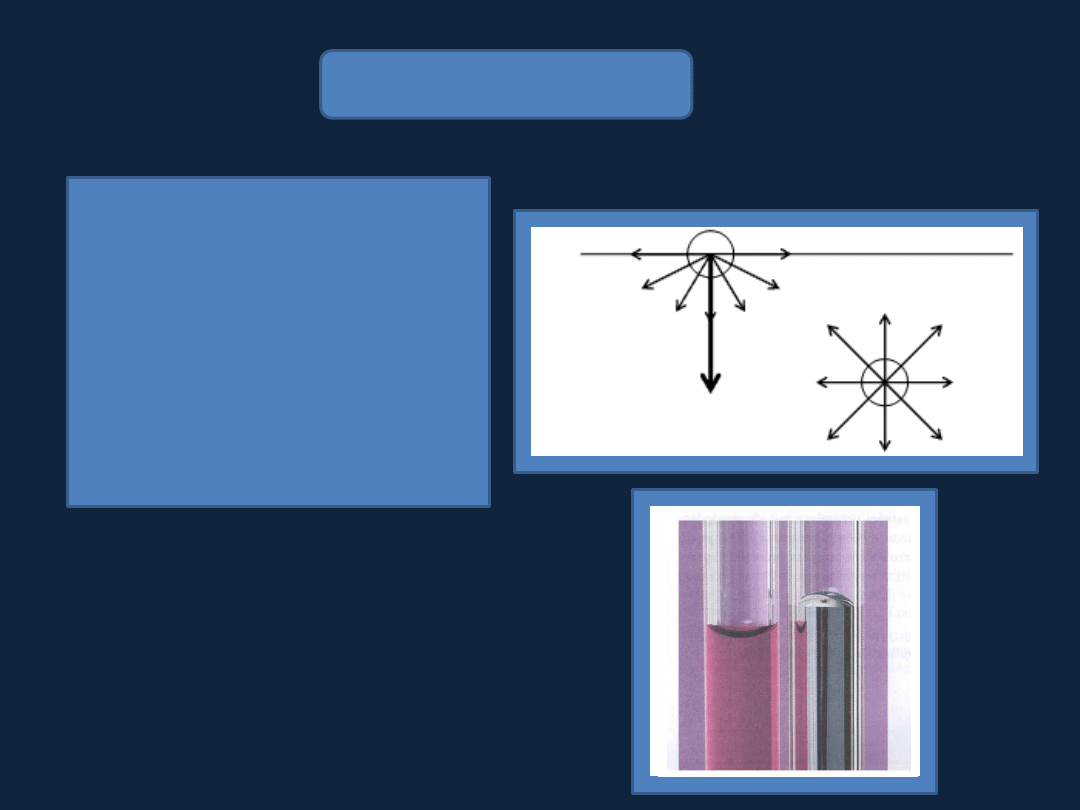
Napięcie powierzchniowe
Może być definiowane jako siła
wypadkowa działająca na
cząsteczkę znajdującą się na
powierzchni cieczy i
zwrócona
do wnętrza cieczy.
Napięcie powierzchniowe
związane jest z istnieniem sił
międzycząsteczkowych.
Siły adhezji – siły wiążące substancję
z
powierzchnią.
Siły kohezji – siły wiążące ze sobą
cząsteczki substancji.
Rys. 10.9 str 450
Atkins
STAN STAŁY
Ciało stałe
– posiada zdefiniowaną objętość i określony kształt, silne
oddziaływania pomiędzy atomami lub cząsteczkami.
Atomy nie przemieszczają się względem siebie.
KLASYFIKACJA CIAŁ STAŁYCH
Ciała krystaliczne
– atomy, jony lub cząsteczki tworzą uporządkowaną
sieć
Ciała bezpostaciowe (amorficzne)
– atomy, jony lub cząsteczki są
rozmieszczone w bezładny sposób
Podział kryształów ze względu na rodzaj wiązań chemicznych, które
w nich występują
Kryształy molekularne
-
mała wytrzymałość, miękkie,
- niska temperatura topnienia,
- izolatory,
-
widmo pochłaniania światła takie samo jak w stanie gazowym i ciekłym,
- tworzone przez tlen, azot, gazy szlachetne lub wodorki kowalencyjne,
-
najczęstszy typ kryształów jaki jest spotykany w przypadku połączeń
organicznych

Kryształy kowalencyjne
-
atomy w krysztale połączone wiązaniami kowalencyjnymi
-
duża wytrzymałość, twarde,
- wysokie temperatury topnienia
-
nie przewodzą prądu lub półprzewodniki,
-
widmo pochłaniania światła inne niż w stanie ciekłym i gazowym,
-
tworzone przez diament, krzem, german, węglik krzemu.
Kryształy jonowe
-
duża wytrzymałość, twarde,
- wysokie temperatury topnienia,
-
w stanie stałym źle przewodzą prąd (dobre przewodnictwo stopione lub
w roztworze),
-
pochłaniają światło w zakresie dalekiej podczerwieni,
- tworzone przez substancje o charakterze jonowym, np. NaCl, CsF.

Kryształy metaliczne
-
różna wytrzymałość, ciągliwość,
-
temperatura topnienia zmienia się w szerokich granicach,
- dobre przewodnictwo elektryczne,
-
nieprzezroczyste, charakterystyczny połysk metaliczny,
- tworzone przez metale.
Pozostałe typy kryształów:
Kryształy condis
– faza pośrednia pomiędzy cieczą i ciałem stałym,
najczęściej tworzone przez długie, cienkie cząsteczki, które mają
swobodę zmiany konformacji, np. teflon
Kwazikryształy
- atomy
układają się w pozornie regularną, ale nie
powtarzającą się strukturę - uniemożliwia to wyróżnienie komórek
elementarnych w tego typu
układzie.

Przykład kwazikryształu

Polimorfizm, izomorfizm, alotropia, ciała anizotropowe
Polimorfizm
– zjawisko występowania tej samej substancji w różnych
strukturach przestrzennych (odmianach krystalograficznych).
Np. węglan wapnia
kalcyt
aragonit
Izomorfizm
– zjawisko występowania różnych substancji w tym samym
typie sieci krystalograficznej.

Alotropia
– zjawisko występowania tej samej substancji prostej
(pierwiastka chemicznego) w tym samym stanie skupienia w
dwóch lub
więcej postaciach (tzw. odmianach alotropowych) charakteryzujących się
odmiennym
składem cząsteczek lub postacią krystaliczną
np.
tlen O
2
, ozon O
3
siarka
jednoskośna i rombowa.
Ciała anizotropowe
– ciała jednorodne wykazujące zależność swych
właściwości od kierunku.
Np. w
niektórych kierunkach atomy mogą być słabiej związane niż
w innych
–większa łupliwość (grafit).

Ciała bezpostaciowe
Powstają, gdy nie następuje krystalizacja.
W miarę obniżania temperatury ruchliwość cząsteczek maleje, co powoduje
wzrost lepkości i zakrzepnięcie cieczy.
Ciała bezpostaciowe nie wykazują zależności swych właściwości od kierunku.
W czasie ogrzewania przejście ich do stanu ciekłego odbywa się
w sposób ciągły, a nie skokowy.
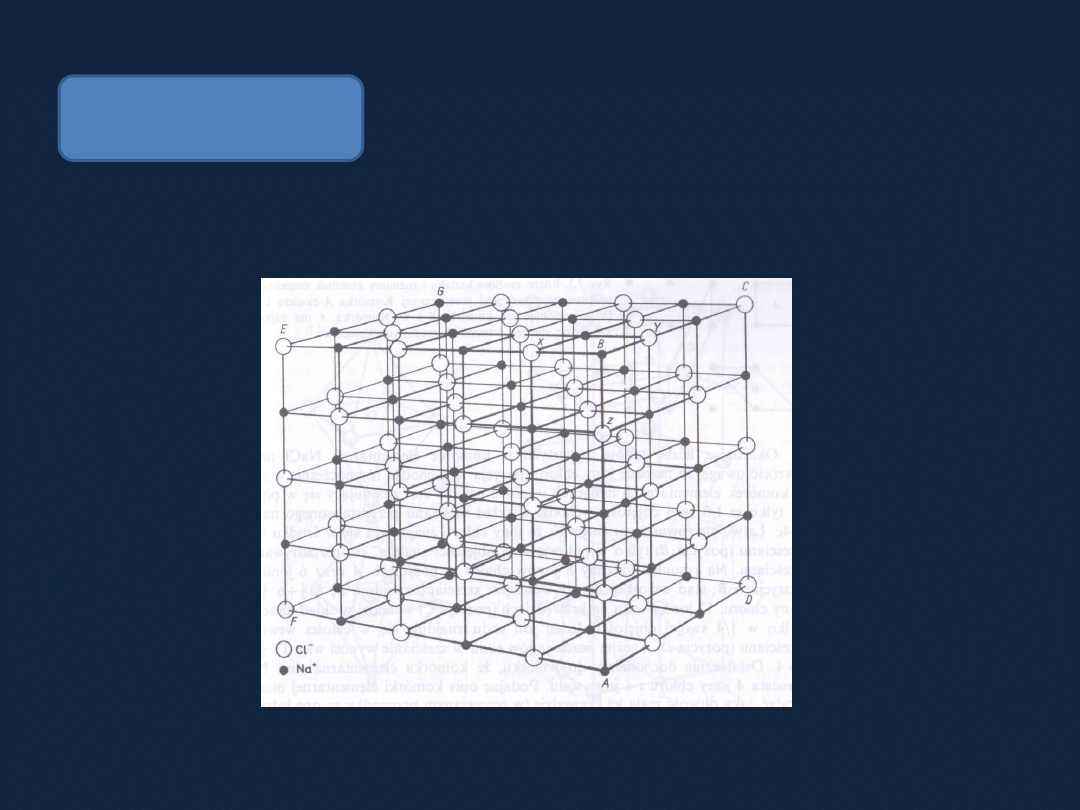
Sieć przestrzenna
W krysztale panuje stan pełnego uporządkowania w każdym kierunku
przestrzeni.
Rys. 7.2 str 209
Bielan
Sieć przestrzenna NaCl
Do opisania
wyglądu sieci przestrzennej nie jest konieczne opisanie położenia
wszystkich
zawartych
w niej
atomów. Wystarcza znajomość komórki
elementarnej.
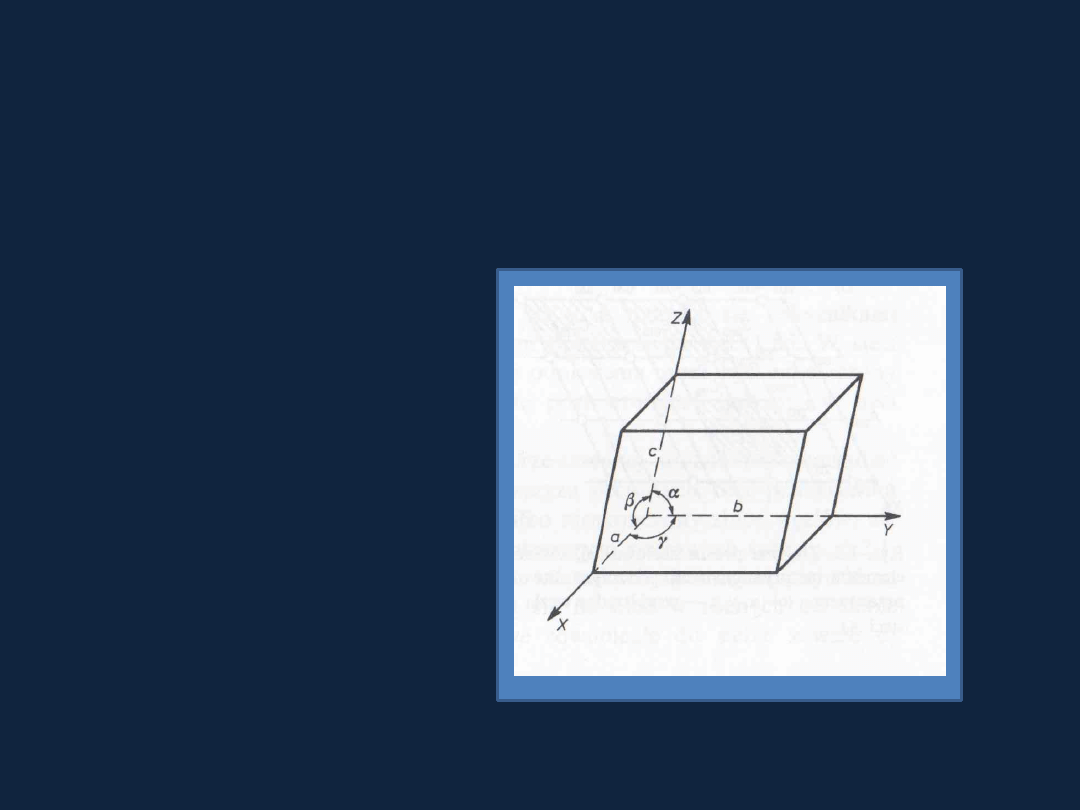
Komórka elementarna
–
najmniejszy wycinek sieci
zachowujący jej wszystkie
cechy.
Komórka elementarna
powtarza się cyklicznie
w przestrzeni.
Jest określana za pomocą
trzech krawędzi a, b, c oraz
trzech kątów α, β, γ
pomiędzy tymi krawędziami
-
są to tzw. parametry sieci
przestrzennej.
Rys 1.4 str 17 Krystalografia
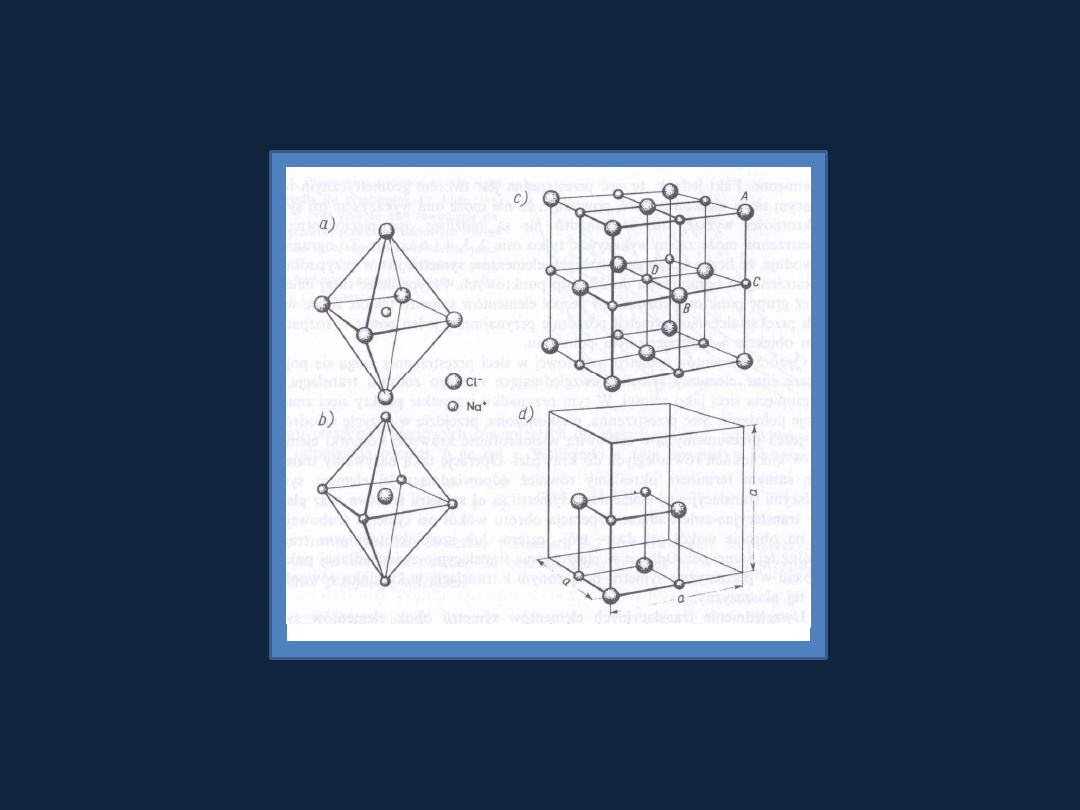
Rys 7.4 str
211 Bielan
Sieć przestrzenna i komórka elementarna kryształu NaCl
Liczba
koordynacyjna
– liczba atomów lub jonów bezpośrednio
sąsiadujących w sieci przestrzennej kryształu z danym atomem lub
jonem.
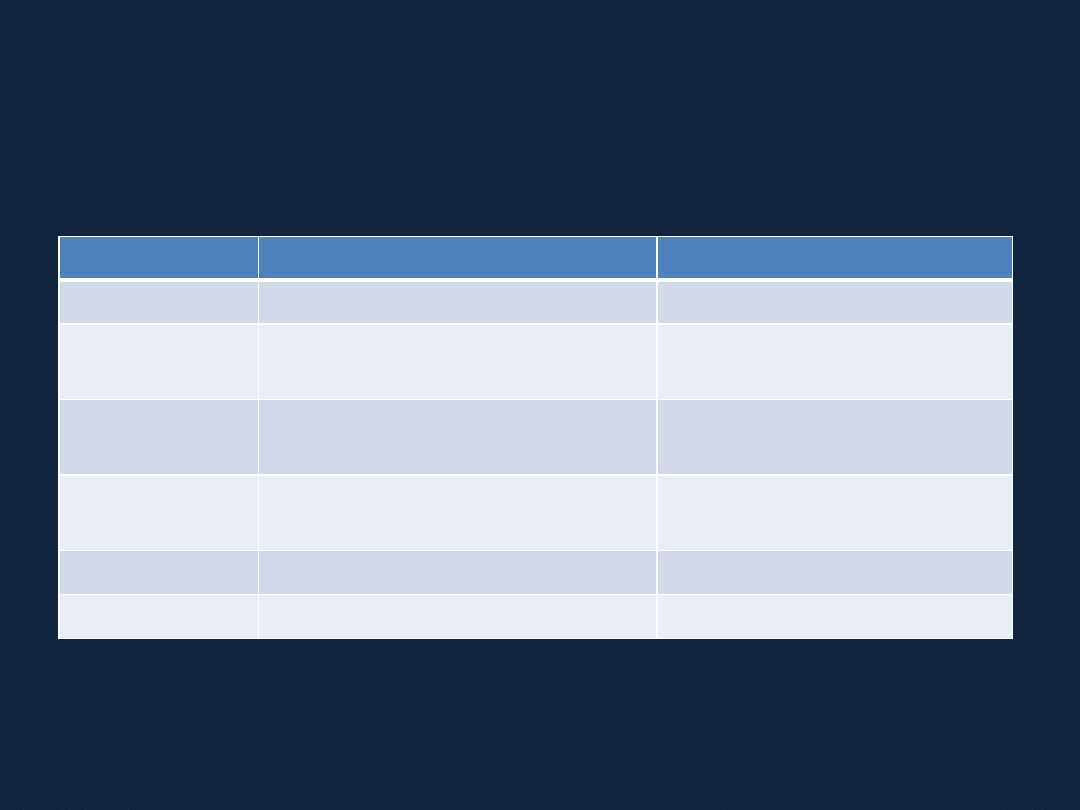
Przykłady komórek elementarnych i układów krystalograficznych
Układ
Kształt komórki elementarnej Parametry sieciowe
Regularny
Sześcian
a = b = c
α = β = γ = 90°
Tetragonalny
Prostopadłościan o podstawie
kwadratowej
a = b ≠ c α = β = γ = 90°
Heksagonalny
Prostopadłościan o podstawie
rombu
a = b
α = β = 90°
γ = 120°
Rombowy
Prostopadłościan o podstawie
prostokątnej
a ≠ b ≠ c α = β = γ = 90°
Jednoskośny
Równoległościan
a ≠ b ≠ c α = β = 90° ≠ γ
Trójskośny
Równoległościan
a ≠ b ≠ c α ≠ β ≠ γ
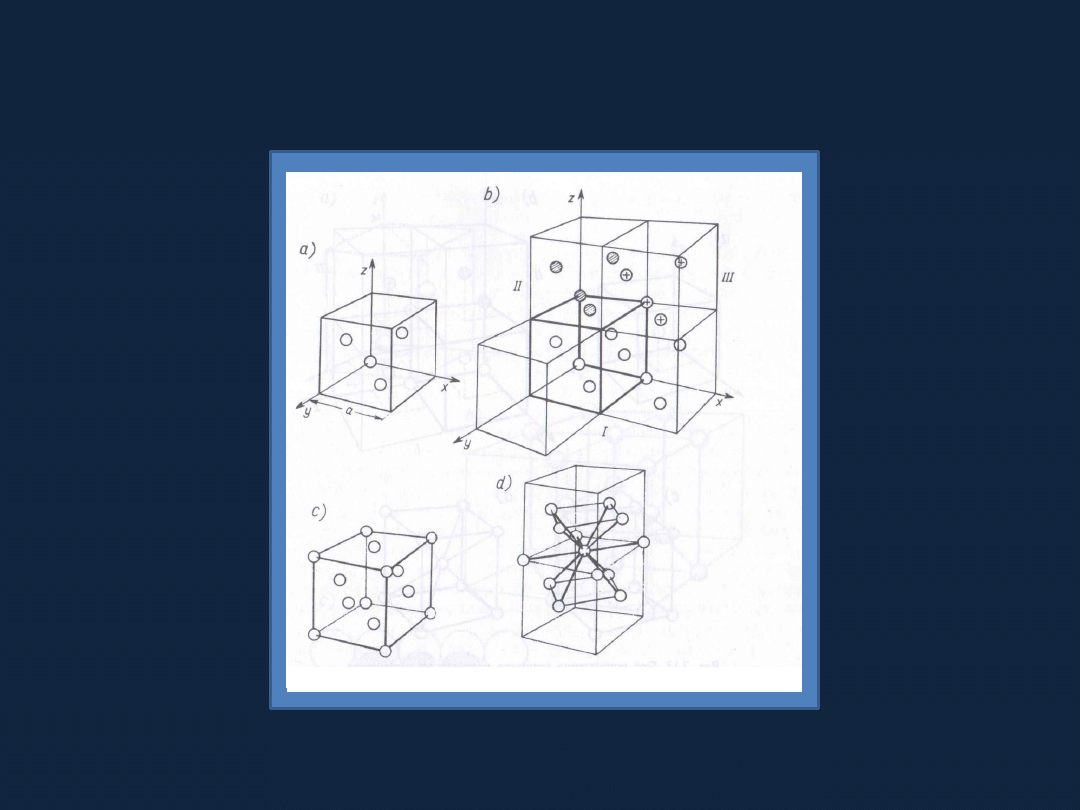
Przykłady sieci przestrzennych w kryształach
Sieć przestrzenna regularna płasko centrowana
Rys. 7.12 str 223 Bielan
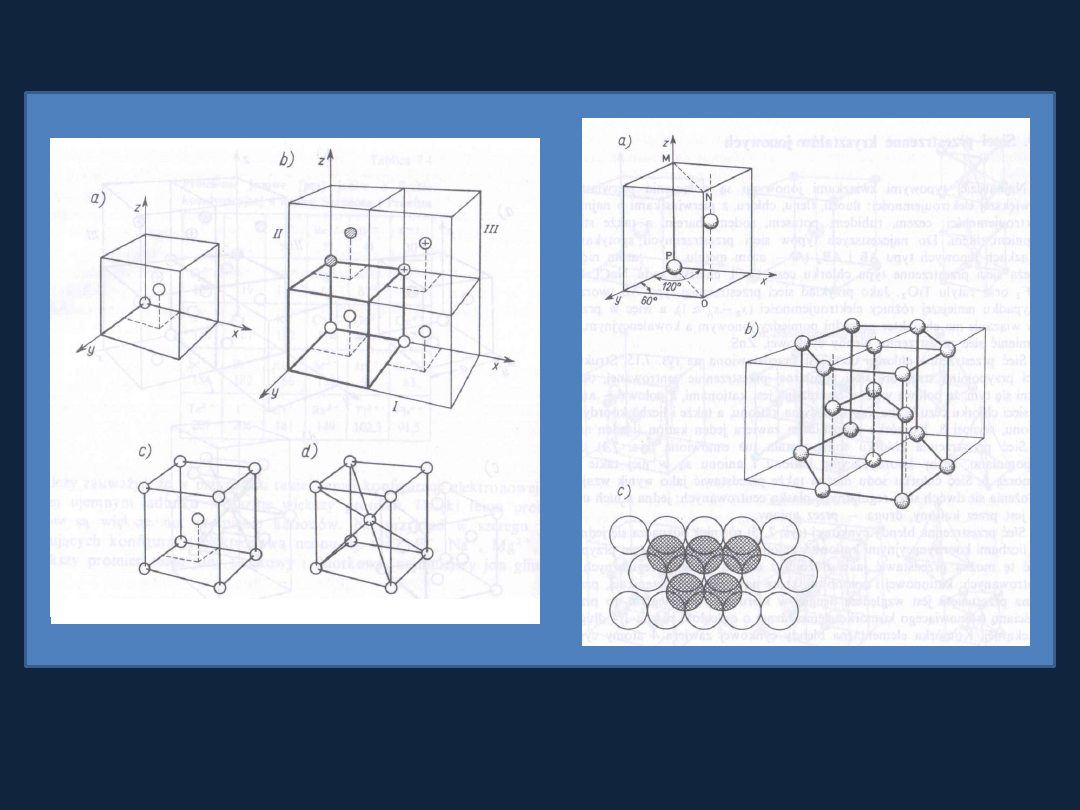
Sieć przestrzenna regularna
przestrzennie centrowana
Sieć przestrzenna heksagonalna
Rys. 7.13 i 7.14 str 224
i 225 Bielan

Energia sieciowa
kryształów jonowych
Energia sieciowa
– ilość energii, którą należy dostarczyć, aby 1 mol
substancji krystalicznej
rozłożyć na jony znajdujące się w nieskończenie
dużych odległościach od siebie.
Energia sieciowa (U
0
)
wyraża całkowitą energię potencjalną kryształu
(zależą od niej trwałość, temp. topnienia, rozpuszczalność, itd.).
N
∙ A ∙ e
2
∙ z
-
∙ z
+
1
U
0
=
1 -
4
∙ π ∙ ε
0
∙ r
0
n
gdzie:
N
– liczba Avogadra, A – stała, e – ładunek elektronu, z- i z+ - liczba
elementarnych
ładunków anionu i kationu, ε
0
– przenikalność elektryczna próżni,
r
0
– odległość dwóch sąsiednich jonów o przeciwnym znaku, n – współczynnik
wyznaczany na podstawie
ściśliwości kryształów.
Np. LiF 1034 kJ/mol
– MgO 3934 kJ/mol – zmiana wartości energii
sieciowej
spowodowana
różnicą ładunku jonów (temp. topnienia
kryształów LiF 845°C, a MgO 2800°C).

PRZEMIANY FAZOWE
Faza
oznacza
fizyczną postać materii (odnosi się nie tylko do trzech
stanów skupienia, ale również do różnych odmian tych stanów).
Układ jednorodny
– każda część układu wykazuje te same cechy co cały
układ.
Układ niejednorodny
– różne części układu wykazują różne właściwości.
Jest to
związane z obecnością różnych faz oddzielonych od innych
powierzchnią rozdziału.
Przemiany fazowe
– parowanie, skraplanie, topnienie, krzepnięcie,
krystalizacja, sublimacja.

Równowagi ciecz – para i ciało stałe - para
Prężność pary
Energia niektórych cząsteczek znajdujących się w fazie ciekłej wystarcza
do oderwania się od cząsteczek sąsiednich i przejścia w stan pary.
Tendencja do parowania zależy od temperatury układu i siły oddziaływań
międzycząsteczkowych.
Proces parowania
W
zamkniętym zbiorniku ustala się stan
równowagi dynamicznej.
szybkość parowania = szybkość kondensacji
H
2
O(c) H
2
O(g)
←
→
Prężność pary nasyconej
Jest to ciśnienie, jakie wywiera para, gdy para i ciecz (lub substancja stała)
znajdują się w równowadze dynamicznej.
Prężność pary jest niska w przypadku substancji o silnych oddziaływaniach
międzycząsteczkowych i wzrasta wraz z temperaturą.
Wrzenie
Gdy ciecz znajduje
się w otwartym zbiorniku, to powstająca para może
odpływać od cieczy.
Wrzenie
następuje gdy prężność pary cieczy jest równa ciśnieniu
atmosferycznemu.
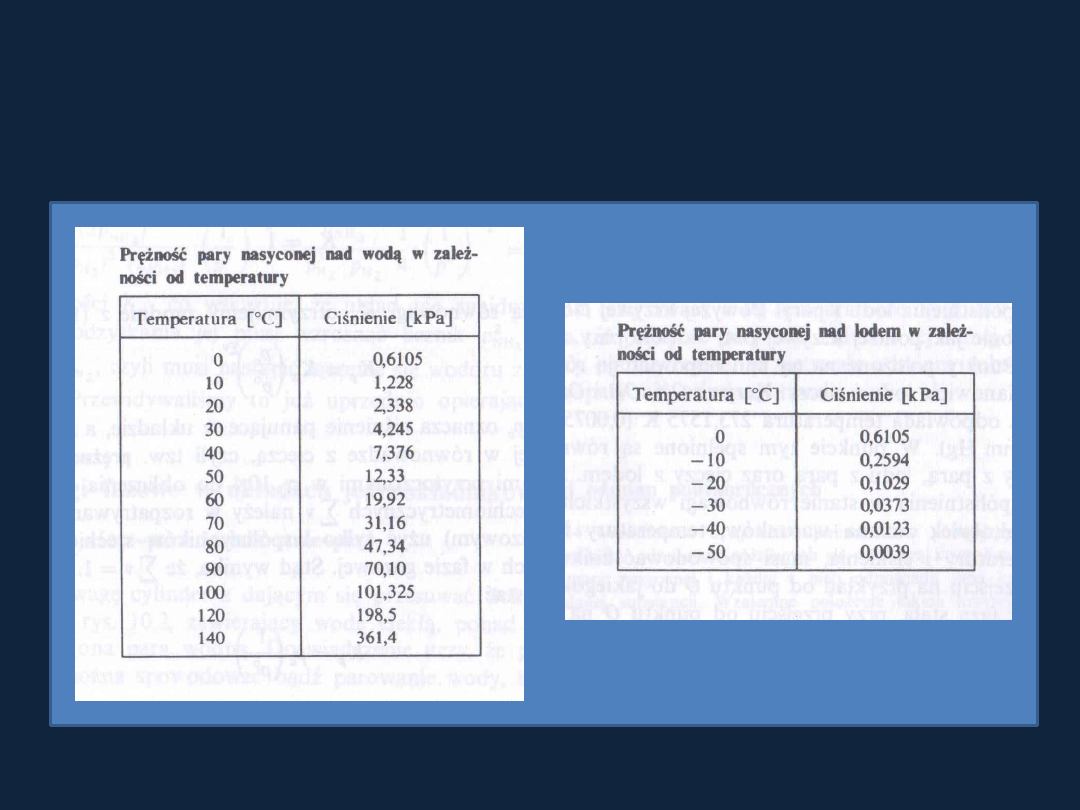
Tablica 10.2 i 10.3 str 322
i 323 Bielan
Prężność pary nasyconej nad wodą (po lewej) i lodem (po prawej) w zależności od temperatury.

Mieszanina
azeotropowa
– mieszanina cieczy wrząca w stałej
temperaturze bez zmiany
składu (udziału każdego ze składników).
Krzepnięcie
Ciecz krzepnie, gdy
cząsteczki mają tak małą energię, że nie mogą się
przemieszczać względem cząsteczek sąsiednich.
W ciele
stałym cząsteczki mogą drgać wokół swoich średnich połączeń,
ale nie
przesuwają się z miejsca na miejsce.
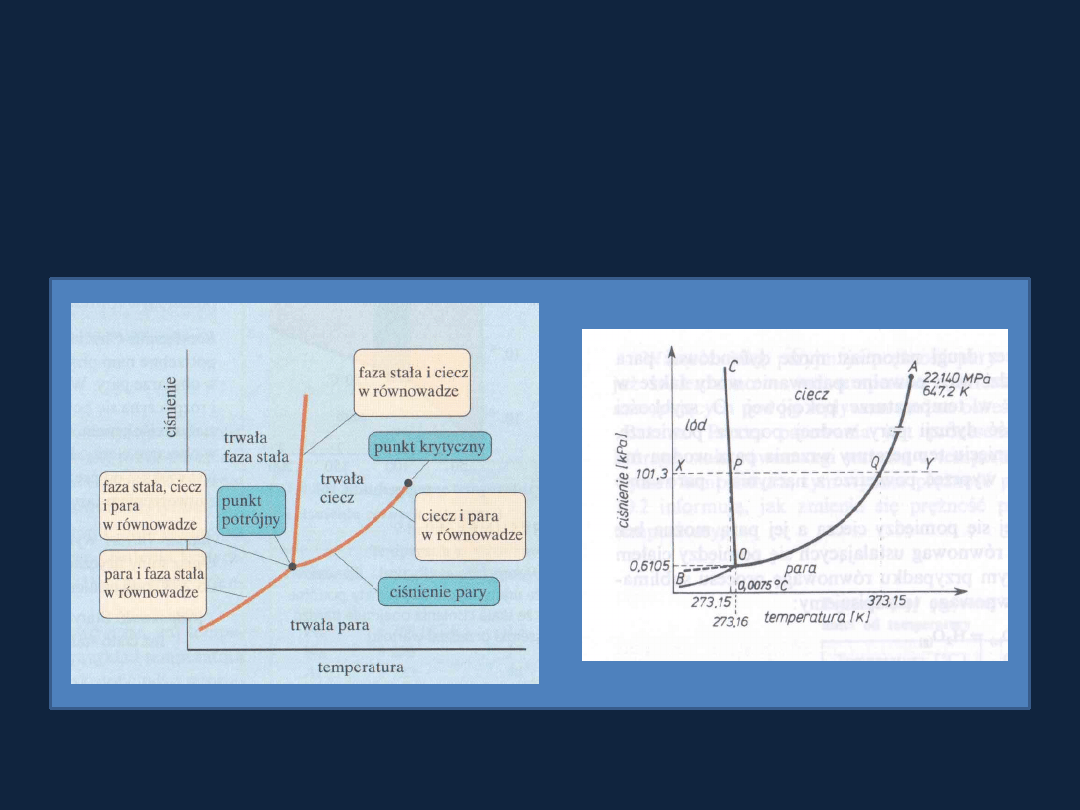
Diagram fazowy
Określa wartości ciśnienia i temperatury, w których każda z faz występujących
w danym
układzie jest najtrwalsza. Granice faz wyznaczają warunki, w których
dwie fazy
mogą istnieć w warunkach równowagi (trzy fazy współistnieją
w punkcie
potrójnym).
Diagram fazowy dla przykładowego
układu jednoskładnikowego
Diagram fazowy wody
Rys 10.48
str 483
Atkins
Rys. 10.3 str
324 Bielan

REGUŁA FAZ GIBBSA
Reguła ta formułuje ogólne warunki równowag fazowych w układach
jedno- i
wieloskładnikowych.
s = n + 2
– f
s
– liczba stopni swobody - czyli liczba czynników wyznaczających
równowagę (temperatura, ciśnienie, stężenia), które można zmieniać
zachowując niezmienioną liczbę faz,
f
– liczba faz obecnych w układzie,
n
– liczba niezależnych składników – najmniejsza liczba substancji
z
których można zbudować układ, np. C + CO
2
↔ 2CO, dwa
niezależne składniki
Prężność pary nasyconej nad roztworami
Zmiany temperatury wrzenia i krzepnięcia roztworów
Prawo Raoulta
Stosunek
prężności pary nasyconej nad roztworem do prężności pary nad
czystym rozpuszczalnikiem jest
równy ułamkowi molowemu rozpuszczalnika
w roztworze.
p
r
/ p
c
= x
c
p
r
– prężność pary nad roztworem
p
c
– prężność pary nad rozpuszczalnikiem
x
c
– ułamek molowy rozpuszczalnika w roztworze

Uwzględniając fakt, że:
x
c
= 1
–x
A
x
A
– ułamek molowy substancji rozpuszczonej
otrzymujemy równanie:
p
c
– p
r
= x
A
p
c
gdzie: (p
c
– p
r
)/p
c
– względne obniżenie prężności pary
W związku z tym można stwierdzić, że względne obniżenie prężności pary
nad roztworem jest równe ułamkowi molowemu substancji rozpuszczonej.

Konsekwencją obniżenia prężności pary pozostającej w równowadze
z
roztworem
jest zmiana temperatury jego
wrzenia i
krzepnięcia
w
porównaniu z czystym rozpuszczalnikiem.
Obniżenie temperatury krzepnięcia i podwyższenie temperatury wrzenia
jest
wprost
proporcjonalne
do
stężenia roztworu (dla roztworów
rozcieńczonych).
ΔT
k
= E
k
∙ c
ΔT
w
= E
w
∙ c
gdzie:
T
k
–temperatura krzepnięcia, T
w
– temperatura wrzenia,
E
k
i E
w
– stała krioskopowa i stała ebulioskopowa
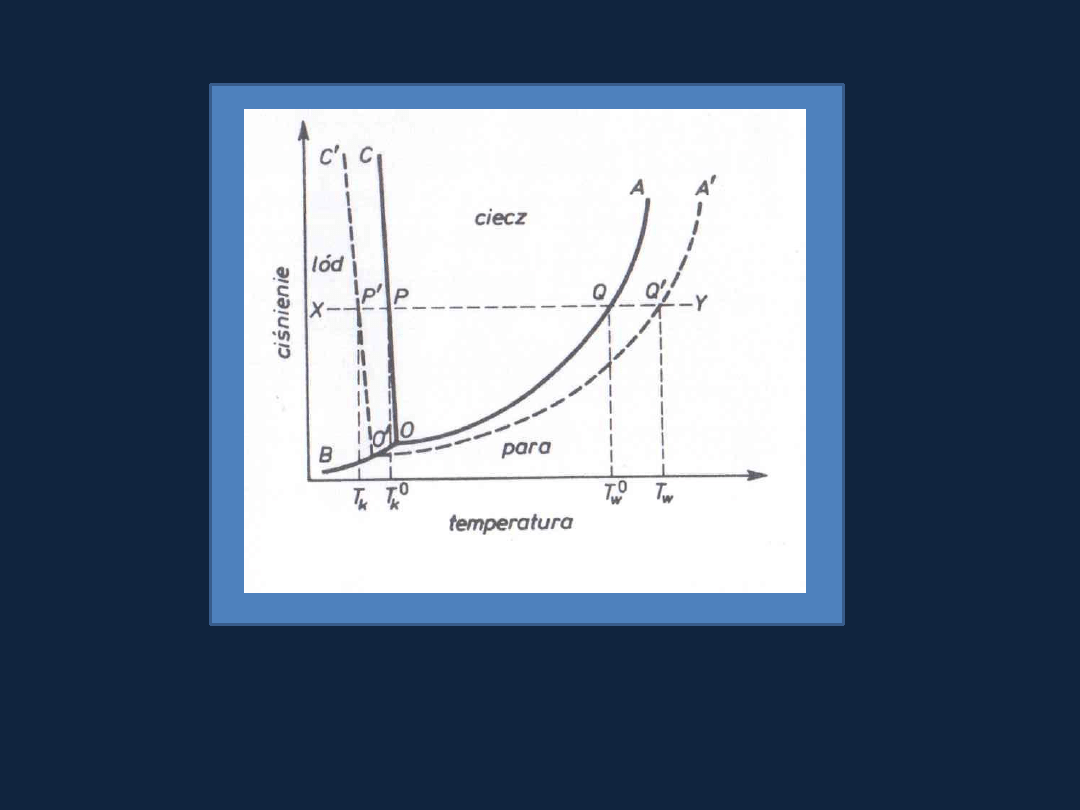
Diagram
prężności
pary
nad
roztworem
wodnym
(linia
przerywana)
i diagram fazowy wody (linia
ciągła).
Rys. 10.7 str 333
Bielan

KRYSTALIZACJA
Krystalizacja polega na utworzeniu
układu przestrzennego, w którym
położenie
tych
samych
jonów
powtarza
się okresowo w każdym
kierunku.
Pierwszym etapem jest tworzenie
zarodków krystalizacji
Roztwory nie
są absolutnie homogeniczne – wykazują pewne zmiany
stężenia rozpuszczonego ciała – prowadzi to do utworzenia niewielkich
skupisk
jonów bądź cząsteczek.

Po utworzeniu zarodków krystalizacji następuje wzrost kryształów
Wielkość kryształów zależy od szybkości wzrostu kryształów i szybkości
tworzenia
się nowych zarodków krystalizacji.
Jeżeli szybkość wzrostu kryształów jest większa powstają osady
grubokrystaliczne.
Jeśli większą jest szybkość tworzenia nowych zarodków krystalizacji,
to
powstają osady drobnokrystaliczne

W dalszych etapach krystalizacji
możemy wyróżnić następujące procesy:
Agregacja
– łączenie się poszczególnych zarodków krystalizacji
w skupienia (tworzenie
agregatów utrzymujących się w dowolny sposób).
Aglomeracja
– tworzenie i wzrost agregatów prowadzący do wydzielenia
osadów złożonych z cząstek o wymiarach większych niż cząstki
koloidalne.
Rekrystalizacja
– przejście nieuporządkowanych pierwotnych skupień
o
nieuporządkowanej budowie w uporządkowaną sieć krystaliczną.
Wyszukiwarka
Podobne podstrony:
więcej podobnych podstron