SHORT COMMUNICATION
DOI: 10.1002/ejoc.201403352
Transition-Metal-Free Oxidative Iodination of 1,3,4-Oxadiazoles
Carl Albrecht Dannenberg,
[a]
Vincent Bizet,
[a]
Liang-Hua Zou,
[a]
and Carsten Bolm*
[a]
Keywords:
Synthetic methods / Iodine / Oxidation / Nitrogen heterocycles / Oxadiazoles
Transition-metal-free oxidative iodination of 2-substituted
1,3,4-oxadiazoles was achieved by using sodium iodide as
the halide source and Selectfluor as the oxidant. Variously
substituted products were obtained in moderate to good
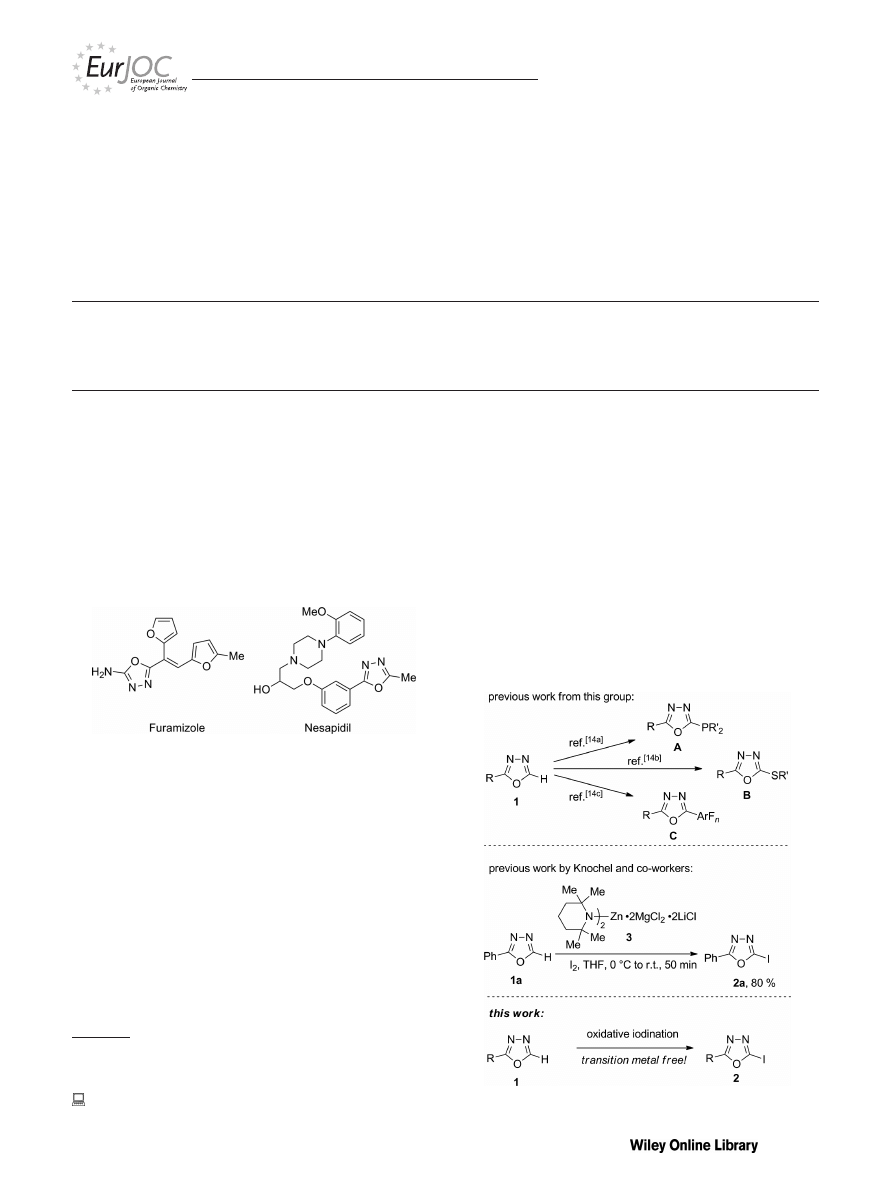
Introduction
1,3,4-Oxadiazoles are important heterocycles in medici-
nal chemistry that exhibit a broad array of bioactivities, and
they are used, for example, as antimicrobial, fungicidal, and
antibacterial agents.
[1–4]
Two representative bioactive com-
pounds are the antibiotic Furamizole and the antihyperten-
sive agent Nesapidil (Figure 1).
[5]
In material science, 1,3,4-
oxadiazoles have extensively been applied in organic light-
emitting diodes.
[6]
Figure 1. 1,3,4-Oxadiazoles with pharmaceutical relevance.
Iodinated heteroarenes are common products in the
pharmaceutical industry, in medicine, and in crop protec-
tion, and furthermore, they serve as useful intermediates
in transition-metal-catalyzed cross-coupling reactions and
allow rapid access to diversified compound libraries.
[7,8]
Several selective iodination methods are known. Owing to
the electron-deficient nature of many heterocycles, electro-
philic iodination reactions are often difficult to achieve;
they require strong, highly reactive iodinating agents such
as N-iodosuccinimide, N-iodosaccharin, iodine mono-
chloride, or iodonium salts such as IPy
2
BF
4
(Barluenga’s
reagent, Py = pyridine).
[7,9,10]
A more effective method is
iododemetalation, which involves initial deprotonation of
the heteroarene with combination of an organometallic rea-
[a] Institute of Organic Chemistry, RWTH Aachen University,
Landoltweg 1, 52056 Aachen, Germany
E-mail: Carsten.Bolm@oc.rwth-aachen.de
http://bolm.oc.rwth-aachen.de/
Supporting information for this article is available on the
WWW under http://dx.doi.org/10.1002/ejoc.201403352.
Eur. J. Org. Chem. 2015, 77–80
© 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
77
yields under operationally straightforward conditions. Com-
pared to existing methods for analogous conversions, the
newly developed protocol appears synthetically attractive.
gent and an alkali salt additive under inert and anhydrous
conditions followed by an iodo–metal exchange.
[11]
Finally,
iodinated heterocycles can also be accessed by Finkelstein-
type substitution reactions of aryl halides
[12]
and Sandme-
yer reactions.
[13]
Recently, we reported the functionalization of 1,3,4-oxa-
diazoles to give products with new C–P, C–S, and C–C
bonds (compounds A–C, Scheme 1).
[14]
To our surprise, we
noted that analogous iodination reactions were essentially
unexplored, although the resulting 2-halogenated products
appeared rather attractive for further functionalization.
[15]
In fact, to the best of our knowledge, only a single example
of the direct iodination of an 1,3,4-oxadiazole has been re-
Scheme 1. Functionalization of 2-substituted 1,3,4-oxadiazoles.
C. A. Dannenberg, V. Bizet, L.-H. Zou, C. Bolm
SHORT COMMUNICATION
ported to date. Therein, Knochel and co-workers obtained
2a
from 2-phenyl-1,3,4-oxadiazole (1a) in 80 % yield by ap-
plying a deprotonative metalation strategy with 3 followed
by halogenation of the resulting metalated intermediate
with molecular iodine (Scheme 1).
[11a]
On the basis of the
expertise gained in our previous studies,
[14]
we wondered if
we could develop an alternative approach towards products
such as 2a by circumventing the use of complex metal- and
salt-rich reagent mixtures such as 3. Herein, we report on
the success of this study and describe the site-selective io-
dination of 2-substituted 1,3,4-oxadiazoles by oxidative
halogenation reactions.
[7,16,17]
Results and Discussion
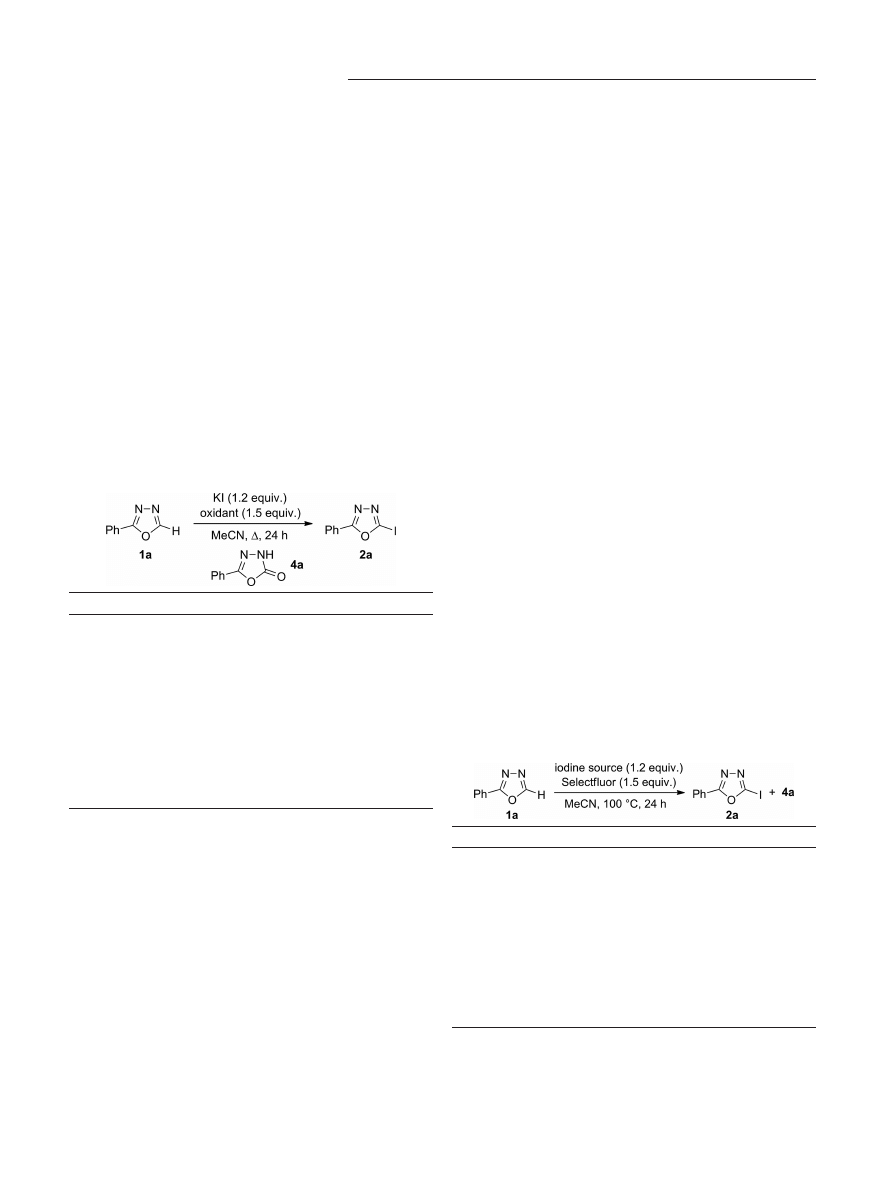
The investigation was initiated by an oxidant and tem-
perature screening with 2-phenyl-1,3,4-oxadiazole (1a) as
the model substrate and potassium iodide as the halogen
source. The results are summarized in Table 1.
Table 1. Oxidant and temperature screening.
[a]
Entry
Oxidant
[b]
Temp. [°C]
Yield of 2a [%]
[c]
1
O
2
130
n.d. (5)
2
PIDA
130
n.d. (n.d)
3
PIFA
130
n.d. (n.d.)
4
DTBP
130
6 (10)
5
K
3
[Fe(CN)
6
]
130
10 (22)
6
NaIO
4
130
8 (24)
7
Oxone
®
130
10 (38)
8
K
2
S
2
O
8
130
45 (11)
9
NFSI
130
47 (5)
10
Selectfluor
130
34 (6)
11
K
2
S
2
O
8
100
7 (17)
12
NFSI
100
51 (1)
13
Selectfluor
100
58 (8)
[a] The reaction was performed in a sealed tube on a 0.2 mmol scale
by using the oxidant (1.5 equiv.) and KI (1.2 equiv.) in acetonitrile
(3 mL). [b] PIDA: [bis(acetoxy)iodo]benzene. PIFA: [bis(trifluoro-
acetoxy)iodo]benzene. DTBP: di-tert-butyl peroxide. NFSI: N-
fluorodibenzenesulfonimide. Selectfluor: 1-chloromethyl-4-fluoro-
1,4-diazoniabicyclo[2.2.2]octane. [c] Determined by
1
H NMR spec-
troscopy. n.d.: not detected. The values in parentheses refer to the
yield of 4a.
As hypothesized, target compound 2a was indeed formed
under these oxidative conditions (at 130 °C) and most oxi-
dants exhibited activity. Unfortunately, however, in many
cases the yields of 2a were low, mainly because of the lack
of conversion of 1a, decomposition of the starting material,
and the formation of hydrolysis product 4a. Dioxygen,
PIFA, and PIDA proved unsuitable oxidants, and at best,
trace amounts of 4a were detected (Table 1, entries 1–3).
The use of DTBP, K
3
[Fe(CN)]
6
, NaIO
4
, and Oxone gave 2a
in very low yields, and the formation of 4a dominated
www.eurjoc.org
© 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Eur. J. Org. Chem. 2015, 77–80
78
(Table 1, entries 4–7). The most promising results were ob-
tained with K
2
S
2
O
8
, NFSI, and Selectfluor, all of which
provided 2a in yields up to 45 % (Table 1, entries 8–10).
[18]
The formation of 4a was still observed, but to a lower ex-
tent. Assuming that the degradation pathways could be
minimized by lowering the reaction temperature, iodination
with the latter three oxidants was also performed at 100 °C
instead of 130 °C (Table 1, entries 11–13). To our delight,
the outcome was positive, and with NFSI and Selectfluor
improved yields (of 51 and 58 %, respectively) of 2a were
observed. Byproduct 4a was still formed, but to an accept-
able extent. As Selectfluor showed the best reactivity, it was
chosen as the oxidant for subsequent optimizations.
A solvent screening confirmed that acetonitrile was the
optimal solvent for the reaction. Only with 1,4-dioxane was
a comparable reactivity observed. Protic, polar solvents
(water, methanol, DMF) decomposed the starting mate-
rial.
[19]
Table 2 summarizes the impact of the halide source on
the oxidative iodination of 1a. As the data show, the coun-
terion had a significant effect on the product yield. Among
ammonium, lithium, sodium, potassium, and cesium, only
the latter three led to moderate yields of 2a (Table 2, en-
tries 1–5). In all cases, 4a was formed as a byproduct. The
use of molecular iodine as the halide source (in varying
amounts) with and without Selectfluor afforded 2a in very
low yields (Table 2, entries 6–9).
[20]
In these reactions, up to
22 % of 4a was obtained. Guided by the results of Jiao and
co-workers,
[17]
the oxidative iodination was performed with
the addition of various bases to improve the yield of 2a
(Table 2, entries 10–12). None of those attempts, however,
were successful. Apparently, the combination of sodium
iodide (1.2 equiv.) and Selectfluor (1.5 equiv. in the absence
of a base) were optimal and provided 2a under straightfor-
Table 2. Screening of the iodine source.
[a]
Entry
Iodine source
Base
[b]
Yield 2a [%]
[c]
1
NH
4
I
–
6 (15)
2
LiI
–
16 (8)
3
NaI
–
69 (8)
4
KI
–
58 (8)
5
CsI
–
62 (12)
6
I
2
–
10 (22)
7
[d]
I
2
–
8 (20)
8
[e]
I
2
–
4 (12)
9
[e]
I
2
NaHCO
3
4 (22)
10
NaI
LiOtBu
7 (23)
11
NaI
NaHCO
3
15 (5)
12
NaI
Et
3
N
n.d. (n.d.)
[a] The reaction was performed in a sealed tube on a 0.2 mmol scale
by using Selectfluor (1.5 equiv.) and the iodine source (1.2 equiv.) in
acetonitrile (3 mL). [b] Use of 1.5 equiv. of base. [c] Determined by
1
H NMR spectroscopy. n.d.: not detected. The values in parenthe-
ses refer to the yield of 4a. [d] Without Selectfluor. [e] Use of
0.6 equiv. of iodine.

Transition-Metal-Free Oxidative Iodination of 1,3,4-Oxadiazoles
ward conditions in 69 % yield (Table 2, entry 3). The
amount of 4a remained at 8 %.
Next, the substrate scope was examined by using 1,3,4-
oxadiazoles with various substituents at the C2 atom. The
results are summarized in Table 3. The use of aryl-substi-
tuted substrates with electron-withdrawing groups led to
the corresponding products (i.e., 2a–e) in yields ranging
from 62 to 69 %. Having electron-donating substituents on
the connected arene lowered the reactivity, and the iodin-
ated oxadiazoles (i.e., 2i–f) were obtained in yields between
32 and 59 %. The position of the substituent had a negligi-
ble effect, as revealed by the results for para- and ortho-
methyl-substituted products 2g and 2h, which were ob-
tained in yields of 57 and 59 %, respectively. 2-Naphthyl-
1,3,4-oxadiazole (1j) reacted well to provide product 2j in
54 % yield. Neither pyridinyl-containing 2k nor para-di-
methylamino-substituted 2l could be obtained by this pro-
cedure, presumably as a result of two factors: one, the pres-
ence of basic nitrogen atoms hampers the iodination pro-
cess; two, their pronounced sensitivity towards the oxidants
present in the reaction mixture. As a representative example
of 2-alkyl-substituted 1,3,4-oxadiazoles, the formation of
Table 3. Substrate scope under optimized reaction conditions.
[a]
[a] Performed in sealed tubes on a 0.5 mmol scale in acetonitrile
(5 mL). [b] Use of NaBr for 5 and NaCl for 6 instead of NaI.
Eur. J. Org. Chem. 2015, 77–80
© 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
www.eurjoc.org
79
2m
was studied. Although the yield of 2m was only 39 %,
this positive result represented a promising basis for future
investigations. To our delight, the iodination of 2-phenyl-
1,3,4-thiadiazole (1n) proceeded well to afford the corre-
sponding product in 87 % yield.
[21]
Performing the oxidative iodination of 1a on a 4 mmol
scale gave 2a in 60 % yield (together with 17 % of unreacted
1a
and 15 % of 4a), which confirmed the scalability of the
process.
[22]
Attempts to use the same methodology for the introduc-
tion of a bromo or chloro substituent onto 1,3,4-oxadiazole
1a
were unsatisfying. With NaBr (instead of NaI), corre-
sponding brominated product 5 was only observed in trace
quantities, and to our surprise, chlorination of 1a occurred.
Apparently, Selectfluor served as a halide source, and as a
result chlorinated 1,3,4-oxadiazole 6 was formed in 30 %
yield.
[23]
Substituting NaI with NaCl led to the same prod-
uct (i.e., 6) in 33 % yield.
[24]
Conclusions
In summary, we developed the oxidative iodination of
1,3,4-oxadiazoles by using NaI as the halogen source and
Selectfluor as the oxidant. Although in some cases the
product yields were only moderate, the protocol is attractive
because it circumvents the use of complex metal mixtures
and does not require an inert atmosphere or anhydrous re-
action conditions. Its applicability to 1,3,4-thiadiazoles was
exemplified. Further iodination reactions of other heterocy-
clic compounds are currently under investigation in our
laboratories.
Experimental Section
Procedure for the Synthesis of 2-Substituted 5-Iodo-1,3,4-oxadi-
azoles:
A sealed tube, equipped with a magnetic stir bar, was
charged
with
2-substituted-1,3,4-oxadiazole
1
(0.5 mmol,
1.0 equiv.),
NaI
(89.9 mg,
0.6 mmol,
1.2 equiv.),
Selectfluor
(265.7 mg, 0.75 mmol, 1.5 equiv.), and acetonitrile (5 mL). Then,
the mixture was stirred at 100 °C for 24 h. After cooling to room
temperature, the mixture was diluted with CH
2
Cl
2
(10 mL) and
washed with a saturated aqueous solution of Na
2
S
2
O
3
(10 mL).
After extracting the aqueous phase with CH
2
Cl
2
(2
⫻ 10 mL) the
organic phases were combined, dried with MgSO
4
, and filtered.
The mixture was evaporated under reduced pressure, and the resi-
due was purified by column chromatography (n-pentane/ethyl acet-
ate, 11:1) to yield iodinated product 2.
Acknowledgments
V. B. and L.-H. Z. acknowledge the Alexander von Humboldt
Foundation and the China Scholarship Council (CSC), respec-
tively, for fellowships. The authors thank Jakob Mottweiler
(RWTH Aachen University) for fruitful discussions and proofread-
ing the manuscript.
[1] G. Majji, S. K. Rout, S. Guin, A. Gogoi, B. K. Patel, RSC Adv.
2014
, 4, 5357–5362.

C. A. Dannenberg, V. Bizet, L.-H. Zou, C. Bolm
SHORT COMMUNICATION
[2] S. Maghari, S. Ramezanpour, F. Darvish, S. Balalaie, F. Rom-
inger, H. R. Bijanzadeh, Tetrahedron 2013, 69, 2075–2080.
[3] S. Vodela, R. V. R. Mekala, R. R. Danda, V. Kodhati, Chin.
Chem. Lett. 2013, 24, 625–628.
[4] For 1,3,4-oxadiazoles as bioisosteres of ester, amide, and acid
functionalities, see: a) M. Rouhani, A. Ramazani, S. W. Joo,
Ultrason. Sonochem. 2014, 21, 262–267; b) D. Leung, W. Du,
C. Hardouin, H. Cheng, I. Hwang, B. F. Cravett, D. L. Boger,
Bioorg. Med. Chem. Lett. 2005, 15, 1423–1428.
[5] S. J. Dolman, F. Gosselin, P. D. O’Shea, I. W. Davies, J. Org.
Chem. 2006, 71, 9548–9551.
[6] J. Wang, R. Wang, J. Yang, Z. Zheng, M. D. Carducci, T.
Cayou, N. Peyghambarian, G. E. Jabbour, J. Am. Chem. Soc.
2001
, 123, 6179–6180.
[7] L. Bedracˇ, J. Iskra, Adv. Synth. Catal. 2013, 355, 1243–1248.
[8] For recent reviews on cross-coupling reactions, see: a) G. A.
Molander, J. P. Wolfe, M. Larhed (Eds.), Science of Synthesis:
Cross Coupling and Heck-Type Reactions, Thieme, Stuttgart,
Germany, 2013, vols. 1–3; b) M. Shimizu, T. Hiyama, Science
of Synthesis: Stereoselective Synthesis (Ed.: P. A. Evans), Thi-
eme, Stuttgart, Germany, 2011, vol. 3, p. 567–614; c) S. Roy, S.
Roy, G. W. Gribble, Tetrahedron 2012, 68, 9867–9923; d) V.
Sarli, Stereoselective Synthesis of Drugs and Natural Products
(Eds: V. Andrushko, N. Andrushko), Wiley, Hoboken, NJ,
2013
, vol. 1, p. 369–393; e) R. Rossi, F. Bellina, M. Lessi, C.
Manzini, Adv. Synth. Catal. 2014, 356, 17–117; f) C.-F. Lee, Y.-
C. Liu, S. S. Badsara, Chem. Asian J. 2014, 9, 706–722; g) J.
Bariwal, E. Van der Eycken, Chem. Soc. Rev. 2013, 42, 9283–
9303.
[9] For examples of electrophilic iodination reactions, see: a) F.
Romanov-Michailidis, L. Guénée, A. Alexakis, Org. Lett. 2013,
15, 5890–5893; b) S. Stavber, M. Jereb, M. Zupan, Synthesis
2008
, 1487–1513; c) J. Barluenga, J. M. González, M. A.
García-Martín, P. J. Campos, G. Asensio, J. Org. Chem. 1993,
58, 2058–2060; d) Y.-L. Ren, H. Shang, J. Wang, X. Tian, S.
Zhao, Q. Wang, F. Li, Adv. Synth. Catal. 2013, 355, 3437–3442;
e) X. Zhang, C. Fu, Y. Yu, S. Ma, Chem. Eur. J. 2012, 18,
13501–13509; f) E. Cleator, J. P. Scott, P. Avalle, M. M. Bio,
S. E. Brewer, A. J. Davies, A. D. Gibb, F. J. Sheen, G. W. Ste-
wart, D. J. Wallace, R. D. Wilson, Org. Process Res. Dev. 2013,
17, 1561–1567.
[10] S. Stavber, P. Kralj, M. Zupan, Synthesis 2002, 11, 1513–1518.
[11] For examples of deprotonative metalation, see: a) S. H. Wund-
erlich, P. Knochel, Angew. Chem. Int. Ed. 2007, 46, 7685–7688;
Angew. Chem. 2007, 119, 7829–7832; b) J.-M. L’Helgoual’ch,
G. Bentabed-Ababsa, F. Chevallier, M. Yonehara, M. Uchi-
yama, A. Derdour, F. Mongin, Chem. Commun. 2008, 5375–
5377; c) E. F. Flegeau, M. E. Popkin, M. F. Greaney, Org. Lett.
2008
, 10, 2717–2720; d) J.-M. L
⬘Helgoual⬘ch, A. Seggio, F.
Chevallier, M. Yonehara, E. Jeanneau, M. Uchiyama, F. Mon-
gin, J. Org. Chem. 2008, 73, 177–183; e) B. M. Partridge, J. F.
Hartwig, Org. Lett. 2013, 15, 140–143; f) F. Chevallier, Y. S.
Halauko, C. Pecceu, I. F. Nassar, T. U. Dam, T. Roisnel, V. E.
Matulis, O. A. Ivashkevich, F. Mongin, Org. Biomol. Chem.
2011
, 9, 4671–4684; g) G. Dayker, A. Sreeshailam, F. Chevall-
ier, T. Roisnel, P. Radha Krishna, F. Mongin, Chem. Commun.
2010
, 46, 2862–2864; h) P. J. Hardford, A. J. Peel, F. Chevallier,
www.eurjoc.org
© 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Eur. J. Org. Chem. 2015, 77–80
80
R. Takita, F. Mongin, M. Uchiyama, A. E. H. Wheatley, Dal-
ton Trans. 2014, 43, 14181–14203.
[12] A. Klapars, S. L. Buchwald, J. Am. Chem. Soc. 2002, 124,
14844–14845.
[13] a) R. Kumar, A. Kumar, S. Jain, D. Kaushik, Eur. J. Med.
Chem. 2011, 46, 3543–3550; b) P. Vachal, L. M. Toth, Tetrahe-
dron Lett. 2004, 45, 7157–7161.
[14] a) L.-H. Zou, Z.-B. Dong, C. Bolm, Synlett 2012, 23, 1613–
1616; b) L.-H. Zou, J. Reball, J. Mottweiler, C. Bolm, Chem.
Commun. 2012, 48, 11307–11309; c) L.-H. Zou, J. Mottweiler,
D. L. Priebbenow, J. Wang, J. A. Stubenrauch, C. Bolm, Chem.
Eur. J. 2013, 19, 3302–3305.
[15] For a recent report on Sonogashira-type cross-coupling reac-
tions starting from 2-bromo-5-aryl-1,3,4-oxadiazoles, see: N.
Salvanna, B. Das, Synlett 2014, 25, 2033–2035.
[16] For another oxidative iodination, see: K. V. V. Krishna Mohan,
N. Narender, S. J. Kulkarni, Tetrahedron Lett. 2004, 45, 8015–
8018.
[17] During the preparation of this manuscript, Jiao and co-workers
reported the oxidative halogenation of indole derivatives by
using Selectfluor, KI, and NaHCO
3
to give 3-iodinated prod-
ucts in good yields. L. Shi, D. Zhang, R. Lin, C. Zhang, X. Li,
N. Jiao, Tetrahedron Lett. 2014, 55, 2243–2245.
[18] For examples of the use of Selectfluor and NFSI as oxidants,
see: a) C. Ye, M. J. Shreeve, J. Org. Chem. 2004, 69, 8561–8563;
b) S. Stavber, Molecules 2011, 16, 6432–6464; c) K. M. Engle,
T.-S. Mei, X. Wang, J.-Q. Yu, Angew. Chem. Int. Ed. 2011, 50,
1478–1491; Angew. Chem. 2011, 123, 1514–1528; d) K. K.
Laali, G. C. Nandi, S. D. Bunge, Tetrahedron Lett. 2014, 55,
2401–2405; e) G. Zhang, Y. Peng, L. Cui, L. Zhang, Angew.
Chem. Int. Ed. 2009, 48, 3112–3115; Angew. Chem. 2009, 121,
3158–3161; f) A. M. Jadhav, S. A. Gawede, D. Vasu, R. B.
Dateer, R.-S. Liu, Chem. Eur. J. 2014, 20, 1813–1817; g) D. V.
Liskin, P. A. Sibbald, C. F. Rosewall, F. E. Michael, J. Org.
Chem. 2010, 75, 6294–6296.
[19] For more details on the optimization of the reaction condi-
tions, see the Supporting Information.
[20] Also, in an attempted electrophilic iodination with N-iodosuc-
cinimide (1.5 equiv.) and trifluoroacetic acid (1.5 equiv.) in
acetonitrile (3 mL) at 50 °C for 5 h, the formation of 2a was
not observed.
[21] For a recent review on the chemistry of 1,3,4-thiadiazole, see:
Y. Hu, C.-Y. Li, X.-M. Wang, Y.-H. Yang, H.-L. Zhu, Chem.
Rev. 2014, 114, 5572–5610.
[22] Performing the reaction starting from 1a in an open flask
(MeCN, reflux) led to a reduction in the yield (35 % of 2a).
[23] For a similar observation in the electrophilic fluorination of
2,4-diarylthiazoles with Selectfluor, see: T. F. Campbell, C. E.
Stephens, J. Fluorine Chem. 2006, 127, 1591–1594.
[24] For standard bromination and chlorination methods of 1,3,4-
oxadiazoles, see: a) M. Golfier, R. Milcent, J. Heterocycl.
Chem. 1973, 10, 989–991; b) E. V. Zarudnitskii, I. I. Pervak,
A. S. Merkulov, A. A. Yurchenko, A. A. Tolmachev, Tetrahe-
dron 2008, 64, 10431–1042; c) K. P. Harish, K. N. Mohana, L.
Mallesha, B. N. Prasanna Kumar, Eur. J. Med. Chem. 2013, 65,
276–283.
Received: October 15, 2014
Published Online: November 6, 2014
Wyszukiwarka
Podobne podstrony:
Hua et al 2009 European Journal of Organic Chemistry
Siebner et al 2001 European Journal of Neuroscience
Lebrini et al 2005 Journal of Heterocyclic Chemistry
Grosser et al A social network analysis of positive and negative gossip
Vandeventer et al 2011 Mechanical disruption of lysis resistant bacterial cells by use of a miniatur
Wartość energetyczna SRWC Stolarski et al 2015
Beconyte, (2014) G , A Eismontaite & J Zemaitiene, Mythical creatures of Europe, Journal of Maps 10
Lester et al 2012 Comparative analysis of strawberry total phenolics via Fast Blue BB vs Folin–Cio
an advanced laboratory manual of organic chemistry Michael Heidelberger
Huang et al 2009 Journal of Polymer Science Part A Polymer Chemistry
Li et al 2010 Chemistry A European Journal
Ionic liquids solvent propert Journal of Physical Organic Che
new media and the permanent crisis of aura j d bolter et al
więcej podobnych podstron