1
Inżynieria genetyczna
Wykład 7, 30.03.2012
Part 1
Ciąg dalszy analizy metod sekwencjonowania.
Ponieważ zaczęto sekwencjonować genom człowieka zaszła potrzeba opracowania zupełnie
nowej metody, nie opartej już na sekwencjonowaniu Sangera. Metoda Sangera wymagała
bardzo dużo czasu i była kosztowna. Nowe technologie są znaczeni szybsze; są tak wydajne,
że przez noc można poznać sekwencje genomu jednego człowieka. Wcześniej to były lata.
PIROSEKWENCJONOWANIE (ang. pyrosequencing)
- metoda w ogóle nie wymaga elektroforezy ( nie analizujemy wielkości fragmentów
powstałych podczas sekwencjonowania)
- procedura jest szybsza niż w Sangerze
- otrzymuje się bardzo krótkie sekwencje (150bp) – takie fragmenty daje jeden
akt sekwencjonowania (co w pewnym sensie jest wadą metody)
- zaleta: można równolegle sekwencjonowac wiele fragmentów składanych w całośc
na końcu
- zautomatyzowana
- technologia w którym otrzymujemy wprost sygnał, jaka zasada jest
wbudowywana.
ISTOTĄ TEJ TECHNOLOGII JEST SYNTEZA, WBUDOWANIE RESZTY
Metoda w zarysie:
Podobnie jak metoda Sangera pirosekwencjonowanie wymaga DNA w postaci pojedynczej
nici. Ta nic jest otrzymywana jako nic powstająca z produktów PCR. Potem po otrzymaniu
pojedynczej nici przyłączany jest starter i polimeraza DNA prowadzi syntezę. Niedodawane
są dideoksyanalogi tylko normalne nukleotydy. Po włączeniu każdej z reszt emitowany jest
sygnał świetlny, rejestrowany przez urządzenie, które służy do analizy.
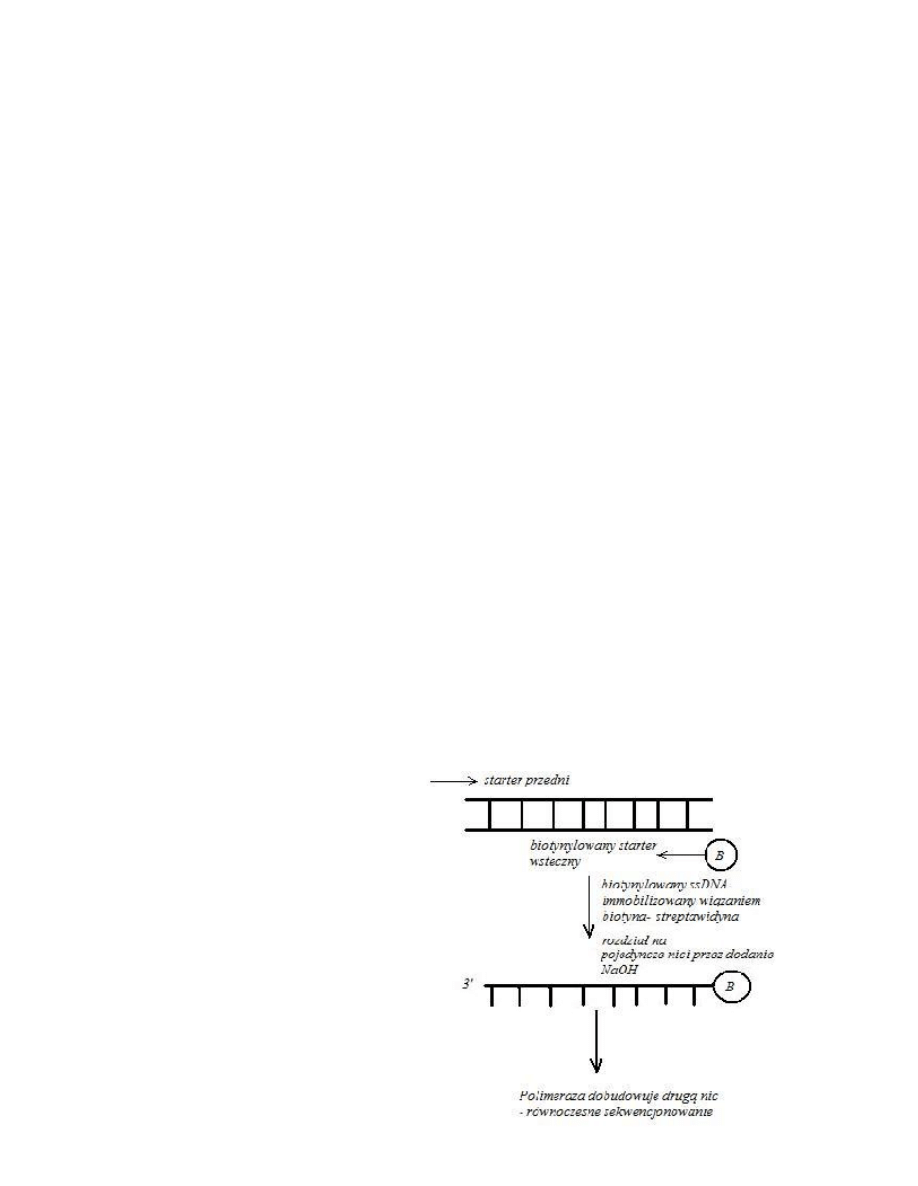
Szczegóły:
Na górze schematu jakaś sekwencja
DNA, którą chcemy poznać.
Oczywiście aby ją poznać musimy
znać sekwencje okalające, ale nie jest
to zawsze potrzebne. Przy pomocy
starterów jest amplifikowana
pojedyncza nic DNA. Jeden ze
starterów jest BIOTYNYLOWANY.
Po amplifikacji PCR biotyna służy do
immobilizowania produktu na nośniku
stałym, po czym w warunkach
alkalicznych (b. wysokiego pH;
działamy NaOH) dochodzi do

2
denaturacji dsDNA, jedna nic (niebiotynylowana) jest odrywana i otrzymujemy pojednynczą
nić DNA związaną ze stałym nośnikiem (biotynylowaną), która służy za matrycę do
sekwencjonowania.
Tu dla przypomnienia schemat syntezy DNA.
(DNA)
n
+dNTP→polimeraza→(DNA)
n+1
+PPi
I tu tkwi klucz do pirosekwencjonowania, ponieważ podczas wbudowywania nukleotydu
uwalnia się PPi (pirofosforan). Nazwa pirosekwencjonowanie nawiązuje do pirofosforanu
właśnie.
Oprócz polimerazy w mieszaninie rekacyjnej są jeszcze inne enzymy. Pierwszym z nich jest
sulfurylaza.
SULFURYLAZA (ang. sulfurylase)
- katalizuje reakcję APS z PPi, powstaje ATP.
LUCYFERAZA (ang. luciferase)
Lucyferaza+ATP -> oksylucyferyna + światło
Każde przekształcenie lucyferyny w oksylucyferynę powoduje emisję światła. Tą drogą
każde wbudowanie nukleotydu skutkuje emisją światła.
Skąd wiadomo jaki nukleotyd został wbudowany?
Najpierw dodajemy ATP. Jeśli jest komplementarne – mamy sygnał światła. Jeśli nie –
sygnału brak. Wtedy usuwany nadmiar ATP i dodajemy następny. Żeby mieć pewność, że
substratu nie ma w układzie jest jeszcze jeden enzym.
APYRAZA (ang. apyrase)
- rozkłada dNTP
dNTP→ dNDP + dNMP + fosforan
np. ATP→ADP+AMP+fosforan
skutkuje brakiem substratu w układzie.
dNTP w nadmiarze → wbudowanie przez polimerazę (jeśli jest w matrycy) → sygnał
świetlny →działanie apyrazy (usuniecie nadmiaru substratu) →wymywanie pozostałości.
Uwaga: te świecące też są wymywane!
Jeśli sygnał jest wysoki to
znaczy, że reszty takie
same następują po sobie w
sekwencji (przedstawione
na schemacie).
SYGNAŁ JEST
PROPORCJONALNY
DO ILOŚCI RESZT.

3
W układzie cztery enzymy: polimeraza, sulfurylaza, lucyferaza i apyraza. Enzymy są
cały czas w niewielkich stężeniach, kluczowe dla reakcji jest dodanie nadmiaru
substratu.
(tu puszczony był film pokazujący urządzenie służące do pirosekwencjonowania)
MASSIVELY PARALLEL DNA SEQUENCING
Prowadzący sam nie wiedział jak to przetłumaczyć Ale ogólnie chodziło o metodę służącą
do sekwencjonowania dużych ilości DNA, takich na miarę całego genomu.
Wykorzystuje reakcje pirosekwencjonowania, ale pozwala sekwencjonować wiele
próbek składających się na genom. Głównie do badan, proste urządzenie jak z filmu tylko
do weryfikacji.
Metody nazywane są różnie, ale dwie nazwy powinniśmy poznać: massively parallel DNA
sequencing, czyli po polsku masowe i równoległe sekwencjonowanie. Sekwencjonujemy
równocześnie wiele próbek w jednym eksperymencie, otrzymujemy informację o całym
genomie. Druga nazwa to metody nowej generacji, gdzie stara generacje to metody
kapilarne.
Ogólnie:
Punktem wyjścia jest genomowe DNA, które poddawane jest fragmentacji (dł ok. 300-
500bp). Każdy z fragmentów poddawany jest ligacji do cząsteczek adaptorowych.
Sekwencje adaptorowe pełnią dwie role:
Jedna z nich posiada przyłączoną biotynę (immobilizacja). Oba adaptery są
sekwencjami, które znamy (potencjalnie pod PCR), otrzymanymi na drodze syntezy
chemicznej.
Biotyna służy do immobilizowania fragmentów na stałym podłożu, na którym jest
streptawidyna. Oddziaływanie streptawidyna-biotyna jest jednym z najsilniejszych do tej
pory opisanych, prawie kowalencyjne. Jest to istotne, bo usuwanie nici
komplementarnej uzyskuje się bardzo wysokim pH, ale wiązanie jest tak trwałe, że
zostaje zachowane, chociaż nie jest kowalencyjne.
W poprzedniej metodzie do jednej „kulki” nośnika było wiązanych kilka fragmentów.
Tutaj dobiera się ilości tak, by do jednej „kulki” przywiązany był tylko jeden fragment
DNA.
Potem każdy z fragmentów poddawany jest amplifikacji poprzez PCR (chodzi o zwiększenie
siły sygnału, by przekroczył wartość progową dla aparatu odczytującego)
Na tym etapie mamy kolekcję (bibliotekę) różnych fragmentów przyłączonych do kulek, a na
końcach sekwencje są identyczne, przez sekwencje adaptorowe. Teraz jak poddać to reakcji
PCR, żeby na końcu otrzymać produkty, które są rozdzielone?
Uzyskuje się taką separację wykorzystując emulsję wody w oleju. Czyli mamy kropelki
wody, które stanowią emulsję w oleju. Emulsja jest przygotowywana w ten sposób, że
każda z tych kuleczek z przywiązanym DNA, znajdzie się w pojedynczej kropli emulsji.
Pozbywamy się problemu mieszania się produktów PCR. W każdej kropli emulsji
przeprowadzana jest reakcja PCR i dostajemy zwielokrotnioną liczbę cząsteczek. Każda z
powstających cząsteczek zaopatrzona jest w biotynę, więc na pojedynczej kulce są też
4
immobilizowane i powstaje sytuacja jak wcześniej: dużo takich samych fragmentów
immobilizowanych na jednej kulce.
Każda z kulek jest następnie umieszczana w dołku płytki i potem następuje już standardowe
pirosekwencjonowanie, tylko na nieco większą skalę.
Zauważmy, że na końcu takiego postępowania mamy w każdym dołku fragment
genomu, który poddawany jest sekwencjonowaniu i na koniec te fragmenty muszą
zostać złożone w całość.
Part 2
Jak można sekwencjonować genom?
Jaką strategię przyjąć, aby pozyskać informacje składające się na genom?
Są dwie strategie, pozwalające na poznanie całej sekwencji.
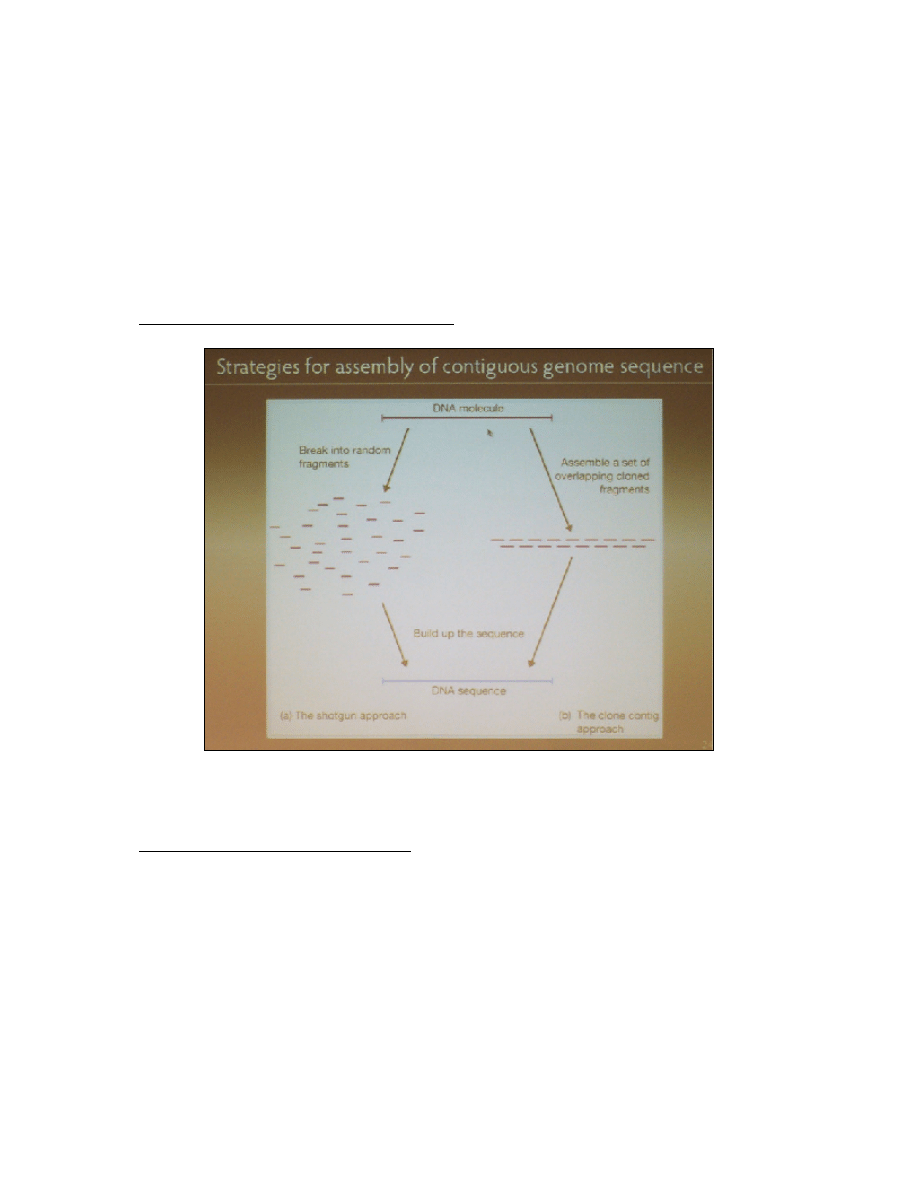
Pierwsza strategia – shotgun approach:
Wychodzimy od genomu DNA. Fragment DNA poddawany jest przypadkowej fragmentacji
na wiele różnych odcinków. Poznawana jest ich sekwencja. Następnie informacje są
składane. Metoda prosta i szybka, ale są różne problemy. Jest najszerzej stosowana,
zwłaszcza dla krótkich genomów.

5
Druga metoda – clone conting approach
Metoda, w której buduje się ciągłe sekwencje, systematycznie postępując i składa się je w
całość. W tej metodzie mamy dwie fazy. W pierwszej fazie przeprowadza się
presekwencjonowanie, czyli poznaje się fragmenty sekwencji dłuższych i informacja o tych
krótkich fragmentach sekwencji dłuższych pozwala na połączenie ze sobą dłuższych i
jeszcze dłuższych. Mamy olbrzymi zbiór różnych sekwencji i próbkujemy zbiór tych różnych
długich sekwencji. Gdy widzimy dwie mające wspólny fragment, tzn. że powinny one
zachodzić na siebie, powinny dawać jakąś większą całość. Patrzymy, czy jest jakaś sekwencja
reprezentowana w jeszcze jakimś innym klonie, mamy trzy, które dają jakąś większą całość i
tak budujemy mozolnie, aż powstanie całość sekwencji. Jeżeli utworzymy mapę, która daje
długą sekwencję, mapę zachodzących fragmentów, które dają długą sekwencję, to
zaczynamy poznawać sekwencje każdego z tych fragmentów wcześniej zidentyfikowanych
jako zachodzące na siebie i składamy z tego całość.
Przykład
Użycie strategii shotgun do poznania prostego genomu.
W pierwszym kroku wyizolowano DNA i poddano go fragmentacji. Użyto ultradźwięków
(zamiast enzymów restrykcyjnych). Ultradźwiękami można też DNA fragmentować.
Otrzymano fragmenty. Otrzymaną mieszaninę naniesiono na żel agarozowy, rozdzielono te
fragmenty i wycięto ten fragment żelu, który odpowiadał fragmentom DNA o dł. 1.6 – 2.0 kb.
Te fragmenty były o przypadkowej sekwencji. One nie pochodziły z jednej komórki, z jednej
cząst. genomu, tylko z wielu. Pewne sekwencje w tych fragmentach musiały się powtarzać,
zachodziły na siebie. Te fragmenty, bibliotekę, sklonowano do odpowiedniego wektora i
otrzymano kolekcję klonów. Ponad 19 000 klonów zostało poddanych sekwencjonowaniu.
Otrzymano sekwencje tych klonów, z czego odrzucono ponad 4 000, bo były za krótkie.
Pozostałe sekwencje poddano analizie komputerowej. Analiza ~30h. Poszukiwano
fragmentów zawierających wspólne sekwencje. Otrzymano 140 fragmentów sekwencji, które
były ciągłe, ale które jeszcze nie składały się na cały genom.
Aby zamknąć luki w sekwencji posłużono się eksperymentem hybrydyzacji. W jaki sposób?

6
Otrzymano 140 contingów – odcinków sekwencji ciągłych. Trzy są pokazane. Sekwencje
znane. Na podstawie tych znanych sekwencji otrzymano krótkie oligonukleotydy. Wybrano
np. oligonukleotyd 2 i przeprowadzono hybrydyzację i znaleziono klon, który hybrydował z
olignolukleotydem 2. Następnie wzięto 3, 4, 5 i 6 i też przeprowadzono hybrydyzację i kiedy
przeprowadzono ją przy pomocy 5, okazało się, że ten sam klon również hybrydował z 5. To
znaczy, że w klonie, który znajdował się w tym miejscu biblioteki musiała być sekwencja
zarówno contingu pierwszego, jak i trzeciego. Na podstawie tych informacji, na podstawie
pierwszego syntezowana była sonda druga, na podstawie trzeciego syntezowana była sonda
piąta. Skoro one hybrydyzują z tym samym klonem, to znaczy, że zarówno jedna, jak i druga
informacja są w tym klonie. Conting I i III są reprezentowane w tym samym klonie, a więc jeśli
ten klon się zsekwencjonuje, to brakująca informacja pomiędzy I i III zostanie zamknięta,
uzupełniona. W ten sposób znaleziono klon, w którym zawarta była brakująca informacja.
Przerwa, a w jej trakcie prognoza pogody na nadchodzący weekend :D
(zapowiadali śnieg, ale wcale nie padał :P)

7
Sekwencjonowanie typu shotgun sprawdza się bardzo dobrze w przypadku niewielkich
genomów, genomów bakteryjnych. Nie tylko z tego powodu, że one są małe i liczba klonów,
liczba informacji, które trzeba ze sobą porównać nie jest tak duża jak w przypadku genomów
eukariotycznych. Dlatego też, że w genomach eukariotycznych są sekwencje nazywane
rozproszonymi, powtarzającymi się. To są sekwencje, które często są praktycznie identyczne.
Popatrzmy na schemat.
Mamy tu pokazany fragment genomu. Są sekwencje rozproszone, powtarzające się
(identyczne), te sekwencje mogą być obecne w sekwencji 1 i 2, które zostały wygenerowane
w technice shotgun. Odnajdujemy jakiś fragment sekwencji 1 i 2 i porównujemy je ze sobą.
Porównanie pokazuje, że w jednym i drugim fragmencie są zawarte sekwencje, które są
identyczne. Skoro są identyczne, to muszą pochodzić z tego samego fragmentu. Zatem
składamy je. I wymyślona, wydedukowana sekwencja byłaby tego rodzaju. To znaczy, że ten
cały fragment jest gubiony w przypadku uproszczonej analizy. Tych sekwencji identycznych,
rozproszonych jest w genomie ludzkim bardzo dużo. Stanowią kilkadziesiąt procent całej
sekwencji genomu. Więc metoda shotgun jest bardzo trudna do zastosowania.
Teraz druga z technik – conting, gdzie nadsekwencjonujemy jakieś fragmenty DNA,
zdobywamy informacje o nich, szukamy podobnych zestawów sekwencji i składamy je w
całość. W odróżnieniu od metody shotgun, ta metoda wymaga znacznie większego nakładu
czasu. Krok po kroku jest składana sekwencja w całość. Na początku sekwencjonowania
otrzymujemy bibliotekę, w której dąży się do tego, by było jak najmniej klonów. To znaczy,
żeby klony zawierały jak największe zespoły informacji i te klony potem są poddawane
analizie i składane w całość.
Jedną z technik, która jest stosowana do poznawania sekwencji genomu w podejściu
contingu jest metoda chromosome walking, czyli chodzenia wzdłuż chromosomu.

8
Mamy schematycznie pokazaną bibliotekę, która zawiera bardzo wiele klonów. W technice
chromosome walking wybiera się przypadkowo jakiś klon w bibliotece. Tu: wybrano A1. Na
podstawie informacji tego klonu przygotowywana jest sonda znakowana radioaktywnie.
Tą sondą poddaje się hybrydyzacji całą bibliotekę, w nadziei, że odnajdziemy inne klony,
zawierające informację komplementarną. Sondą A1 hybrydyzowano bibliotekę i otrzymano
pozytywne sygnały. Dla klonu A1, bo z niego pochodziła sonda, ale i dla B4 oraz I4. To znaczy,
że informacja z A1 musi też być zawarta w klonach B4 i I4. W ten sposób po jednym
eksperymencie otrzymano sekwencję ciągłą, czyli conting, obejmującą klony A1, B4, I4. Żeby
to zrobić nie trzeba było znać pełnej sekwencji. Wiemy, że w tych trzech klonach jest zawarta
pewna ciągła informacja. Jeżeli chcemy ją poznać w szczegółach, trzeba te klony
zsekwencjonować i złożyć w całość. Następnie wykonano kolejną hybrydyzację przy pomocy
sondy I4. Otrzymano kolejną informację. I4 hybrydyzował z A1, I4, F2. W ten sposób
wykonując kolejne hybrydyzacje, kolejne sekwencjonowania, można poznać całą
sekwencję jakiegoś wybranego genomu.
Aby przyspieszyć tę metodę, zaproponowano, żeby zamiast postępować systematycznie i
szukać klonów przez hybrydyzację, można szukać par klonów, które zawierają sekwencje
zachodzące na siebie. W jaki sposób? Poprzez fingerprinting.

9
Fingerprinting jest to odcisk palca. Poprzez poszukiwanie par, które zawierają
charakterystyczne cechy wspólne. Jednym z pomysłów było to, żeby szukać klonów, które
mają podobne produkty trawienia endonukleazą. Bierzemy dwa klony, trawimy je np. EcoRI
i jeżeli w obu klonach jest taki fragment, który jest wspólny, to po trawieniu EcoRI
powinien dać też wspólne produkty, identyczne produkty.
Alternatywnym pomysłem jest PCR, którego startery skierowane są do sekwencji
powtarzających się, rozproszonych.
W genomach eukariotycznych, w szczególności w genomie człowieka te sekwencje są bardzo
powszechne. Jeżeli użyjemy starterów do PCR, to możemy takie sekwencje amplifikować.
Sekwencje, do których startery są kierowane, są znane. Te sekwencje mają określone
lokalizacje. Czyli jeżeli wybierzemy jakąś specyficzną sekwencję rozproszoną, to długość
produktu dla tej sekwencji, która powstanie po PCR, będzie bardzo charakterystyczna. Jeżeli
wybierzemy wiele klonów do analizy i wykonamy analizę PCR, to pojawienie się
charakterystycznego produktu w jednym i drugim klonie, który analizujemy, mówi nam o
tym, że klon jeden i drugi zawiera fragment takiej samej sekwencji.
Przykładem takiej analizy jest IRE-PCR (od Interspersed repeat element). Startery są
kierowane do jakiejś wybranej sekwencji i następnie wykonujemy PCR.

10
Wybrano trzy klony przypadkowe i przy pomocy tych samych starterów wykonano PCR. Co
się okazało? W klonie pierwszym otrzymano dwa produkty o określonej długości, w klonie
drugim też dwa produkty o różnej długości, w klonie trzecim - trzy produkty o różnej długości.
Elementy pojawiają się w wielu miejscach w genomie i w różnych odległościach. Sekwencje
są identyczne, ale rozproszone przypadkowo, zatem produkty mogą być różne, ale okazało
się, że w tych dwóch klonach otrzymano produkt identyczny. One są klonami, które na
siebie zachodzą. Ich sekwencja jest po części wspólna. W ten sposób bez hybrydyzacji
możemy szybko odnajdywać cechy wspólne.
Są też inne metody, np. metoda, w której wykorzystuje się tzw. STS. Co to jest STS? Jest to
sequence tagged site, czyli miejsce znakowane przez sekwencje. Wyobraźmy sobie, że
poznaliśmy wcześniej bardzo dokładnie jakąś sekwencję. I ta sekwencja określana jest jako
STS. Czyli dysponujemy jakąś bardzo precyzyjną informacją o sekwencji. To może być gen,
może być fragment genu, sekwencja międzygenowa, ale zawsze musi to być sekwencja,
która jest już znana. Do takiej sekwencji możemy skonstruować startery i wykonać PCR.
Popatrzmy na schemat.

11
Mamy kolekcję klonów. Mamy też wiedzę na temat STS. Dla tej kolekcji wykonujemy PCR,
posługując się starterami kierowanymi do STS, czyli do tej znanej sekwencji. Po wykonaniu
PCR otrzymujemy informację, że specyficzny produkt pojawił się dla klonu IV i II. Jeżeli STS
było sekwencją wyjątkową, to mamy dużą pewność, że klon II i IV są klonami, które zawierają
wspólną sekwencję, a dokładnie sekwencję, która jest sekwencją STS. Ta metoda jest bardzo
efektywna.
Analiza metod, które służą badaniu ekspresji i funkcji genu
Metody pozwalające na analizę transkryptu
Pierwszą z metod jest hybrydyzacja Northern. Eksperyment składa się z dwóch etapów.
Pierwszy to elektroforeza, w której rozdziałowi podlega RNA, następnie RNA jest
przenoszony na błonę – może być nitrocelulozowa, błona nylonowa i w kolejnym etapie po
transferze zachodzi hybrydyzacja przy pomocy sondy specyficznej. W ten sposób
wykrywamy cząsteczki RNA, które są komplementarne do sondy.
Hybrydyzacja jest konieczna. Po wykonaniu elektroforezy praktycznie w każdej grupce, która
jest rozdzielana, mamy taki sam obraz. Dlatego, że RNA w komórce jest dużo. To są różne
RNA. Hybrydyzacja służy analizie obecności RNA w danym preparacie. W ten sposób można
dużo powiedzieć na temat ekspresji genów, czyli na temat istnienia lub braku transkryptu.
Koniec :-)
Wyszukiwarka
Podobne podstrony:
więcej podobnych podstron