
PRAWIDŁOWY KARIOTYP
CZŁOWIEKA I ANOMALIE
AUTOSOMÓW

PRAWIDŁOWY KARIOTYP CZŁOWIEKA
Kryteria klasyfikacji chromosomów:
• Wielkość chromosomów wyrażona w procentach w
odniesieniu do długości wszystkich chromosomów
haploidalnych i chromosomu X przyjętej jako 100%
• Położenie centromeru
• Rozmieszczenie prążków w chromosomach

• Kariotyp-suma chromosomów występujących w
komórce somatycznej, właściwa organizmowi lub
grupie organizmów o charakterystycznej liczbie i
morfologii
• Kariogram- sfotografowany zestaw chromosomów
typowych dla danego organizmu, grupy
organizmów lub gatunku, uszeregowany wg
umownych zasad.

Chromosomy, najważniejsze składniki jąder
komórek roślinnych i zwierzęcych będące
siedliskiem czynników dziedzicznych, czyli
genów. Chromosomy zbudowane są głównie
z silnie barwiącej się chromatyny, w której
skład wchodzą długie cząsteczki kwasu
dezoksyrybonukleinowego (DNA, kwasy
nukleinowe) oraz białka (głównie histonowe)
i kwas rybonukleinowy (RNA).

Chromosomy mają
zdolność
do
samoodtwarzania się
w
trakcie podziałów
komórki. Widoczne są po wybarwieniu tylko
w czasie podziałów komórki (mitoza, mejoza),
kiedy ulegają silnej kondensacji, w okresach
między podziałami (interfaza) ulegają silnej
despiralizacji
i
stają
się
niewidoczne.

Liczba chromosomów danego gatunku (określana jako
podstawowa liczba chromosomów = 2n) jest stała
i charakterystyczna i może wynosić od 2 do kilkuset (u
niektórych roślin), najczęściej od 10 do 40 (u człowieka
46). Każdemu organizmowi odpowiada zespół
chromosomów o określonej liczbie (jednakowej we
wszystkich jądrach komórkowych) i różniących się między
sobą morfologią oraz składem występujących w nich
genów.Liczba chromosomów, ich wielkość i kształt są dla
danego gatunku stałe i charakterystyczne. Tak więc np.:
u człowieka występuje 23 pary chromosomów z których 22
pary to chromosomy homologiczne, jednakowe
(autosomy), oraz 1 para chromosomów płciowych,
heterochromosomów, różniących się od siebie (allosomy).

W komórkach rozrodczych (gamety)
w wyniku mejozy liczba chromosomów
zredukowana jest do połowy, po
połączeniu się dwóch gamet odtwarzana
jest dzięki temu liczba chromosomów
charakterystyczna dla gatunku.
Chromosomy organizmów
prokariotycznych, do których należą
bakterie, sinice oraz wirusy, są pod
względem strukturalnym mało
skomplikowane w
porównaniu
z
chromosomami organizmów
eukariotycznych.

METODY BADAŃ CHROMOSOMÓW
W celu określenia liczby
chromosomów bada się komórki:
Szpiku kostnego, gonad męskich,
fibroblasty z hodowli wycinków
skóry, limfocyty krwi obwodowej.
Najczęstszą
metoda badań
chromosomów jest krótkotrwała
hodowla limfocytów krwi
obwodowej

Obecnie w rutynowych badaniach
cytogenetycznych stosuje się metody
barwienia pozwalające stwierdzić na
chromosomach obecność prążków G,Q
lub R. Charakterystyczny dla każdego
chromosomu układ prążków umożliwia
identyfikację poszczególnych par oraz
analizę nieprawidłowości ich struktury.

BARWIENIE DIASTAMYCYNĄ A/DAPI
- barwienie chromosomów fluoorochromem 4,6-
dwuamino-2-fenyloindolem (DAPI) wykazującym
powinowactwo do par zasad A-T pozwala na uzyskanie
wzoru prążkowego podobnego do wzoru prążkowego Q.
Zastosowanie DAPI i antybiotyku diastamycyny A, która
wykazuje również powinowactwo do zasad A-T pozwala
uzyskać bardziej kontrastowy obraz prążkowy.
Metodą tą uzyskujemy silnie fluoryzujące prążki
odpowiadające regionom heterochromatyny
konstytutywnej chromosomów 1, 9, 16, dystalnej części
ramion długich chromosomu Y oraz proksymalnej części
ramion krótkich chromosomu 15.

AUTORADIOGRAFIA
Metoda badania struktury chromosomów
oraz badania kinetyki i asynchronii
replikacji DNA. Często wykorzystywana w
hybrydyzacji kwasów nukleinowych z
użyciem sond znakowaych takimi
izotopami jak:
3
H,
32
P,
35
S,
14
C.
Autoradiografia jest metodą stosowaną do
badania czasu trwania poszczególnych faz
cyklu komórkowego.

CYTOMETRIA PRZEPŁYWOWA
Wykorzystanie pomiaru ugięcia i rozproszenia światła oraz
wzbudzenia fluorescencji w zawiesinie komórkowej.
Można zmierzyć zawartość DNA w jądrze komórkowym.
Ocenia się intensywność fluorescencji w cytometrze
przepływowym. Na podstawie wyników odczytywanych
na monitorze mikrokomputera sporządza się wykresy
zawartości DNA w poszczególnych populacjach komórek
czyli histogramy.

HYBRYDYZACJA IN SITU (In situ
Hybrydyzation- ISH)
Pozwala zlokalizować specyficzne sekwencje DNA lub
RNA bezpośrednio w materiale biologicznym(preparat
cytogenetyczny, rozmazy komórkowe, skrawki tkanek)
znajdującym się na szkiełku podstawowym lub rzadziej
w zawiesinie.


ZAPISYWANIE WYNIKÓW BADAŃ
CYTOGENETYCZNYCH
• 46,XX
• 46,XY
• 46,XX,1qh+ ( prawidłowy kariotyp żeński z obecnością
większego odcinka heterochromatyny konstytutywnej (h+)
w okolicy centromerowej ramienia długiego q
chromosomu pary 1
• 46,XX,15ps+ (duże satelity (s+) na krótkich ramionach (p)
chromosomu pary 15.
Są to cechy polimorficzne chromosomów

Wykaz skrótów
+ (plus) - nadmiar , dodatek
-(minus) niedobór, ubytek
: (dwukropek) – pęknięcie
:: (podwójny dwukropek) – pęknięcie i połączenie
, (przecinek) - rozdziela liczbę chromosomów od chromosomów płci i
ewentualnego opisu aberracji
; (średnik) - rozdziela zapisy dotyczące różnych chromosomów
. (kropka) – oddziela zapis subprążków od cyfry określającej prążek
(strzałka – od do (określa zakres regionu chromosomu)
( ) nawiasy – obejmują strukturalnie zmienione chromosomy lub
miejsca pęknięć za skrótem określającym typ aberracji.
/ (ukośnik) – oddziela różne linie w mozaikach

WYKAZ SKRÓTÓW
• cen-centromer
• del-delecja
• der-chromosom pochodny
• dic-chromosom dicentryczny
• dup- duplikacja
• h-heterochromatyna konstytutywna
• i-izochromosom
• ins-insercja
• inv-inwersja
• kpz-kilo par zasad
• mar-chromosom markerowy

Wykaz skrótów
• mat-pochodzenia matczynego
• NOR –organizator jąderkowy
• p-krótkie ramię chromosomu
• pat – pochodzenia ojcowskie
• PAR – region pseudoautosomalny
• PCR-łańcuchowa reakcja polimerazy
• q- długie ramię chromosomu
• r-chromosom pierścieniowy
• s-satelity
• t-translokacja
• tel-telomer
• ter- koniec chromosomu (fragment terminalny)
• UPD- jednorodzicielska (uniparentalna) disomia
• UPHD – uniparentalna heterodisomia
• UPID – uniparentalna izodisomia

KOMÓRKI POLIPLOIDALNE
KOMÓRKI POLIPLOIDALNE POWSTAJĄ W
WYNIKU ZWIELOKROTNIENIA CAŁEGO
HAPLIDALNEJ GARNITURU CHROMOSOMÓW
• KOMÓRKA TRIPLOIDALNA ;
• 69,XXY
• Tetraploidalna
• 92,XXXX

KOMÓRKI ANEUPLOIDALNE
• Komórki aneuploidalne to takie, w których
brakuje jednego chromosomu lub jest obecny
dodatkowy jeden, dwa lub rzadziej więcej
chromosomów
• 45,X - monosomia X
• 47,XXX - polisomia X
• 47,XYY - polisomia Y
• 47,XY,+21 - trisomia 21

Translokacja robertsonowska (der)
• Powstaje w wyniku pęknięć w pobliżu cenromeru dwóch
chromosomów akrocentrycznych i połączenia się ich
ramion długich z jednoczesną utratą ramion krótkich w
wyniku czego powstaje jeden chromosom pochodny
zapisywany skrótem der
Translokacja zrównoważona
• 45,XX,der(13;21)(q10;q10)-zapis skrócony
• 45,XX,der(13;21)(13qter 13q10::21q10 21qter) –
zapis rozszerzony
• Pęknięcie w prążku q10 chromosomu pary 13 i 21.

Translokacja niezrównoważona
46,XY,der(21;21)(q10;q10),+21
do translokacji niezrównoważonej doszło między
chromosomami 21 pary w wyniku czego powstał
chromosom utworzony z ramion długich dwóch
chromosomów 21.

Translokacja wzajemna (t)
Wymiana odcinków chromosomów leżących dystalnie od pęknięć
w obu chromosomach.
Zjawisko dotyczy zarówno ramion długich, jak i krótkich
wszystkich autosomów i heterochromosmów
46,XX,t(2;5)(q21;q31) – zapis skrócony,
46,XX,t(2;5)(2pter (2p21::5q31
5qter;5pter 5q31::2q21 2qtr) - zapis rozszerzony
Kariotyp żeński z 46 chromosomami, w którym doszło do
wzajemnej zrównoważonej translokacji pomiędzy chromosomami
pary 2 i 5. Punkty złamań występują w prążku q21 chromosomu 2
i prążku q31 chromosomu 5. Podwójny dwukropek obrazuje
pęknięcie w jednym chromosomie i przyłączenie fragmentu z
drugiego chromosomu.

DELECJA (DEL)
Utrata fragmentu chromosomu
Wyróżniamy:
a)
Delecję terminalną
– gdy utracony został końcowy fragment ramion
długich lub krótkich.
46,XY,del(5)(q13)-zapis skrócony
46,XY,del(5)(pter q13)- zapis rozszerzony
-delecji uległ końcowy fragment ramion długich chromosomu pary 5
prążka q13, co obrazuje pojedynczy dwukropek
b) delecje interstycjalną
–gdy doszło do utraty środkowego fragmentu
chromosomu.
46,XX,del(5)(q13q33)-zapis skrócony
46,XX,del(5)(pter q13::q33 qter) – zapis rozszerzony
Utraty wewnętrznego fragmentu ramienia długiego chromosomu pary 5
pomiędzy prążkami q13 i q33

• DUPLIKACJA (dup)
Podwojenie określonego fragmentu chromosomu w sposób
bezpośredni lub z jego odwróceniem o o 180 ° (inwersja)
Duplikacja fragmentu chromosomu w sposób bezpośredni
46,XX,dup(1)(q22q25)- zapis skrócony
46,XX,dup(1)(pter q25::q22 qter)- zapis rozszerzony
Jest to podwojenie fragmentu chromosomu pary 1 zawartego między
prążkami q22 i q25
Duplikacja tego samego fragmentu chromosomu, ale z jego inwersją
(odwróceniem o 180 ° )
46,XX,dup(1)(q25q22)-zapis skrócony
46,XX,dup(1)(pter q25::q25 q22::q25 qter) -
Zapis rozszerzony

Inwersja (inv)
• Odwrócenie o 180 ° fragmentu chromosomu
• Inwersja paracentryczna – w obrębie ramion długich lub krótkich nie
obejmujący centromeru
• 46,XX,inv(3)(q21q26) – zapis skrócony
• 46,XX,inv(3)(ptr q21::q26 q21::q26 qter) - zapis
rozszerzony
• Inwersja fragmentu pomiędzy prążkami q21 i q26 w długim ramieniu
chromosomu 3
inwersja paricentryczna – odwrócenie fragmentu chromosomu obejmujące
centromer
46,XX,inv(3)(p13q21)- zapis skrócony
46,XX,inv(3)(pter p13::q21 p13::q21 qter) - zapis
rozszerzony
Inwersja fragmentu chromosomu pary 3 pomiędzy prążkami w ramieniu
krótkim p13 i q21 w ramieniu długim, a więc obejmującego centromer

• Insercja (ins)
Włączenie fragmentu chromosomu w jego ramię długie
lub krótkie
46,XX,ins(2)(p13q21q31) –zapis skrócony
46,XX,ins(2)(pter p13::q31 q21::p13
q21::q31 qter) - zapis rozszerzony
Insercja fragmentu ramion długich chromosomu pary 2
zawartego między prążkami q21 i q31 w ramię krótkie
tego samego chromosomu, w prążek p13

• Izochromosomy
• Powstają w wyniku nieprawidłowego, poprzecznego
podziału chromosomu w obrębie centromeru, czego
skutkiem jest duplikacja jednego a delecja drugiego
ramienia.
• Izochromosom składa się z dwóch identycznych krótkich
lub długich ramion.
• 46,X,i(X)(q10) - zapis skrócony
• 46,Xi(X)(qter q10::q10qter) – zapis rozszerzony
• 46,XX,i(17)(q10) – zapis skrócony
• 46,XX,i(17)(qter q10::q10 qter) – zapis rozszerzony
• Najczęściej spotyka się izochromosom ramion długich

• Chromosomy dicenrtyczne (dic)
• Powstają w wyniku translokacji, inwersji i innych aberacji
chromosomowych. Zawierają dwa centromery
45,XX,dic(13;15)(q22;q24) –zapis skrócony
45,XX,dic(13;15)(13pter 13q22::15q24
15pter) - zapis rozszerzony
• Chromosomy pierścieniowe (r)
• Powstają w wyniku pęknięcia i ponownego połączenia
złamanych końców jednego lub kilku chromosomów.
• Fragmenty dystalne od miejsc pęknięcia ulegają delecji
• 46,XX,r(7)(p22q36)-zapis skrócony
• 46,XX,r(7)(::p22 q36::) - zapis rozszerzony

• Chromosomy markerowe
Są to dodatkowe chromosomy , których pochodzenie i
mechanizm powstawania są nieznane. Najczęściej sa to
chromosomy meta lub akrocentryczne.
Zawierają jeden lub dwa centromery
47,XX, +mar
47,XX,t(12;16)(q13;p11), + mar
Kariotyp mozaikowy
To kariotyp w którym obecne są dwie lub więcej linie
komórkowe u tego samego osobnika Jedna zawiera
prawidłową liczbę prawidłowych chromosomów, drugi lub
kolejne nieprawidłową liczbą chromosomów.
45,X/46,XX/47,XXX
46,XY(70%)/47,XY,+21(30%)
46,X(40%)/46,XY(60%)
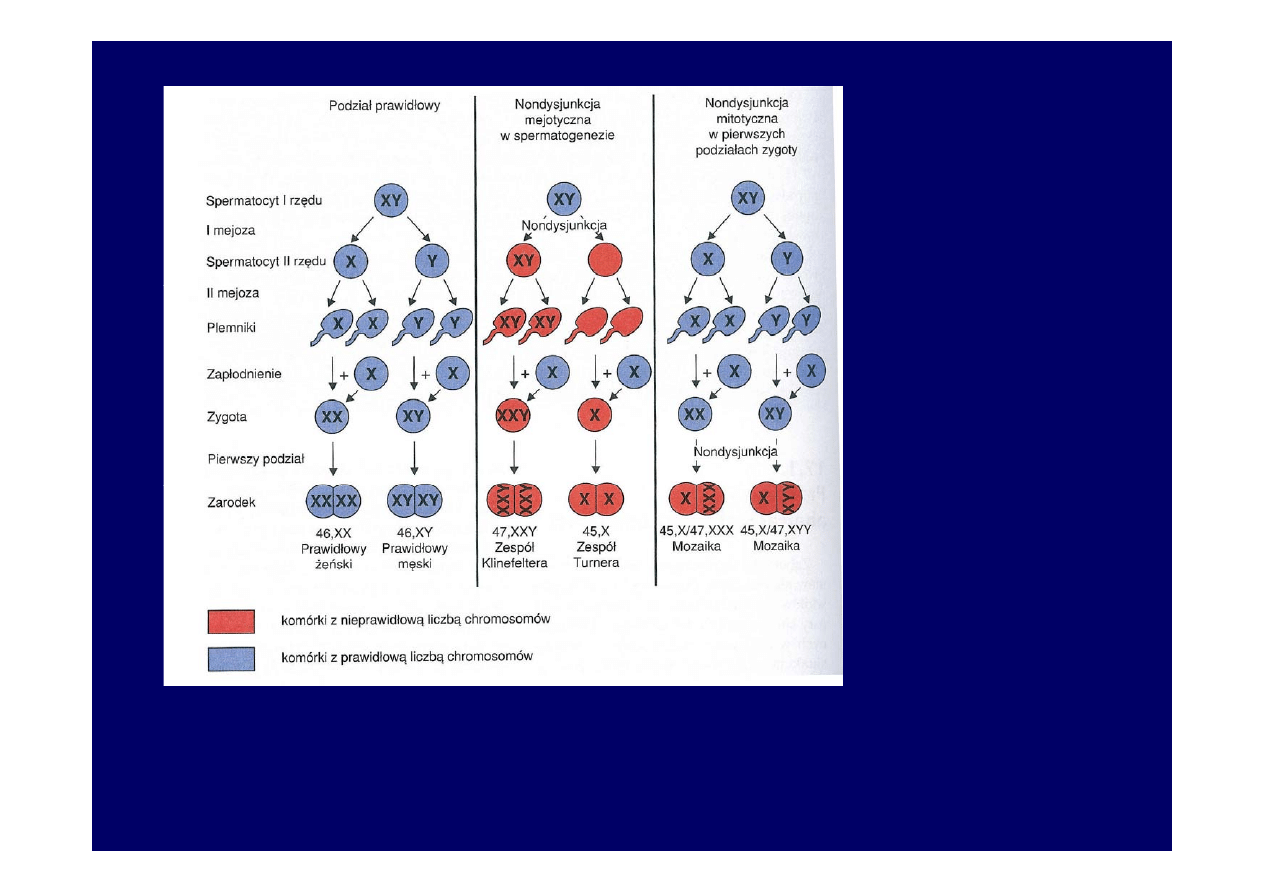
Nieprawidłowe
rozchodzenie się
chromosomów
płciowych w
mejozie podczas
pierwszych
podziałów
zygoty
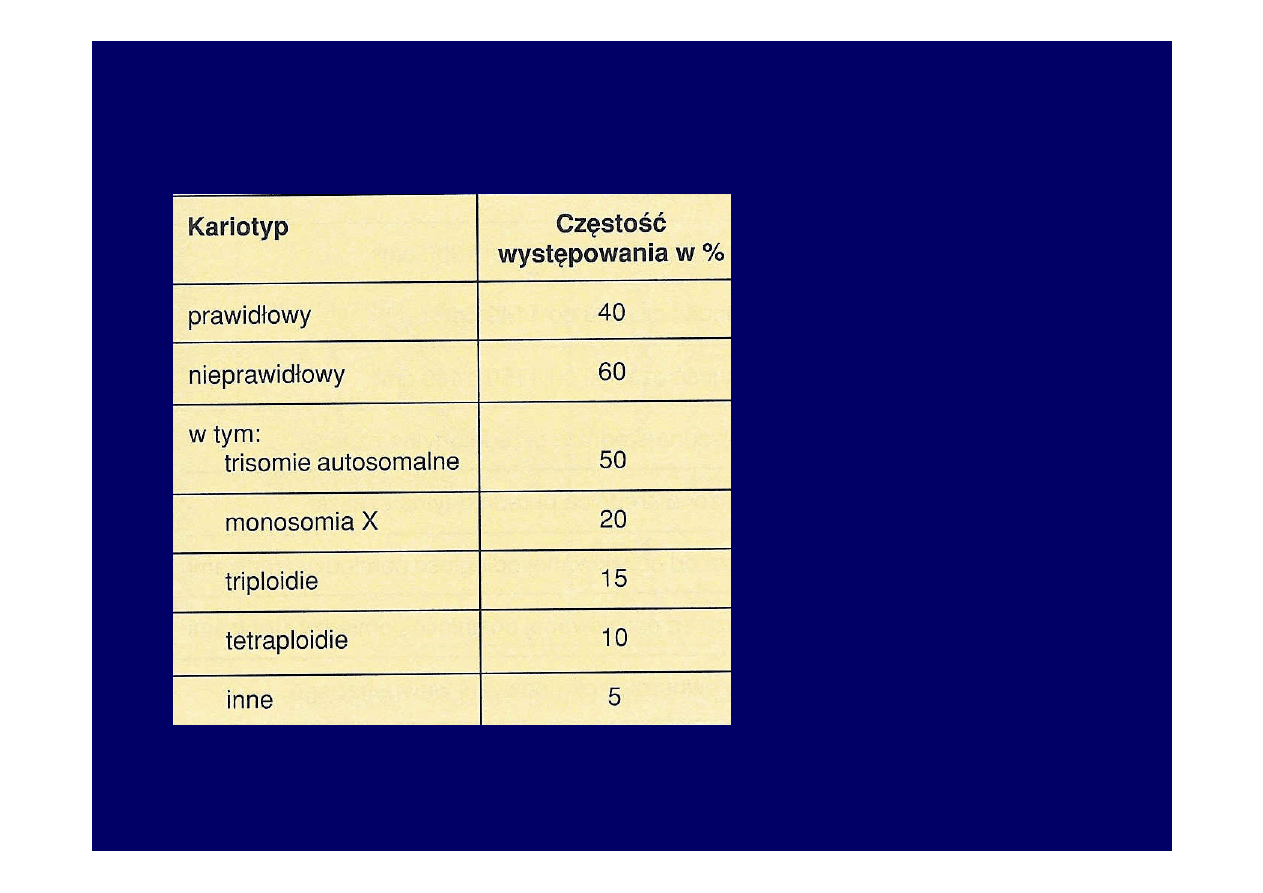
Rodzaje kariotypów
u zarodków we
wczesnych
poronieniach

Najczęstsze aberracje
chromosomowe stwierdzane u
noworodków

Nazwy opisowe stosowane w dysmorfologii

ZESPOŁY ABERACJI LICZBOWYCH
CHROMOSOMÓW SOMATYCZNYCH
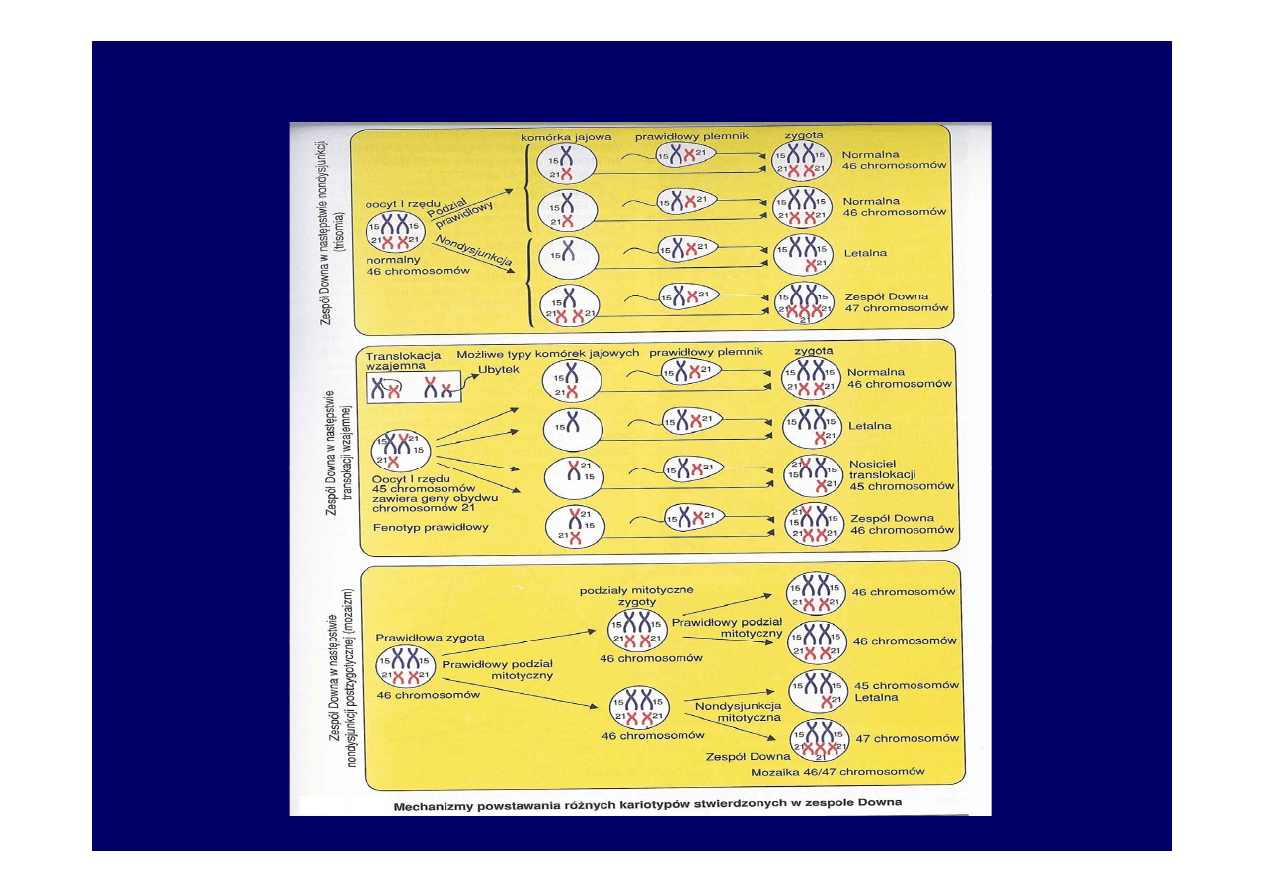
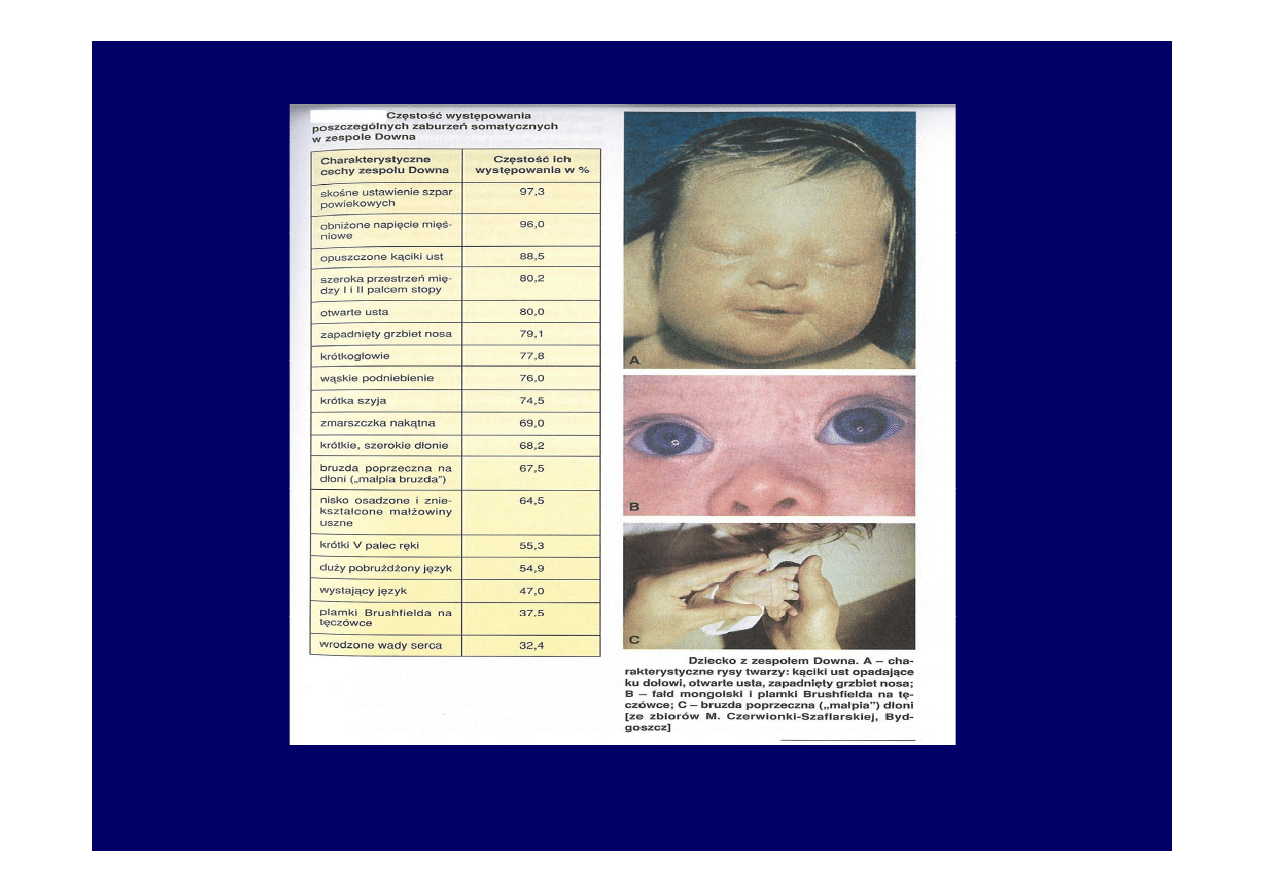
• Zespół Downa
1:700 urodzeń. Dodatkowy chromosom 21 pary.
Do wystąpienia zespołu prowadzą:
-trisomia 21 pary(95%) – 47,XX, + 21 lub 47,XY,+21
-translokacja niezrównoważona (4%) 46,XX,der(21;21)(q10;q10), +21
lub 46,XY,der(21;21)(q10;q10) +21
-kariotyp mozaikowy (1%)
46,XX/47,XX, +21 lub 46,XY/47,XY, +21
Chromosom pary 21 może ulec translokacji na jeden z chromosomów
grupy D (13,14,15) lub grupy G (21,22)

Zespół Downa
• W przypadku trisomii 21 nondysjunkcja zachodziła
najczęściej w pierwszym podziale mejotycznym u matek
(80%). Ponad 60% zarodków i płodów z trisomią 21 ulega
samoistnemu poronieniu
• Prawdopodobieństwo urodzenia dziecka z Zespołem
Downa wzrasta z wiekiem. Kobieta 35 letnia- 1:300
• 45 letnia -
1:22
• Ryzyko wystąpienia zespołu 1:700 żywo urodzonych
dzieci

ZESPÓŁ PATAU
• Częstość występowania-1:8000-10 000 urodzeń.
• Przyczyna-dodatkowy chromosom pary 13.
• Kariotypy
- 47,XX,+13 i 47XY, +13 (75%)
- - translokacja niezrównoważona w obrębie pary
chromosomu 13
- - kariotypy mozaikowe

ZEPÓŁ PATAU
Cechy zespołu:
Mikrocefalia, ubytki skóry na głowie, wystające czoło,
rozszczep wargi i podniebienia, wady gałek ocznych
(częściowy ubytek siatkówki i naczyniówki), hipoteloryzm,
nisko osadzone uszy, anomalie palców (polidaktylia,
syndaktylia). Wrodzone wady rozwojowe narządów
wewnętrznych (nerek, serca, macicy, anomalie w budowie
mózgu. Hipotonia mięśni i głuchota.
70% UMIERA W PIERWSZYM PÓŁROCZU ŻYCIA
10% PRZEŻYWA DO 1 ROKU ŻYCIA.
Ryzyko ponownego urodzenia dziecka z zespołem Patau
mniejsze niż 1%

ZESPÓŁ EDWARDSA
Trsomia chromosomu 18 (47,XX,+18 lub 47,XY,+18
Częstość – 1:5000
Duży wpływ na wystąpienie aberracji ma wiek matki
Cechy :
stopa cepowata z wystającą kością piętową, krótkim
paluchem i zrostami palców, wrodzone wady
rozwojowe serca, nerek przewodu pokarmowego,
niedorozwój narządów płciowych u dziewczynek,
niezstąpienie jąder u chłopców.
30% - zgon w okresie noworodkowym.
Ok. 10% przeżywa 1 rok.


TRSOMIA CHROMOSOMU 8
47,XX,+8 lub 47,XY,+8, kariotyp mozaikowy 46,XX/47,XX,+8
lub 46,XY/47,XY,+8
Cechy:
Zahamowanie wzrostu, zaburzenia w budowie twarzoczaszki,
anomalie kostno-stawowe
W obrębie głowy:- wysokie czoło, mała cofnieta żuchwa,
małżowiny uszne duże i odstające oraz nisko osadzone, nos
duży i zadarty, nieznaczny hiperteloryzm i czasami zez.
Obserwuje się skrzywienie kręgosłupa, nadliczbowe kręgi,
rozszczep kręgów, brak rzepki, wodonercze, spodziectwo,
niezstąpienie jąder. Cechą charakterystyczna zespołu jest
pogłębienie bruzdy w obrębie dłoni i stóp. Występuje
upośledzenia umysłowe niewielkiego stopnia.

ZESPOŁY DELECJI CHROMOSOMOWYCH
ZESPÓŁ WOLFA –HIRSCHHORNA
Częstość- 1:50 000 żywo urodzonych
Przyczyna - delecja terminalna części krótkiego ramienia
chromosomu 4.
Krytyczne miejsce pęknięcia - region 4p16.3
46,XY,del(4)(p16.3) i 46,XX,del(4)(p16.3)
Badanie wykazały obecność innych aberracji chromosomu 4;
Translokacja,
Delecja interstycjalna ramienia krótkiego
Chromosom pierścieniowy


DELECJA KRÓTKICH RAMION
CHROMOSOMU 5.
(ZESPÓŁ CRI DU CHAT)
• Delecja terminalna części ramion krótkich chromosomu 5 z
krytycznym miejscem pęknięcia w regionie 5p15.
• Inaczej „zespół miauczenia kota”
• Kariotyp: 46,XX,del(5)(p15 lub 46,XY,del(5)(p15)
• Częstość- 1:50 000 do 1:100 000
• Cechy:
- u niemowląt małogłowie, okrągła twarz, (księżyc w pełni), gałki
oczne szeroko rozstawione, zez zbieżny, małżowiny uszne małe,
nisko osadzone,
- -u starszych dzieci- powiększenia żuchwy, wydłużenie części
twarzowej żuchwy,
- płacz noworodka przypomina miauczenia kota(zaburzenia
budowy krtani)
- - brak zdolności mówienia

DELECJA DŁUGICH RAMION
CHROMOSOMU 13.
Częściowa, rzadziej całkowita delecja ramion długich chromosomu 13.
-najczęściej delecja interstycjalna chromosomu 13(13g14-q22)
-kariotyp: 46,XX,del(13)(q14q22) lub 46,XY,del(13)(q14q22)
Cechy:
Mikrocefalia, zniekształcenia twarzoczaszki, (głowa kształtu trójkątnego,
zaburzenia rozwoju komór mózgowia, hiperteloryzm, i małoocze), wąskie
szpary powiekowe, zmarszczka nakątna, wady tęczówki zaćma, szeroki
grzbiet nosa, duże małżowiny uszne, nisko osadzone, szyja krótka.
Inne wady:
zarośnięcie odbytu, spodziectwo, niezstąpienie jąder,
zaburzenia budowy moszny, wrodzone zwichnięcie stawu biodrowego,
stopa końsko-szpotawa, niedorozwój kciuka.

WYBRANE ZESPOŁY MIKRODELECJI
Mikrodelecja to aberracja strukturalna na
poziomie cytogenetycznym tak mała, że nie
uwidocznienia się w mikroskopie świetlnym.
Najmniejsza mikrodelecja widoczna w
mikroskopie świetlnym o dużej rozdzielczości
obrazu obejmuje około 4000 kb.

ZESPÓŁ PRADERA-WILLEGO
• Częstość- 1:10 000 – 1: 15 000 żywo urodzonych.
• W ok. 75 % przyczyna zespołu jest delecja interstycjalna
długiego ramienia chromosomu 15 (15q11-q13), który jest
pochodzenia ojcowskiego.
• Kariotyp:
46,XX,del(15)(q11q13) lub 46,XY,del (15)(q11q13)
W 20% -uniparentalna disomia pochodzenia matczynego
U około 1-2% prawidłowe dziedziczenie regionu
15q11q-q13.
Cechy zespołu zmieniają się z wiekiem pacjenta


ZESPÓŁ ANGELMANA
• 1:25 000 urodzeń
• U 70% -delecja interstycjalna długiego ramienia
chromosomu 15 w regionie 15q11-13q, który jest
pochodzenia matczynego
• Kariotyp:
• 46,XX,del(15)(q11q13) lub 46,XY,del(15)(q11q13)
• 3-5% uniparentalna disomia chromosomu 15 pochodzenia
ojcowskiego
• 8% imprinting genomowy


ZASPÓŁ DiGEORGE
• mikrodelecja w obrębie ramion długich chromosomu22.
• kariotyp
• 46,XX,del(22)(q11.21q11.23) lub
46,XY,del,(22)(q11.21q11.23)
• Cechy:
• Niska waga urodzeniowa, rozszczep podniebienia, małe
szpary powiekowe, hiperteloryzm, antymongoidalne
ustawienie szpar powiekowych, „rybie usta”, czworokątny
czubek nosa., wrodzone wady serca, łuku aorty, brak
grasicy
• Z powodu niedoboru limfocytów T częste infekcje
Document Outline
- PRAWIDŁOWY KARIOTYP CZŁOWIEKA I ANOMALIE AUTOSOMÓW
- PRAWIDŁOWY KARIOTYP CZŁOWIEKA
- Chromosomy mają zdolność do samoodtwarzania się w trakcie podziałów komórki. Widoczne są po wybarwieniu tylko w czasie p
- METODY BADAŃ CHROMOSOMÓW
- AUTORADIOGRAFIA
- CYTOMETRIA PRZEPŁYWOWA
- HYBRYDYZACJA IN SITU (In situ Hybrydyzation- ISH)
- ZAPISYWANIE WYNIKÓW BADAŃ CYTOGENETYCZNYCH
- Wykaz skrótów
- WYKAZ SKRÓTÓW
- Wykaz skrótów
- KOMÓRKI POLIPLOIDALNE
- KOMÓRKI ANEUPLOIDALNE
- ZESPOŁY ABERACJI LICZBOWYCH CHROMOSOMÓW SOMATYCZNYCH
- Zespół Downa
- ZESPÓŁ PATAU
- ZEPÓŁ PATAU
- ZESPÓŁ EDWARDSA
- TRSOMIA CHROMOSOMU 8
- ZESPOŁY DELECJI CHROMOSOMOWYCH
- DELECJA KRÓTKICH RAMION CHROMOSOMU 5. (ZESPÓŁ CRI DU CHAT)
- DELECJA DŁUGICH RAMION CHROMOSOMU 13.
- WYBRANE ZESPOŁY MIKRODELECJI
- ZESPÓŁ PRADERA-WILLEGO
- ZESPÓŁ ANGELMANA
- ZASPÓŁ DiGEORGE
Wyszukiwarka
Podobne podstrony:
PRAWIDŁOWY KARIOTYP CZŁOWIEKA, Fizjoterapia, biologia medyczna
4 Prawidłowy kariotyp człowieka Anomalie auto i heterochromosomów
PRAWIDŁOWY KARIOTYP CZŁOWIEKA I ANOMALIE AUTOSOMÓW
Prawidłowy kariotyp człowieka
Prawidłowy kariotyp człowieka
Prawidłowy kariotyp człowieka (A g)
wpływ prawidłowego odżywiania człowieka (4 str), Prawo Administracyjne, Gospodarcze i ogólna wiedza
Diabelskie zło nerwicy Nerwica jako przeszkoda uniemożliwiająca prawidłowy rozwój człowieka
Anatomia człowieka Układ mięśniowy ćw. 4, Anatomia
Sery - ćw z mleka, Technologia żywności i żywienia człowieka, Mleczarstwo, Technologia mleczarstwa
opracowania, Mikro JU cw 5, • Flora fizjologiczna człowieka ze szczególnym uwzględnieniem
Poloaczenia ruchomej czesci kregoslupa, Anatomia Prawidłowa człowieka
KREGOSLUP BUDOWA I POLACZENIA, Anatomia Prawidłowa człowieka
Każdy dorosły człowiek posiada większą lub mniejszą wiedzę na temat czynników warunkujących prawidło
Ćw 9 Fenomen sportu w życiu człowieka
CW 2011 czlowiek 1 3
ŻYCIE CZŁOWIEKA NIEROZERWALNIE ZWIĄZANE Z WYCHOWANIEM ćw, pedagogika, semestr I, teoria wychowania
więcej podobnych podstron