
1
Warszawa, listopad 2007
ENZYMOLOGIA
Materia³y do æwiczeñ
opracowa³ zespó³ Zak³adu Regulacji Metabolizmu Instytutu Biochemii
Wydzia³u Biologii UW
Æwiczenia s¹ przeznaczone dla studentów biologii i biotechnologii Wydzia³u Biologii oraz dla
studentów MISMaP.
Zestaw zadañ praktycznych
strona
1. Wyznaczanie sta³ej Michaelisa i szybkoœci maksymalnej dla
dehydrogenazy L-mleczanowej
2
2. Otrzymywanie dehydrogenazy glukozo-6-fosforanowej z dro¿d¿y piwnych.
Sporz¹dzenie bilansu oczyszczania bia³ka enzymatycznego
7
3. Wykorzystanie tyrozynazy otrzymywanej z pieczarki dwuzarodnikowej
do produkcji L-DOPA
12
4. Izolacja i oznaczanie aktywnoœci fosforylazy z ziemniaków
18
5. Oznaczanie aktywnoœci wieloenzymowego uk³adu fotosyntetycznego
21
6. Oznaczenie aktywnoœci immobilizowanej inwertazy
25
1
7. Analiza elektroforetyczna produktów trawienia histonu H papain¹
28
2
WYZNACZANIE STA£EJ MICHAELISA I SZYBKOŒCI MAKSYMALNEJ
DLA DEHYDROGENAZY L-MLECZANOWEJ
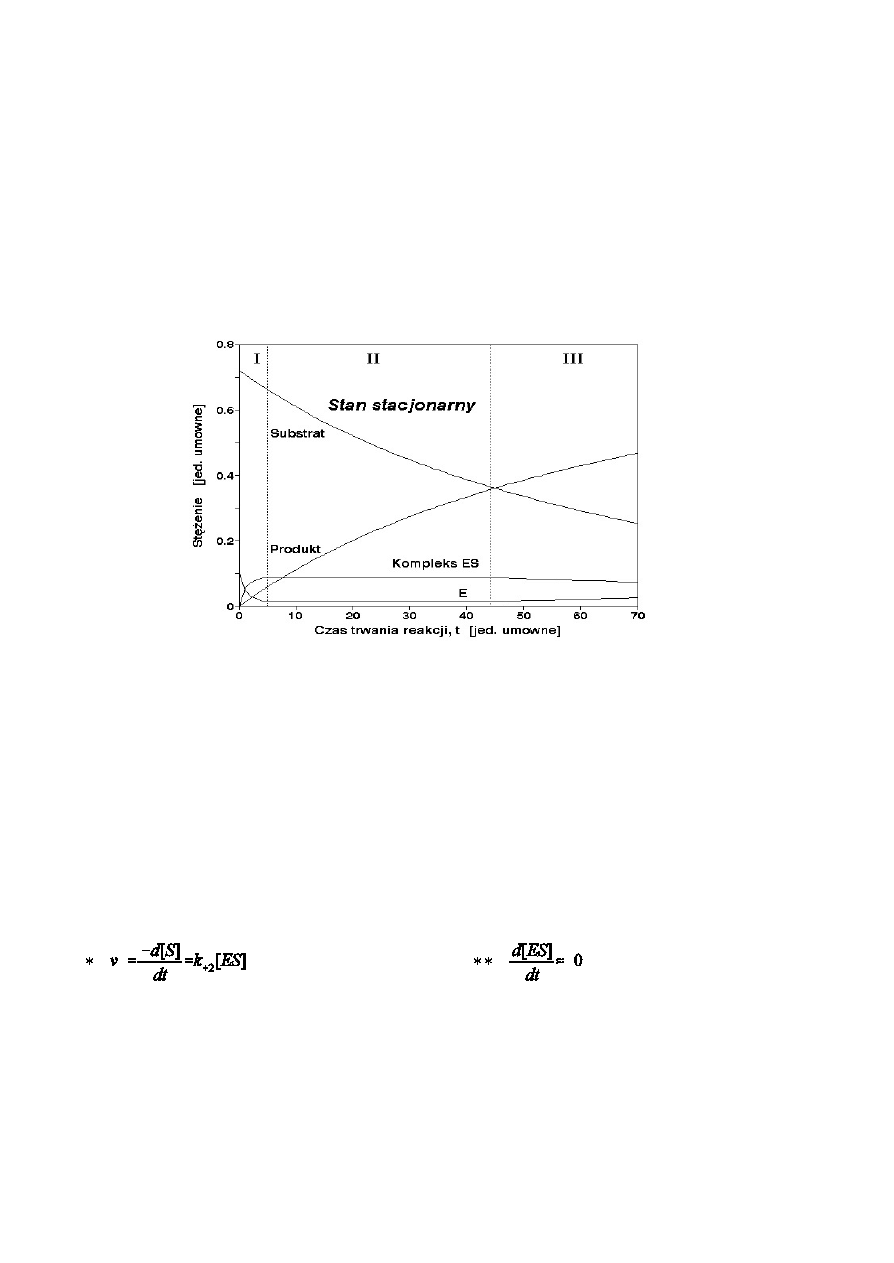
W czasie trwania ka¿dej reakcji enzymatycznej mo¿na wyró¿niæ trzy fazy. Pierwsza z nich, faza prestacjonarna
(I) trwa kilka milisekund i rozpoczyna siê z chwil¹ dodania enzymu do mieszaniny inkubacyjnej. Nastêpuje
wtedy szybkie tworzenie kompleksu enzym-substrat ES i spadek stê¿enia wolnego enzymu E. Druga faza,
stacjonarna (II) trwa wystarczaj¹co d³ugo, aby mo¿liwy by³ pomiar szybkoœci reakcji metodami powszechnie
stosowanymi w laboratoriach biochemicznych. Charakterystyczne cechy fazy stacjonarnej to sta³a szybkoœæ
reakcji, sta³e stê¿enie kompleku ES i enzymu E. Gdy stê¿enie substratu obni¿y siê na tyle, ¿e zacznie obni¿aæ
siê stê¿enie kompleku ES i spada szybkoœæ reakcji, zaczyna siê trzecia, poststacjonarna (III) faza reakcji.
Zmiany ztê¿enia substratu [S], produktu [P], wolnego enzymu [E] i kompleksu enzym-substrat [ES] w
czasie trwania jednosubstratowej reakcji enzymatycznej.
Fazy reakcji enzymatycznej: I - prestacjonarna, II - stacjonarna, III - poststacjonarna.
Za³o¿enia teorii Briggsa-Haldena, rozszerzaj¹cej postulaty Michaelisa i Menten, dotycz¹ w³aœnie fazy
stacjonarnej reakcji enzymatycznej. W fazie stacjonarnej przyrostowi stê¿enia produktu towarzyszy
równomolowy spadek stê¿enia substratu a szybkoœæ reakcji zale¿y od stê¿enia kompleksu ES (wzór *), gdzie
+2
[S] i [ES] oznacza stê¿enia substratu i kompleksu enzym-substrat a k jest sta³¹ rozpadu kompleksu ES.
W czasie trwania fazy stacjonarnej ustala siê stan równowagi dynamicznej rozpadu i tworzenia kompleksu ES
(wzór **).
Równanie kinetyczne reakcji chemicznej mo¿na przedstawiæ jako zale¿noœæ opisuj¹c¹ szybkoœæ reakcji od
zmian stê¿enia reaktantów. Równanie Michaelisa-Menten oparte jest na tej zasadzie opisu procesów
kinetycznych. Wyznaczenie sta³ych kinetycznych wymaga zmierzenia, w fazie stacjonarnej reakcji,

3
pocz¹tkowych szybkoœci reakcji enzymatycznej (V) przy znanych wartoœciach pocz¹tkowych stê¿eñ substratu
([S]).
max
m
Szybkoœæ maksymaln¹ reakcji V
i sta³¹ Michaelisa K wyznacza siê przy u¿yciu metod przekszta³caj¹cych
równanie Michaelisa-Menten z zale¿noœci hiperbolicznej w prostoliniow¹. Najczêœciej stosowana jest metoda
Lineweavera-Burke'a, gdzie zastosowano odwrotnoœæ równania podstawowego (I). Istnieje wiele przekszta³ceñ
równania Michaelisa-Menten umo¿liwiaj¹cych proste graficzne wyznaczenie sta³ych kinetycznych np. metoda
Hofstee-Eadie'go (II), czy metoda Hanesa (III). Dok³adne wyznaczenie sta³ej Michaelisa (Km) i szybkoœci
maksymalnej (Vmax) na podstawie pocz¹tkowych szybkoœci reakcji (V) i pocz¹tkowych stê¿eñ substratu [S]
umo¿liwia metoda Eisenthala i Cornish-Bowdena (IV). W tej metodzie nale¿y wykreœliæ proste ³¹cz¹ce wartoœci
szybkoœci pocz¹tkowych (V) od³o¿one na dodatniej osi 0Y z wartoœciami pocz¹tkowych stê¿eñ substratu [S]
od³o¿onych na ujemnej osi 0X. Wspó³rzêdne punktu ich przeciêcia to wartoœci Vmax (oœ 0Y) i Km (oœ 0X).
(I)
(II)
(III)
(IV)
Równanie kinetyczne reakcji chemicznej mo¿na tak¿e przedstawiæ jako zale¿noœæ opisuj¹c¹ zmiany stê¿enia
reaktantów w czasie. Jednym ze sposobów analizy kinetycznej ca³ej krzywej przebiegu reakcji enzymatycznej
0
s¹ metody pos³uguj¹ce siê sca³kowan¹ w przedziale czasowym [t =0, t] postaci¹ równania szybkoœci reakcji
enzymatycznej, tak jak zaproponowana przez Walkera i Schmidta:
(V)
0
gdzie [P] to stê¿enie produktu w czasie t, zaœ [S ] to pocz¹tkowe stê¿enie substratu.
m
max
Wzór (V) mo¿na ³atwo przekszta³ciæ do postaci umo¿liwiaj¹cej ³atwe wyznaczenie wartoœci K i V
:

4
(VI)
0
gdzie [S ] to stê¿enie pocz¹tkowe substratu, zaœ [S] to stê¿enie substratu po czasie t.
U¿ywana w doœwiadczeniu dehydrogenaza L-mleczanowa (EC 1.1.1.27 ) jest jednym z enzymów glikolizy
katalizuj¹cym reakcjê:
CH3 CH3
*
*
H
)
C
)
OH + NAD
W
C
4
O + NADH + H
+
+
*
*
COOH COOH
Materia³y i odczynniki (sporz¹dziæ na wodzie dejonizowanej o przewodnioœci nie wiêkszej ni¿ 3 ìS)
1. 0,07 M potasowy bufor fosforanowy pH 6,6. Przygotowaæ 100 ml buforu.
2. 3,2 M roztwór siarczanu amonowego.
3. 20 mM roztwór pirogronianu sodu. Przygotowaæ 10 ml roztworu. Oznaczyæ dok³adnie stê¿enie roztworu
pirogronianu sodu metod¹ enzymatyczn¹ (patrz Wykonanie).
2
4. Roztwór NADH zawieraj¹cy 20 mg nukleotydu rozpuszczonego w 1 ml H O (przygotowana nawa¿ka
nukleotydu).
5. Roztwór dehydrogenazy L-mleczanowej w 3,2 M siarczanie amonowym (preparat handlowy).
Materia³y pomocnicze i aparatura
Kuwety szklane 2 ml, ma³e bagietki plastikowe, regulowane pipety automatyczne, ³aŸnia lodowa, stoper.
Spektrofotometr.
Wykonanie
Roztwory enzymu, NADH i pirogronianu nale¿y umieœciæ w ³aŸni lodowej, a wszystkie pomiary
przeprowadzaæ w sta³ej temperaturze 25 C gdy spektrofotometr posiada termostat lub w zanotowanej
o
temperaturze pokojowej.
A. Oznaczanie stê¿enia pirogronianu
Do kuwety pomiarowej odmierzyæ :
- 1,4 ml potasowego buforu fosforanowego
- tak¹ iloœæ roztworu NADH aby absorpcja mieszaniny reakcyjnej wynios³a od 0,600 do 0,900 (10- 20 ìl)
- 0,01 ml roztworu pirogronianu, którego pocz¹tkowe stê¿enie okreœlono orientacyjnie na 20 mM
2
- H O do objêtoœci ca³kowitej 2 ml
Zawartoœæ kuwety wymieszaæ bagietk¹, nastêpnie preinkubowaæ przez okres 1 minuty, po czym zmierzyæ
absorpcjê przy d³ugoœci fali 340 nm u¿ywaj¹c wody jako próby odniesienia (materia³owej). Nale¿y zwróciæ
uwagê, aby absorpcja przy d³ugoœci fali 340 nm wynosi³a od 0,600 do 0,900. Je¿eli absorpcja odbiega od tej

5
wartoœci to nale¿y zwiêkszyæ lub zmniejszyæ iloœæ dodawanego roztworu NADH. Zapisaæ pocz¹tkow¹ wartoœæ
absorpcji. Nastêpnie dodaæ 5 ìl roztworu enzymu. Roztwór enzymu odmierzyæ pipet¹ automatyczn¹ na ³opatkê
bagietki i przenieœæ do kuwety energicznie mieszaj¹c. Zmierzyæ absorpcjê po zakoñczeniu reakcji. Je¿eli
koñcowa absorpcja spadnie poni¿ej 0,200 nale¿y oznaczenie powtórzyæ, u¿ywaj¹c odpowiednio mniejszej iloœci
pirogronianu.
Do osobnej kuwety odmierzyæ wszystkie odczynniki, za wyj¹tkiem pirogronianu i zmierzyæ absorpcjê przed
i po dodaniu enzymu. Zmiana absorpcji stanowi poprawkê, któr¹ nale¿y dodaæ do pocz¹tkowej wartoœci
absorpcji zmierzonej w próbie badanej (jest to miara zmêtnienia roztworu w kuwecie po dodaniu bia³ka
enzymatycznego). Znaj¹c milimolowy wspó³czynnik absorpcji å dla NADH przy d³ugoœci fali 340 nm
wynosz¹cy 6,22 mM cm , wartoœæ absorpcji pocz¹tkowej i koñcowej (po zakoñczeniu reakcji enzymatycznej)
-1
-1
oraz rozcieñczenie badanego roztworu pirogronianu w kuwecie pomiarowej, obliczyæ stê¿enie pirogronianu w
roztworze wyjœciowym.
B. Dobieranie rozcieñczenia enzymu
Wyjœciowy roztwór enzymu rozcieñczyæ orientacyjnie (wg. wskazañ asystenta) w 3,2 M roztworze siarczanu
amonu. Nastêpnie do kuwety odmierzyæ:
- 1,4 ml potasowego buforu fosforanowego
- odpowiedni¹ iloœæ NADH daj¹c¹ absorpcjê od 0,600 do 0,900
2
- H O do objêtoœci ca³kowitej 2 ml
Zmierzyæ absorpcjê (po wymieszaniu zawartoœci kuwety) przy d³ugoœci fali 340 nm. Nastêpnie dodaæ 5 ìl
rozcieñczonego enzymu i zanotowaæ zmiany absorbcji do 60 s preinkubacji (co 15 s). Reakcjê rozpocz¹æ
dodaniem w 60 s (czas 0 trwania reakcji) roztworu pirogronianu. Pomiary przeprowadziæ przy koñcowym
stê¿enie pirogronianu równym 0,3 mM i 0,01 mM (wykonaæ dwa powtarzalne pomiary). Roztwór pirogronianu
nale¿y dodawaæ w objêtoœci nie wiêkszej ni¿ 50 ìl. Pomiary przeprowadzaæ co 15 sekund przez 4 minuty.
Nale¿y pamiêtaæ, aby zanotowaæ wszystkie wartoœci absorbcji; wyjœciow¹, po dodaniu enzymu i po rozpoczêciu
reakcji dodaniem substratu. Rozcieñczenie enzymu jest odpowiednie, je¿eli spadek absorpcji w pierwszych 60-
100 sekundach pomiaru jest liniowy. Ustalone stê¿enie enzymu u¿ywaæ do badania kinetyki reakcji
enzymatycznej. Rozcieñczonego enzymu nie nale¿y u¿ywaæ d³u¿ej ni¿ 5 godzin. Przy du¿ych rozcieñczeniach
dehydrogenazy uwzglêdnianie poprawki na zmiany absorpcji przez bia³ko enzymu nie jest konieczne.
C. Oznaczenia szybkoœci reakcji przy ró¿nych stê¿eniach substratu
Do kuwet odmierzyæ:
- 1,4 ml potasowego buforu fosforanowego
- odpowiedni¹ iloœæ roztworu NADH daj¹c¹ absorpcjê od 0,600 do 0,900
2
- H O do objêtoœci ca³kowitej 2 ml
Oznaczenia nale¿y przeprowadziæ tak, jak opisano w punkcie 2. Reakcjê rozpocz¹æ tak¹ iloœci¹ pirogronianu,
aby stê¿enie koñcowe w kuwecie wynosi³o odpowiednio: 0,3; 0,2; 0,1; 0,075; 0,05; 0,02; i 0,01 mM
(ewentualnie wg. wzkazañ asystenta). Pomiary nale¿y wykonywaæ co 15 sekund przez 4 minuty. Nale¿y
wykonaæ trzy powtarzalne pomiary przy ka¿dym podanym stê¿eniu pirogronianu.

6
D. Wyznaczanie przebiegu ca³ej krzywej reakcji enzymatycznej
Do kuwet odmierzyæ:
- 1,4 ml potasowego buforu fosforanowego
- odpowiedni¹ iloœæ roztworu NADH daj¹c¹ absorpcjê od 0,900 do 1,000
2
- H O do objêtoœci ca³kowitej 2 ml
Oznaczenia nale¿y przeprowadziæ tak, jak opisano w punkcie 2, przy czym u¿yte rozcieñczenie enzymu
powinno byæ od pó³tora do dwóch razy wiêksze ni¿ wyznaczone w punkcie B. Reakcjê rozpocz¹æ tak¹ iloœci¹
pirogronianu, aby stê¿enie koñcowe w kuwecie wynosi³o 0,3 mM (ewentualnie wg. wzkazañ asystenta). Pomiary
nale¿y wykonywaæ co 15 sekund a¿ do ca³kowitego zakoñczenia reakcji. Nale¿y wykonaæ trzy powtarzalne
pomiary.
Obliczenia nale¿y przeprowadziæ wykorzystuj¹c arkusz kalkulacyjny (QPRO 8.0). Na podstawie otrzymanych
wyników statystycznych przygotowaæ dyskusjê b³êdów przeprowadzonego doœwiadczenia.
RACHUNKOWE WYZNACZANIE WARTOŒCI SZYBKOŒCI POCZ¥TKOWYCH (V),
max
m
SZYBKOŒCI MAKSYMALNEJ (V
) ORAZ STA£EJ MICHAELISA (K ) DLA
DEHYDROGENAZY MLECZANOWEJ (LDH) PRZY POMOCY ARKUSZA
KALKULACYJNEGO, ZAPROJEKTOWANEGO W PROGRAMIE QUATRO PRO 6.0
Arkusz kalkulacyjny jest graficzn¹ metod¹ zapisywania i przetwarzania danych liczbowych. U¿ytkownik ma
do dyspozycji obszar z³o¿ony z pól (komórek) z których ka¿da posiada nazwê (adres) sk³adaj¹c¹ siê z litery i
cyfry, np. adres A5 oznacza, ¿e komórka po³o¿ona jest w pierwszej kolumnie i pi¹tym rzêdzie arkusza. W ka¿d¹
komórkê mo¿na wpisaæ liczbê, która w ten sposób zostaje przyporz¹dkowana okreœlonemu adresowi. Na
przyk³ad, wpisuj¹c liczbê 0.897 w komórkê znajduj¹c¹ siê w pierwszej kolumnie i pi¹tym rzêdzie arkusza
nadajemy tej liczbie adres A5. W komórkê mo¿na tak¿e wpisaæ wzór matematyczny lub statystyczny,
przeliczaj¹cy liczby wpisane w inne komórki. I tak wpisany w komórkê C5 wzór (A5+B5) dodaje liczby wpisane
w komórki A5 i B5 a wynik zapisuje w komórce C5. W komórki arkusza mo¿na tak¿e wpisywaæ dowolny tekst
lub inne elementy graficzne zwiêkszaj¹ce czytelnoœæ zawartych w nim danych. Wskazanie odpowiednich
komórek (adresów) pozwala tak¿e na automatyczne tworzenie wykresów ró¿nego typu. Nale¿y pamiêtaæ, ¿e
wpisanie wartoœci lub wzoru do nieodpowiedniej “komórki” powoduje otrzymanie b³êdnego rozwi¹zania.
Przed wprowadzeniem danych sprawdziæ format zapisu liczb.
Do wykonania obliczeñ sta³ych kinetycznych zosta³ przygotowany arkusz kalkulacyjny o nazwie 2007_KIN(n),
gdzie n jest liczb¹ przyporz¹dkowan¹ u¿ytkownikowi, poniewa¿ ka¿dy student zapisuje i zapamiêtuje wyniki
swoich doœwiadczeñ w osobnym zbiorze. Nale¿y wprowadziæ wartoœci absorpcji w komórki odpowiadaj¹cych
czasom i powtórzeniom pomiarów. Na podstawie wartoœci odchylenia standardowego oceniæ powtarzalnoœæ
wyników. Nastêpnie z odcinka prostoliniowego wykresu absorpcja/czas policzyæ metod¹ regresji liniowej
szybkoœæ reakcji. Otrzymane wartoœci wpisaæ do odpowiednich “komórek” arkusza i obliczyæ wartoœci sta³ych
kinetycznych. Na podstawie wykresów i wartoœci b³êdów pomiarów przeprowadziæ dyskusjê otrzymanych
wyników.

7
OTRZYMYW ANIE DEHYDROGENAZY GLUKOZO-6-FOSFORANOW EJ Z
DRO¯D¯Y PIWNYCH
Dehydrogenaza glukozo-6-fosforanowa, której koenzymem jest NADP, katalizuje reakcjê utleniania
glukozo-6-fosforanu do kwasu 6-fosfoglukonowego:
Glukozo-6-fosforan + NADP
W
kwas 6-fosfoglukonowy + NADPH + H
+
+
Preparaty dehydrogenazy glukozo-6-fosforanowej s¹ stosowane do enzymatycznego oznaczania
glukozo-6-fosforanu i NADP lub, w obecnoœci heksokinazy, do oznaczania ATP:
heksokinaza
ATP + glukoza <
4444444444444
> ADP + glukozo-6-fosforan
dehydrogenaza glukozo-6-fosforanowa
Glukozo-6-fosforan+NADP <
44444444444444444444444444444
+
44444444444444444444444
> kwas 6-fosfoglukonowy+NADPH+ H
+
Z reakcji tej wynika, ¿e 1 mol utworzonego NADPH odpowiada 1 molowi glukozo-6-fosforanu lub ATP.
Preparat enzymu oczyszcza siê z autolizatu dro¿d¿y piwnych poprzez wysalanie siarczanem amonowym i
adsorpcjê na ¿elu fosforanowym.
Materia³y i odczynniki
1. Autolizat dro¿d¿owy, przygotowany z wysuszonych dro¿d¿y piwnych.
3
2. 0,1 M NaHCO . Przygotowaæ 100 ml roztworu.
2
2
2
3. ¯el fosforanowy. 50 ml roztworu CaCl (132g CaCl x6H O w 1l) rozcieñczyæ wod¹ do koñcowej objêtoœci
3
4
2
500 ml i dodaæ 50 ml roztworu fosforanu trójsodowego (152g Na PO x12H O w 1 dcm ). pH mieszaniny
3
doprowadziæ 1N kwasem octowym do wartoœci 7,4. Powsta³y osad fosforanu wapnia przemyæ szeœcioma
pó³litrowymi porcjami wody destylowanej o temperaturze pokojowej. Nastêpnie odwirowaæ przy 3000
obr/min przez 10 min. Okreœliæ such¹ masê (zawartoœæ fosforanu wapnia w ¿elu) poprzez wysuszenie w
temperaturze 105 C próbki ¿elu o znanej masie.
o
4. Siarczan amonowy, wysuszony w 50 C i zmielony. Przygotowany na pocz¹tku æwiczeñ.
o
5. 0,1 M potasowy bufor fosforanowy pH 7,5. Przygotowaæ 150 ml buforu.
6. 0,1 M bufor Tris-HCl pH 7,5. Przygotowaæ 50 ml buforu.
2
7. 0,2 M MgCl . Przygotowaæ 25 ml roztworu.
8. 10 mM glukozo-6-fosforan, bardzo nietrwa³y (przygotowany w jednorazowych porcjach, przechowywaæ w
lodzie).
9. 1,5 mM roztwór NADP (przygotowany w jednorazowych porcjach, przechowywaæ w lodzie)

8
Wykonanie
A. Otrzymywanie enzymu z dro¿d¿y piwnych.
Uwaga; studenci rozpoczynaj¹ oczyszczanie enzymu pocz¹wszy od etapu III.
ETAP I. Przygotowanie suchych dro¿d¿y
Gêstwê dro¿d¿y piwnych zalaæ równ¹ jej objêtoœci¹ zimnej wody z kranu i po 2-3 godzinach zdekantowaæ.
Czynnoœci te powtarzaæ wielokrotnie w ci¹gu dnia, przez trzy kolejne dni. Nastêpnie zdekantowan¹ gêstwê
odwirowaæ przez 60 min. przy 3500 x g . Supernatant odrzuciæ, a osad roz³o¿yæ porcjami na tacy wy³o¿onej
bibu³¹ i pozostawiæ do wysuszenia pod wentylatorami w temperaturze pokojowej, stopniowo rozdrabniaj¹c
grudy dro¿d¿owe. Po wysuszeniu dro¿d¿e rozdrobniæ w m³ynku elektrycznym i przesiaæ przez sito 0,6 mm.
ETAP II. Autolizat dro¿d¿owy
3
25 g suchych, zmielonych dro¿d¿y zawiesiæ w 75 ml 0,1 M NaHCO i pozostawiæ w ³aŸni wodnej o
temperaturze 40 C przez 4 godz., delikatnie mieszaj¹c. Zawiesinê odwirowaæ przez 50 min. przy 18 000 x g.
o
Supernatant przechowywaæ zamro¿ony w temp. -20 C.
o
ETAP III. Wysalanie siarczanem amonu
Pobraæ 0,5 ml autolizatu do oznaczenia bia³ka i aktywnoœci enzymatycznej. Ze wzglêdu na oszczêdnoœæ czasu
nie nale¿y wykonywaæ oznaczeñ na bie¿¹co. Próby z poszczególnych etapów oczyszczania (tak¿e próby
autolizatu) przechowywaæ w zamra¿alniku lodówki i wszystkie oznaczyæ jednoczeœnie.
Frakcjonowanie siarczanem amonu. Wszystkie nastêpne czynnoœci wykonywaæ w temp. 2 C (³aŸnia lodowa).
o
3
Otrzymany autolizat (30 ml) rozcieñczyæ czterokrotnie w 0,1 M NaHCO , a nastêpnie powoli, ci¹gle mieszaj¹c
(mieszad³o magnetyczne), dodaæ tak¹ iloœæ sta³ego suchego siarczanu amonu aby otrzymaæ roztwór o stê¿eniu
4 2
4
(NH ) SO równym 55% roztworu nasyconego. Mieszaninê odwirowaæ przez 10 minut przy 15000 x g, osad
odrzuciæ a supernatant wysoliæ dodaj¹c siarczan amonu do wartoœci równej 64% roztworu nasyconego.
Zawiesinê odwirowaæ jak poprzednio i otrzymany osad rozpuœciæ w 34 ml wody destylowanej. Pobraæ 0,5 ml
do oznaczenia bia³ka i aktywnoœci.
ETAP IV. Adsorpcja na ¿elu fosforanowym
Do 33 ml ¿elu rozcieñczonego wod¹ destylowan¹ (0.02 g fosforanu wapnia w 1 ml roztworu) dodaæ, ci¹gle
mieszaj¹c, roztwór preparatu enzymatycznego otrzymany w poprzednim etapie (33 ml). Delikatnie mieszaæ
zawiesinê przez 15 min. ¯el z zaadsorbowanym enzymem odwirowaæ (10 min, 1000 x g), supernatant odrzuciæ,
a do osadu dodaæ, stale mieszaj¹c 66 ml 0,1 M buforu fosforanowego o pH 7,5. Po 10 min. ¿el odwirowaæ jak
wy¿ej, osad odrzuciæ. Supernatant po pobraniu próby do oznaczeñ (1 ml) poddaæ dalszemu oczyszczaniu.
ETAP V. Frakcjonowanie siarczanem amonu
Do eluatu powoli dodaæ, ci¹gle mieszaj¹c, siarczan amonu do wartoœci równej 60% ca³kowitego nasycenia
roztworu. Zawiesinê odwirowaæ przez 10 min. przy 15000 x g, osad odrzuciæ, a supernatant wysoliæ dodaj¹c
siarczan amonu do wartoœci równej 75% nasycenia roztworu. Zawiesinê odwirowaæ jak wy¿ej a otrzymany osad
rozpuœciæ w 0.5-1 ml oziêbionej wody destylowanej. W roztworze oznaczyæ bia³ko i aktywnoœæ enzymu.

9
Obliczenia rachunkowe
Wzór umo¿liwiaj¹cy przeliczenie przyœpieszenia liniowego wyra¿onego w wielokrotnoœci g na liczbê obrotów
wirówki wyra¿on¹ w rpm (liczba obrotów na minutê):
z - przyœpieszenie liniowe (wielokrotnoœæ przyœpieszenia ziemskiego, g = 9,81 m s )
-2
ù - przyœpieszenie k¹towe
r - promieñ rotora (minimalny, œredni, maksymalny) (w cm)
rpm - liczba obrotów na minutê dla danej wirówki i rotora
w wirówkach z przek³adni¹ firmy Janetzki otrzyman¹ wartoœæ rpm dla rotora nale¿y podzieliæ przez 4.7.
Wzór umo¿liwiaj¹cy wyliczenie iloœci siarczanu amonu potrzebn¹ do otrzymania roztworu o stê¿eniu bêd¹cym
u³amkiem roztworu nasyconego:
g - iloœæ gramów siarczanu amonu
V - objêtoœæ pocz¹tkowa (ml)
1
S - nasycenie pocz¹tkowe (u³amek dziesiêtny)
2
S - nasycenie koñcowe (u³amek dziesiêtny)
Z - iloœæ gramów w roztworze nasyconym (53.3 g dla siarczanu amonu)
p - przyrost objêtoœci (0.3 dla siarczanu amonu)
B. Oznaczanie aktywnoœci dehydrogenazy glukozo-6-fosforanowej
Oznaczenie polega na pomiarze szybkoœci wzrostu absorpcji przy d³ugoœci fali 340 nm spowodowanym
redukcj¹ NADP w obecnoœci dehydrogenazy glukozo-6-fosforanowej. Do kuwety o przekroju 1 cm i pojemnoœci
2 ml odmierzyæ nastêpuj¹ce iloœci roztworów (w ml):
2
Bufor Tris-HCl
M gCl
NADP
Glukozo-6-fosforan
W oda
0,75
0,1
0,2
0,1
0,35
Nastêpnie dodaæ pipet¹ automatyczn¹ od 5 do 30 ìl enzymu (wg. wskazañ asystenta) i po wymieszaniu
odczytywaæ wartoœci absorpcji co 15 sekund przez 3 minuty. Jako próbê odniesienia stosowaæ wodê
destylowan¹. Oznaczenie powtórzyæ z tak¹ sam¹ iloœci¹ enzymu. Sporz¹dziæ wykres zmiany absorpcji w czasie
i obliczyæ zmianê absorpcji w ci¹gu 1 minuty. Na tej podstawie obliczyæ iloœæ przereagowanego
glukozo-6-fosforanu.
Milimolowy wspó³czynnik absorpcji å dla NADPH przy d³ugoœci fali 340 nm wynosi 6,22 mM cm .
-1 -1

10
C. Opracowanie wyników
Po oznaczeniu stê¿enia bia³ka i aktywnoœci w próbach z poszczególnych etapów oczyszczania sporz¹dziæ
bilans oczyszczania enzymu wg wzoru:
ETAP
Objêtoœæ
Stê¿enie ca³kowite
bia³ka
*
Ca³kowita zawartoœæ
bia³ka
AktywnoϾ
ca³kowita
AktywnoϾ
w³aœciwa
W ydajnoϾ
Stopieñ
oczyszczenia
ml
mg/ml
mg
U
U/mg
%
%
Orientacyjne, ca³kowite stê¿enia bia³ka po kolejnych etapach oczyszczania enzymu (w mg/ml) wynosi: autolizat
10-100, pierwsze wysalanie 10-100, adsorpcja na ¿elu 1-10, drugie wysalanie 1-10.

11
OZNACZANIE BIA£KA METOD¥ BRADFORD
Metoda oznaczenia polega na pomiarze wzrostu absorpcji przy d³ugoœci faki 595 nm, wywo³anego powstaniem
kompleksu barwnika Coomassie Blue G-250 z bia³kiem.
Odczynniki
1. Odczynnik Bradford (przygotowany). 100 mg Coomassie Blue G-250 rozpuœciæ w 50 ml 96% etanolu.
Do tego roztworu dodaæ 100 ml 85% (wag/obj) kwasu ortofosforowego. Otrzymany roztwór rozcieñczyæ
wod¹ destylowan¹ do objêtoœco koñcowej 1 dcm . Przechowywaæ w ciemnej butelce w lodówce.
3
2. Roztwór wzorcowy bia³ka 0,250 mg/ml w 1 M NaOH
3. 50% metanol lub aceton
4. 1 M roztwór NaOH
Wykonanie
A. Wyznaczanie krzywej wzorcowej (przygotowana)
Przygotowaæ 11 czystych, suchych i ponumerowanych probówek na 10 ml. Do probówki nr 1 dodaæ pipet¹
automatyczn¹ 100 ìl 1 M NaOH. Do pozosta³ych probówek dodaæ, w dwóch powtórzeniach, 20, 40, 60, 80 i
100 ìl wzorcowego roztworu bia³ka w 1M NaOH. Roztwory bia³ka w probówkach uzupe³niæ 1 M NaOH do
objêtoœci 100 ìl. Nastêpnie do ka¿dej probówki dodaæ po 1,5 ml odczynnika Bradforda i wymieszaæ zawartoœæ
na mikromieszadle. Po up³ywie 15 min. zmierzyæ absorpcjê prób przy d³ugoœci fali 595 nm, stosuj¹c roztwór
z probówki nr 1 jako próbê odniesienia. Nale¿y u¿ywaæ czystych, przep³ukanych 50% metanolem kuwet.
Pomiarów dokonywaæ pocz¹wszy od prób o najmniejszym stê¿eniu bia³ka. Na podstawie uzyskanych wyników
wykreœliæ krzyw¹ wzorcow¹, odk³adaj¹c na osi odciêtych wartoœci absorpcji (A), a na osi rzêdnych zawartoœæ
bia³ka w próbie (w ìg). Obliczyæ metod¹ najmniejszych kwadratów wspó³czynnik nachylenia prostej regresji
dla odcinka prostoliniowego.
B. Oznaczanie bia³ka w próbie
Próbê rozcieñczyæ 1 M NaOH tak aby zawartoœæ bia³ka wynosi³a od 5 do 25 ìg bia³ka, a koñcowa objêtoœæ
roztworu nie by³a wiêksza ni¿ 100 ìl. Rozcieñczon¹ próbê odmierzyæ za pomoc¹ pipety automatycznej do
suchej probówki na 10 ml. W razie potrzeby objêtoœæ doprowadziæ do 100 ìl roztworem 1 M NaOH, dodaæ 1,5
ml odczynnika Bradford, zamieszaæ i po 15 minutach zmierzyæ absorpcjê przy 595 nm wobec próby nie
zawieraj¹cej bia³ka (tzw. materia³owej lub odniesienia). Je¿eli iloœæ bia³ka wziêta do oznaczania by³a
odpowiednia (absorpcja 0,1 - 0,5), oznaczenie powtórzyæ jeszcze 2 razy. Z krzywej wzorcowej odczytaæ iloœæ
bia³ka w próbie badanej i na tej podstawie obliczyæ jego zawartoœæ w próbie wyjœciowej.
12
W Y K O R ZY ST A N IE T Y R O ZY N A ZY O TR ZY M Y W A N E J Z P IE C ZA R K I
DWUZARODNIKOWEJ (AGARICUS BISPORUS) DO PRODUKCJI L-DOPA
L-DOPA (L-3,4-dihydroksyfenyloalanina) jest naturalnym prekursorem dopaminy, jednego z najwa¿niejszych
neurotransmitterów wykorzystywanych przez neurony ssaków, a tak¿e adrenaliny i noradrenaliny,
syntetyzowanych zarówno w oœrodkowym uk³adzie nerwowym jak i w tkankach obwodowych [Carlsson, 2001].
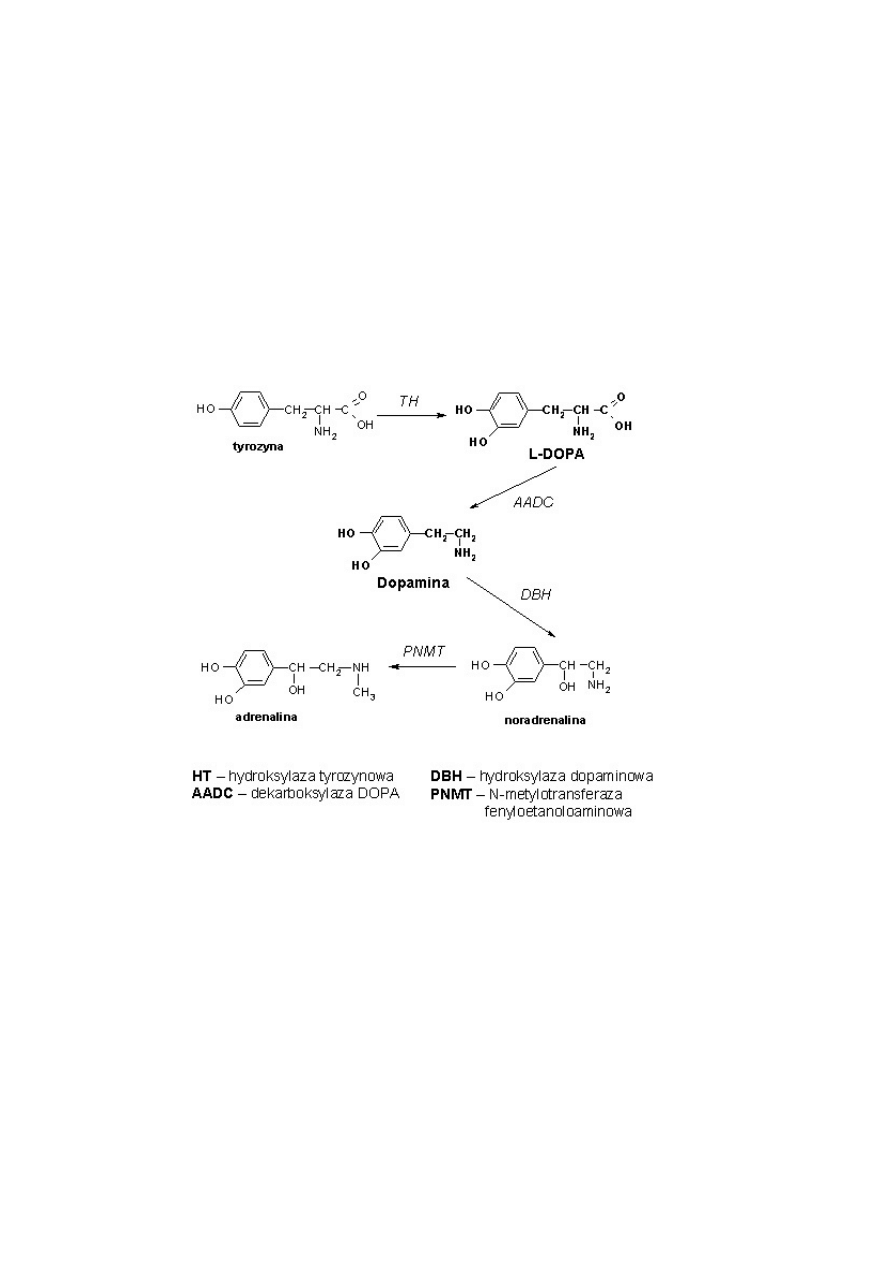
W wiêkszoœci komórek zdolnych do syntezy amin katecholowych, L-DOPA jest wytwarzana z tyrozyny w
reakcji katalizowanej przez hydroksylazê tyrozynow¹ (TH, Rys. 1), a nastêpnie przekszta³cana w dopaminê
przez dekarboksylazê DOPA (AADC). Dopamina mo¿e byæ nastêpnie wykorzystana do syntezy noradrenaliny
i adrenaliny w reakcjach katalizowanych przez hydroksylazê dopaminow¹ oraz N-metylotransferazê
fenyloetanoloaminow¹ (PNMT).
Rys. 1. Schemat biosyntezy amin katecholowych w organizmie cz³owieka
L-DOPA jest podstawowym i najbardziej skutecznym œrodkiem stosowanym w terapii choroby Parkinsona –
schorzenia zwyrodnieniowego uk³adu nerwowego, na które cierpi oko³o 1,5 % osób w wieku powy¿ej 65 lat.
Przyczyny choroby Parkinsona nie zosta³y dotychczas w pe³ni wyjaœnione, aczkolwiek wiadomo, ¿e
obserwowane objawy: dr¿enie r¹k, ramion, nóg ¿uchwy i twarzy, sztywnoœæ koñczyn i tu³owia, spowolnienie
ruchów, upoœledzenie koordynacji ruchowej i równowagi s¹ wynikiem obumieranie neuronów istoty czarnej
mózgu (tzw. neuronów dopaminergicznych) odpowiedzialnych za produkujê dopaminy [Samii i wsp., 2004].
Podstawow¹ metod¹ produkcji L-DOPA w przemyœle farmaceutycznym jest synteza chemiczna [Ho i wsp.,
2003]. W latach 90-tych minionego wieku nasili³y siê badania nad wykorzystaniem mikroorganizmów lub
preparatów enzymatycznych w produkcji tego leku. Jednak biosynteza L-DOPA z u¿yciem bakterii lub grzybów
okaza³a siê doœæ kosztowna, g³ównie ze wzglêdu na koniecznoœæ usuwania du¿ych iloœci zanieczyszczeñ
powstaj¹cych w procesie produkcyjnym, a przez to niekonkurencyjna z syntez¹ chemiczn¹. Z drugiej strony,
13
prace nad wykorzystaniem enzymu – tyrozynazy, jako biokatalizatora reakcji przekszta³cania aminokwasu
tyrozyny w L-DOPA wskaza³y na wyraŸn¹ op³acalnoœæ ekonomiczn¹ takiego podejœcia, szczególnie w œwietle
du¿ej trwa³oœci enzymu i ³atwoœci jego oddzielenia od mieszaniny reakcyjnej.
Tyrozynaza (EC 1.14.18.1) jest enzymem szeroko rozpowszechnionym w organizmach ¿ywych. Obecnoœæ tego
enzymu stwierdza siê zarówno w mikroorganizmach jak i w komórkach roœlin. Natomiast w organizmach
ssaków najwiêksz¹ aktywnoœæ tyrozynazy wykrywa siê w melanocytach, gdzie jest ona kluczowym enzymem
szlaku syntezy melaniny, barwnika nadaj¹cego charakterystyczne zabarwienie skóry.
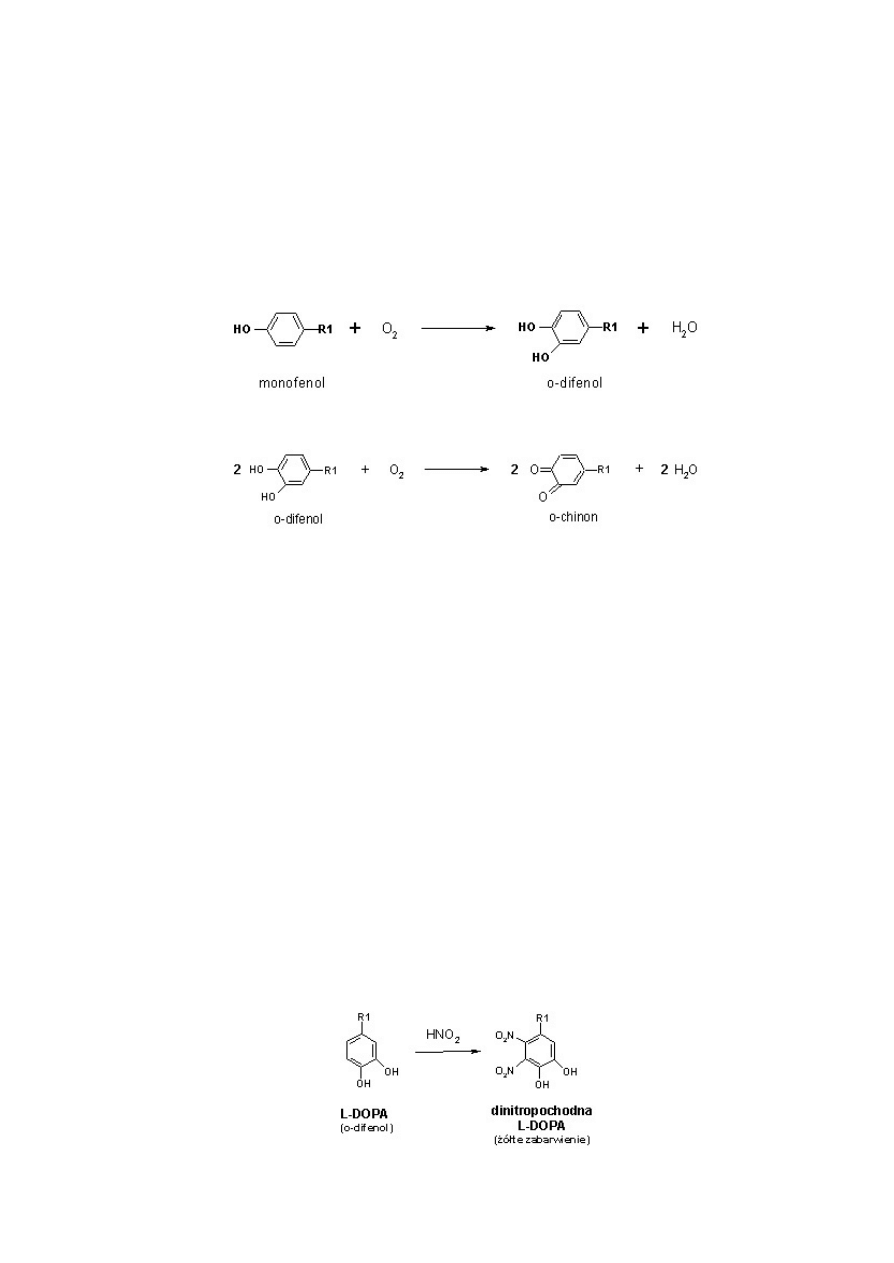
Tyrozynaza mo¿e katalizowaæ dwie ró¿ne reakcje chemiczne:
a) hydroksylacjê monofenoli do o-difenoli:
b) utlenianie o-difenoli do o-chinonów:
Spoœród ró¿norodnych zwi¹zków chemicznych o charakterze monofenoli, tyrozyna wydaje siê byæ
najwa¿niejszym substratem tyrozynazy. Enzym ten wydajnie hydroksyluje tyrozynê do L-DOPA (o-difenol,
por. powy¿ej), a nastêpnie umo¿liwia przekszta³cenie cz¹steczek L-DOPA do dopachinonów (o-chinon).
Proces powstawania dopachinonów mo¿na zablokowaæ przez dostarczenie do œrodowiska reakcji zwi¹zków
chemicznych o w³aœciwoœciach silnie redukuj¹cych np. askorbinianu lub NADH, co sprawia, ¿e jedynym
produktem reakcji katalizowanej przez tyrozynazê jest L-DOPA. Mo¿liwoœæ tê wykorzystuje siê w procesie
enzymatycznej produkcji L-DOPA, a pomiar iloœci powstaj¹cej L-DOPA w czasie pozwala okreœliæ aktywnoœæ
u¿ytej tyrozynazy.
Powszechnie stosowan¹, czu³¹ i prost¹ metod¹ detekcji oraz iloœciowego oznaczania L-DOPA w próbach nie
zawieraj¹cych innych o-difenoli (np. adernaliny, 2,3-dihydroksybenzoesanu etc.) jest chemiczne przekszta³cenie
tego aminokwasu do dinitrochinonu. Poniewa¿ powsta³y zwi¹zek charakteryzuje siê intensywnym, czerwono-
pomarañczowym zabarwieniem mo¿liwe jest oznaczenie jego zawartoœci metodami kolorymetrycznymi (l=460
nm). Proces derywatyzacji L-DOPA do dinitrochinonu przebiega w dwóch kolejnych reakcjach chemicznych
(Waite i Benedict, 1984; Arnow, 1937):
a) nitrowania L-DOPA do dinitropochodnej o charakterystycznym ¿ó³tym zabarwieniu:

14
b) utlenienia dinitropochodnej L-DOPA do dinitrochinonu w obecnoœci jonów wodorotlenowych:
Celem æwiczenia jest zapoznanie siê z enzymatyczn¹ metod¹ produkcji L-DOPA z wykorzystaniem
tyrozynazy uzyskanej z pieczarki dwuzarodnikowej (Agaricus bisporus).
Materia³y
200 g œwie¿ych pieczarek, mro¿onych w temperaturze ok. -20°C przez 12 godzin.
1. Lejek Büchnera o œrednicy ok. 20 cm oraz kr¹¿ek bibu³y filtracyjnej Whatman No. 1 dopasowany rozmiarem
do lejka, pompka wodna, homogenizator no¿owy, waga do równowa¿enia probówek wirówkowych, wirówka
z ch³odzeniem, woreczek do dializy (ok. 40 cm), spektrofotometr Spekol, kuwety spektrofotometryczne.
Odczynniki
1. 1000 ml acetonu sch³odzonego do temperatury ok. -20°C
2. 1500 ml 0,1 M sodowego buforu fosforanowego o pH 7,0 sch³odzonego do temperatury ok. +4°C
3. 80 ml mieszaniny reakcyjnej do oznaczania aktywnoœci tyrozynazy zawieraj¹cej 0,1 M sodowy bufor
fosforanowy pH 7,0, 7,5 mM askorbinian sodu, 2,5 mM tyrozynê
4. siarczan amonu, wysuszony w 50°C przez 24 godz. i zmielony
5. 35 ml 2 M NaOH
6. 30 ml 2 M HCl
2
7. 20 ml 15 % (w/obj.) roztwóru molibdenianu sodu zmieszanego z 20 ml 15 % (w/obj.) azotynu sodu (NaNO ),
8. 10 mM roztwór wzorcowy L-DOPA. Odwa¿yæ bardzo dok³adnie nawa¿kê L-DOPA (na 5 ml). Tu¿ przed
u¿yciem, rozpuœciæ w ogrzanej do temp. oko³o 60°C mieszaninie reakcyjnej do oznaczania aktywnoœci
tyrozynazy.
9. Odczynnik Bradford do oznaczania bia³ka (przygotowany)
Wykonanie
A. Otrzymywanie enzymu z pieczarki dwuzarodnikowej
Pokroiæ 250 g pieczarek na drobne kawa³ki i umieœciæ w zamra¿alniku lodówki lub zamra¿arce na okres 12
godzin.
Etap I. Ekstrakcja bia³ek acetonem
Wszystkie nastêpne czynnoœci wykonywaæ w temp. 2-4°C (w ch³odni). Pokrojone i mro¿one pieczarki
umieœciæ w homogenizatorze no¿owym, a nastêpnie dodaæ 300 ml acetonu sch³odzonego do temp. ok. -20°C.
Homogenizowaæ 4 impulsami po 15 sekund z krótkimi przerwami (5 sekund), przy niskich („Low”) obrotach
no¿y homogenizatora. Uzyskany homogenat przes¹czyæ przez bibu³ê filtracyjn¹ umieszczon¹ w lejku Büchnera.
Osiad³¹ na bibule papkê zebraæ i przenieœæ ponownie do homogenizatora dodaj¹c kolejn¹ porcjê 300 ml
sch³odzonego acetonu i homogenizowaæ jak wy¿ej, a nastêpnie powsta³y homogenat przes¹czyæ na lejku

15
Büchnera. Osiad³¹ papkê zebraæ, zwa¿yæ i pobraæ oko³o 10 g proszku acetonowego do probówki wirówkowej.
Pozosta³oœæ papki umieœciæ w homogenizatorze i dodaæ 220 ml 30% (obj./obj.) roztworu acetonu w sch³odzonej,
destylowanej wodzie. Homogenizowaæ 8 impulsami po 15 sekund (2 min.).
Homogenat przenieœæ do probówek wirówkowych. Wszystkie probówki zrównowa¿yæ i wirowaæ przez 25 minut
przy 10 000 x g w temp. 0°C. Osad proszku acetonowego w probówce wirówkowej zawiesiæ w 15 ml 0,1 M
buforu fosforanowego o pH 7,0. Z pozosta³ych probówek pobraæ do cylindra miarowego otrzymany supernatant,
zanotowaæ objêtoœæ i przelaæ do zlewki o odpowiedniej objêtoœci. Nastêpnie dodaæ, ci¹gle mieszaj¹c, 1,5
objêtoœci acetonu sch³odzonego do temp. -20°C i umieœciæ tak sporz¹dzony roztwór w zamra¿arce do czasu
utworzenia siê wyraŸnego osadu (1 -2 godz.). Powsta³¹ zawiesinê bia³ka oraz wczeœniej oddzielony proszek
acetonowy w buforze fosforanowym odwirowaæ jak wy¿ej. Supernatant odrzuciæ, a osad rozpuœciæ w 18 ml 0,1
M sodowego buforu fosforanowego (pH 7,0) sch³odzonego do temp. +4°C. Pobraæ do oddzielnej probówki
3 ml roztworu w celu oznaczenia bia³ka i aktywnoœci tyrozynazy. Próbkê przechowywaæ w stanie
zamro¿onym do czasu wykonywania oznaczeñ.
Etap II. Frakcjonowanie bia³ek siarczanem amonu
Wszystkie nastêpne czynnoœci wykonywaæ w temp. ok. 2°C (³aŸnia lodowa). Otrzymany ekstrakt rozcieñczyæ
dwukrotnie dodaj¹c 15 ml sch³odzonego 0,1 M sodowego buforu fosforanowego (pH 7,0). Nastêpnie do
ekstraktu (30 ml) powoli dodawaæ, ci¹gle mieszaj¹c, sta³y suchy siarczan amonowy do otrzymania roztworu
o stê¿eniu koñcowym równym 30 % roztworu nasyconego*. Roztwór pozostawiæ na okres 45 minut w ³aŸni
lodowej stale mieszaj¹c. Nastêpnie odwirowaæ powsta³¹ zawiesinê przez 30 minut przy 10 000 x g w temp. 0°C.
Osad odrzuciæ, a supernatant wysoliæ dodaj¹c siarczan amonu do wartoœci równej 70 % nasycenia roztworu. Po
45 minutach mieszania w ³aŸni lodowej, zawiesinê odwirowaæ jak wy¿ej, otrzymany osad rozpuœciæ w 8 ml
oziêbionego 0,1 M sodowego buforu fosforanowego (pH 7,0). Ponownie pobraæ do oddzielnej probówki 3
ml roztworu w celu oznaczenia bia³ka i aktywnoœci tyrozynazy. Próbkê przechowywaæ w stanie
zamro¿onym do czasu wykonywania oznaczeñ.
*Iloœci siarczanu amonu potrzebn¹ do otrzymania roztworu o stê¿eniu bêd¹cym u³amkiem roztworu nasyconego
obliczyæ z nastêpuj¹cego wzoru:
2
1
2
g
= [z×V/100] × [(S -S )/1-(p×S )]
g - iloœæ gramów siarczanu amonu
V - objêtoœæ pocz¹tkowa (ml)
1
S – nasycenie pocz¹tkowe (u³amek dziesiêtny)
2
S – nasycenie koñcowe (u³amek dziesiêtny)
Z – iloœæ gramów w roztworze nasyconym (53,3 g dla siarczanu amonu)
p – przyrost objêtoœci (0,3 dla siarczanu amonu)
Etap III. Dializa
Pozosta³¹ iloœæ roztworu otrzymanego w poprzednim etapie przelaæ do woreczka dializacyjnego. Dializê
prowadziæ wzglêdem 0,1 M sodowego buforu fosforanowego (pH 7,0, ok. 1000 ml) przez 6-12 h w temp. 2-4°C
przy ci¹g³ym mieszaniu buforu (mieszad³o magnetyczne). Preparat zamroziæ.
16
B. Oznaczanie aktywnoœci tyrozynazy - produkcja L-DOPA
Wyznaczanie krzywej wzorcowej do oznaczeñ L-DOPA
Do 7 jednorazowych, plastykowych i ponumerowanych probówek odmierzyæ nastêpuj¹ce iloœci roztworów
(w ìl):
Probówka
0
1
2
3
4
5
6
10 mM
L-DOPA
0
10
20
40
60
80
100
*Mieszanina
1000 990
980
960
940
920
900
*Mieszanina reakcyjna do oznaczania aktywnoœci tyrozynazy
Przy u¿yciu pipety automatycznej dodaæ do ka¿dej probówki po: 500 ìl 2 M HCl oraz 1000 ìl mieszaniny 15
% molibdenianu sodu z 15 % azotynem sodu. Nastêpnie zawartoœæ probówek zamieszaæ na mikrowytrz¹sarce,
dodaæ po 1000 ìl 2 M NaOH i ponownie zamieszaæ. Po 5 minutach zmierzyæ wartoœci absorbancji przy ë=460
nm wzglêdem próby odniesienia (probówka „0”). Na podstawie uzyskanych wyników wykreœliæ krzyw¹
wzorcow¹, odk³adaj¹c na osi rzêdnych wartoœci absorpcji (A), a na osi odciêtych zawartoœæ L-DOPA w próbie
(w ìmol).
Oznaczanie aktywnoœci tyrozynazy
4 zlewki o poj. 25 ml (podpisane I, II, III oraz IV) nape³niæ 15 ml mieszaniny reakcyjnej do oznaczania
aktywnoœci tyrozynazy. W zlewkach umieœciæ mieszade³ko magnetyczne, zapewniæ regularne i jak najwolniejsze
obroty(u¿yæ mieszad³a czteropozycyjnego). Przygotowaæ 4 zestawy probówek po 7 sztuk wg schematu i
odpipetowaæ do ka¿dej probówki po 500 ìl 2 M HCl. Reakcjê rozpoczyna siê dodaniem preparatów enzymów:
1) proszek acetonowy do zbiornika I (2 ml); 2) przed wysoleniem do zbiornika II (1 ml); 3) po wysoleniu
do zbiornika III (1 ml) oraz 4) po dializie do zbiornika IV (1 ml)
Zestaw probówek dla prób
pobieranych ze zbiornika X
0
20
40
60
80
100
120
X
X
X
X
X
X
X
Jako X opowiednio oznaczono I, II, III lub IV
W celu oznaczenia produkcji L-DOPA pobieraæ co 20 minut po 1000 ìl próby z ka¿dej zlewki i przenosiæ do
uprzedni przygotowanych probówek zawieraj¹cych 2 M HCl. Po zakoñczeniu reakcji dodaæ po 1000 ìl
mieszaniny 15 % molibdenianu amonu z 15 % azotynem sodu. Zawartoœæ probówek zamieszaæ na
mikrowytrz¹sarce i do ka¿dej dodaæ po 1000 ìl 2 M NaOH i ponownie zamieszaæ. Po 5 minutach zmierzyæ
wartoœci absorbancji przy ë=460 nm wzglêdem odpowiedniej próby pobranej w czasie „0”.
C. Oznaczanie zawartoœci bia³ka metod¹ Bradford
Zawartoœæ bia³ka w pobranych próbach oznaczyæ metod¹ Bradford (patrz opis przygotowania krzywej
wzorcowej i przeprowadzenia oznaczeñ str. 11). Orientacyjne, ca³kowite stê¿enia bia³ka po kolejnych etapach
oczyszczania enzymu (w mg/ml) wynosi: Etap I : 1,5 - 5, Etap II: 5 -10, Etap III: 5 - 10, Etap IV: 5 - 10.
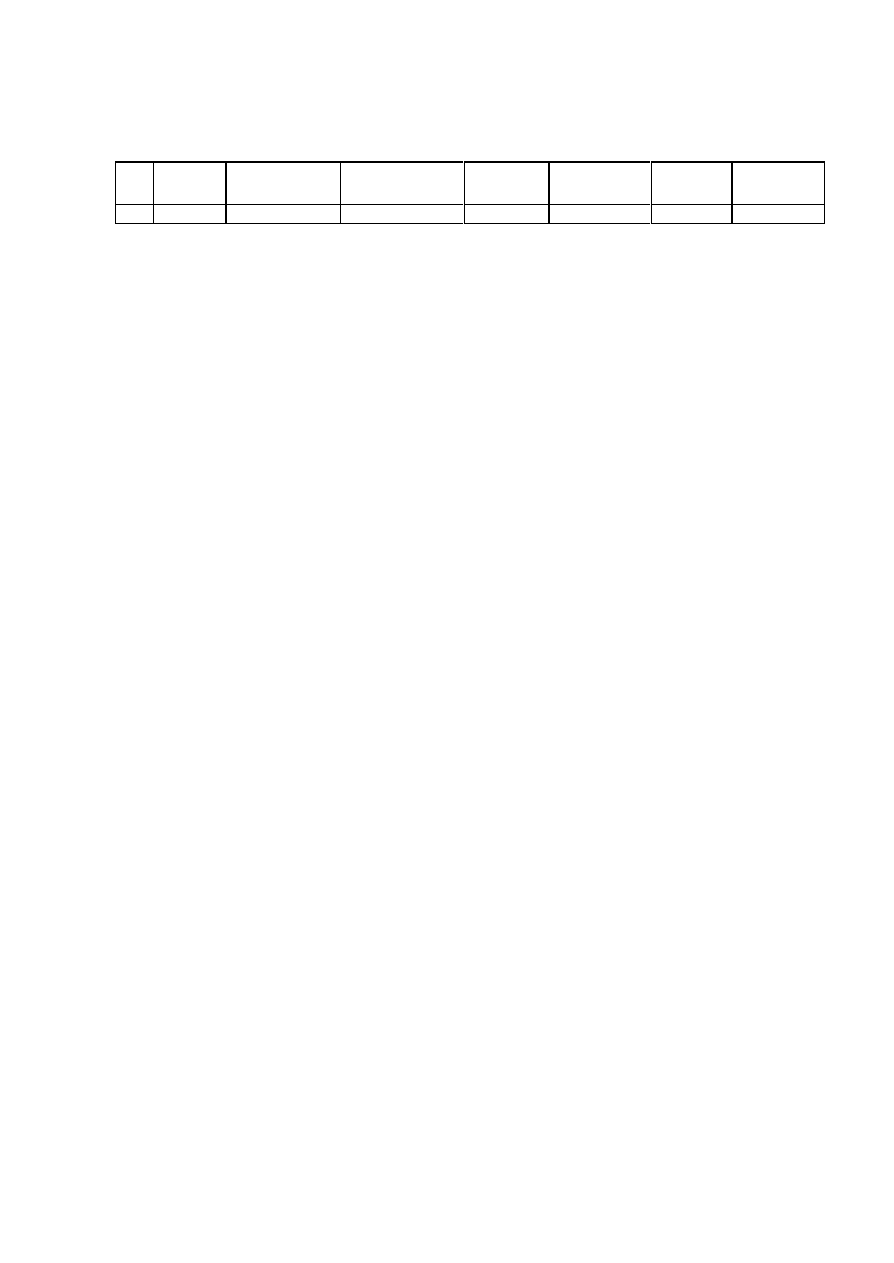
17
D. Opracowanie wyników
Na podstawie wykonanych pomiarów zawartoœci bia³ka i aktywnoœci w próbach z poszczególnych etapów
oczyszczania sporz¹dziæ bilans oczyszczania enzymu wg wzoru:
Etap Objêtoœæ
Stê¿enie
ca³kowite bia³ka
Ca³kowita
zawartoœæ bia³ka
AktywnoϾ
ca³kowita
AktywnoϾ
w³aœciwa
WydajnoϾ
Stopieñ
oczyszczenia
ml
mg/ml
mg
U
U/mg
%
%

18
IZOLOWANIE I OZNACZANIE AKTYWNOŒCI FOSFORYLAZY Z ZIEMNIAKÓW
Polisacharydy s¹ zwi¹zkami bardzo powszechnie wystêpuj¹cymi w przyrodzie. Zbudowane s¹ z powtarzalnych
jednostek sk³adaj¹cych siê z jednego do siedmiu monosacharydów. Pocz¹tkowo polisacharydy by³y postrzegane
jedynie jako materia³ strukturalny i zapasowy. Gdy okaza³o siê, ¿e modyfikowane polimery wêglowodanów
mog¹ mieæ ró¿ne zastosowania komercyjne, (np. we w³ókiennictwie, rolnictwie, czy kosmetyce) zainteresowanie
nimi znacznie wzros³o. Najprostsze polimery s¹ utworzone przez identyczne, nie modyfikowane reszty pentoz
lub heksoz po³¹czonych takimi samymi wi¹zaniami. Przyk³adami takich polimerów s¹: amyloza zbudowana z
reszt D-glukozy po³¹czonych wi¹zaniami a-1,4-glikozydowymi oraz laminaryna zbudowana z reszt D-glukozy
po³¹czonych wi¹zaniami b-1,3-glikozydowymi. Amylozê mo¿na syntetyzowaæ w reakcji katalizowanej przez
fosforylazê z ziemniaka, gdy¿ substratem tej reakcji jest glukozo-1-fosforan, a jako starter s³u¿y maltoheptoza.
Celem æwiczenia jest otrzymanie fosforylazy z ziemniaka, oznaczenie jej aktywnoœci i sporz¹dzenie bilansu
oczyszczania.
Odczynniki
2
2
4
1. Roztwór do homogenizacji: 0,5% podsiarczyn sodowy (Na S O ) zawieraj¹cy 0,5% cytrynianu sodowego
2. Siarczan amonowy, wysuszony w 50°C przez 24 godz. i zmielony
3. 0,5 M Tris-HCl, pH 8
4. 0,1 M bufor cytrynianowy, pH 6
5. 1 M bufor TRA (13.32 ml czystego TRA w koñcowej objêtoœci 100 ml), pH=6
6. 20 ml 2,5 M kwasu siarkowego
7. 5% roztwór molibdenianu amonowego
8. odczynnik fosforanowy (przygotowaæ tu¿ przed wykonaniem oznaczenia):
0.2 g p-metyloaminofenolu rozpuœciæ w 100 ml wody, dodaæ 1.04 ml wodorosiarczynu sodu, 20 ml
2.5 M kwasu siarkowego i 20 ml 5% molibdenianu amonowego
9. Glukozo-1-fosforan
10. Maltoheptoza
11. Kwaœny fosforan sodowy
12. Odczynnik Bradford do oznaczania bia³ka (przygotowany)
A. Otrzymywanie i oczyszczanie preparatu fosforylazy
Etap I. Homogenat z ziemiaka
Oko³o 1 kg obranych i op³ukanych ziemniaków zhomogenizowaæ w 100 ml roztworu do homogenizacji w
temperaturze 4 C. Homogenat przes¹czyæ przez 4 warstwy gazy i odwirowaæ przy 4 000 x g przez 30 min. w
o
temperaturze 4 C. pH otrzymanego supernatantu doprowadziæ do wartoœci 7 u¿ywaj¹c 0,5 M buforu Tris-HCl
o
o pH=8. Do probówki pobraæ 10 ml roztworu w celu oznaczenia bia³ka i aktywnoœci fosforylazy. Próbkê
przechowywaæ w stanie zamro¿onym do czasu wykonywania oznaczeñ.

19
Etap II. Denaturacja cieplna
Supernatant ogrzaæ do temperatury 55 C i inkubowaæ w takich warunkach przez 40 min., a nastêpnie
o
odwirowaæ przy 4 000 x g przez 30 min (4 C). Do oddzielnej probówki pobraæ 10 ml supernatantu w celu
o
oznaczenia bia³ka i aktywnoœci fosforylazy. Próbkê przechowywaæ w stanie zamro¿onym do czasu
wykonywania oznaczeñ.
Etap III. Frakcjonowanie siarczanem amonowym
Otrzymany w poprzednim etapie supernatant nale¿y wysoliæ siarczanem amonu. W tym celu do otrzymanego
roztworu dodaæ ma³ymi porcjami 1/10 (wag./obj., 10 g na 100 ml) siarczanu amonu i mieszaæ w temperaturze
4 C przez 30 minut, a nastêpnie odwirowaæ przy 4 000 x g przez 30 min. Do otrzymanego supernatantu dodaæ
o
1/4 (wag./obj., 25 g na 100 ml roztworu) siarczanu amonowego i mieszaæ przez 30 minut a nastêpnie odwirowaæ
jak wy¿ej. Otrzymany osad rozpuœciæ w niewielkiej iloœci wody destylowanej (2-5 ml) i zamroziæ.
B. Oznaczanie aktywnoœci fosforylazy
W 50 ml buforu cytrynianowego rozpuœciæ 11 mg glukozo-1-fosforanu i 7 mg maltoheptozy. Tak
przygotowanej mieszaniny reakcyjnej odmierzyæ po 6 ml do próbówek plastikowych o objêtoœci 10 ml i wstawiæ
na oko³o 5 minut do ³aŸni wodnej nastawionej na temp. 45 C. Nastêpnie rozpocz¹æ reakcjê syntezy amylozy
o
dodaniem 1 lub 2 ml prób pobieranych w kolejnych etapach oczyszczania fosforylazy. Zawartoœæ probówek
dobrze wymieszaæ. Natychmiast po dodaniu ekstraktów pobraæ 1 ml próby w czasie 0. Kolejne próby o objêtoœci
1 ml pobieraæ z mieszaniny reakcyjnej w 5, 10, 15, 20, 25 i 30 minutach inkubacji. Reakcjê przerwaæ poprzez
wstawienie prób do ³aŸni wodnej o temp. 100 C na 10 min. Nastêpnie nale¿y niezw³ocznie oznaczyæ we
o
wszystkich próbach zawartoœæ fosforanu. Gdy oznaczenie wykonuje siê poŸniej, próby natychmiast zamroziæ.
C. Oznaczanie fosforanu
Pobrane w trakcie inkubacji poszczególnych preparatów fosforylazy próby (punkt B) ogrzewaæ przez 10 min.
w temperaturze 100 C. Do kolejno ponumerowanych probówek odmierzyæ: 50 - 300 ìl badanej próby, 3,5 ml
o
odczynnika fosforanowego oraz bufor TRA do koñcowej objêtoœci 5 ml. Ca³oœæ wymieszaæ i pozostawiæ przez
15 minut. Pomiary zawartoœci fosforanu prowadziæ przy d³ugoœci fali 578 nm wzglêdem próby odniesienia
(1.5 ml TRA i 3.5 ml odczynnika fosforanowego).
D. Przygotowanie krzywej wzorcowej do oznaczenia fosforu
Przygotowaæ szeœæ roztworów kwaœnego fosforanu sodowego o objêtoœci 10 ml zawieraj¹cych odpowiednio
nastêpuj¹ce iloœci fosforu: 125 ìg, 250 ìg, 500 ìg, 750 ìg, 1 m g oraz 1,25 mg. Do o ponumerowanych
probówek odmierzyæ po 200 ìl roztworu fosforanu sodowego zawieraj¹cego odpowiedni¹ iloœæ fosforu, 3,5 ml
odczynnika fosforanowego oraz buforu TRA do koñcowej objêtoœæ 5 ml. Ca³oœæ wymieszaæ i inkubowaæ przez
15 minut. Pomiary stê¿enia fosforanu prowadziæ przy d³ugoœci fali 578 nm wzglêdem próby odniesienia (1.5
ml TRA i 3.5 ml odczynnika fosforanowego). Wykreœliæ krzyw¹ wzorcow¹ odk³adaj¹c na osi odciêtych iloœci
fosforu w próbie, a na osi rzêdnych odpowiadaj¹ce im wartoœci absorbancji.
E. Oznaczanie zawartoœci bia³ka metod¹ Bradford
Zawartoœæ bia³ka w pobranych próbach oznaczyæ metod¹ Bradford (patrz opis przygotowania krzywej
wzorcowej i przeprowadzenia oznaczeñ na str. 11).
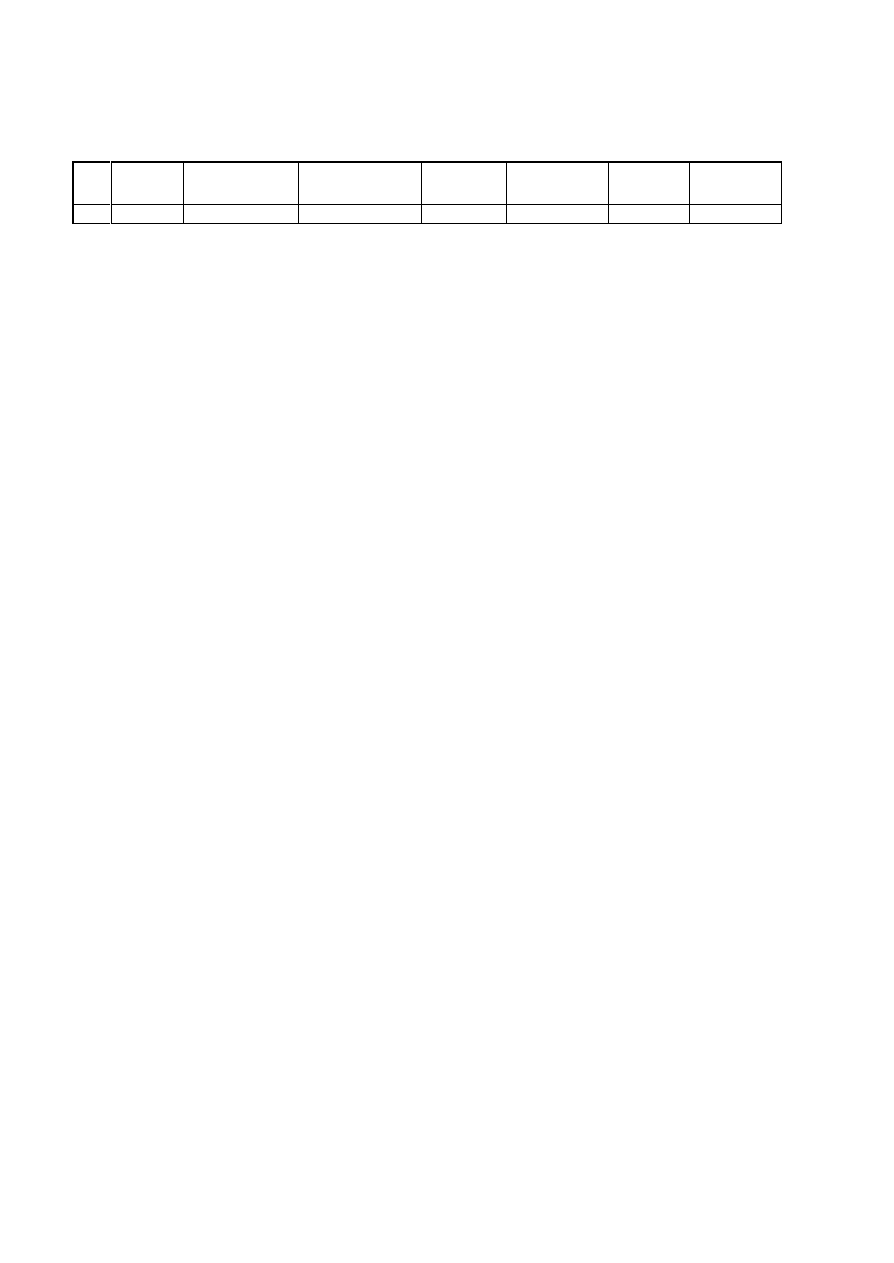
20
F. Opracowanie wyników
Na podstawie wykonanych pomiarów zawartoœci bia³ka i aktywnoœci w próbach pobranych w poszczególnych
etapach oczyszczania sporz¹dziæ bilans oczyszczania enzymu wg wzoru:
Etap Objêtoœæ Stê¿enie bia³ka
Ca³kowita
zawartoœæ bia³ka
AktywnoϾ
ca³kowita
AktywnoϾ
w³aœciwa
WydajnoϾ
Stopieñ
oczyszczenia
ml
mg/ml
mg
U
U/mg
%
%

21
O Z N A C Z A N I E A K T Y W N O Œ C I W I E L O E N Z Y M O W E G O U K £ A D U
FOTOSYNTETYCZNEGO
A. Izolowanie chloroplastów
W badaniach uk³adów fotosyntetycznych stosuje siê powszechnie izolowane chloroplasty. Wybór jednej
spoœród wielu metod preparatywnych zale¿y od rodzaju planowanych badañ. W zale¿noœci od stopnia
strukturalnej i funkcjonalnej integralnoœci otrzymanych chloroplastów wyró¿nia siê kilka ich klas (klasyfikacja
Halla). W wielu badaniach wystarczaj¹ca jest prosta technika wirowania ró¿nicowego homogenatu liœci w
izotonicznym zbuforowanym roztworze o odpowiedniej sile jonowej i pH.
Materia³y i odczynniki.
1. Œwie¿e liœcie grochu lub szpinaku (20-50g)
2. 50 mM bufor Tris-HCl o pH 7,5 zawieraj¹cy 0,4 M sacharozê, 40 mM kwas askorbinowy, 20 mM chlorek
sodowy (bufor A). Odwa¿yæ sacharozê, kwas askorbinowy, NaCl, odpowiedni¹ iloœæ roztworu Tris
odmierzyæ ze stê¿onego roztworu. Po rozpuszczeniu sk³adników, doprowadziæ pH kwasem solnym do
wartoœci 7,5. Sporz¹dziæ 200 ml roztworu.
3. Bufor B o sk³adzie takim samym jak roztwór A, za wyj¹tkiem askorbinianu. pH tego roztworu doprowadziæ
roztworem HCl do wartoœci 7,0. Przygotowaæ 100 ml roztworu.
2
4. 20 mM bufor Hepes-NaOH o pH 7,0 zawieraj¹cy 0,33 M sorbitol, 5 mM NaCl, 4 mM MgCl (bufor C)
(przygotowany).
5. 80% wodny roztwór acetonu.
Materia³y pomocnicze i aparatura
Fizelina, homogenizator no¿owy, wirówka z ch³odzeniem, homogenizator Pottera-Elvenhjema,
mikrowytrz¹sarka, spekol, kuwety szklane.
Wykonanie
Ca³¹ preparatykê nale¿y wykonywaæ szybko w temperaturze nie wy¿szej ni¿ 4 C (ch³odnia) i w przyæmionym
o
œwietle.
20-50 g liœci grochu lub pozbawionych nerwów g³ównych liœci szpinaku przemyæ wod¹ destylowan¹. Liœcie
zhomogenizowac w oko³o 200 ml oziêbionego do temperatury ok. 2 C buforu A przez 10 s, przy maksymalnych
o
obrotach no¿y homogenizatora. Nastêpnie przes¹czyæ homogenat przez cztery warstwy fizeliny do zlewki
znajduj¹cej siê w ³aŸni lodowej.
Przes¹cz odwirowaæ w wirówce z ch³odzeniem przez 2 minuty przy przyœpieszeniu 225 x g. Osad odrzuciæ,
a supernatant wirowaæ przez 10 minut przy przyœpieszeniu 1000 x g. Supernatant odrzuciæ, a osad chloroplastów
zawiesiæ w oko³o 100 ml och³odzonego do ok. 2 C buforu B i odwirowaæ przez 10 minut przy przyœpieszeniu
o
1000 x g. Supernatant zdekantowaæ, a osad chloroplastów zawiesiæ w homogenizatorze Pottera-Elvenhjema w
jak najmniejszej objêtoœci (0,5-1,5 ml) zimnego buforu C.
Zawiesinê chloroplastów przechowywaæ w ciemnoœci w ³aŸni lodowej. Oznaczyæ stê¿enie chlorofilu oraz
aktywnoœæ fotosyntetycznego transportu elektronów. Je¿eli oznaczeñ nie przeprowadza siê w dniu preparatyki,
nale¿y zamroziæ chloroplasty w plastikowej probówce w temperaturze -80 C. Preparat rozmra¿aæ w 4 C
o
o
wytrz¹saj¹c delikatnie przy pomocy mikrowytrz¹sarki.

22
B. Oznaczenie stê¿enia chlorofilu
Do 0,025 ml zawiesiny chloroplastów w probówce dodaæ 10 ml 80% (v/v) wodnego roztworu acetonu.
Zawartoœæ probówek wytrz¹saæ przez oko³o 30 sekund w celu wyekstrahowania chlorofilu i ekstrakt przes¹czyæ
przez bibu³ê filtracyjn¹ do kuwet szklanych o d³ugoœci drogi œwietlnej 1 cm. Niezw³ocznie zmierzyæ absorpcjê
otrzymanych roztworów przy d³ugoœci fali 652 nm, wobec próby odniesienia zawieraj¹cej 80% wodny roztwór
acetonu. W celu obliczenia stê¿enia chlorofilu (w mg/ml zawiesiny) nale¿y pomno¿yæ wartoœæ absorpcji przez
wspó³czynnik 11,6.
C. Oznaczenie aktywnoœci fotosyntetycznego transportu elektronów
Izolowane chloroplasty katalizuj¹ reakcj oksydoredukcyjn¹ uwarunkowan¹ dzia³aniem œwiat³a, w której woda
jest naturalnym donorem elektronów. W fotosyntezie roœlinnej zachodz¹ dwa rodzaje reakcji œwietlnych :
krótkofalowa wynikaj¹ca z aktywnoœci II uk³adu fotosyntezy (PSII) i d³ugofalowa wynikaj¹ca z aktywnoœci I
uk³adu fotosyntezy (PSI). Aktywnoœæ PS II i PS I mierzy siê stosuj¹c sztuczne donory i akceptory elektronów
odpowiednio dostarczaj¹ce lub pobieraj¹ce elektrony w ró¿nych miejscach ³añcucha fotosyntetycznego:
2
H O
))))))))
> PS II
)))))))))))))))))))))
> PS I
))))))))))
> NADP
+
*
*
*
*
sztuczny sztuczny sztuczny sztuczny
donor e akceptor e donor e akceptor e
-
-
-
-
1 1 2 2
Aktywnoœæ PSII mo¿na mierzyæ jako szybkoœæ wydzielania tlenu przez chloroplasty. Tlen jest bowiem
produktem reakcji zachodz¹cej w PSII. Pomiary przeprowadza siê u¿ywaj¹c elektrody tlenowej Clarka. Po
zahamowaniu transportu elektronów pomiêdzy sztucznym akceptorem 1 a sztucznym donorem elektronów 2,
w obecnoœci zredukowanej formy donora elektronów 2 mo¿na badaæ aktywnoœæ PSI równie¿ u¿ywaj¹c elektrody
tlenowej Clarka. Wykorzystuje siê fakt, ¿e zredukowana forma akceptora elektronów 2 ulega samoutlenieniu
z równoczesnym wytworzeniem rodnika ponadtlenkowego, co wymaga udzia³u tlenu. Reakcja ta nosi nazwê
reakcji Mehlera.
Zastosowanie egzogennego akceptora elektronów umo¿liwia badanie fotofosforylacji
zwi¹zanej z niecyklicznym transportem elektranów w chloroplastach, mimo ¿e w czasie izolowania organelli
jest usuwana z nich ferredoksyna oraz NADP. W celu okreœlenia stopnia sprzê¿enia otrzymanego preparatu
chloroplastów mierzy siê stymulacjê szybkoœci niecyklicznego transportu elektronów pod wp³ywem
egzogennego ADP i fosforanu.
Przez analogiê do kontroli oddechowej mitochondriów, stosunek szybkoœci transportu elektronów przed
dodaniem ADP i fosforanu, do szybkoœci transportu elektronów po ich dodaniu, nazwano kontrol¹
fotosyntetyczn¹. Oprócz zwi¹zków rozprzêgaj¹cych mitochondrialn¹ i fotosyntetyczn¹ fosforylacjê niecykliczn¹,
jak 2,4-dinitrofenol i FCCP (karbonylocyjanek p-trifluorome-toksyfenylohyd-razonu), znane s¹ te¿ zwi¹zki
rozprzêgaj¹ce specyficznie fosforylacjê fotosyntetyczn¹. Nale¿¹ do nich jony amonowe.
Do szczegó³owego scharakteryzowania ³añcucha fotosyntetycznego transportu elektronów, przyczyni³o siê
zastosowanie szeregu inhibitorów np. Tris-HCl w wysokich stê¿eniach ( >0,8 M; >pH 8,3), kwasów
t³uszczowych, hydroksyloaminy, herbicydów, aminotriazyn, 1,10-fenantroliny, DBMIB
(2,5-dibromo-3-metylo-6-izopropylo-p-benzochinonu), chlorku rtêciowego, cyjanku, DSIP-SA (kwasu di-
salicyloino-denopropanodiamino-1,2- disulfonowego).

23
Materia³y i odczynniki
1. Œwie¿o otrzymane lub rozmro¿one chloroplasty ze szpinaku lub grochu o stê¿eniu chlorofilu 4-6 mg/ml
2. Mieszanina podstawowa do pomiarów aktywnoœci PS II i ca³ego ³añcucha fotosyntetycznego zawieraj¹ca:
2
0.8 M sacharozê, 40 mM Tris, 40 mM NaCl, 10 mM MgCl . Mieszaninê przygotowaæ przez zmieszanie
stê¿onych roztworów wyjœciowych i po doprowadzeniu pH roztworem HCl do wartoœci 7,6 uzupe³niæ
wod¹ dejonizowan¹ do ¿¹danej objêtoœci. Przygotowaæ 50 ml mieszaniny.
3. 50 mM alkoholowy roztwór fenylo-p-benzochinonu, akceptora elektronów z II uk³adu fotosyntezy
(odczynnik przygotowany).
4. Mieszanina reakcyjna do pomiarów aktywnoœci PSII. Nale¿y j¹ przygotowaæ, tu¿ przed pomiarem,
poprzez 2-krotne rozcieñczenie mieszaniny podstawowej i 125-krotne rozcieñczenie roztworu fenylo-p-
benzochinonu. Aby rozpuszczony w alkoholu zwi¹zek nie wytr¹ci³ siê w czasie dodawania do roztwóru
wodnego, nale¿y roztwór akceptora dodawaæ ma³ymi kroplami, ca³y czas mieszaj¹c roztwór wodny.
5. 1 mM wodny roztwór metylowiologenu, akceptora elektronów z I uk³adu fotosyntezy. Uwaga -
metylowiologen jest œmierteln¹ trucizn¹.
6. Mieszanina reakcyjna do pomiarów aktywnoœci ca³ego ³añcucha fotosyntetycznego. Nale¿y j¹
przygotowaæ, tu¿ przed pomiarem, poprzez 2-krotne rozcieñczenie mieszaniny podstawowej (2) i 7.5-
krotne rozcieñczenie roztworu metylowiologenu.
7. Mieszanina reakcyjna do pomiarów aktywnoœci PSI o pH 8,0 zawieraj¹ca: 50 mM Tris, 16 mM
askorbinian sodu, 66 ìM TMPD (N,N,N,N-tetrametylo-p-fenylodiamina), 133 ìM MV (metylowiologen)
(przygotowana).
8. Etanolowy roztwór kwasu linolenowego o stê¿eniu 10 mg/ml (przygotowany).
9. 0,025 mM roztwór DCMU (przygotowany).
10. 5,0 mM etanolowy roztwór DBMIB (przygotowany).
11. 8 mM etanolowy roztwór DCMU (przygotowany).
12. 0,96 M roztwór metyloaminy (przygotowany).
Materia³y pomocnicze i aparatura
£aŸnia lodowa, pipety automatyczne, elektroda Clarka umieszczona w termostatyzowanym naczynku z
urz¹dzeniami towarzysz¹cymi i rejestratorem, Ÿród³o œwiat³a o natê¿enie 900 ìmoli m s , pH-metr.
-2 -2
Wykonanie pomiarów
Czynnoœci przygotowawcze. Ustawiæ temperaturê termostatu na 25 C lub wykonywaæ pomiar w stabilnej
o
temperaturze pokojowej. Uruchomiæ urzadzenia towarzysz¹ce. Umyæ dok³adnie, pos³uguj¹c siê pompk¹ wodn¹,
naczynko elektrodowe (wod¹ destylowan¹ i etanolem). Ustaliæ szybkoœæ mieszania roztworu w naczynku
elektrodowym tak, aby nie tworzy³y siê pêcherzyki powietrza. Nale¿y wykalibrowaæ uk³ad pomiarowy, w tym
celu do naczynka elektrodowego odmierzyæ odpowiedni¹ objêtoœæ napowietrzonej w temperaturze 25 C
o
mieszaniny reakcyjnej uzyskan¹ wartoœæ uznaj¹c za 100% nasyceniu mieszaniny tlenem. Po usuniêciu z
naczynka mieszaniny zawierajêcej tlen, odmierzyæ do naczynka odpowiedni¹ objêtoœæ mieszaniny reakcyjnej
nasyconej azotem w temperaturze 25 C. Otrzyman¹ wartoœæ uznaæ za odpowiadaj¹c¹ zerowemu nasyceniu
o
mieszaniny reakcyjnej tlenem. Nale¿y zanotowaæ wartoœci odpowiadaj¹ce szerokoœæ skali pomiarowej.
Pomiary wydzielania i poch³aniania tlenu przeprowadziæ wykorzystuj¹c komputerowy uk³ad rejestracji danych,
z³o¿ony z karty analogowo-numerycznej i odpowiedniego programu. Ustalenie maksymalnego i minimalnego
stê¿enia tlenu oraz zasad obliczania wartoœci szybkoœci poch³aniania czy wydzielania tlenu nale¿y wykonaæ
korzystaj¹c z porad prowadz¹cego.

24
Pomiary wydzielania tlenu
Oznaczenie aktywnoœci PSII. Do naczynka z mieszanin¹ reakcyjn¹ (4) nasycon¹ azotem dodaæ w ciemnoœci tak¹
iloœæ chloroplastów aby otrzymaæ prostoliniowy przebieg reakcji wydzielania tlenu. Jeœli jest inaczej powtórzyæ
oznaczenie odpowiednio zwiêkszaj¹c lub zmniejszaj¹c iloœæ dodawanego preparatu.Stê¿enie chloroplastów w
naczynku elektrodowym powinno wynosiæ oko³o10 ìg Chl/ml mieszaniny. Przeprowadziæ dalsze oznaczenia,
dodaj¹c w po 1-2 minucie oœwietlania:
1.
Kolejno porcje po 1-5 ìl 0,025 mM DCMU
2.
10-15 ìl kwasu linolenowego o stê¿eniu 10 mg/ml.
Pomiary pobierania tlenu.
Oznaczenie aktywnoœci ca³ego ³añcucha fotosyntetycznego. Do naczynka pomiarowego dodaæ natlenowan¹
mieszaninê reakcyjn¹ (6). Po ustabilizowaniu siê uk³adu pomiarowego, dodaæ do naczynka w ciemnoœci preparat
chlorolastów w iloœci takiej samej lub 2-krotnie mniejszej ni¿ w czasie pomiarów aktywnoœci PSII. W³¹czyæ
Ÿród³o œwiat³a. Przeprowadziæ oznaczenia, dodaj¹c w kolejnych próbach po 1-2 minucie oœwietlania:
1.
tak¹ iloœæ 0.98 M metyloaminy, aby jej stê¿enie koñcowe wynios³o 3 mM (jeœli do oznaczeñ stosuje siê
chloroplasty uprzednio zamro¿one i przechowywane w - 80 C, pomin¹æ to oznaczenie). Reakcje
o
przeprowadzaæ do momentu wyczerpania siê tlenu w naczynku elektrodowym.
2.
1-5 ìl 5,0 mM DBMIB.
Oznaczenie aktywnoœci PSI. Do umytego wod¹ i etanolem naczynka elektrodowego odpipetowaæ dwukrotnie
rozcieñczon¹ mieszaninê reakcyjn¹ (7) i 3 ìl 8 mM DCMU. Dodaæ zawiesinê chloroplastów w iloœci ustalonej
poprzednio i po ustabilizowaniu siê uk³adu w³¹czyæ Ÿród³o œwiat³a. Reakcjê przeprowadziæ do momentu
wyczerpania siê tlenu.
D. Opracowanie wyników
Otrzymane dane pomiarowe w formacie ASCII zaimportowaæ do arkusza kalkulacyjnego. Opracowaæ wykresy
przedstawiaj¹ce zmiany napiêcia (mV) w funkcji czasu. Na podstawie prostoliniowych przebiegów zmian
2
napiêcia elektrodowego obliczyæ szybkoœæ reakcji poch³aniania lub wydzielania O w mV/min, a nastêpnie
2
przeliczyæ szybkoœæ reakcji na iloœæ ìmoli O /min. Do obliczeñ potrzebna jest znajomoœæ skali pomiarowej (0%
i 100% stê¿enia tlenu) oraz informacja, ¿e w temperaturze 25 C stê¿enie tlenu w mieszaninie reakcyjnej wynosi
o
0,25 mM.
2
Aktywnoœæ PSII wyraziæ w ìmolach O wydzielonego na godzinê na mg chlorofilu. Obliczyæ stymulacjê
aktywnoœci przez zwi¹zek rozprzêgaj¹cy. Wykreœliæ krzyw¹ hamowania wydzielania tlenu przez DCMU
odk³adaj¹c na osi odciêtych stê¿enie DCMU w mieszaninie reakcyjnej, a na osi rzêdnych szybkoœæ wydzielania
tlenu (mo¿na j¹ wyraziæ w % kontroli). Wyznaczyæ przez interpolacjê stê¿enie inhibitora, przy którym
obserwuje siê hamowanie reakcji w 50%. Obliczyæ, w jakim stopniu reakcja jest hamowana przez kwas
2
t³uszczowy, a w jakim przez DBMIB. Aktywnoœæ PSI wyraziæ w ìmolach O pobranego na godzinê na mg
chlorofilu.
Wyniki zestawiæ w tabeli.
25
ZASTOSOW ANIE IM M OBILIZACJI ENZYM ÓW W BIOTECHNOLOGII.
OZNACZENIE AKTYWNOŒCI IMMOBILIZOWANEJ INWERTAZY
Immobilizacja enzymów. Immobilizacjê mo¿na okreœliæ jako technikê, która znacznie ogranicza swobodn¹
dyfuzjê cz¹stek enzymu lub komórek. Metody immmoblizacji enzymów maj¹ zastosowanie w biochemii,
biotechnologii, mikrobiologii, in¿ynierii chemicznej, g³ównie ze wzglêdu na mo¿liwoœæ ich wykorzystania
technologicznego i handlowego. Enzymy mo¿na immobilizowaæ metodami chemicznymi, np. kowalencyjnie
wi¹¿¹c z noœnikami rozpuszczalnymi lub nierozpuszczalnymi w wodzie, lub metodami fizycznymi, np. poprzez
adsorpcjê na nierozpuszczalnych noœnikach, inkluzjê w sieci polimerowej czy mikrokapsu³kowanie wewn¹trz
pó³przepuszczalnych membran. Stosowana procedura nie powinna wp³ywaæ na aktywnoœæ biologiczn¹ enzymu.
Od rodzaju matrycy i sposobu wi¹zania zale¿y iloœæ, aktywnoœæ, stabilnoœæ, optimum pH i temperatury dzia³ania
immobilizowanego enzymu.
Przyk³adem immobilizacji jest wykorzystanie jako z³o¿a kwasu alginowego do inkluzji inwertazy, enzymu
hydrolizuj¹cej sacharozê. Alginian jest to polisacharyd zbudowany z reszt kwasów D-mannuronowego (M) i
L-guluronowego (G) po³¹czonych wi¹zaniami 1,4. Wystêpuj¹ w nim bloki sk³adaj¹ce siê z M, z G oraz
x
y
z
mieszane: (M–M) –(G–G) –(M–G)
Kwas alginowy
Alginian otrzymuje siê z wodorostów lub niektórych szczepów bakteryjnych. Ma masê cz¹steczkow¹ oko³o
250 000, jest nierozga³êziony, d³ugo³añuchowy. Rozpuszczony w wodzie daje lepkie roztwory, które mo¿na
sterylizowaæ (20 min w temp. 121°C). Cech¹ charakterystyczn¹ alginianu jest jego pêcznienie, a z jonami metali
dwu- i trójwartoœciowych (np. Ca , Zn , Mn , Sr , Ba , Al , Fe , lecz nie z Mg ) tworzenie ¿eli. ¯el
2+
2+
2+
2+
2+
3+
3+
2+
alginianowy jest doœæ stabilny, obojêtny chemicznie i ma strukturê mikroporowat¹. W czasie immobilizacji
enzymów w ¿elu alginianowym nie obserwuje siê istotnej denaturacji enzymu, poniewa¿ nie towarzysz¹ temu
procesowi zmiany temperatury i pH.
Celem æwiczenia jest zbadanie aktywnoœci immobilizowanej inwertazy (â-fruktofuranozydazy, EC 3.2.1.26)
oraz okreœlenie wydajnoœci zamkniêtego, cyklicznego reaktora, w którym zachodzi reakcja hydrolizy sacharozy.
Odczynniki
1. 66 mM pirofosforan sodu (pH 8,5) (50 ml)
2. Alginian sodowy (1,5% zawiesina) (10 ml)
2
3. 0,1 M CaCl (50 ml)
4. 0,6 M roztwór sacharozy (30 ml)
5. 2% kwas 3,5-dinitrosalicylowy (DNS) w 0,4 M NaOH (50 ml)
6. 0,05 M bufor octanowy (pH 4,7) (20 ml)
7. Roztwór wzorcowy glukozy (2 mg/ml) (10 ml)
26
Materia³y pomocnicze i aparatura
Kolumna (strzykawka) ok. 1–3 ml, termostatyzowany zbiornik 100–150 ml, pompa perystaltyczna – przep³yw
od 1 do 6 ml/min (zale¿nie od przekroju rurki), mieszad³o magnetyczne, termostat 37°C.
Wykonanie
A. Ekstrakcja inwertazy z dro¿d¿y piekarniczych
10 g suchych (lub 30 g œwie¿ych) dro¿d¿y piekarskich zawiesiæ w 30 ml 66 mM pirofosforanu sodu (pH 8,5)
i pozostawiæ na noc w 20°C (lub termostacie w 37°C). Odwirowaæ przy 5000 x g przez 20 min. Otrzymany
supernatant, zawieraj¹cy m.in. inwertazê, przechowywaæ zamro¿ony w –20°C.
Studenci otrzymuj¹ przygotowany ekstrakt z dro¿d¿y piekarniczych
B. Immobilizacja inwertazy
Ekstrakt inwertazy (2 ml) miesza siê z 10 ml 1,5% zawiesiny alginianu sodu. Nastêpnie zawiesinê
2.
alginian-enzym pipet¹ dodaje siê kroplami do roztworu 0,1 M CaCl Powstaj¹ce granulki immobilizowanego
2
enzymu o œrednicy od 0,1–4 mm pozostawia siê na 20 min w roztworze CaCl w celu ich stwardnienia.
2
Nastêpnie zlewa siê roztwór CaCl , a otrzymane granulki immobilizowanego enzymu przep³ukuje wod¹
destylowan¹ i formuje z nich z³o¿e kolumny reaktora.
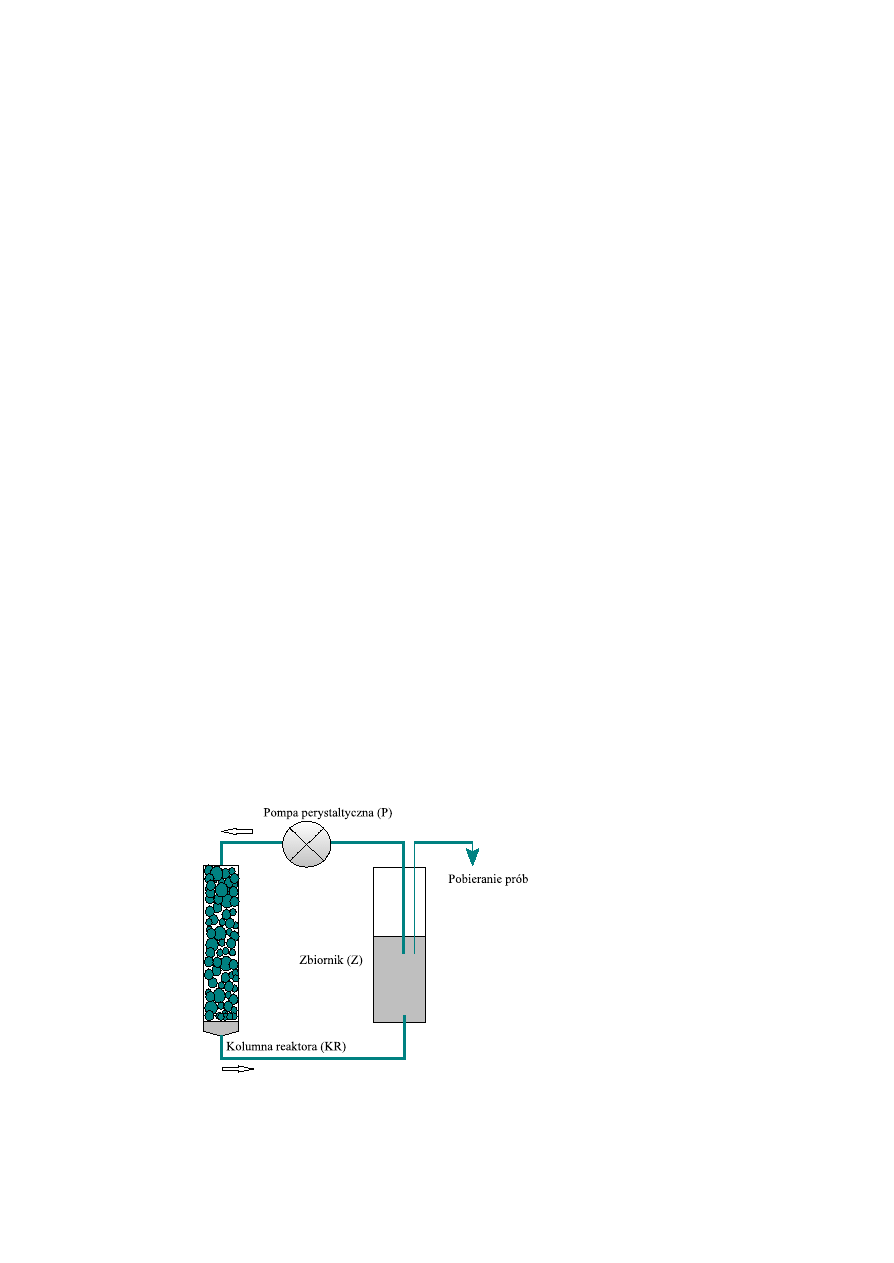
C. Budowa zamkniêtego, cyklicznego uk³adu reaktora
Uk³ad reaktora sk³ada siê z trzech czêœci: kolumny reaktora (KR), zbiornika (Z) i pompy perystaltycznej (P).
Kolumna reaktora ma objêtoœæ 1–3 ml, a zbiornik 45 ml. Szybkoœæ przep³ywu, rzêdu 1–5 ml min , zale¿y od
–1
przekroju rurki i wydajnoœci pompy.
Kolumn¹ reaktora mo¿e byæ np. strzykawka, zamkniêta zwitkiem waty szklanej. Wyp³yw z kolumny ³¹czy siê
elastyczn¹ rurk¹ ze zbiornikiem. Bufor zawieraj¹cy sacharozê jest podawany ze zbiornika na kolumnê za
pomoc¹ pompy perystaltycznej (P). Kolumnê wype³nia siê ma³ymi ziarnami immobilizowanego enzymu.
Wewnêtrzny przekrój rurki ³¹cz¹cej kolumnê ze zbiornikiem powinien byæ ma³y, by zawiera³ niewielk¹ objêtoœæ
roztworu.
Zawartoœæ termostatyzowanego zbiornika (37°C) powinna byæ mieszana. Kolumna reaktora mo¿e byæ
termostatowana, co przyspiesza reakcjê, ale nie jest to konieczne, je¿eli szybkoœæ przep³ywu ogrzanego roztworu
jest na tyle szybka, aby zapobiec jego sch³odzeniu.
Budowa zamkniêtego, cyklicznego uk³adu reaktora
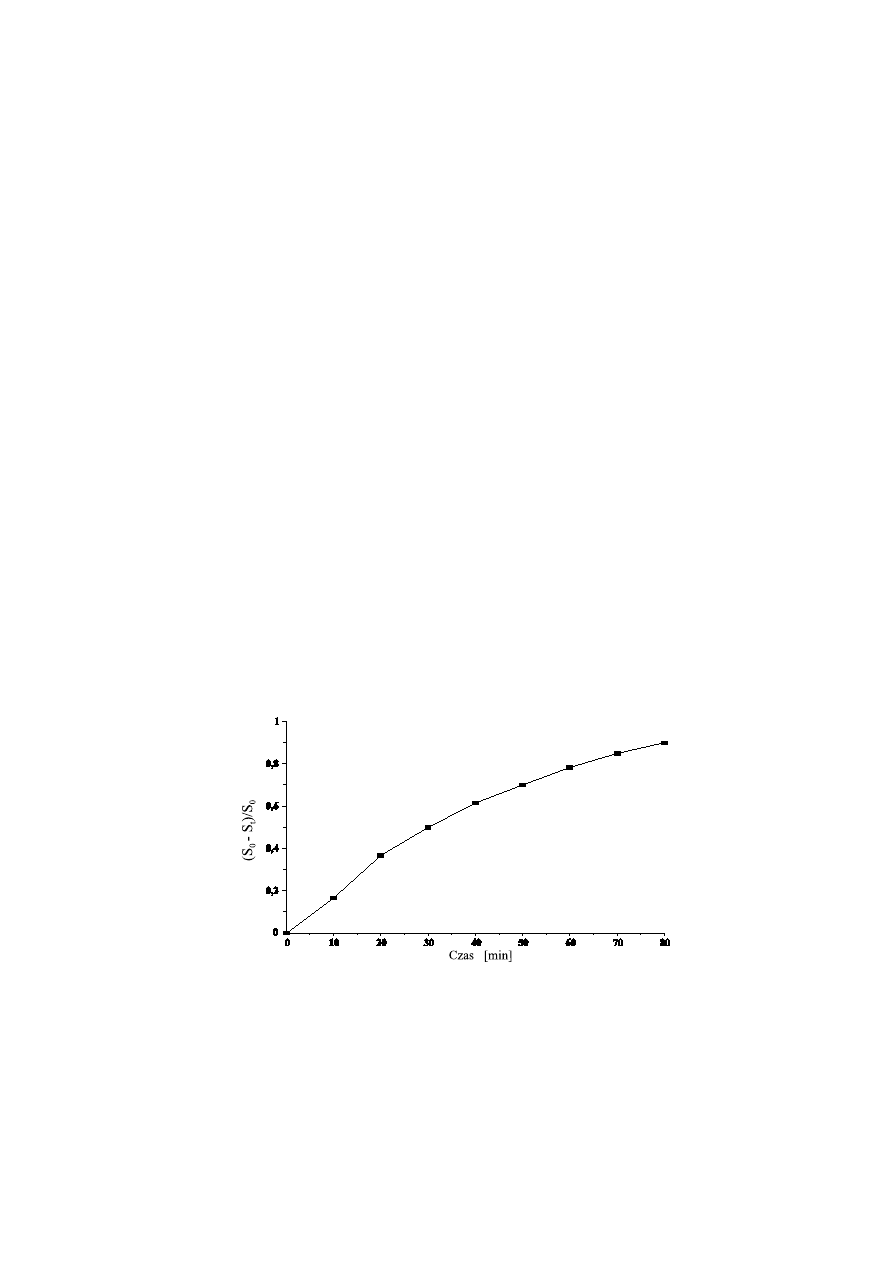
27
D. Sporz¹dzanie krzywej wzorcowej do oznaczania cukrów redukuj¹cych
Krzyw¹ wzorcow¹ przygotowuje siê, dodaj¹c do odpowiednio ponumerowanych probówek 0,1 ; 0,2; 0,3; 0,4;
0,6; 0,8 ml roztworu wzorcowego glukozy o stê¿eniu 2 mg/ml. Próby uzupe³nia siê wod¹ do objêtoœci 2 ml i
dodaje 0,2 ml odczynnika DNS. Próbê odniesienia przygotowuje siê dodaj¹c 0,2 ml odczynnika DNS do 2 ml
wody. Nastêpnie próby po wymieszaniu umieszcza siê na 15 min we wrz¹cej ³aŸni wodnej, po czym ch³odzi i
mierzy ich absorpcjê przy 540 nm. Krzyw¹ wzorcow¹ sporz¹dza siê, odk³adaj¹c na osi 0X iloœæ glukozy w
poszczególnych próbach, a na osi 0Y odpowiadaj¹c¹ im wartoœæ absorbancji, a nastêpnie wykreœlaj¹c
przechodz¹c¹ przez punkt (X:Y) = 0, prost¹ regresji dla wyznaczonych punktów. Na podstawie pomiarów
6
12
6
cz
absorbancji dla wzorcowych roztworów glukozy (C H O , M = 180,16 g mol ) wykreœliæ krzyw¹ wzorcow¹.
–1
E. Hydroliza roztworu sacharozy
Zbiornik nape³nia siê 20 ml roztworu sacharozy (0,6 M) i 10 ml 0,05 M buforu octanowego (pH 4,7). Od
momentu w³¹czenia mieszad³a i pompy liczy siê czas 0. Kontrolê przebiegu hydrolizy sacharozy przeprowadza
siê przez 80–100 minut, pobieraj¹c do ponumerowanych probówek co 10 minut okreœlone objêtoœci roztworu
(50–300 ìl) do oznaczania powstaj¹cych cukrów redukuj¹cych. Nale¿y pamiêtaæ o pobraniu próby w czasie 0.
Nastêpnie po³owê objêtoœci pobranej próby nale¿y przenieœæ do nowej probówki i uzupe³niæ wod¹ do 2 ml,
dodaæ 0,2 ml roztworu DNS i wstawiæ do wrz¹cej ³aŸni wodnej na 15 min. Nastêpnie zch³odziæ, wymieszaæ i
zmierzyæ absorbancjê przy 540 nm. W ten sposób œledzi siê przebieg hydrolizy sacharozy w ci¹gu godziny.
Szybkoœæ tej reakcji zale¿y od aktywnoœci immobilizowanego enzymu i jego kontaktu z substratem.
F. Opracowanie wyników, krzywa hydrolizy sacharozy
Z krzywej wzorcowej wyznaczyæ zawartoœæ cukrów redukuj¹cych (w ìmolach) w poszczególnych próbkach
12
22
11
pobranych w czasie trwania reakcji. Z tych wartoœci wyliczyæ ca³kowite stê¿enia (mM) sacharozy (C H O ,
cz
t
M = 342,3 g mol ) w zbiorniku w kolejnych czasach (S ), pamiêtaj¹c, ¿e z jednej cz¹steczki sacharozy
–1
0
powstaje cz¹steczka glukozy i cz¹steczka fruktozy. Znaj¹c pocz¹tkowe stê¿enie sacharozy (S ), wykreœliæ
0
t
0
krzyw¹ konwersji: f(t) = (S – S )/S , informuj¹c¹ o wydajnoœci reaktora.
Krzywa konwersji (przyk³ad)

28
ANALIZA ELEKTROFORETYCZNA PRODUKTÓW TRAWIENIA
HISTONU H1 PAPAIN¥
Papaina (EC 3.4.22.2) jest enzymem proteolitycznym z mleczu roœliny tropikalnej Carica papaya. Jest to
zasadowe bia³ko, sk³adaj¹ce siê z jednego ³añcucha peptydowego zawieraj¹cego 4 mostki dwusiarczkowe i
istotn¹ dla funkcji katalitycznej cysteinê. Do osi¹gniêcia pe³ni aktywnoœci enzymatycznej papaina musi mieæ
woln¹ grupê -SH i lekko kwaœne œrodowisko. Czynniki blokuj¹ce grupy tiolowe, jak np. kwas jodooctowy, s¹
silnymi inhibitorami papainy natomiast zwi¹zki tiolowe, jak merkaptoetanol, jej aktywatorami. Papaina jest
enzymem o wzglêdnie ma³ej specyficznoœci, na któr¹ sk³adaj¹ siê: aktywnoœæ endopeptydazy, esterazy i
amidazy. Z uwagi na budowê centrum aktywnego papaina wykazuje jednak preferencje do pewnych sekwencji
aminokwasów. Centrum aktywne papainy sk³ada siê z 7 miejsc, z których ka¿de wi¹¿e jedn¹ resztê
aminokwasow¹ substratu.
H2N-R1-R2-R3-R4-R5-R6-R7-COOH
8
hydroliza
Miejsce R3 wykazuje szczególne powinowactwo do fenyloalaniny. Dlatego obecnoœæ Phe w pozycji 3 lub dalej
od C-koñca zwiêksza podatnoœæ polipeptydu na hydrolizê, natomiast peptydy zawieraj¹ce Phe w pozycji 2, np
Ala-Ala-Phe-Ala, s¹ inhibitorami papainy.
W
æwiczeniu substratem papainy jest histon H1 wystêpuj¹cy w postaci dwóch wariantów sekwencyjnych o
masie cz¹steczkowej 21000 i 22000. Cz¹steczka histonu H1 sk³ada siê z trzech domen: fragmentu œrodkowego
o d³ugoœci 80 aminokwasów, który tworzy stabiln¹, oporn¹ na dzia³anie proteaz, strukturê globularn¹ w
stê¿onych roztworach soli, oraz dwóch luŸnych, niesfa³dowanych koñców.
Materia³y i odczynniki
1. 30% akrylamid, 0,8% bisakrylamid. 30 g akrylamidu i 0,8 g bisakrylamidu rozpuœciæ w oko³o 50 ml wody
destylowanej. Uzupe³niæ objêtoœæ do 100 ml i mieszaæ przez 0,5 godz. z 10 g wymieniacza jonowego.
Przes¹czyæ przez bibu³ê i przechowywaæ w ciemnej butelce. Uwaga! akrylamid jest groŸn¹ trucizn¹
(odczynnik przygotowany).
2. 1,5 M Tris-HCl, pH 8,8 (potrzeba 50 ml buforu).
3. 0,625 M Tris-HCl, pH 6,8 (potrzeba 25 ml buforu).
4. 1% SDS (odczynnik przygotowany).
5. 10% nadsiarczan amonowy. Bezpoœrednio przed u¿yciem rozpuœciæ 30 mg nadsiarczanu w 300 ìl wody
destylowanej. Przechowywaæ w lodzie.
6. TEMED. Przechowywaæ w lodówce (odczynnik firmowy).
7. 5-ciokrotnie stê¿ony bufor elektrodowy Tris-glicyna, pH 8,3 (0,125 M Tris, 0,96 M glicyna) zawieraj¹cy
0,5% SDS (odczynnik przygotowany). Przed u¿yciem rozcieñczyæ wod¹ dejonizowan¹! Przygotowaæ 0,8 l
buforu.
8. 1M Tris-HCl, pH 6,8 (potrzeba 25 ml buforu).
9. Mieszanina do denaturacji bia³ka o sk³adzie: 20% glicerol, 0,2% b³êkit bromofenolowy, 4% SDS, 20%
merkaptoetanol (odczynnik przygotowany).
10. 5 M NaCl. Przygotowaæ 10 ml roztworu.
11. Roztwór soli rtêci (Hg) 2mg/ml (odczynnik przygotowany).
12. 5% merkaptoetanol (MET) (odczynnik przygotowany).
29
13. Roztwór papainy 0,2 mg/ml. Odwa¿yæ bardzo dok³adnie nawa¿kê na 10 lub 20 ml. Rozpuœciæ w oziêbionej
wodzie dejonozowanej tu¿ przed u¿yciem. Przechowywaæ w lodzie.
14. Roztwór Histonu H1 (4mg/ml) (przygotowany).
15. Roztwór barwi¹cy. 1 g Coomassie Blue R-250 rozpuœciæ w 1000 ml mieszaniny 25% alkoholu
izopropylowego i 10% kwasu octowego (odczynnik przygotowany).
16. Roztwór odbarwiaj¹cy. 100 ml izopropanolu zmieszaæ ze 100 ml kwasu octowego i uzupe³niæ wod¹
destylowan¹ do objêtoœci 1 l.
17. n-Butanol (odczynnik gotowy).
Materia³y pomocnicze i aparatura
Forma do ¿elu (2 p³ytki szklane, listewki plastikowe, "grzebieñ", zaciskacze 6 szt.), smar, pipety serologiczne,
pipety automatyczne, nasadka na pipety, aparat do elektroforezy, zasilacz pr¹du sta³ego, termostat (37 C),
o
rêkawiczki chirurgiczne, szpatu³ki plastikowe, mikrowirówka, mikromieszad³o, wentylator, probówki plastikowe
typu Eppendorfa 20 szt, kuweta porcelanowa.
Wykonanie
Uwaga!
Niespolimeryzowany akrylamid jest siln¹ neurotoksyn¹. Wszystkie czynnoœci zwi¹zane z przygotowaniem ¿elu
wykonaæ na arkuszu bibu³y, pod opiek¹ asystenta. U¿ywaæ rêkawiczek i nasadki na pipety.
A. Przygotowanie ¿elu poliakrylamidowego
Dwie czyste, odt³uszczone metanolem, suche p³ytki szklane (jedna pe³na, druga z wyciêciem) z³o¿yæ równo,
wk³adaj¹c miêdzy nie plastikowe listewki nasmarowane smarem silikonowym tak, aby po dociœniêciu
zaciskaczami utworzy³y szczeln¹ formê. Tak przygotowan¹ formê ustawiæ pionowo.
Przygotowaæ roztwory monomerów wg tabeli:
Odczynnik
6% ¿el zatê¿aj¹cy
15% ¿el rozdzielaj¹cy
30% akrylamid 0,8% bis
2 ml
15 ml
1% SDS
1 ml
3 ml
0,625 M Tris-HCl pH 6,8
2 ml
--------
1,5 M Tris-HCl pH 8,8
--------
7,5 ml
Woda dejonizowana
5 ml
4,5 ml
Objêtoœæ ca³kowita
10 ml
30 ml
Roztwory dok³adnie wymieszaæ i oziêbiæ w ³aŸni lodowej. Z roztworu 15% pobraæ do osobnej zlewki 5 ml,
dodaæ najpierw 20 ìl TEMED, a nastêpnie 100 ìl roztworu nadsiarczanu amonu, zamieszaæ. Z przygotowanego
roztworu utworzyæ "stopkê" uszczelniaj¹c¹ formê od do³u. Po ok. 20 min, kiedy ¿el stê¿eje, do pozosta³ej
czêœci 15% roztworu dodaæ 20 ìl TEMED i 100 ìl roztworu nadsiarczanu amonu (nale¿y zachowaæ kolejnoœæ
dodawania roztworów). Po zamieszaniu, wylaæ roztwór miêdzy szyby formy, tak aby górna granica p³ynu
znajdowa³a siê w odleg³oœci oko³o 2.5 cm od górnej, wyciêtej krawêdzi szyby. Na wierzch roztworu nanieœæ 0,5
ml butanolu i pozostawiæ oko³o godziny do czasu spolimeryzowania ¿elu.
Po zakoñczeniu polimeryzacji, co sygnalizuje powstanie wyraŸnej granicy faz miêdzy butanolem a wypchniêt¹
z ¿elu wod¹, zlaæ butanol a woln¹ przestrzeñ miêdzy szybami przep³ukaæ dok³adnie wod¹ destylowan¹ i
starannie osuszyæ bibu³¹. Zamontowaæ "grzebieñ" i wlaæ w woln¹ przestrzeñ, za pomoc¹ pipety, 6% roztwór
monomerów po uprzednim dodaniu 20 ìl TEMED i 50 ìl nadsiarczanu amonu. Po oko³o 30 minutach, kiedy
30
¿el spolimeryzuje, zdj¹æ zaciskacze i szczelnie owin¹æ formê foli¹ aluminiow¹. ¯el przechowywaæ w lodówce
(nie zamra¿aæ).
B. Trawienie histonu H1 papain¹
2
Z roztworu HgCl o stê¿eniu 2 mg/ml sporz¹dziæ, poprzez kolejne rozcieñczenia, po 1 ml roztworów Hg o
+
stê¿eniach 10 , 10 i 10 w odniesieniu do roztworu wyjœciowego. W probówce Eppendorfa wymieszaæ 200 ìl
-1
-2
-3
roztworu histonu H1 z 200 ìl 1 M Tris-HCl (pH 6,8). Nastêpnie dodaæ po 30 ìl rozcieñczonego roztwóru
histonu do 12 ponumerowanych probówek Eppendorfa.
Pozosta³e odczynniki (w ìl) dodaæ wed³ug schematu:
Numer próby
1
2
3
4
5
6
7
8
9
10
11
12
5% M ET
---
---
---
---
---
---
10
---
---
---
---
---
5 M NaCl
---
---
---
---
---
---
---
10
---
---
---
---
Hg
+
Stê¿enie
wyjœciowe
mg/ml
---
---
---
---
---
---
---
---
10
2
10
0,2
10
0,02
10
0,002
2
H O
20
20
---
10
15
18
8
---
---
---
---
---
Papaina
---
---
20
10
5
2
2
10
10
10
10
10
*
Roztwór papainy o stê¿eniu 0,2 mg/ml sporz¹dziæ dopiero po odpipetowaniu wszystkich pozosta³ych roztworów i dok³adnym ich
*
wymieszaniu. Roztwór papainy przechowywaæ w lodzie.
Roztwór papainy nale¿y dodaæ jako ostatni i próby natychmiast wymieszaæ. Po odwirowaniu przez 5 sekund
w mikrowirówce inkubowaæ przez 20 minut w temperaturze 37 C. Niezw³ocznie po zakoñczeniu inkubacji do
o
ka¿dej probówki dodaæ po 50 ìl roztworu do denaturacji, wymieszaæ zawartoœæ i wstawiæ probówki do gotuj¹cej
siê wody na 1-2 minut. Po ostudzeniu próby odwirowaæ przez 5 sekund w mikrowirówce. Próby mo¿na
przechowaæ zamro¿one w temp. -10 C.
o
C. Elektroforeza
Ostro¿nie wyj¹æ "grzebieñ" z formy do ¿elu, nastêpnie przy pomocy zaciskaczy zainstalowaæ formê w aparacie
do elektroforezy, tak aby jej tylna szyba (wyciêta) przylega³a do œcianki. Nalaæ rozcieñczonego buforu
elektrodowego do górnego naczynia aparatu, tak aby "studzienki" powsta³e po wyjêciu "grzebienia" by³y
ca³kowicie zalane buforem i przep³ukaæ je przy pomocy 1 ml pipety automatycznej. Nalaæ buforu do dolnego
naczynia, nastêpnie po³¹czyæ elektrody aparatu z zasilaczem pr¹du sta³ego: górn¹ z (-) a doln¹ z (+) (bardzo
wa¿ne, nie pomyliæ siê).
Pierwsz¹ i ostatni¹ "studzienkê" zostawiæ niewype³nion¹, a do pozosta³ych 12 wprowadziæ ostro¿nie, za
pomoc¹ pipety automatycznej, po oko³o 40 ìl zdenaturowanych roztworów bia³ka, zwracaj¹c uwagê na
kolejnoœæ nanoszonych prób. W³¹czyæ wentylator i skierowaæ strumieñ powietrza na aparat do elektroforezy.
W³¹czyæ zasilacz pr¹du sta³ego nastawiwszy uprzednio napiêcie na 0 V (0 mA). Doprowadziæ natê¿enie pr¹du
do wartoœci 10 mA. Natê¿enie pr¹du nale¿y utrzymaæ na sta³ym poziomie 10-20 mA przez ca³y czas trwania
elektroforezy. Elektroforezê przerwaæ po oko³o 4 godzinach, gdy barwnik dojdzie do koñca p³ytki. Doprowadziæ
napiêcie do 0 V (0 mA), wy³¹czyæ zasilacz i od³¹czyæ przewody ³¹cz¹ce zasilacz z aparatem do elektroforezy.
31
D. Barwienie ¿elu
Znanych jest wiele metod barwienia ¿eli elektroforetycznych. Przedstawiona poni¿ej metoda (patrz
odczynniki) nie wymaga stosowania utrwalania ¿elu poliakrylamidowego przed zabarwieniem roztworem
barwnika.
Po wylaniu buforu z aparatu zdj¹æ zaciskacze i ostro¿nie, pod nadzorem asystenta, wyj¹æ ¿el z formy. ¯el
zagêszczaj¹cy odci¹æ a rozdzielaj¹cy przenieœæ do kuwety porcelanowej i zalaæ roztworem barwnika (Coomassie
R-250). Roztwór barwnika z ¿elem pozostawiæ w temperaturze pokojowej przez noc, nie dopuszczaj¹c do
przywarcia ¿elu do dna naczynia. Nastêpnie zlaæ barwnik z powrotem do butelki, a ¿el przemywaæ kolejnymi
porcjami odbarwiacza a¿ do momentu ukazania siê wyraŸnych pr¹¿ków bia³ka. Zu¿yty odbarwiacz zlewaæ do
osobnej zlewki. ¯el przechowywaæ w roztworze 50% metanolu lub etanolu (ulega wtedy czêœciowemu
odwodnieniu).
E. Opracowanie wyników
Wybarwiony ¿el nale¿y pozostawiæ przez dwie doby w 50% metanolu, nastepnie mo¿na zeskanowaæ w celu
dokumentacji doswiadczenia.
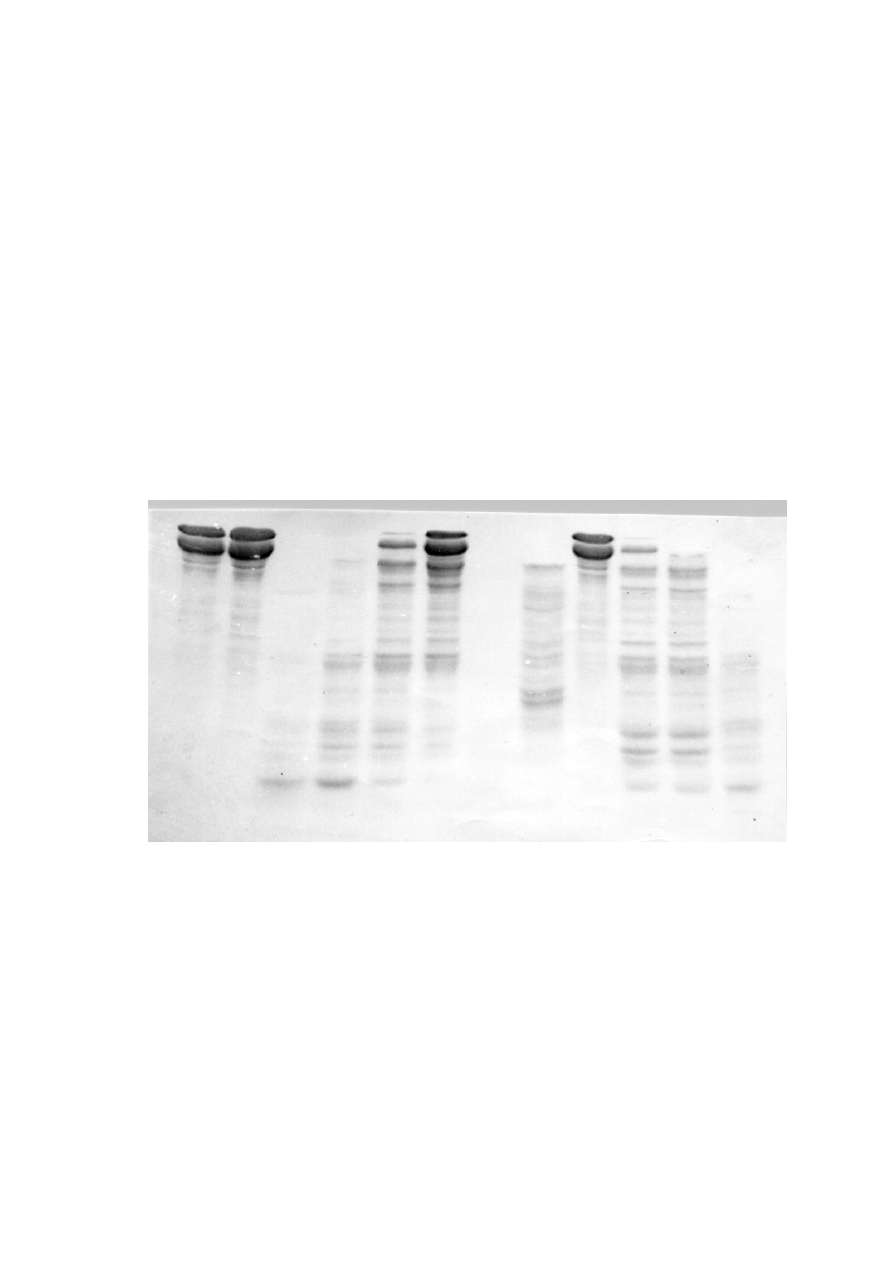
Na podstawie w³asnego elektroforogramu opisaæ efekty trawienia histonu H1 papain¹ w ró¿nych warunkach
i wyt³umaczyæ istniej¹ce ró¿nice.
1
Elektroforetyczny rozdzia³ produktów trawienia histonu H papain¹. Próby naniesiono na ¿el w
kolejnoœci podanej w tabeli

32
Literatura
Zagadnienia podstawowe, metody badañ i obliczeñ biochemicznych.
Stryer L., Biochemia. PWN, Warszawa 2005
Zgirski A., Gondko R., Obliczenia biochemiczne, PWN, Warszawa 1998 i wydania póŸniejsze.
K³yszejko-Stefanowicz L. (red.), Æwiczenia z biochemii, PWN, Warszawa 1980 i wydania póŸniejsze.
Nicholls D.G., Ferguson S.J. Bioenergetyka 2. PWN, Warszawa 1995.
D. Hall, K. Rao, Fotosynteza, WNT, Warszawa 1999
Cornish-Bowden A. Fundamentals of Enzyme Kinetics. Buttrrworths, London 1995
strona WWW Cornish-Bowdena:
http://bip.cnrs-mrs.fr/bip10/mcainfo.htm
Witwicki J., Ardelt W. (red), Elementy enzymologii, PWN, Warszawa 1984.
dowolny podrêcznik chemii fizycznej, np. Pigoñ K., Ruziewicz Z. Chemia Fizyczna, PWN, Warszawa
Strona g³ówna NC-IUBMB, nomenklatura enzymów:
http://www.chem.qmul.ac.uk/iubmb/enzyme/
Baza enzymatyczna Brenda:
http://www.brenda.uni-koeln.de/
Baza enzymatyczna Expasy:
Strona g³ówna programu Gepasi:
Strona g³ówna programu Copasi:
http://www.copasi.org/tiki-index.php
Wymagania (nale¿y zwróciæ szczególn¹ uwagê na praktyczn¹ stronê wykonywanych æwiczeñ oraz na
zwi¹zek za³o¿eñ teoretycznych ze sposobem zaplanowania i wykonania konkretnych doœwiadczeñ).
Kinetyka chemiczna. Kinetyka enzymatyczna - hiperboliczna z jednym substratem, z wieloma substratami.
Kinetyka niehiperboliczna. Enzymy regulatorowe, allosteria. Kinetyka hamowania reakcji enzymatycznych.
Metody badania aktywnoœci enzymów. Jednostki enzymatyczne. Budowa bia³ka enzymatycznego (struktura
molekularna enzymu). Oddzia³ywania miêdzy enzymem a substratem. Energetyka reakcji enzymatycznych.
Mechanizm reakcji enzymatycznych. Podzia³ enzymów. Kofaktory. Przyk³adowe szlaki metaboliczne.
Metody izolowania bia³ek enzymatycznych. Oczyszczanie enzymów. Bilans oczyszczania. Charakterystyka
oczyszczonych enzymów. Wykorzystanie metod elektroforetycznych i spektrofotometrycznych w badaniach
enzymów i kompleksów b³onowych.
B³onowe kompleksy enzymatyczne bior¹ce udzia³ w procesach bioenergetycznych. Budowa kompleksów
fotosyntetycznych i mitochondrialnych. Procesy biochemiczne wykorzystuj¹ce elektrochemiczny potencja³
jonowy. Komórkowe procesy utlenienia i redukcji. Termodynamika a procesy bioenergetyczne. Izolowanie
struktur komórkowych. Metody badania fotosyntezy i oddychania mitochondrialnego.

33
“Enzymologia II" - wykonanie i zaliczanie æwiczeñ.
1. Rozpoczynaj¹c æwiczenia nale¿y zaplanowaæ bie¿¹c¹ pracê na podstawie informacji zawartych w
przepisach do æwiczeñ. W przypadkach w¹tpliwoœci nale¿y niezw³ocznie skorzystaæ z pomocy
prowadz¹cych.
Warunkiem rozpoczêcia zajêæ jest dostarczenie prowadz¹cemu planu pracy na dany dzieñ
æwiczeñ.
2. Szk³o potrzebne do wykonania æwiczeñ znajduje siê w szafkach studenckich. Dodatkowy drobny
sprzêt wydawany jest przez prowadz¹cych.
3. Niektóre odczynniki (oznaczone w tekœcie jako "przygotowane") s¹ przygotowane i wydawane
przez prowadz¹cych. Pozosta³e odczynniki nale¿y przygotowaæ samodzielnie.
4. Student ma obowi¹zek prowadziæ dziennik laboratoryjny oraz wype³niaæ na bie¿¹co
arkusze sprawozdañ, do których wpisuje wyniki, obliczenia rachunkowe, uwagi i wnioski
dotycz¹ce wykonywanego æwiczenia oraz opracowanie wyników.
5. Zaliczenie æwiczeñ odbywa siê na podstawie:
a) poprawnego wykonania æwiczeñ i opracowania wyników
b) zdania przewidzianych sprawdzianów
6. Student mo¿e wykonywaæ jednoczeœnie jedynie dwa æwiczenia. Wydanie kolejnego æwiczenia
mo¿e nast¹piæ po zaliczeniu jednego z dwóch poprzednio wykonanych.

34
Regulamin pracy w laboratorium.
1. Podczas pracy w laboratorium konieczny jest fartuch i odpowiednie obuwie.
2. W pracowni nie wolno jeœæ ani paliæ.
3. Ubranie wierzchnie nale¿y pozostawiæ w szatni, natomiast torby, teczki itp. sk³adaæ w miejscu do tego
przeznaczonym.
4. Ka¿dy student jest odpowiedzialny za utrzymanie porz¹dku w pracowni. Nie utrudniaj pracy sobie,
kolegom i prowadz¹cym.
5. Nale¿y specjalnie dbaæ o aparaturê. Przed przyst¹pieniem do pracy nale¿y zapoznaæ siê z jej obs³ug¹. Po
zakoñczeniu pracy nale¿y pozostawiæ aparat w takim stanie, aby móg³ byæ bez przeszkód u¿yty przez
nastêpn¹ osobê.
6. Z rozpuszczalnikami organicznymi niskowrz¹cymi mo¿na pracowaæ wy³¹cznie w pomieszczeniach
przystosowanych do tego celu.
7. Alkohol u¿ywany w laboratoriach ska¿ony jest w dawkach niebezpiecznych dla zdrowia.
8. Stê¿one kwasy, zasady i inne substancje ¿r¹ce u¿ywaæ w wyznaczonych miejscach. Rozlane ¿r¹ce p³yny
nale¿y natychmiast wytrzeæ. Skórê oparzon¹ kwasem lub zasad¹ nale¿y sp³ukaæ dok³adnie bie¿¹c¹
3
3
wod¹, nastêpnie przemyæ 5% roztworem NaHCO (kwas) lub 2% CH COOH (zasada).
9. Maj¹c do czynienia z silnie truj¹cymi zwi¹zkami nale¿y zachowaæ szczególn¹ czystoœæ r¹k i miejsca
pracy. Do pipetowania trucizm u¿ywaæ specjalnych gruszek lub nasadek na pipety.
10. O ka¿dym, nawet drobnym wypadku nale¿y natychmiast powiadomiæ prowadz¹cego æwiczenia.
Document Outline
- Page 1
- Page 2
- Page 3
- Page 4
- Page 5
- Page 6
- Page 7
- Page 8
- Page 9
- Page 10
- Page 11
- Page 12
- Page 13
- Page 14
- Page 15
- Page 16
- Page 17
- Page 18
- Page 19
- Page 20
- Page 21
- Page 22
- Page 23
- Page 24
- Page 25
- Page 26
- Page 27
- Page 28
- Page 29
- Page 30
- Page 31
- Page 32
- Page 33
- Page 34
Wyszukiwarka
Podobne podstrony:
Materiały do ćwiczeń z geologii
Materiały do ćwiczeń nr 1
Materiały do cwiczenia nr 5
Materiały do ćwiczeń nr 2
Materiały do ćwiczeń z geologii te co umieć
Materialy do cwiczen, biochemia
materialy do cwiczen 1, Studia FIR, Podstawy zarządzania
XX materiały do ćwiczeń z historii wych 2
Materiały do cwiczenia 10
MATERIALY DO CWICZENIA BIOLOGIA CYTOMETR
Materiały do cwiczenia nr 11
Kula K, Słowikowska Hilczer J Medycyna rozrodu z elementami seksuologii Materiały do ćwiczeń
KiK materiały do ćwiczeń
39 A Formułki materiały do ćwiczeń
Materiały do ćwiczeń nr 3
Cw Materialy do cwiczen z elektrot
materialy do cwiczenia 6 id 285 Nieznany
więcej podobnych podstron