
1
Numer
Д‡wiczenia:
11
DziaЕ‚ analizy i temat Д‡wiczenia:
Analiza instrumentalna – spektrofotometria.
Oznaczanie Ејelaza(III) metodД… rodankowД….
Data wykonania
Д‡wiczenia:
20.05.13 r.
Data oddania
sprawozdania:
27.05.13 r.
Grupa:
A3
ImiД™ i nazwisko:
PrzemysЕ‚aw KoЕ‚oczek
Nazwisko
sprawdzajД…cego:
Uwagi:
Ocena:

2
1. WstД™p.
Spektrofotometria wykorzystuje pomiary stopnia absorpcji promieniowania
elektromagnetycznego w zakresie UV, Vis i bliskiej podczerwieni, w zaleЕјnoЕ›ci od stД™Ејenia
analitu w próbce. Metodą tą można oznaczać zarówno substancje organiczne
(posiadajД…ce wiД…zania ПЂ lub grupy chromoforowe) jak i nieorganiczne (barwne, lub
absorbujД…ce w zakresie UV). CzД™sto do uzyskania barwnych zwiД…zkГіw wykorzystuje siД™
reakcje kompleksowania. Analiza iloЕ›ciowa w spektrofotometrii UV-Vis wykorzystuje
prawo Lamberta-Beera:
рќђґ
рќњ†
= рќњЂ
рќњ†
в€™ рќ‘™ в€™ рќ‘ђ
gdzie:
A
О»
– absorbancja (ekstynkcja) badanej próbki przy danej długości fali,
Оµ
О»
– molowy współczynnik absorpcji przy danej długości fali [dm
3
/molВ·cm],
l – grubość warstwy absorbującej [cm],
c – stężenie analitu w próbce [mol/dm
3
].
Z kolei absorbancja okreЕ›lona jest wzorem:
рќђґ = log
рќђј
0
рќђј
gdzie:
I
0
– natężenie promieniowania monochromatycznego padającego na próbkę
I – natężenie promieniowania monochromatycznego po przejściu przez próbkę.
Czasami w spektrofotometrii (w szczegГіlnoЕ›ci IR) uЕјywa siД™ transmitancji danej wzorem:
𝑇 =
рќђј
рќђј
0
⇒ 𝐴 = − log 𝑇
Ponadto absorbancja jest wielkoЕ›ciД… addytywnД… (prawo addytywnoЕ›ci absorbancji):
𝐴 = ∑ 𝐴
рќ‘–
рќ‘›
рќ‘–=1
= 𝑙 ∙ ∑ 𝜀
рќ‘–
рќ‘ђ
рќ‘–
рќ‘›
рќ‘–=1
JeЕјeli badany ukЕ‚ad speЕ‚nia prawo Lamberta-Beera to zaleЕјnoЕ›Д‡ absorbancji od stД™Ејenia
analitu jest prostoliniowa, co wykorzystuje siД™ w metodzie kalibracji oznaczenia.
NajczД™Е›ciej stosowanД… metodД… kalibracji jest metoda serii wzorcГіw, ktГіra polega na
sporzД…dzeniu serii roztworГіw o znanych stД™Ејeniach analitu, dokonaniu pomiarГіw sygnaЕ‚u
dla kaЕјdego z nich i wykreЕ›leniu zaleЕјnoЕ›ci sygnaЕ‚u w funkcji stД™Ејenia analitu. NastД™pnie
postД™puje siД™ analogicznie z prГіbkД… i sygnaЕ‚ dla niej zmierzony odnosi siД™ do krzywej
kalibracyjnej, wyznaczajД…c w ten sposГіb zawartoЕ›Д‡ analitu w prГіbce. Do pomiarГіw
absorbancji uЕјywa siД™ spektrofotometru. Jego elementy, to: ЕєrГіdЕ‚o promieniowania
(lampa wolframowa i halogenowa do Vis; deuterowa, wodorowa, ksenonowa do UV),
monochromator (pryzmat, siatka dyfrakcyjna), komora pomiarowa (uchwyt i kuweta),
detektor (fotokomГіrka, fotopowielacz, fotodioda) i miernik (galwanometr, ukЕ‚ad scalony).

3
Oznaczanie Ејelaza(III) metodД… rodankowД… opiera siД™ na omГіwionej wczeЕ›niej metodzie
serii wzorcГіw. Fe
3+
reaguje z anionami tiocyjanianowymi, dajД…c zwiД…zki kompleksowe o
barwie czerwonej, wykazujД…ce maksimum absorpcji przy dЕ‚ugoЕ›ci fali rГіwnej 480 nm. W
zaleЕјnoЕ›ci od stД™ЕјeЕ„ reagentГіw i pH powstaje zwiД…zek kompleksowy [FeSCN]
2+
, przy czym
reakcja zachodzi juЕј przy mikrogramowych stД™Ејeniach Fe
3+
.
2. CzД™Е›Д‡ doЕ›wiadczalna.
a) SprzД™t i odczynniki:
– 2 kolby miarowe 100 cm
3
,
– Podstawowy roztwór wzorcowy
– 9 kolb miarowych 50 cm
3
,
Fe
3+
1 mg/cm
3
,
– biureta 10 cm
3
,
– 0,1% roztwór HCl,
– pipeta półautomatyczna 1 cm
3
,
– 0,1M roztwór HCl,
– pipeta jednomiarowa 20 cm
3
,
– 20% roztwór KSCN,
– zlewka 400 cm
3
,
– woda destylowana.
– lejek,
– spektrofotometr LAMBDA XLS,
– 2 polietylenowe kuwety pomiarowe.
b) Wykonanie.
PrzepЕ‚ukano kilkakrotnie 0,1% roztworem HCl kolbД™ na 100 cm
3
. Do tak przygotowanej
kolby dodano Е›ciЕ›le 1 cm
3
podstawowego roztworu wzorcowego Fe
3+
o stД™Ејeniu 1
mg/cm
3
za pomocД… pipety automatycznej. KolbД™ uzupeЕ‚niono 0,1% roztworem HCl do
kreski, zamkniД™to korkiem i wymieszano, odwracajД…c kolbД™ kilkakrotnie do gГіry dnem,
otrzymujД…c w ten sposГіb roboczy roztwГіr wzorcowy Fe
3+
o stД™Ејeniu 10 Ојg/cm
3
.
NastД™pnie przepЕ‚ukano 0,1M roztworem HCl 6 kolb miarowych 50 cm
3
i do kaЕјdej
wprowadzono odpowiednio: 0,00; 2,00; 4,00; 6,00; 8,00 i 10,00 cm
3
roboczego
roztworu wzorcowego Fe
3+
, za pomocД… biurety 10 cm
3
, otrzymujД…c w ten sposГіb
roztwory o stД™Ејeniach Fe
3+
(po pГіЕєniejszym uzupeЕ‚nieniu do kreski) odpowiednio: 0,0;
0,4; 0,8; 1,2; 1,6 i 2,0 Ојg/cm
3
. WЕ‚Д…czono spektrofotometr, wybrano funkcjД™ pomiaru
absorbancji i ustawiono dЕ‚ugoЕ›Д‡ fali na 480 nm. Do pierwszej kolby (prГіba Е›lepa)
dodano 5 cm
3
20% roztworu KSCN, uzupeЕ‚niono 0,1M roztworem HCl do kreski,
dokЕ‚adnie wymieszano. W komorze pomiarowej spektrofotometru, umieszczono
kuwetД™ z roztworem Е›lepej prГіby i wyzerowano urzД…dzenie. Do kolejnej kolby dodano
roztworu KSCN, uzupeЕ‚niono roztworem HCl i wymieszano. NastД™pnie dokonano
pomiaru absorbancji dla tego roztworu w drugiej kuwecie pomiarowej. Analogicznie
postД…piono z pozostaЕ‚ymi roztworami wzorcowymi, za kaЕјdym razem zerujД…c
urzД…dzenie na prГіbД™ Е›lepД…, a takЕјe za kaЕјdym razem przepЕ‚ukujД…c kilkakrotnie drugД…
kuwetД™ odpowiednim roztworem. Otrzymany do oznaczenia roztwГіr przeniesiono
iloЕ›ciowo z pomocД… lejka do skalibrowanej kolby miarowej 100 cm
3
, przepЕ‚ukano lejek,
a roztwГіr uzupeЕ‚niono wodД… destylowanД… do kreski i wymieszano. Trzykrotnie pobrano
skalibrowanД… pipetД… odpowiedniД… objД™toЕ›Д‡ przygotowanego roztworu do oznaczenia
do trzech kolb miarowych 50 cm
3
, przemytych wczeЕ›niej 0,1% roztworem HCl. Dalej
postД™powano analogicznie jak w przypadku roztworГіw wzorcowych.

4
3. Wyniki.
Tabela 1. Wyniki pomiarГіw.
рќђ¶
рќђ№рќ‘’
3+
[
рќњ‡рќ‘”
рќ‘ђрќ‘љ
3
]
рќђґ
1
рќђґ
2
рќђґ
3
рќђґ
0,0
0,000
0,000
0,000
0,000
0,4
0,063
0,062
0,059
0,061
0,8
0,144
0,142
0,139
0,142
1,2
0,208
0,206
0,204
0,206
1,6
0,277
0,274
0,271
0,274
2,0
0,338
0,335
0,333
0,335
C
1
0,179
0,176
0,175
0,177
C
2
0,186
0,183
0,180
0,183
C
3
0,180
0,177
0,175
0,177
4. Opracowanie wynikГіw.
a) Obliczenia.
Obliczono Е›redniД… absorbancjД™ dla kaЕјdego roztworu wzorcowego i prГіbki, na
podstawie wzoru:
𝐴 = ∑ 𝐴
рќ‘›
рќ‘›
рќ‘–=1
gdzie:
𝑛 – liczba wyników,
рќђґ
рќ‘›
– n-ty wynik absorbancji.
Otrzymane wyniki wprowadzono do Tabeli 1.
PoniewaЕј wynik Е›redniej absorbancji dla prГіbki oznaczonej symbolem C
2
odbiega od
pozostałych, przeprowadzono dla niego test Grubbsa aby sprawdzić, czy jest on
obarczony bЕ‚Д™dem grubym. W tym celu skorzystano ze wzoru:
рќђє
рќ‘љрќ‘Ћрќ‘Ґ
=
|рќ‘Ґ
рќ‘љрќ‘Ћрќ‘Ґ
— 𝑥|
рќ‘
рќ‘Ґ
gdzie:
рќђє
рќ‘љрќ‘Ћрќ‘Ґ
– wartość krytyczna testu Grubbsa dla największego wyniku,
рќ‘Ґ
рќ‘љрќ‘Ћрќ‘Ґ
– największy wynik,
𝑥 – średnia arytmetyczna wyników,
рќ‘
рќ‘Ґ
– odchylenie standardowe wyników.
рќђє
рќ‘љрќ‘Ћрќ‘Ґ
= 1,15
WartoЕ›Д‡ tablicowa dla 3 wynikГіw oraz poziomu istotnoЕ›ci 5% wynosi 1,15. Wynika
stąd, że wynik ten jest obarczony błędem grubym i należy go odrzucić.
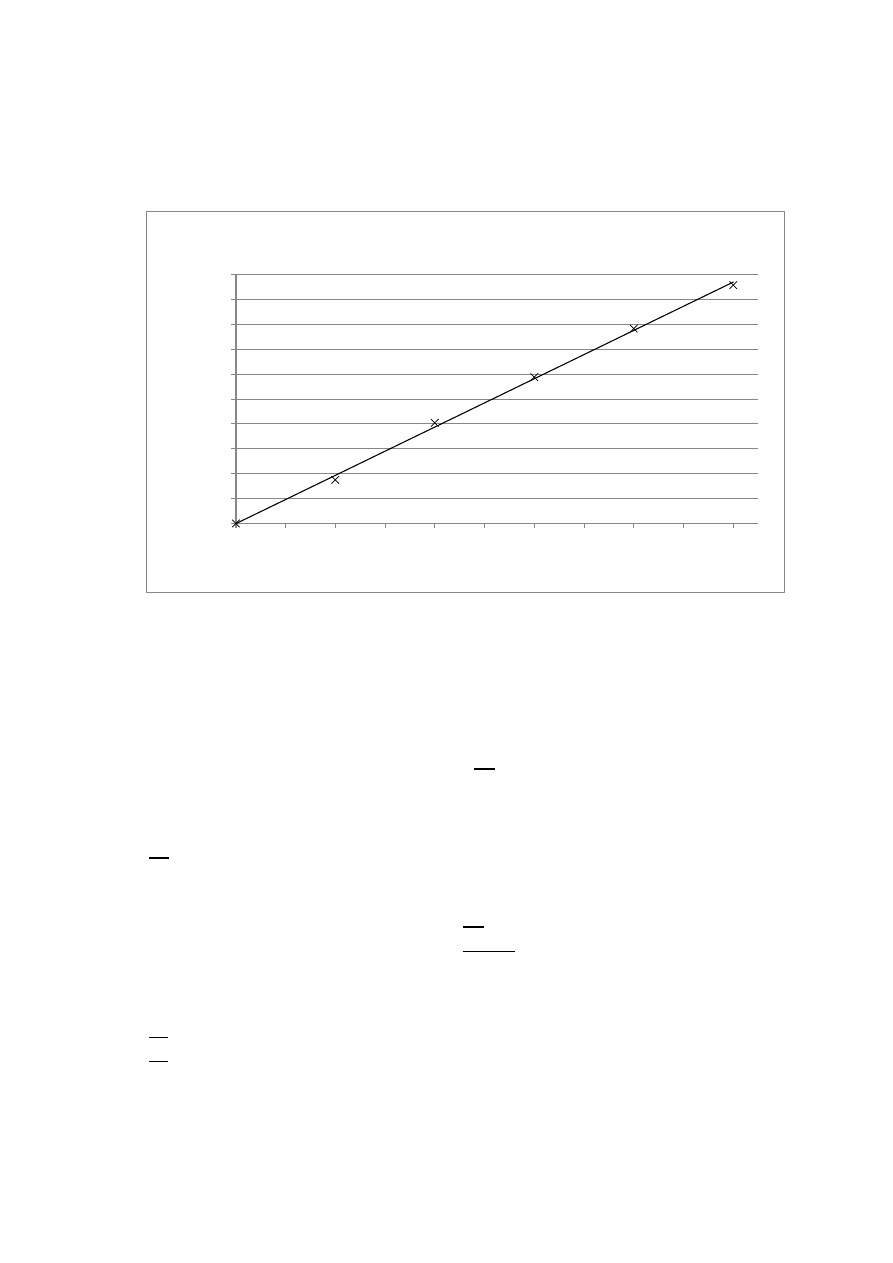
5
Wykres 1. Krzywa kalibracji.
SporzД…dzono wykres zaleЕјnoЕ›ci absorbancji od stД™Ејenia Fe
3+
, dopasowano do niego
liniД™ trendu i wyЕ›wietlono jej rГіwnanie, za pomocД… arkusza kalkulacyjnego Excel oraz
danych zawartych w Tabeli 1.:
Obliczono stД™Ејenie Ејelaza(III) dla danych prГіbek na podstawie otrzymanej zaleЕјnoЕ›ci
liniowej:
𝑦 = 𝑎𝑥 + 𝑏
oraz nastД™pujД…cych zaleЕјnoЕ›ci:
𝑦 = 𝐴
рќ‘›
рќ‘Ґ = рќђ¶
рќ‘›
gdzie:
рќђґ
рќ‘›
– średnia absorbancja danego roztworu,
рќђ¶
рќ‘›
– stężenie żelaza(III) danego roztworu [μg/cm
3
].
рќђ¶
рќ‘›
=
рќђґ
рќ‘›
в€’ рќ‘Џ
рќ‘Ћ
рќ‘Ћ = 0,1699
рќ‘Џ = в€’0,0002
рќђґ
1
= 0,177
рќђґ
3
= 0,177
рќђ¶
1
= 1,04 рќњ‡рќ‘”/рќ‘ђрќ‘љ
3
рќђ¶
3
= 1,04 рќњ‡рќ‘”/рќ‘ђрќ‘љ
3
y = 0.1699x - 0.0002
RВІ = 0.9987
0.000
0.035
0.070
0.105
0.140
0.175
0.210
0.245
0.280
0.315
0.350
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
A
C
Fe3+
[Ојg/cm
3
]
Krzywa wzorcowa roztworu Fe
3+
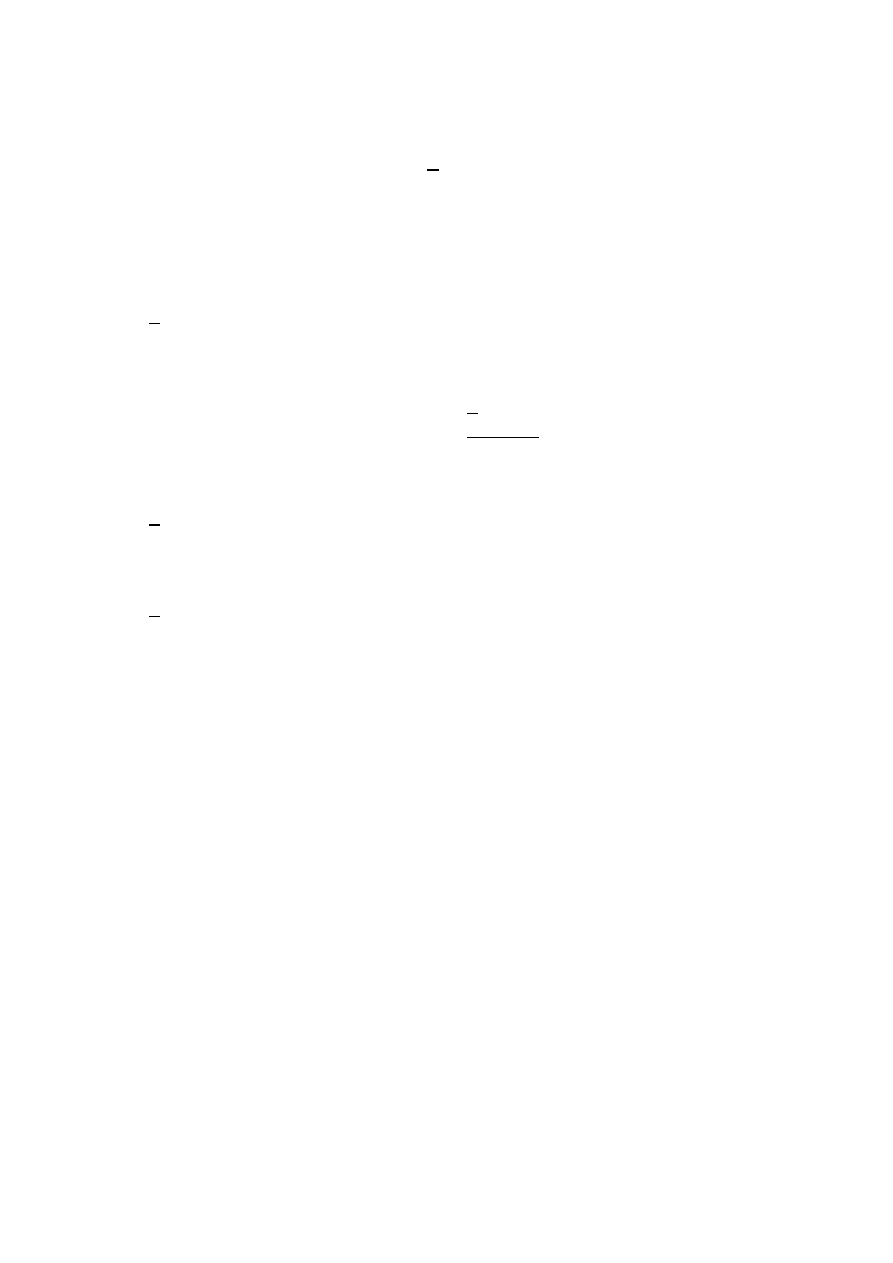
6
Obliczono Е›rednie stД™Ејenie Ејelaza(III) w badanym roztworze na podstawie wzoru:
𝐶 = ∑ 𝐶
рќ‘›
рќ‘›
рќ‘–=1
gdzie:
𝑛 – liczba wyników,
рќђ¶
рќ‘›
– n-ty wynik stężenia żelaza(III) w badanym roztworze [μg/cm
3
].
рќђ¶ = 1,04 рќњ‡рќ‘”/рќ‘ђрќ‘љ
3
Obliczono masД™ Ејelaza(III) w prГіbce na podstawie wzoru:
рќ‘љ
рќђ№рќ‘’
3+
=
𝐶 ∙ 𝑉
рќ‘ѓ
в€™ рќ‘Љ
1000
gdzie:
рќ‘љ
рќђ№рќ‘’
3+
– masa Fe
3+
zawarta w badanej prГіbce [mg],
𝐶 – średnie stężenie Fe
3+
w prГіbce [Ојg/cm
3
],
𝑉
рќ‘ѓ
– objętość próbki [cm
3
],
𝑊 – współmierność kolby i pipety.
рќђ¶ = 1,04 рќњ‡рќ‘”/рќ‘ђрќ‘љ
3
𝑉
рќ‘ѓ
= 50 рќ‘ђрќ‘љ
3
рќ‘Љ = 4,976
рќ‘љ
рќђ№рќ‘’
3+
= 0,2594 рќ‘љрќ‘”
b) Wynik koЕ„cowy.
Masa Fe
3+
w badanej prГіbce: 0,2594 mg.
5. Podsumowanie.
Metoda zastosowana do oznaczenia Fe
3+
jest bardzo czuła – pozwala oznaczyć jego
zawartoЕ›Д‡ w iloЕ›ciach mikrogramowych. UЕјyty spektrofotometr ma dobrД… czuЕ‚oЕ›Д‡, daje
powtarzalne i stabilne wyniki – największa różnica między kolejno zmierzonymi
absorbancjami wynosi 0,003 jednostek absorbancji. WartoЕ›Д‡ ta, nie przekracza
niepewnoЕ›ci kalibracji spektrofotometru, znalezionej na stronie
http://www.perkinelmer.com/CMSResources/Images/4474452BRO_LAMBDAXLSandXLSPl
us.pdf (dostД™p: 27.05.13 r.), ktГіra wynosi 0,003 jednostek absorbancji dla pomiarГіw z
zakresu 0 – 0,5 jednostek absorbancji. Kolejno mierzone absorbancje zmniejszają się.
Przyczyną tego zjawiska może być nietrwałość [FeSCN]
2+
. NajwiД™kszy wpЕ‚yw na wynik
koЕ„cowy miaЕ‚o przygotowanie roztworu roboczego. NieprawidЕ‚owe odmierzenie i
rozcieЕ„czenie roztworu podstawowego generuje staЕ‚y bЕ‚Д…d systematyczny. BЕ‚Д…d ten jest
pГіЕєniej niemoЕјliwy do skorygowania. RГіwnie waЕјne byЕ‚o utrzymanie odpowiedniej
czystoЕ›ci uЕјytego sprzД™tu laboratoryjnego.
Wszystkie obliczenia wykonano za pomocД… arkusza kalkulacyjnego Excel.
Wyszukiwarka
Podobne podstrony:
Laboratorium sprawozdania cz. 3, Mechanika III semestr, Fizyka, Laboratoria i sprawozdania
Laboratorium sprawozdania cz. 3, Mechanika III semestr, Fizyka, Laboratoria i sprawozdania
sprawozdanie 11 5
sprawozdanie M6, Fizyka, Laboratoria, Sprawozdania, Sprawozdania cd, 1
CHEMIA - LABORATORIUM - SPRAWOZDANIE - Klasyfikacja poЕ‚Д…czeЕ„ nieorganicznych - wersja 2, STUDIA
Spr. 4-MateriaЕ‚oznawstwo, Politechnika PoznaЕ„ska ZiIP, II semestr, nom, Laboratoria-sprawozdania NOM
Filtracja - sprawozdanie 1, Biotechnologia PWR, Semestr 7, Separacje i oczyszczanie bioproduktГіw - L
Д†w[1]. 04 - Stale narzД™dziowe, Politechnika PoznaЕ
wiД™cej podobnych podstron