
Chromosomopatie

Chromosomopatie
Zespoły chorobowe spowodowane
aberracjami chromosomowymi
zarówno strukturalnymi jaki i
liczbowymi.

Chromosomopatie - Aneuploidie
Chromosomopatie - Aneuploidie
•
Choroby człowieka spowodowane aberracjami
liczbowymi pojedynczych chromosomów:
– Trisomie:
tylko trzy rodzaje dają szansę na
narodziny – chromosomu 13, 18 i 21
– Monosomie
– letalne w przypadku autosomów
i chromosomu Y (brak chromosomu X)

Aberracje liczbowe autosomów:
Trisomie:
•
chromosomu
21
- zespół Downa
•
chromosomu
18
- zespół Edwardsa
•
chromosomu
13
- zespół Patau

•
średnia częstość 1/700, wzrasta z wiekiem matki (np.u
kobiety 35 letniej wynosi 1/300, u 40 letniej 1/22)
•
wykrywalny w badaniach prenatalnych USG: torbiel
pęcherzowa okolicy szyjnej, obrzęk, wady serca,
zwężenie lub zarośnięcie dwunastnicy
•
wysoki odsetek płodów z trisomią 21 ulega poronieniu
(60%)
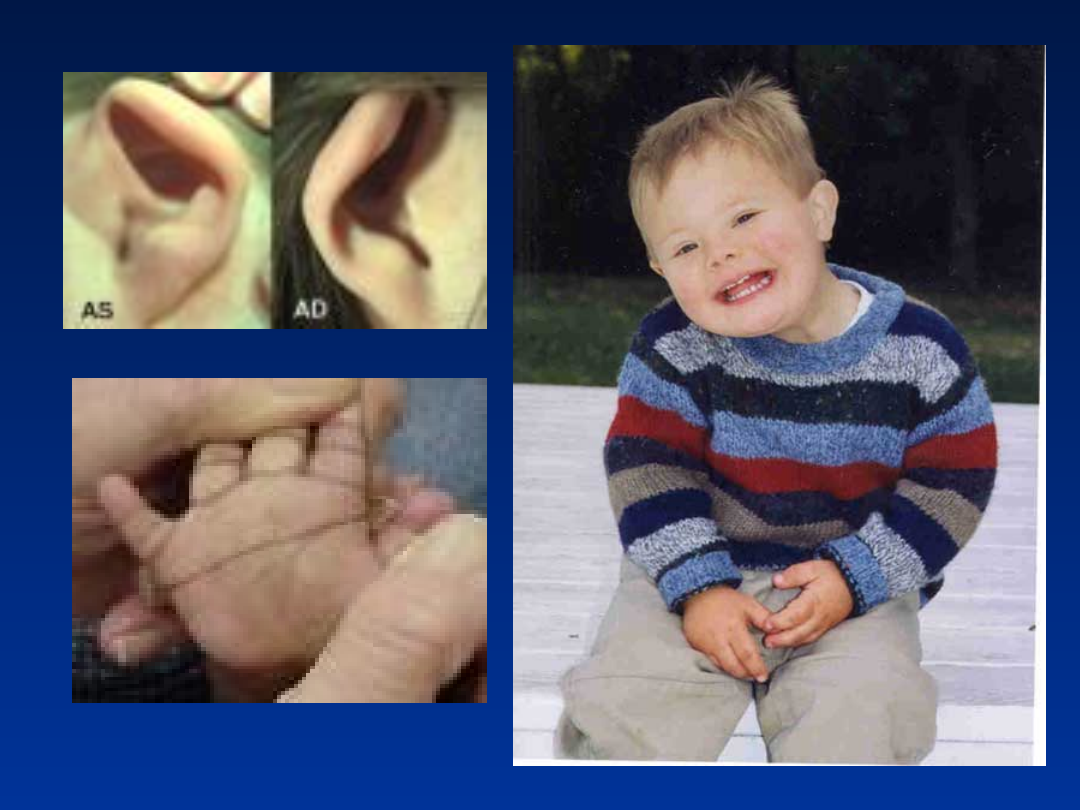
Zespół Downa – trisomia 21

• upośledzenie umysłowe
• mongoloidalne ustawienie szpar powiekowych
• zmarszczka nakątna
• dysplastyczne uszy
• wystający, duży język
• „małpia bruzda”
• płaska potylica
• często towarzyszące wady serca
Zespół Downa – fenotyp
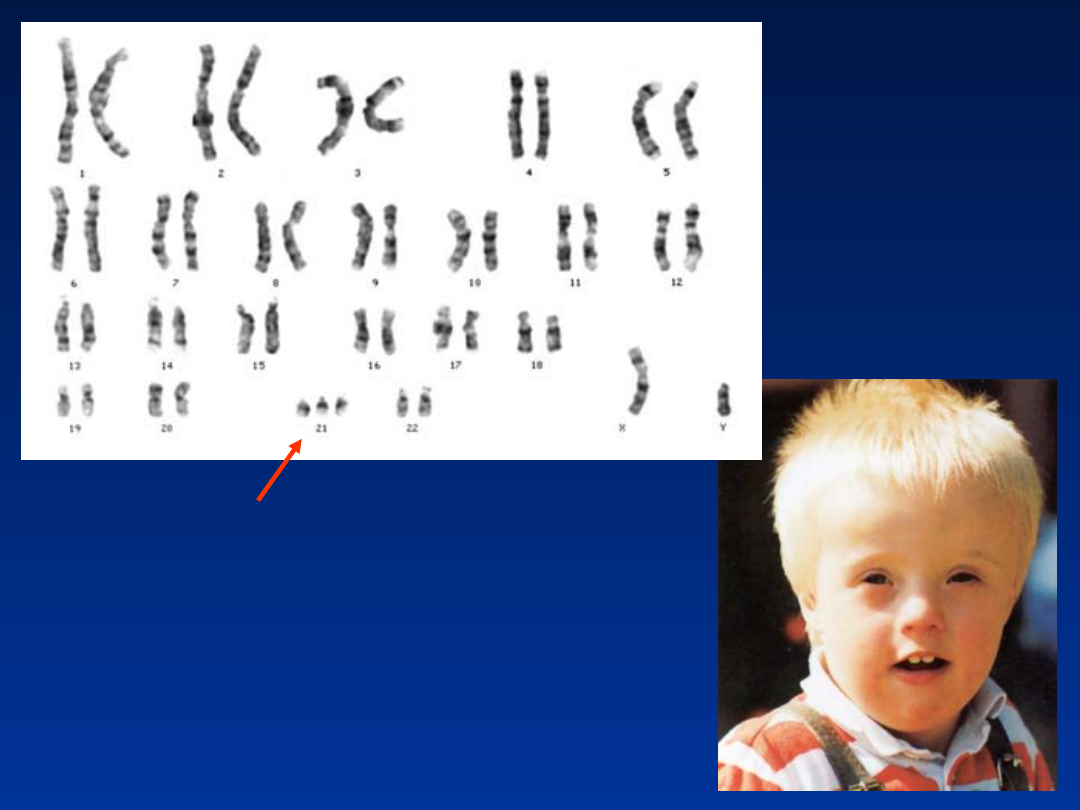
Zespół Downa
(regularny)
47,XY,+21

• Prosta trisomia:
47,XY,+21 (96%)
• Zespoły translokacyjne (3%):
46,XX,der(14;21)(q10;q10),+21
46,XX,der(21;21)(q10;q10),+21
• Kariotyp nosiciela translokacji zrównoważonej:
45,XX,der(14;21)(q10;q10)
45,XY,der(21;21)(q10;q10)
• Kariotypy mozaikowe (1%):
mos 46,XX/47,XX,+21
Zespół Downa – kariotypy
Zespół Downa – kariotypy
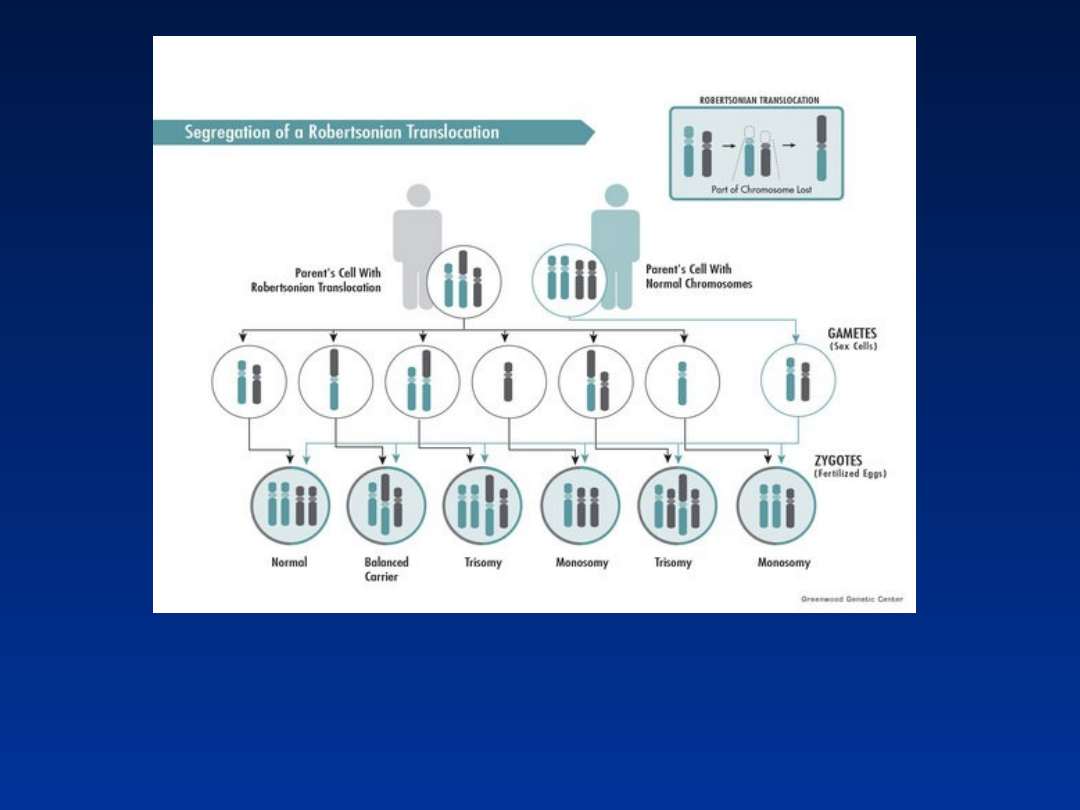
Ryzyko teoretyczne
urodzenia dziecka z
zespołem Downa, niezależnie od płci nosiciela
translokacji, wynosi 1:3

Ryzyko empiryczne
urodzenia dziecka z
zespołem Downa w przypadku nosicielstwa
translokacji robertsonowskiej
wynosi:
• 10-15% gdy nosicielem jest kobieta
• 2-5% gdy nosicielem jest mężczyzna

Zespół Edwardsa – trisomia 18
•
częstość około 1/3000
•
przewaga płci żeńskiej
•
większość płodów ulega poronieniu (95%) –
wysoka umieralność we wczesnym okresie
niemowlęcym (30% dzieci umiera w pierwszym
miesiącu życia)
Kariotypy:
47,XX,+18
mos46,XX/47,XX,+18
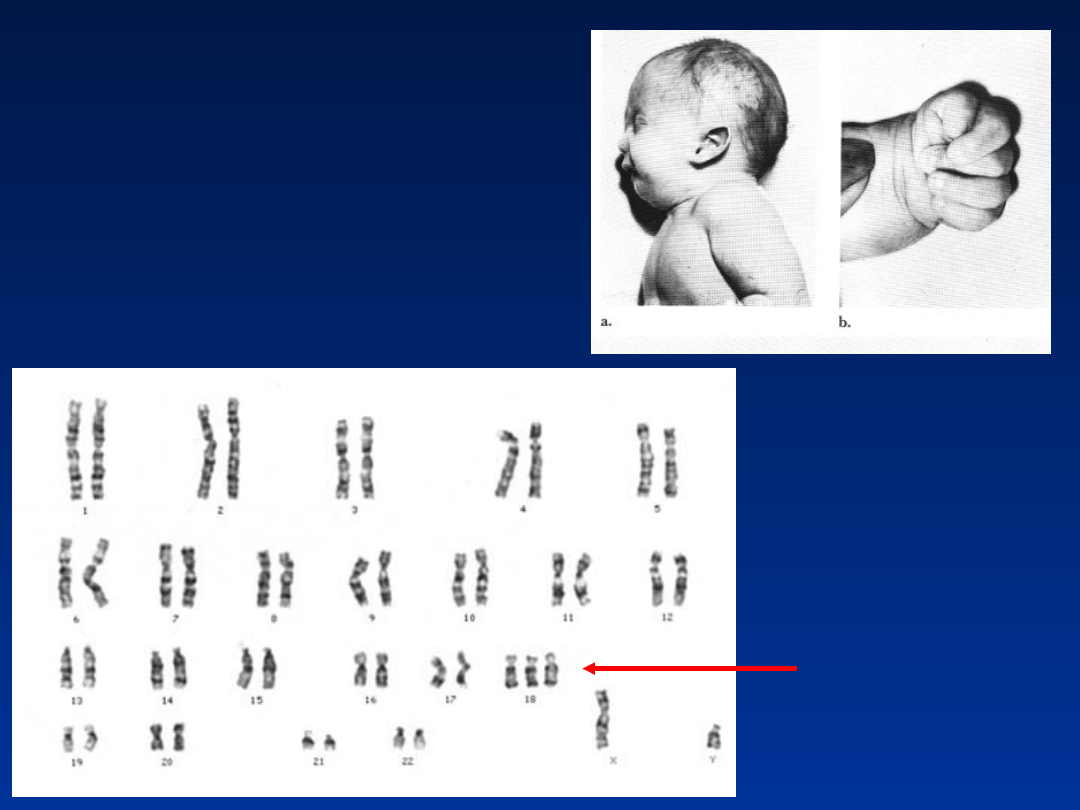
Zespół Edwardsa
47,XY,+18

Zespół Edwardsa – trisomia 18
•
retrognatia, wydatna potylica
•
zniekształcone, nisko osadzone uszy
•
syndaktylia
•
stopy o łukowato wygiętych podeszwach
•
krótki mostek
•
u chłopców zwykle występuje wnętrostwo
•
znaczne opóźnienie rozwoju
•
często towarzyszące wady narządów
wewnętrznych
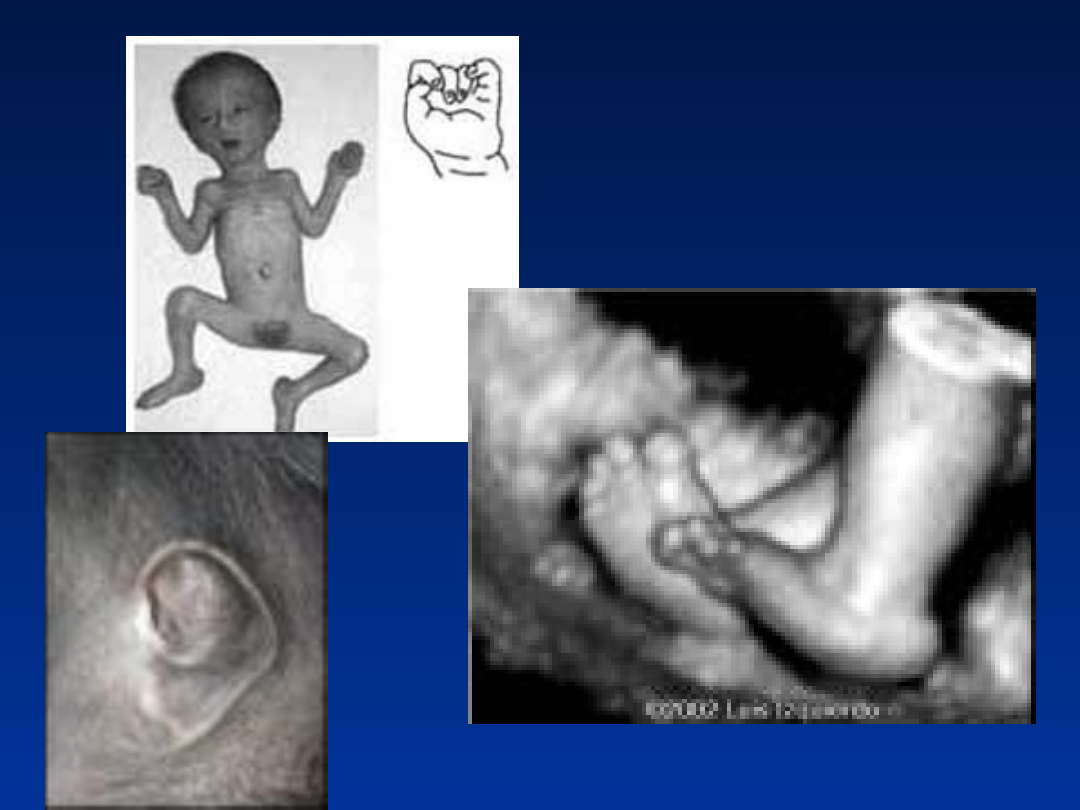

Zespół Patau – trisomia 13
• częstość około 1/5000 urodzeń
• większość płodów ulega poronieniu
• około 50% dzieci z zespołem Patau umiera
w ciągu pierwszego miesiąca życia
• tylko 10% dzieci przeżywa pierwszy rok
życia - wykazują głębokie upośledzenie
fizyczne i umysłowe
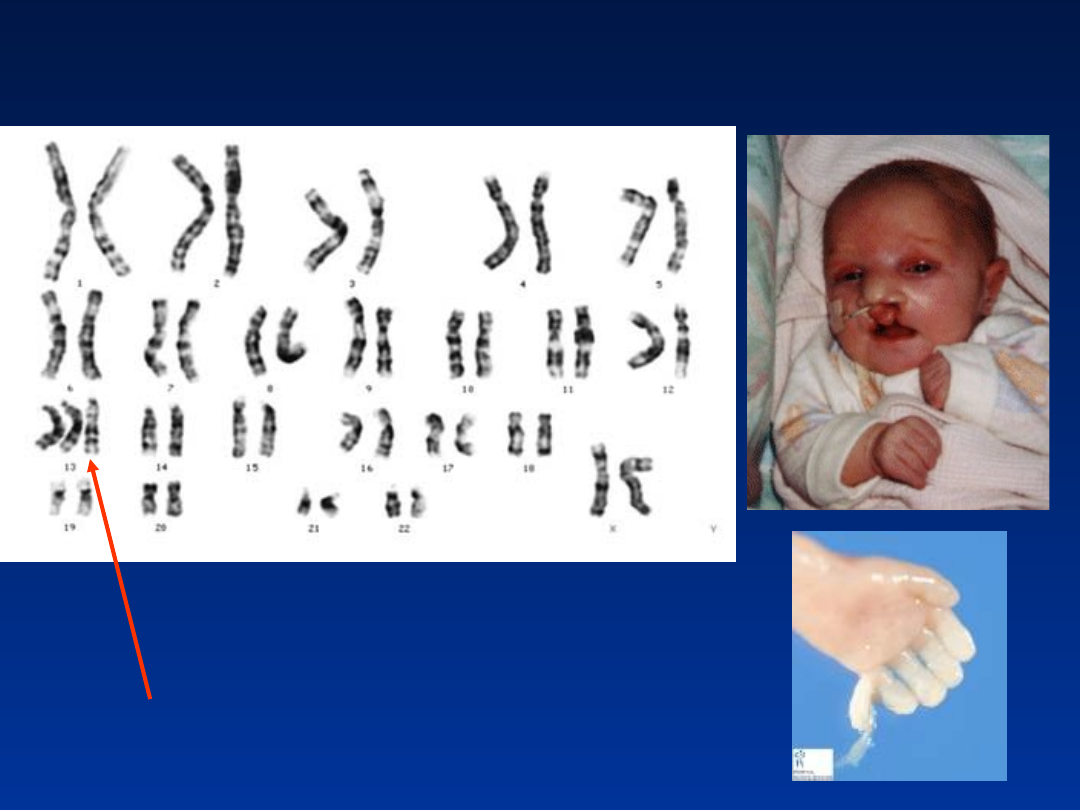
Zespół Patau’a
47,XX,
+13

•
wady OUN – holoprosencefalia (brak podziału
przodomózgowia na półkule)
•
wady wrodzone twarzoczaszki
•
hipoteloryzm
•
rozszczepienie wargi i podniebienia
•
dysplastyczne uszy
•
zmarszczka nakątna
•
często towarzyszące wady narządów
wewnętrznych
Zespół Patau – trisomia 13
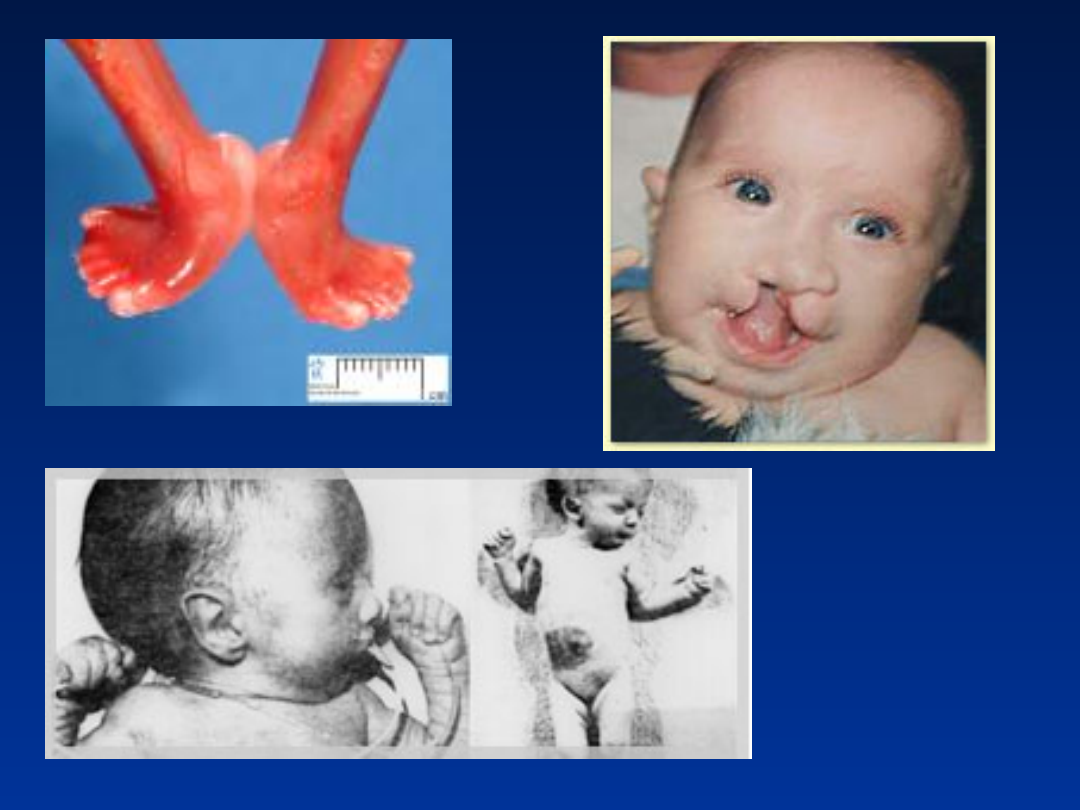

Aberracje strukturalne
Zespół Cri du chat (częściowa
monosomia chromosomu 5)
• częstość około 1/50 000 urodzeń
• około 75% chorych umiera w pierwszych
miesiącach życia, a około 90% - przed ukończeniem
1-go roku życia
• wśród noworodków przeważają dziewczynki
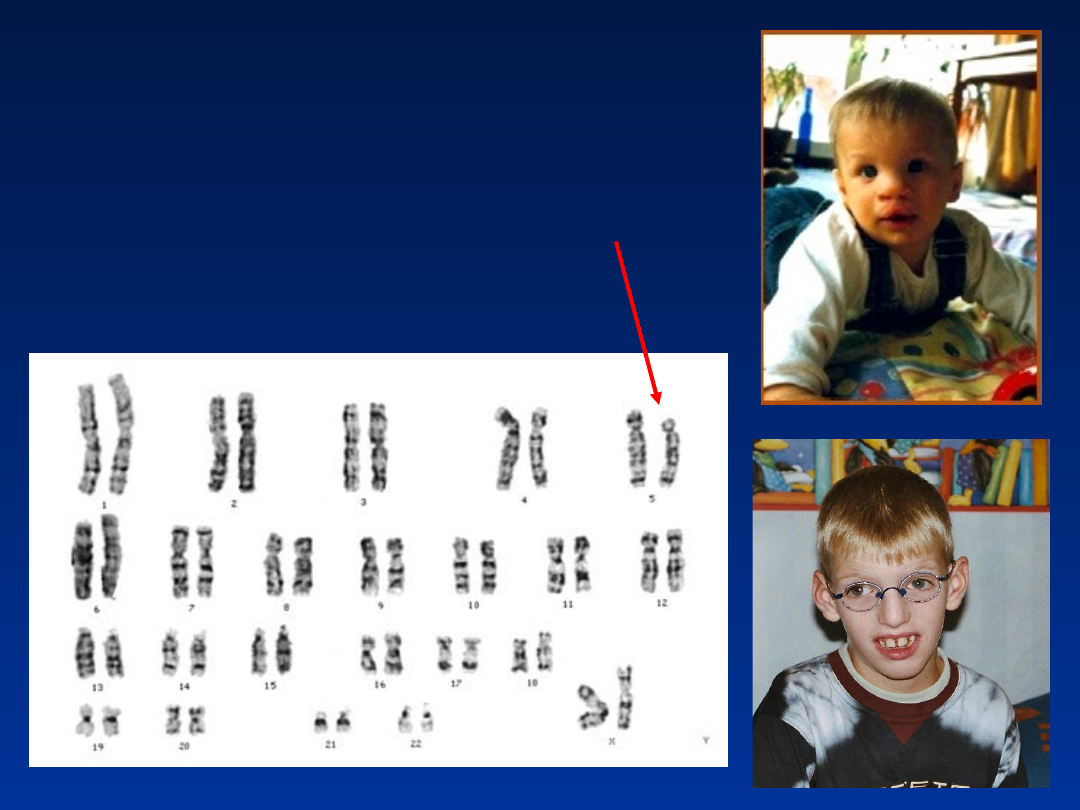
Zespół Cri du chat
46,XX,del(5)(p13)

Zespół Cri du chat (częściowa
monosomia chromosomu 5)
•
płacz przypominający miauczenie kota
•
małogłowie
•
hiperteloryzm
•
nisko osadzone uszy
•
małożuchwie

• znaczne opóźnienie rozwoju ruchowego i
umysłowego
• 10-15% przypadków to potomstwo nosicieli
translokacji zrównoważonej
Kariotypy:
46,XX, del(5)(p14)
46,XY, del(5)(p15)

Zespoły chorobowe człowieka
spowodowane
aberracjami chromosomów
płciowych
Zespół Turnera
Monosomia
chromosomu X
45,X

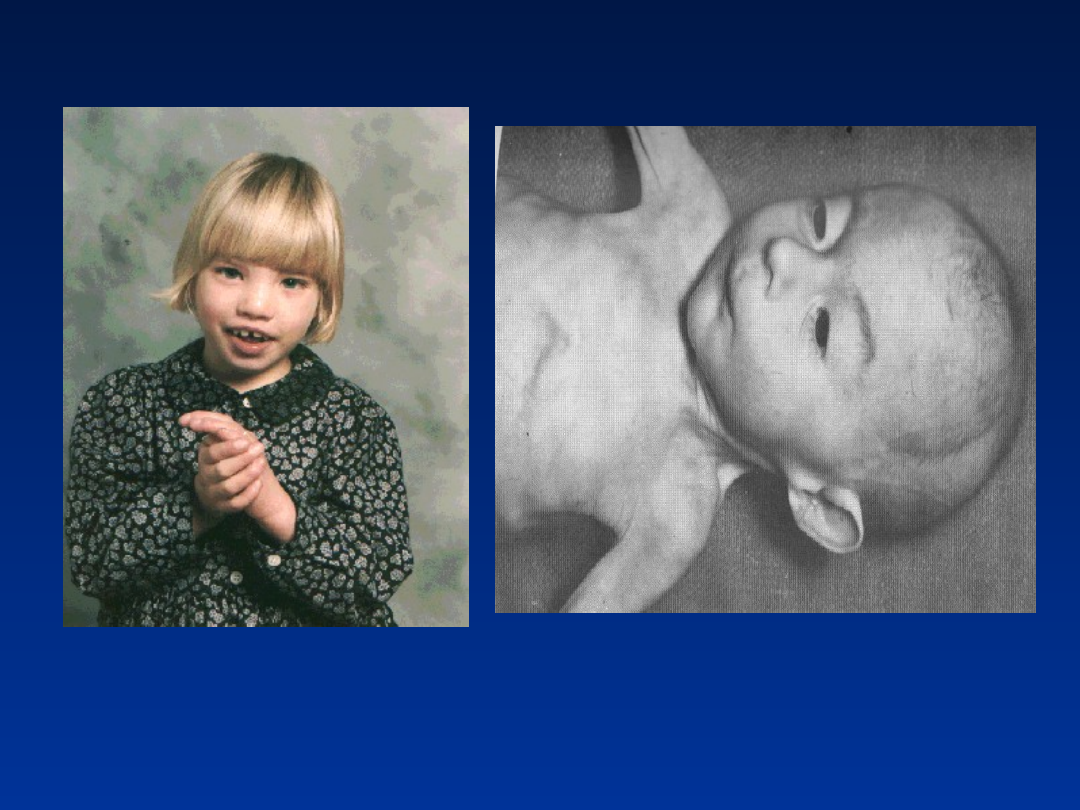
Zespół Turnera
•częstość 1/5000 urodzeń dziewczynek
•fenotyp żeński
• 99% przypadków ulega samoistnemu poronieniu
• w okresie prenatalnym i u noworodków
występuje: nadmiar skóry w okolicy karku,
obwodowe obrzęki limfatyczne

Zespół Turnera
Zespół Turnera
•
niski wzrost przy zachowaniu proporcji ciała, brak
skoku pokwitaniowego - wzrost ostateczny przy
braku terapii ok. 145 cm.
•
płetwiasta szyja
•
szeroka klatka piersiowa
•
szeroko rozstawione gruczoły sutkowe
•
nisko schodząca linia włosów
•
koślawość łokci
•
niedorozwój wtórnych cech płciowych

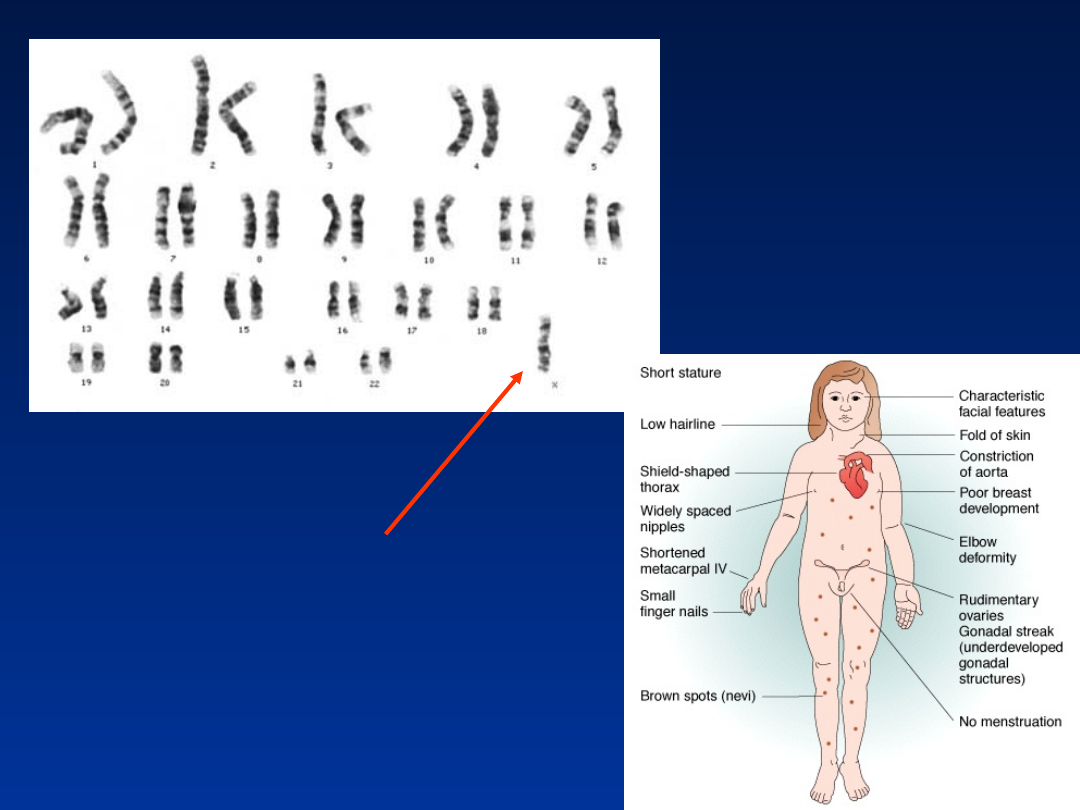
Zespół Turnera
Zespół Turnera
•
pasma łącznotkankowe w jajnikach
•
wcześnie rozwijająca się osteoporoza
•
brak upośledzenia umysłowego
Kariotypy:
•
monosomia chromosomu X
: 45,X
•
aberracje strukturalne chromosomu X:
46,X,del(X)(p11),
46,X,del(X)(q13),
46,X,i(X)(q10),
46,X,idic(X)(p11),
46,X,r(X)(p22;q28)
•
kariotypy mozaikowe:
mos 46,XX/45,X lub mos 46,XY/45,X,
mos 45,X/46,X,der(X)t(X;X)(q28;q28)
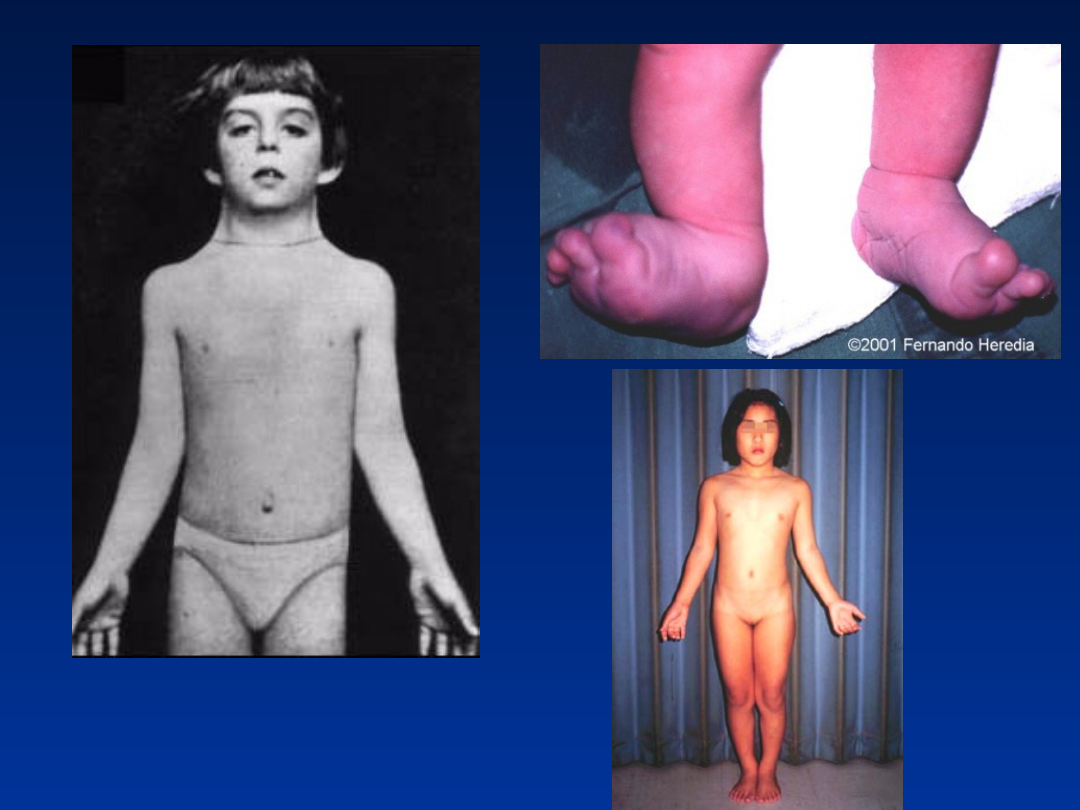
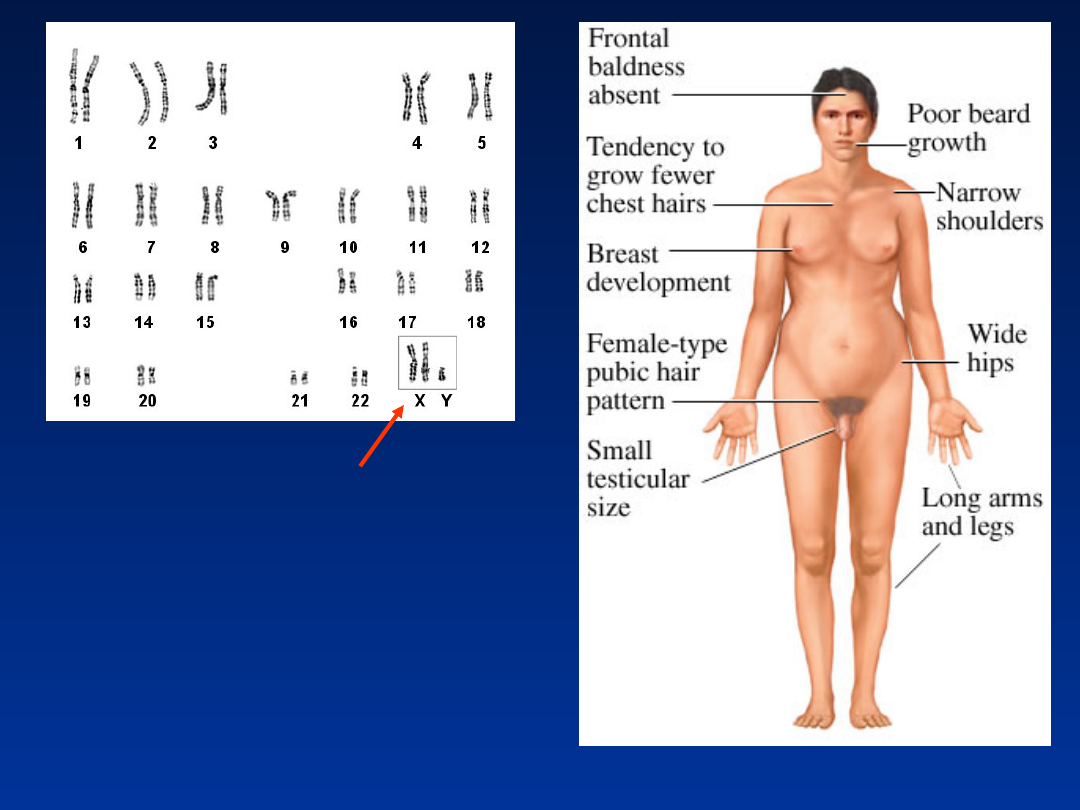
DODATKOWY CHROMOSOM
X
Zespół Klinefeltera
47,XXY

Zespół Klinefeltera
•
częstość 1/1000 urodzeń chłopców
•
fenotyp męski
•
wysoki wzrost
•
bezpłodność
•
atrofia jąder
•
słaby rozwój drugorzędowych cech płciowych
•
ginekomastia
•
brak upośledzenia umysłowego, czasem lekkie obniżenie IQ
Kariotypy:
47,XXY
mos 46,XY/47,XXY
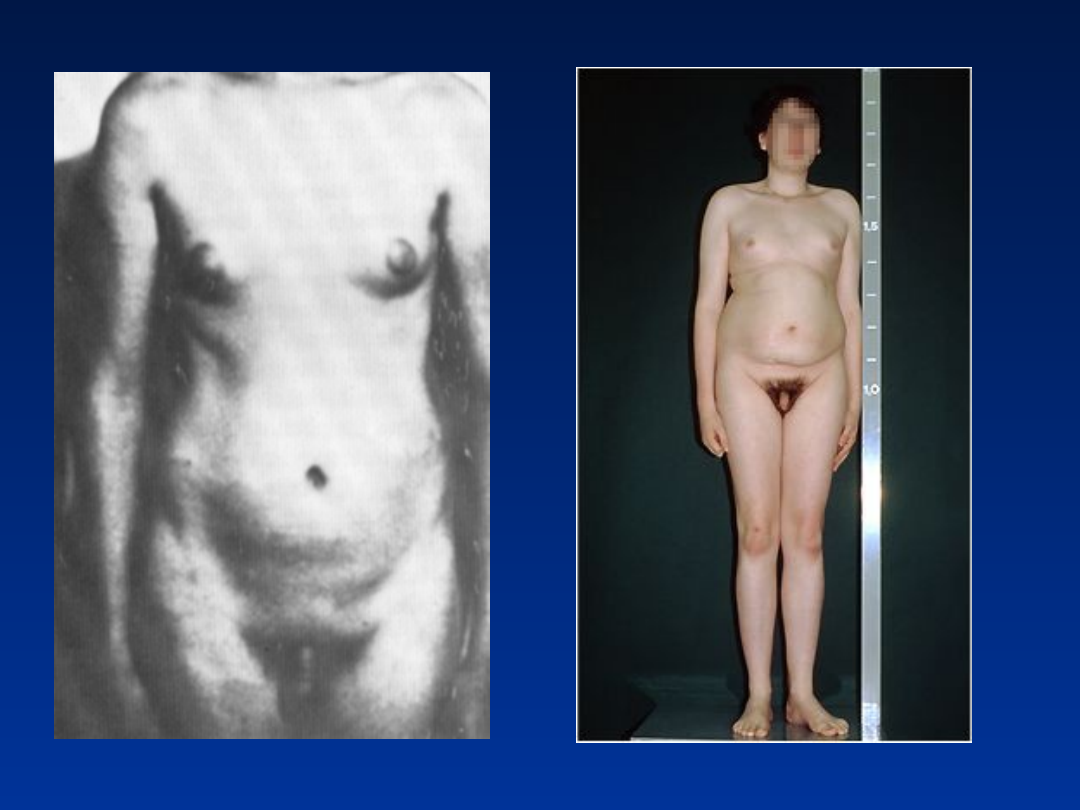

47, XXX
•
1/1000 urodzeń dziewczynek
•
fenotyp żeński
•
czasami wtórny brak miesiączki i
przedwczesna menopauza
•
łatwiej zapadają na choroby psychiczne
•
75% płodnych

47, XYY
•
1/1000 urodzeń chłopców
•
fenotyp męski
•
wysoki wzrost i prawidłowa budowa
ciała
•
płodni
•
nie są to mężczyźni o większej agresji!

Chromosomopatie - Euploidie
Chromosomopatie - Euploidie
Case Report
A newborn baby was transferred to our hospital from a peripheral center for
further assessment because of the "unusual appear-ance". The baby was the
second born child to the non-consanguineous Caucasian couple. The mother
was aged 30 years. They have a 3-year- old boy, who is healthy and well. The
second pregnancy had spontaneously aborted at 14 weeks of gestation.
There was no family history of any significant illnesses or any other disorders.
The baby was small for gestational age with severe intrauterine
growth deficiency with a birth weight of 1600 g at 41 weeks of
gestation.
He had a large anterior fontanel and a posterior fontanel,
low set
ears, hypertelorism, pointed nose and a high arched palate
. There
was
syndactaly
of third and fourth digits in both upper and lower limbs.
Simian crease was noted on both hands. Testes were undescended on both
sides and penile length was less than 0.5 cm. Only 2 umbilical vessels were
noted. He also had
rocker bottom feet
on both sides. He also had a small
ventricular septal defect confirmed on 2-dimensional echocardiogram. In view
of all the above,
he was suspected to have trisomy 18
.
His renal
ultrasound scan was reported to be normal.
His chromosomal study
revealed 18p deletion, but otherwise no evi-dence of any trisomy. In all the
cells analyzed, one chromosome 18 homologue appeared atypical within the
centrometric region of the short arm. In view of this, parental chromosomal
study was performed revealing maternal chromosomes with 18p deletion,
exactly same as the baby. But the mother is phenotypically normal with no
evidence of dysmorphism and no mental handicap. Hence, this was
attributed by the geneticists to be an incidental finding.
The baby was
susequently reviewed by our senior clinical geneticist, who
diagnosed triploidy syndrome and a skin biopsy was performed. The
skin fibroblast culture revealed 46XY/69XXY mosaic karyotyping
,
consistent with the diagnosis of Mosaic Triploidy syndrome.
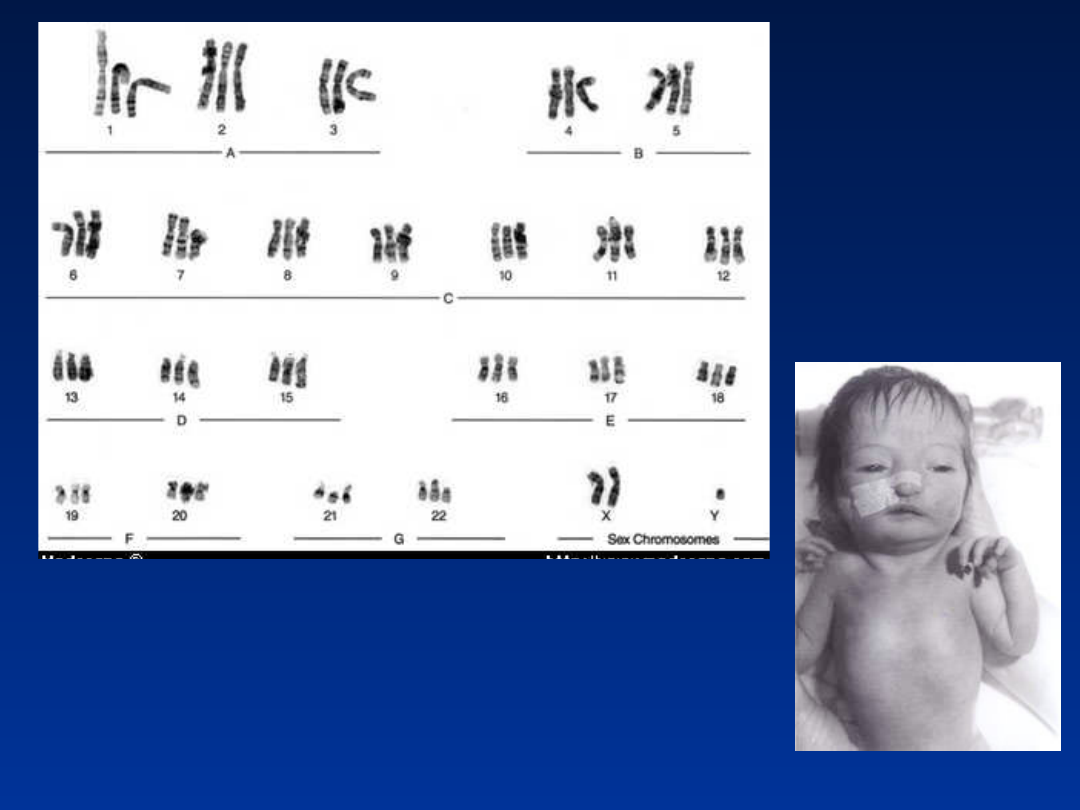
Kariotyp
triploidalny
69,XXY
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
Wyszukiwarka
Podobne podstrony:
Rodzaje aberracji chromosomowych pop
Mutacje chromosomowe strukturalne
Chromoterapia, edukacja specjalna
Zespół łamliwego chromosomu X – problem dziecka i rodziców
Aberracje chromosomowe i mutacje
2006 topologia chromosomow w jadrze kom dipl kom som PHMD
ABERRACJE CHROMOSOMOWE, BIOLOGIA MEDYCZNA
Prelekcja 10 - cz 2 - Mutacje chromosomowe człowieka, Genetyka
L. Matela - ABC leczenia kolorami (fragmenty), Chromoterapia
Brazowanie Galwanizernie chromowanie niklowanie anodowanie cynkowanie
Chromosomowa teoria Morgana
zaburzeania chromosonalne
Barwienie cynku i cyny Galwanizernie chromowanie niklowanie anodowanie cynkowanie
Cynowanie Galwanizernie chromowanie niklowanie anodowanie cynkowanie
więcej podobnych podstron