
Analiza sekwencji amino
kwasowych na podstawie
ich podobieństwa
Analiza sekwencji amino
kwasowych na podstawie
ich podobieństwa

Podobieństwo a
Homologia
Podobieństwo a
Homologia
Podobieństwo –
oznacza że dwie sekwencje
aminokwasowe są podobne w swojej strukturze
pierwszorzędowej, jakkolwiek analiza nie wnosi żadnych
informacji co do ich pokrewieństwa czy ewolucji
molekularnej.
Podobieństwo
–
oznacza że dwie sekwencje
aminokwasowe są podobne w swojej strukturze
pierwszorzędowej, jakkolwiek analiza nie wnosi żadnych
informacji co do ich pokrewieństwa czy ewolucji
molekularnej.
Homologia –
odnosi się do podobieństwa włączając w to
pokrewieństwo ewolucyjne
- Ortolog –
podobieństwo ewolucyjne i zarazem
funkcjonalne. Geny/białka pochodzące od wspólnego
przodka, które rozdzieliły się z powodu dywergencji
organizmów, pełnią podobną funkcje.
- Paralog -
podobieństwo ewolucyjne ale nie
funkcjonalne. To geny/białka pochodzące od wspólnego
przodka, który został zduplikowany w danym organizmie, a
następnie ich funkcja uległa zróżnicowaniu.
-
Ksenolog
– to geny/białka homologiczne powstałe w
wyniku transferu horyzontalnego genów między dwoma
organizmami. Zwykle funkcja jest podobna.
Homologia
–
odnosi się do podobieństwa włączając w to
pokrewieństwo ewolucyjne
- Ortolog –
podobieństwo ewolucyjne i zarazem
funkcjonalne. Geny/białka pochodzące od wspólnego
przodka, które rozdzieliły się z powodu dywergencji
organizmów, pełnią podobną funkcje.
- Paralog -
podobieństwo ewolucyjne ale nie
funkcjonalne. To geny/białka pochodzące od wspólnego
przodka, który został zduplikowany w danym organizmie, a
następnie ich funkcja uległa zróżnicowaniu.
-
Ksenolog
– to geny/białka homologiczne powstałe w
wyniku transferu horyzontalnego genów między dwoma
organizmami. Zwykle funkcja jest podobna.

Dopasowanie sekwencji
- globalne
- lokalne
Dopasowanie sekwencji
- globalne
- lokalne
Dopasowanie sekwencji:
- globalne – odnosi się do sekwencji homologicznych które nie
zróżnicowały się w znacznym stopniu w czasie ewolucji. Zazwyczaj
białka składające się z pojedynczej domeny można analizować
za pomocą dopasowania globalnego.
- lokalne – dotyczy białek modularnych, składających się z kilku
domen, które powstały w wyniku np., wymiany całych eksonów.
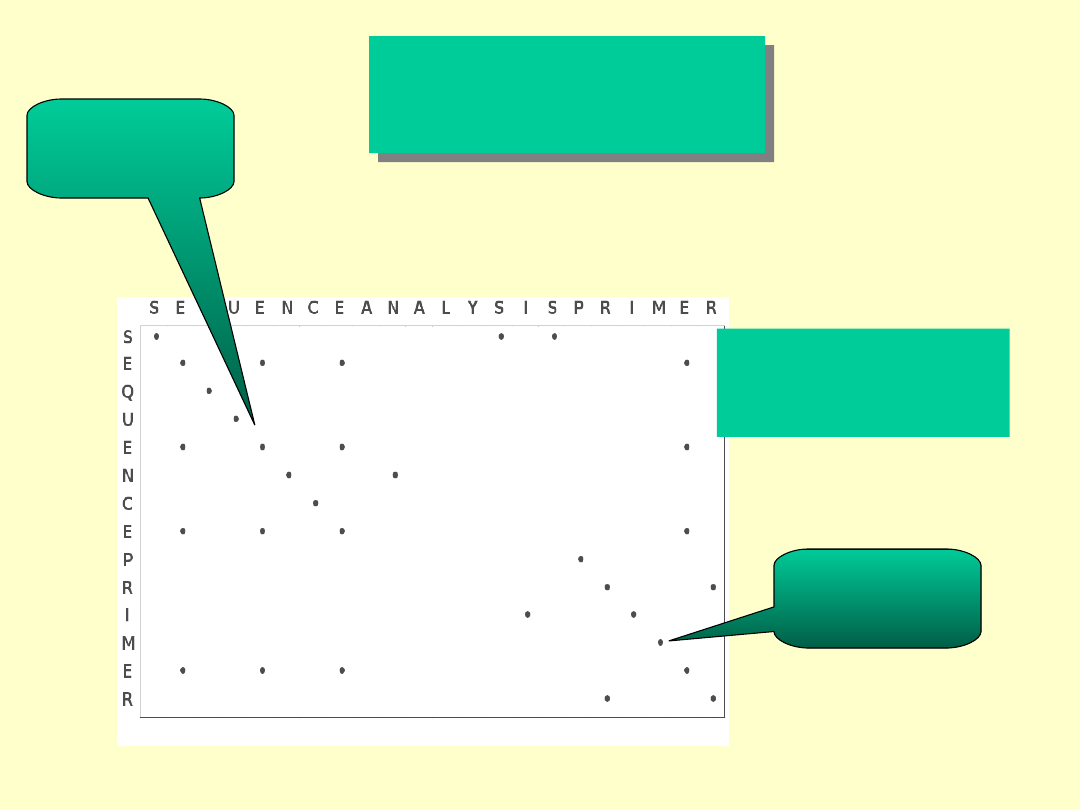
Dot
MatriX
Dot
MatriX
SEQUENCE ANALYSIS PRIMER
SEQUENCE ANALYSIS PRIMER
S E Q U E N C E A N A L Y S
I
S P R
I M E R
S
•
•
•
E
•
•
•
•
Q
•
U
•
E
•
•
•
•
N
•
•
C
•
E
•
•
•
•
A
•
•
N
•
•
A
•
•
L
•
Y
•
S
•
•
•
I
•
•
S
•
•
•
P
•
R
•
•
I
•
•
M
•
E
•
•
•
•
R
•
•
S E Q U E N C E A N A L Y S
I
S P R
I M E R
S
•
•
•
E
•
•
•
•
Q
•
U
•
E
•
•
•
•
N
•
•
C
•
E
•
•
•
•
A
•
•
N
•
•
A
•
•
L
•
Y
•
S
•
•
•
I
•
•
S
•
•
•
P
•
R
•
•
I
•
•
M
•
E
•
•
•
•
R
•
•
Ścieżka
podobieństwa
Dot MatriX
Dot MatriX
SEQUENCE ANALYSIS PRIMER
SEQUENCE PRIMER
Analiza sekwencji
gdzie zaszła delecja
fragmentu bialka
Ścieżka
podobieńst
wa
Ścieżka
podobieńst
wa
Dot
MatriX
Dot
MatriX
S
E
Q U
E
N C
E
A
N A
L
Y
S
I
S
P
R
I
M E
R
S
•
•
•
E
•
•
•
•
Q
•
U
•
E
•
•
•
•
N
•
•
C
•
E
•
•
•
•
S
•
•
•
E
•
•
•
•
Q
•
U
•
E
•
•
•
•
N
•
•
C
•
E
•
•
•
•
S
•
•
•
E
•
•
•
•
Q
•
U
•
E
•
•
•
•
N
•
•
C
•
E
•
•
•
•
S
E
Q U
E
N C
E
A
N A
L
Y
S
I
S
P
R
I
M E
R
S
•
•
•
E
•
•
•
•
Q
•
U
•
E
•
•
•
•
N
•
•
C
•
E
•
•
•
•
S
•
•
•
E
•
•
•
•
Q
•
U
•
E
•
•
•
•
N
•
•
C
•
E
•
•
•
•
S
•
•
•
E
•
•
•
•
Q
•
U
•
E
•
•
•
•
N
•
•
C
•
E
•
•
•
•
SEQUENCE
ANALYSIS PRIMER
SEQUENCE
SEQUENCE
SEQUENCE
Analiza sekwencji
gdzie zaszła duplikacja
fragmentu białka
Ścieżka
podobieńst
wa
Dot
MatriX
Dot
MatriX
SEQUENCE ANALYSIS PRIMER
PRIMER ON HOW TO ANALYZE DERIVED AMINO ACID SEQUENCES
PRIMER
IIIIII
PRIMER
1
ANALYSIS
IIIIIII.
ANALYZED
2
SEQUENCE
IIIIIIII.
SEQUENCES
3
ANALYSISPRIMER
IIIII II I
ANALYZEDERIVED
4

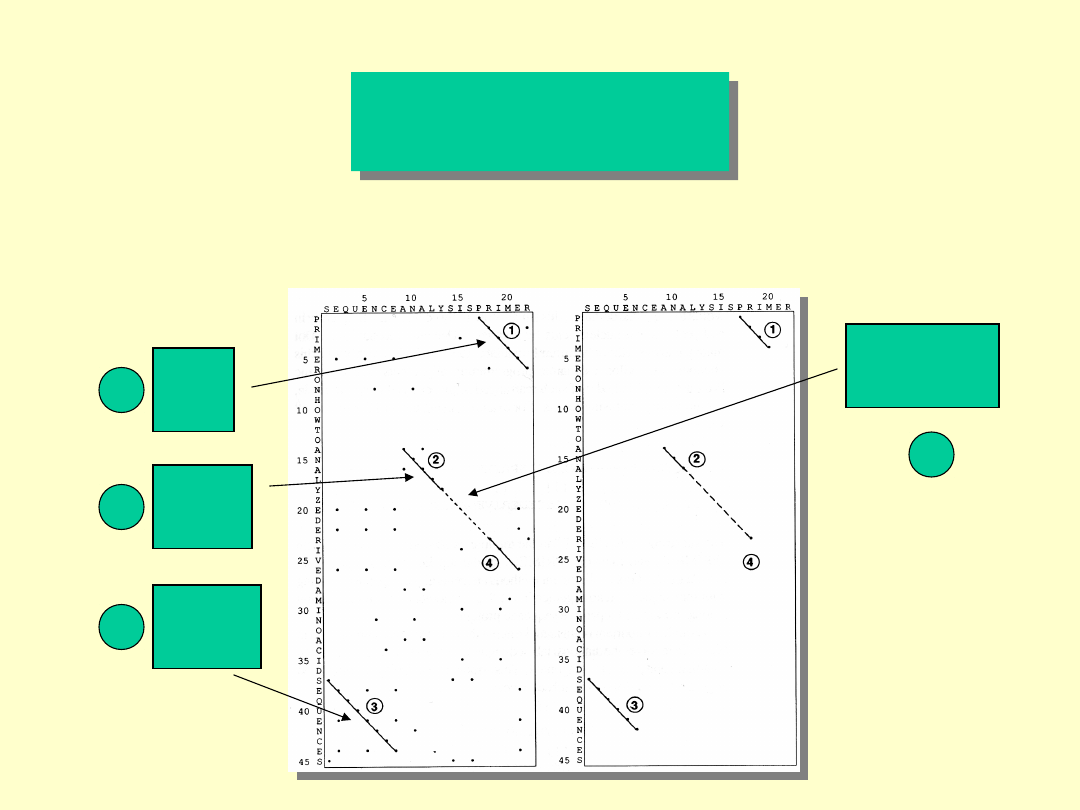
Dot MatriX
Scoring system (metody wartościowania
oparte na optymalizacji dopasowań lokalnych)
Dot MatriX
Scoring system (metody wartościowania
oparte na optymalizacji dopasowań lokalnych)
SEQUENCE ANALYSIS PRIMER
----I
PRIME
1/5 podobieństw
(20%)
SEQUENCE ANALYSIS PRIMER
-----
PRIME
0/5 podobieństw
(0%)
SEQUENCEANALYSISPRIMER
IIIII
PRIME
5/5 podobieństw
(100%)
0/5 podobieństw
(0%)
SEQUENCEANALYSISPRIMER
-----
PRIME
Analiza sekwencji AA w formie ramek
Analiza sekwencji AA w formie ramek
aminokwasowych
aminokwasowych
Analiza sekwencji AA w formie ramek
Analiza sekwencji AA w formie ramek
aminokwasowych
aminokwasowych
Matryce Dopuszczalnych Mutacji
- macierze substytucji konserwatywnych
Matryce Dopuszczalnych Mutacji
- macierze substytucji konserwatywnych
W białkach spokrewnionych część aminokwasów może być
zastąpiona innymi aminokwasami o podobnej strukturze i funkcji.
Wartości w macierzach substytucji identyczne aminokwasy
powinny dawać najwyższe wartości, zaś zmiany konserwatywne
powinny dawać wartości wyższe nic zmiany niekonserwatywne.

Matryce Dopuszczalnych Mutacji
- macierze substytucji konserwatywnych
Matryce Dopuszczalnych Mutacji
- macierze substytucji konserwatywnych
Pierwsze macierze opierały się na modelu ewolucyjnym
akceptowanych mutacji punktowych PAM - (Percent Accepted
Mutations) - Akceptowalna punktowa mutacja to taka, która albo nie
zmieniła funkcji białka, lub była korzystna dla organizmu (co najmniej
nie spowodowała śmierci organizmu).
Istnieje kilka macierzy PAM oznaczonych od 1 do 250, np., 100PAM,
250PAM
Zastosowanie: jeśli analizowane sekwencje charakteryzują się
wysokim stopniem dywergencji ewolucyjnej najlepsze wyniki dają
macierze o wysokich wartościach, zaś sekwencje o wysokim stopniu
podobieństwa są analizowane z wykorzystaniem macierzy o małych
wartościach
Macierze substytucyjne BLOSUM (Blocks Substitution Matrix)
zostały opracowane jako efekt krytyki macierzy PAM, które nie oddają
dobrze wpływu czasu na zmiany sekwencji kodujących białka.
Macierze BLOSUM zostały oparte na białkowej bazie danych
BLOCKS.
Istnieje kilka macierzy BLOSUM, o różnych indeksach, ale wartość
indeksu określa maksymalny stopień identyczności jaki możemy
napotkać w czasie analizy sekwencji białkowych, np., BLOSUM62,
BLOSUM30.

Matryce Dopuszczalnych Mutacji
- macierze substytucji konserwatywnych
Matryce Dopuszczalnych Mutacji
- macierze substytucji konserwatywnych
250PAM

Dot
MatriX
Scoring system
Dot
MatriX
Scoring system
S E Q U ENCE ANALYSIS PRIMER
1-1-2+0+4
P R I M E
+2
S E Q U E N CE ANALYSIS PRIMER
-1+0+0-2+1
P R I M E
0
SEQUENCEANALYSISP R I M E R
+6+6+5+6+4
P R I M E
27
-1
SEQUENCEANALYSISPR I M E R
0-2+4-2-1
P R I M E
Analiza sekwencji AA w formie ramek
Analiza sekwencji AA w formie ramek
aminokwasowych z wykorzystaniem analizy
aminokwasowych z wykorzystaniem analizy
jakościowo/ilościowe
jakościowo/ilościowe
Analiza sekwencji AA w formie ramek
Analiza sekwencji AA w formie ramek
aminokwasowych z wykorzystaniem analizy
aminokwasowych z wykorzystaniem analizy
jakościowo/ilościowe
jakościowo/ilościowe

Dot MatriX
Scoring system
Dot MatriX
Scoring system
SEQUENCE ANALYSIS PRIMER
PRIMER ON HOW TO ANALYZE DERIVED AMINO ACID SEQUENCES
ExPASy
ExPASy
ExPASy
-sequence
alignment
ExPASy
-sequence
alignment
Dotlet
Dot Matrix
program
Dotlet
Dot Matrix
program
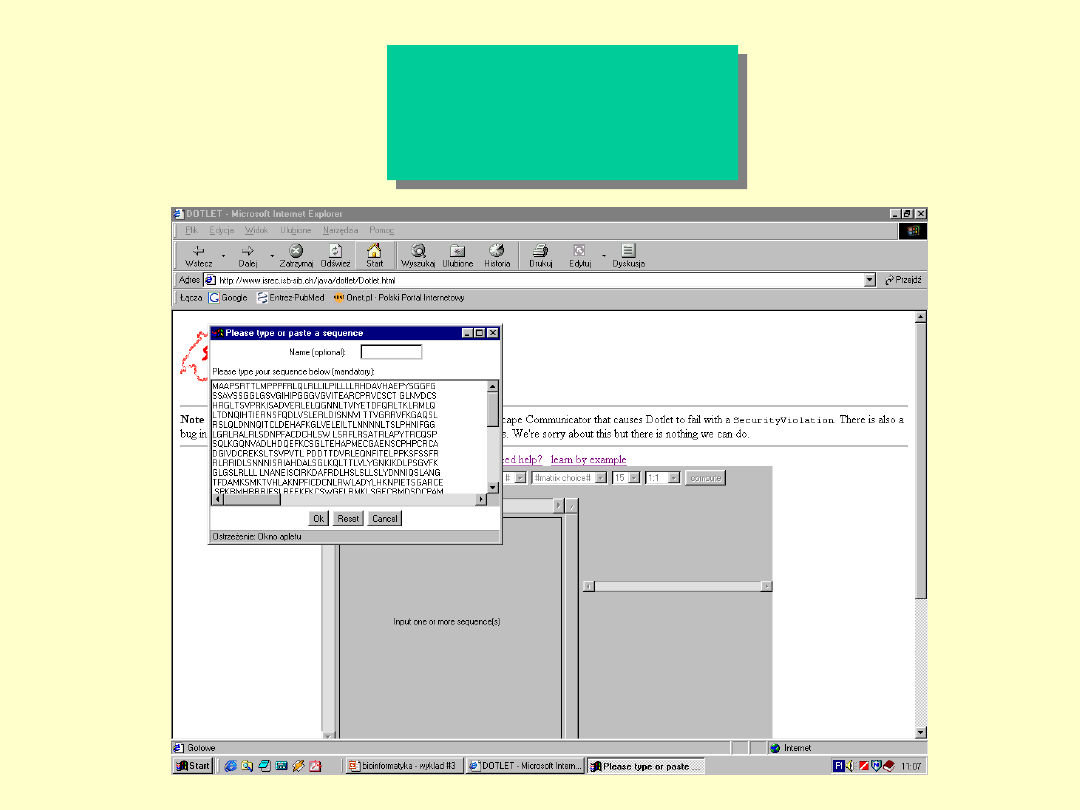
Dotlet
Dot Matrix
program
Dotlet
Dot Matrix
program

Dotlet
Dot Matrix
program
Dotlet
Dot Matrix
program
Analiza sekwencji AA bez zastosowania
Analiza sekwencji AA bez zastosowania
filtrów
filtrów
Analiza sekwencji AA bez zastosowania
Analiza sekwencji AA bez zastosowania
filtrów
filtrów
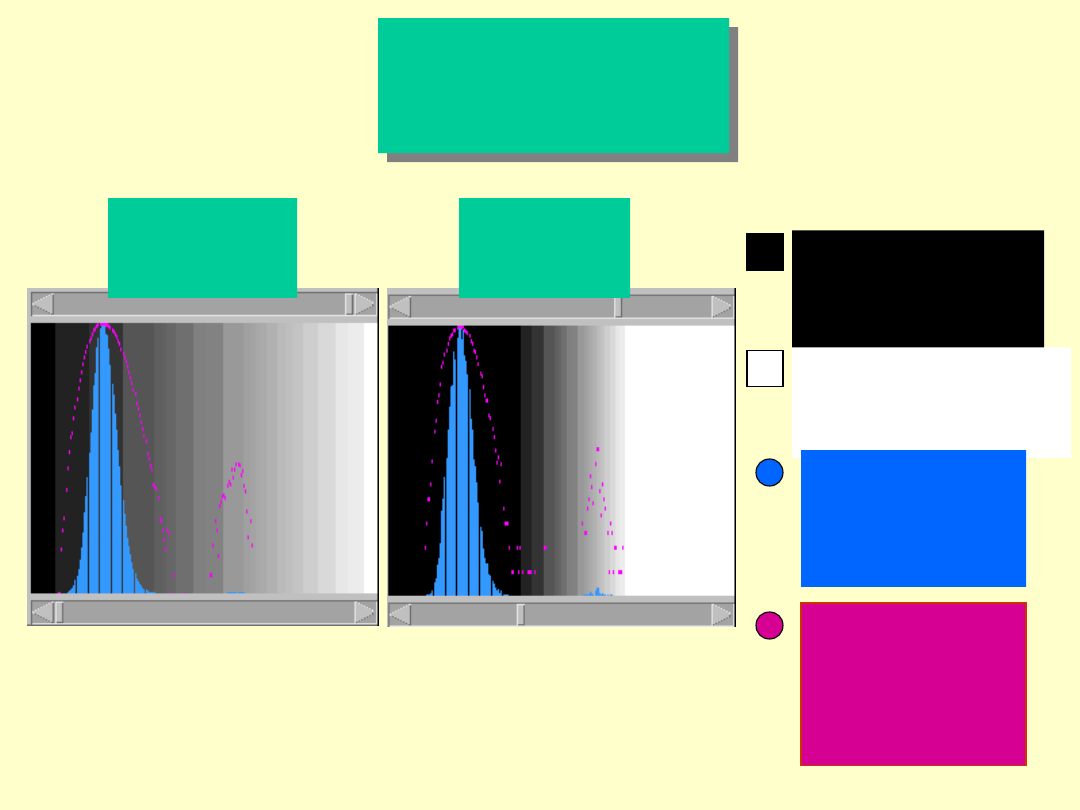
Dotlet
Dot Matrix
program
Dotlet
Dot Matrix
program
Czarne piksele
określają
niski stopień
podobieństwa
Białe piksele określają
wysoki stopień
podobieństwa
niebieski określa
częstotliwość
różnych stopni
podobieństwa w
skali liniowej
Różowy określa
częstotliwość
różnych stopni
podobieństwa w
skali
logarytmicznej
Przed
określeniem
progu czułości
Po
określeniu
progu
czułości
Dotlet
Dot Matrix
program
Dotlet
Dot Matrix
program
Analiza sekwencji AA z zastosowaniem
Analiza sekwencji AA z zastosowaniem
filtrów
filtrów
Analiza sekwencji AA z zastosowaniem
Analiza sekwencji AA z zastosowaniem
filtrów
filtrów
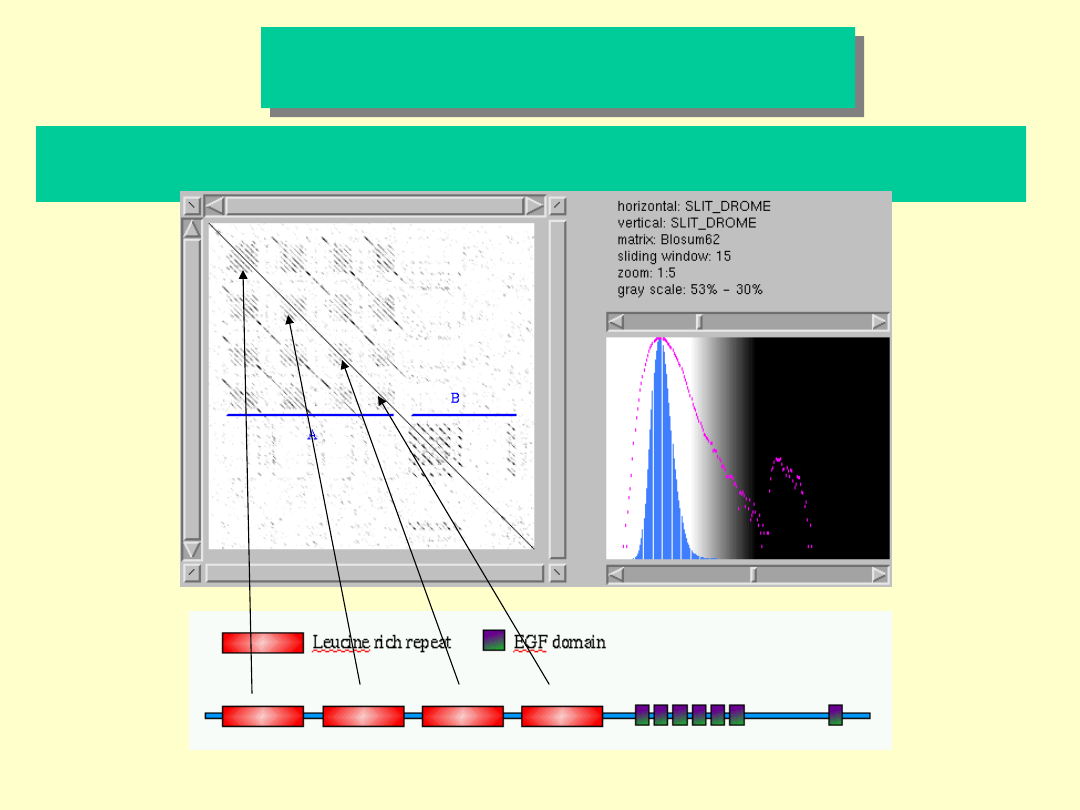
Drosophila melanogaster
białko SLIT
Drosophila melanogaster
białko SLIT
Analiza sekwencji białka SLIT gdzie zaszła duplikacja fragmentów
aminokwasowych
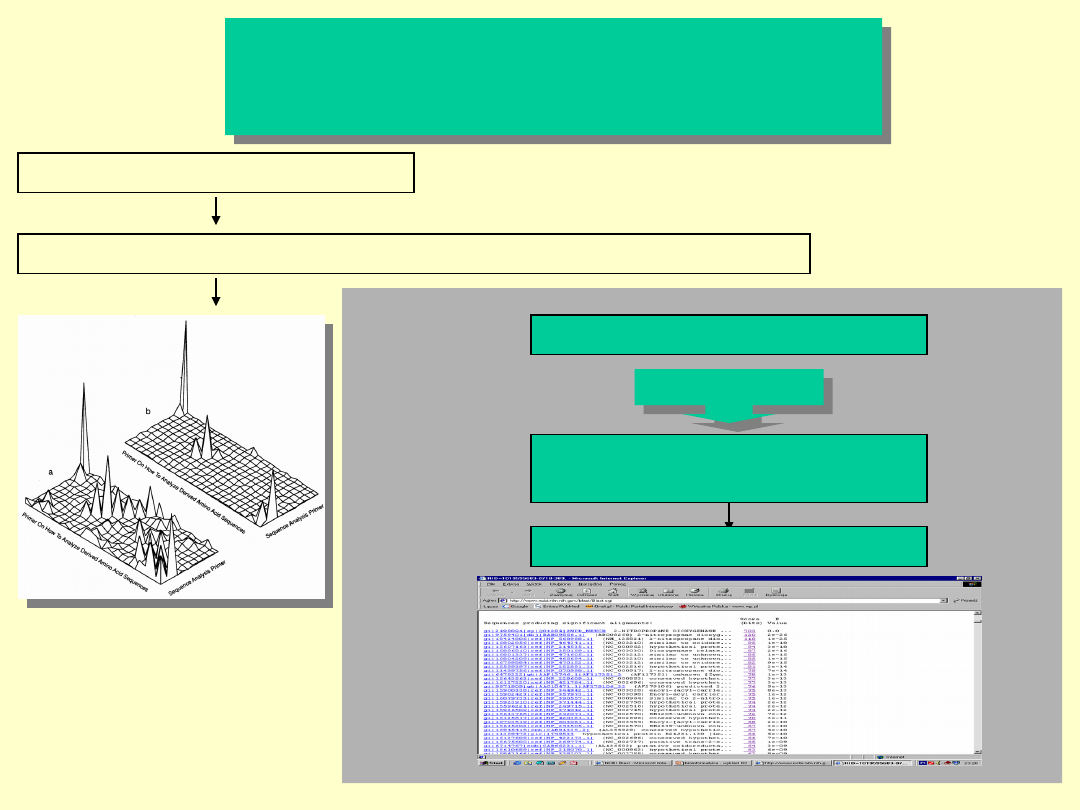
Przeszukiwanie baz danych
Przeszukiwanie baz danych
SEQUENCE ANALYSIS PRIMER
PRIMER ON HOW TO ANALYZE DERIVED AMINO ACID SEQUENCES
SEQUENCE ANALYSIS PRIMER
Baza Danych:
- GeneBank ...
Rankingowa lista trafień
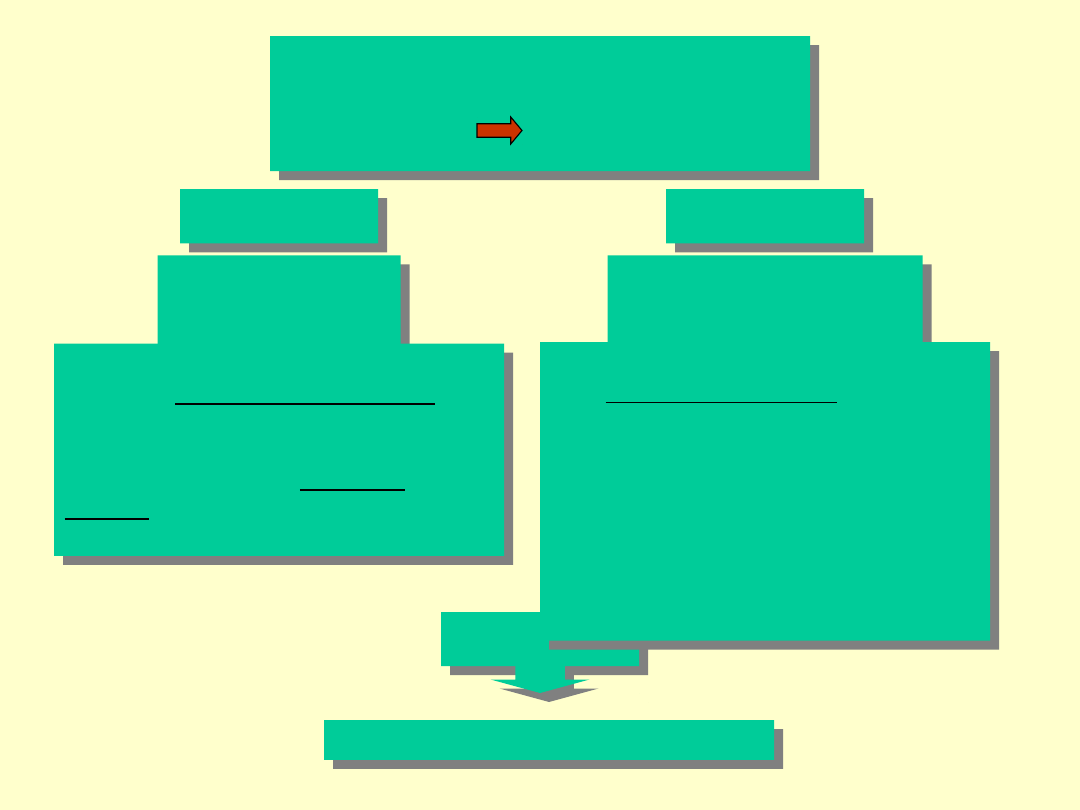
Programy służące do
analizy
białko baza danych
Programy służące do
analizy
białko baza danych
FASTA
FASTA
BLAST
BLAST
Fast Alignment
with Statistical
Analysis
F
ast
A
lignment
with
St
atistical
A
nalysis
Basic Local
Alignment Search
Tool
B
asic
L
ocal
A
lignment
S
earch
T
ool
Baza Danych
Baza Danych
Poszukiwanie odbywa się na
zasadzie dopasowań lokalnych
opartych na macierzach
substytucji PAM lub BLOSUM,
wykorzystując tzw. szablony
słowne uzyskane w uzyskanych
trafieniach.
Poszukiwanie odbywa się na
zasadzie dopasowań lokalnych
opartych na macierzach
substytucji PAM lub BLOSUM,
wykorzystując tzw. szablony
słowne uzyskane w uzyskanych
trafieniach.
Program posługuje się algorytmem
tzw. sąsiadujących słów, gdzie
napotkanie poszukiwanego słowa
prowadzi do znalezienia
optymalnego dopasowania
lokalnego (które spełnia kryterium
opisane przez wartość T) a
następnie wydłużanie
dopasowanych sekwencji z lewej
jak i z prawej strony.
Program posługuje się algorytmem
tzw. sąsiadujących słów, gdzie
napotkanie poszukiwanego słowa
prowadzi do znalezienia
optymalnego dopasowania
lokalnego (które spełnia kryterium
opisane przez wartość T) a
następnie wydłużanie
dopasowanych sekwencji z lewej
jak i z prawej strony.
Rankingowa lista trafień
Rankingowa lista trafień

BLAST
Basic Local Alignment
Search Tool
BLAST
B
asic
L
ocal
A
lignment
S
earch
T
ool
BLASTP
- Analiza porównawcza –
białko/białko
BLASTN
- Analiza porównawcza –
DNA/DNA
BLASTX
- Analiza porównawcza –
tłumaczone DNA na
białko/białko
TBLASTN
- Analiza porównawcza –
białko/ tłumaczone
DNA na białko
TBLASTX
- Analiza porównawcza –
tłumaczone DNA na
białko/
tłumaczone
DNA na białko

Bazy danych zawierające
sekwencje DNA lub AA
Bazy danych zawierające
sekwencje DNA lub AA
Database
Description
nr
All non-redundant GenBank CDS translations+PDB+SwissProt+PIR+PRF
month
All new or revised GenBank CDS translation+PDB+SwissProt+PIR released in the
last 30 days.
swissprot
The last major release of the SWISS-PROT protein sequence database (no
updates). These are uploaded to our system when they are received from EMBL.
patents
Protein sequences derived from the Patent division of GenBank.
yeast
Yeast (Saccharomyces cerevisiae) protein sequences. This database is not to be
confused with a listing of all Yeast protein sequences. It is a database of the
protein translations of the Yeast complete genome.
E. coli
E. coli (Escherichia coli) genomic CDS translations.
pdb
Sequences derived from the 3-dimensional structure Brookhaven Protein Data
Bank.
kabat
[kabatpr
o]
Kabat's database of sequences of immunological interest. For more information
Białka
Białka

DNA
DNA
Database
Description
nr
All non-redundant GenBank+EMBL+DDBJ+PDB sequences (but no EST, STS, GSS, or
HTGS sequences).
month
All new or revised GenBank+EMBL+DDBJ+PDB sequences released in the last 30 days.
dbest
Non-redundant database of GenBank+EMBL+DDBJ EST Divisions.
dbsts
Non-redundant database of GenBank+EMBL+DDBJ STS Divisions.
mouse
ests
The non-redundant Database of GenBank+EMBL+DDBJ EST Divisions limited to the
organism mouse.
human
ests
The Non-redundant Database of GenBank+EMBL+DDBJ EST Divisions limited to the
organism human.
other
ests
The non-redundant database of GenBank+EMBL+DDBJ EST Divisions all organisms except
mouse and human.
yeast
Yeast (Saccharomyces cerevisiae) genomic nucleotide sequences. Not a collection of all
Yeast nucelotides sequences, but the sequence fragments from the Yeast complete
genome.
E. coli
E. coli (Escherichia coli) genomic nucleotide sequences.
pdb
Sequences derived from the 3-dimensional structure of proteins.
kabat
[kabatnu
c]
Kabat's database of sequences of immunological interest. For more information
patents
Nucleotide sequences derived from the Patent division of GenBank.
vector
Vector subset of GenBank(R), NCBI,
mito
Database of mitochondrial sequences (Rel. 1.0, July 1995).

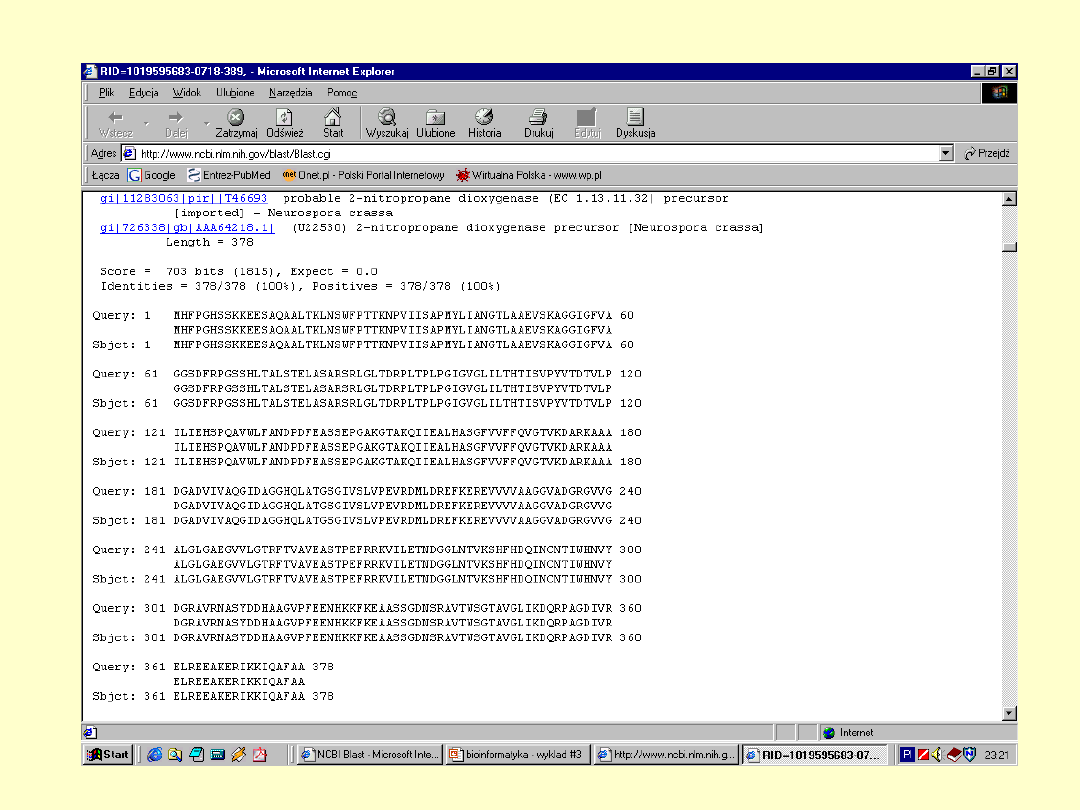
2-nitropropane
dioxygenase
-precursor, FMN
dependent
2-nitropropane
dioxygenase
-precursor, FMN
dependent
MHFPGHSSKK EESAQAALTK LNSWFPTTKN PVIISAPMYL IANGTLAAEV
SKAGGIGFVA GGSDFRPGSS HLTALSTELA SARSRLGLTD RPLTPLPGIG
VGLILTHTIS VPYVTDTVLP ILIEHSPQAV WLFANDPDFE ASSEPGAKGT
AKQIIEALHA SGFVVFFQVG TVKDARKAAA DGADVIVAQG IDAGGHQLAT
GSGIVSLVPE VRDMLDREFK EREVVVVAAG GVADGRGVVG ALGLGAEGVV
LGTRFTVAVE ASTPEFRRKV ILETNDGGLN TVKSHFHDQI NCNTIWHNVY
DGRAVRNASY DDHAAGVPFE ENHKKFKEAA SSGDNSRAVT WSGTAVGLIK
DQRPAGDIVR ELREEAKERI KKIQAFAA
BLAST
Server
BLAST
Server
http://www.ncbi.nlm.nih.gov/
http://www.ncbi.nlm.nih.gov/
BLAST

BLAST
Server
BLAST
Server
BLAST
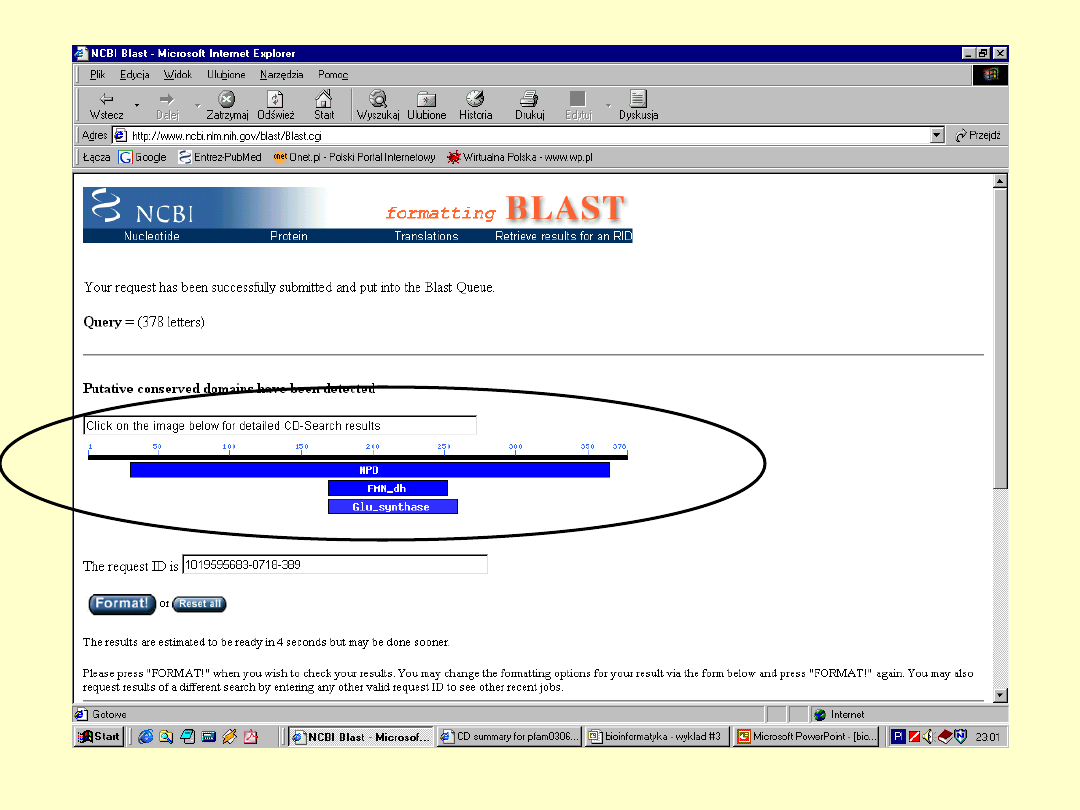
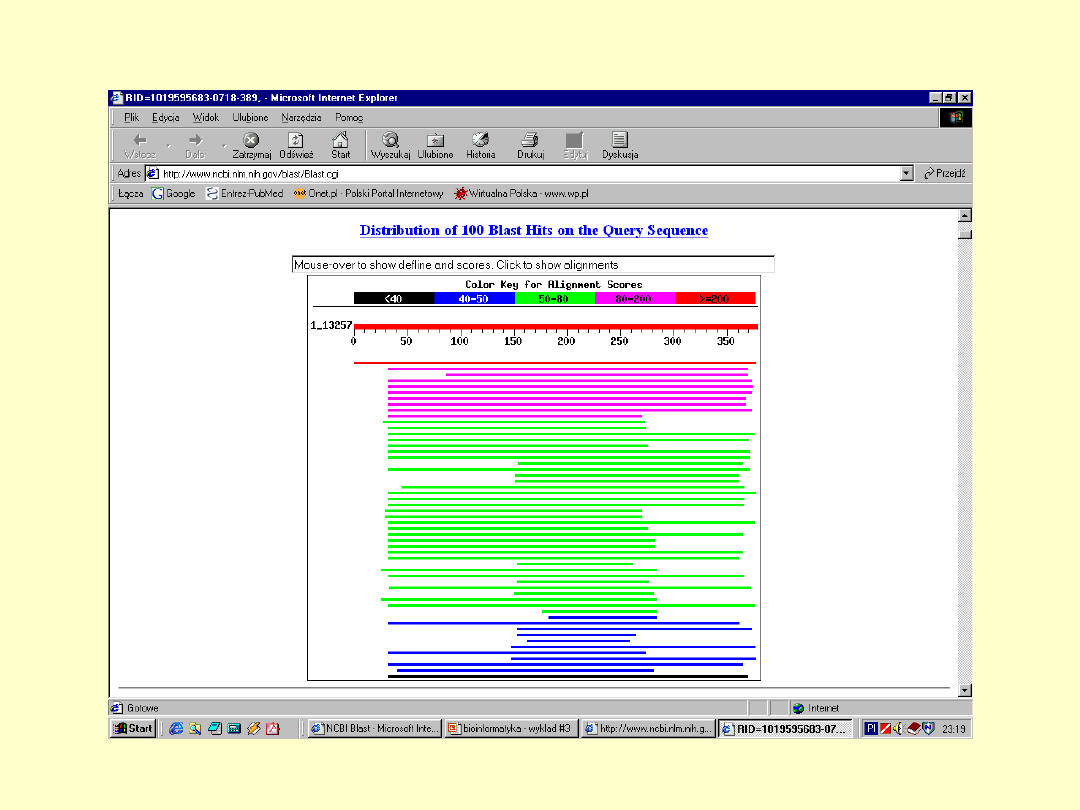
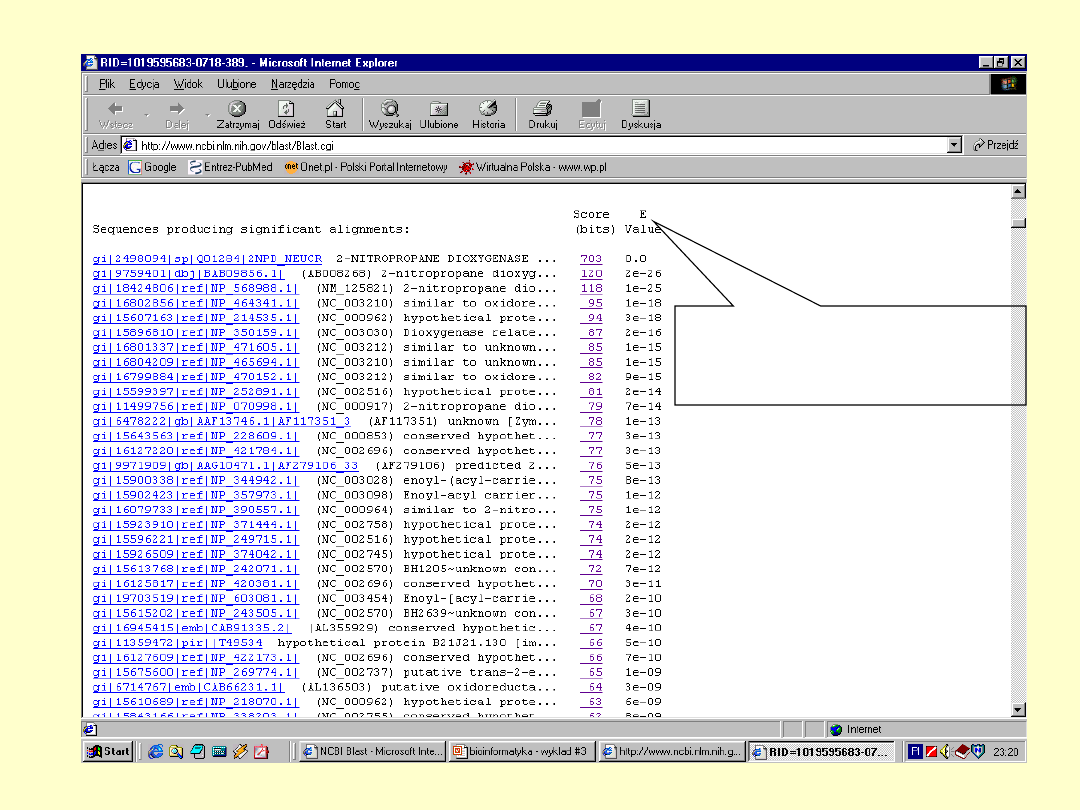
E – określa
spodziewaną liczbę
losowych dopasowań
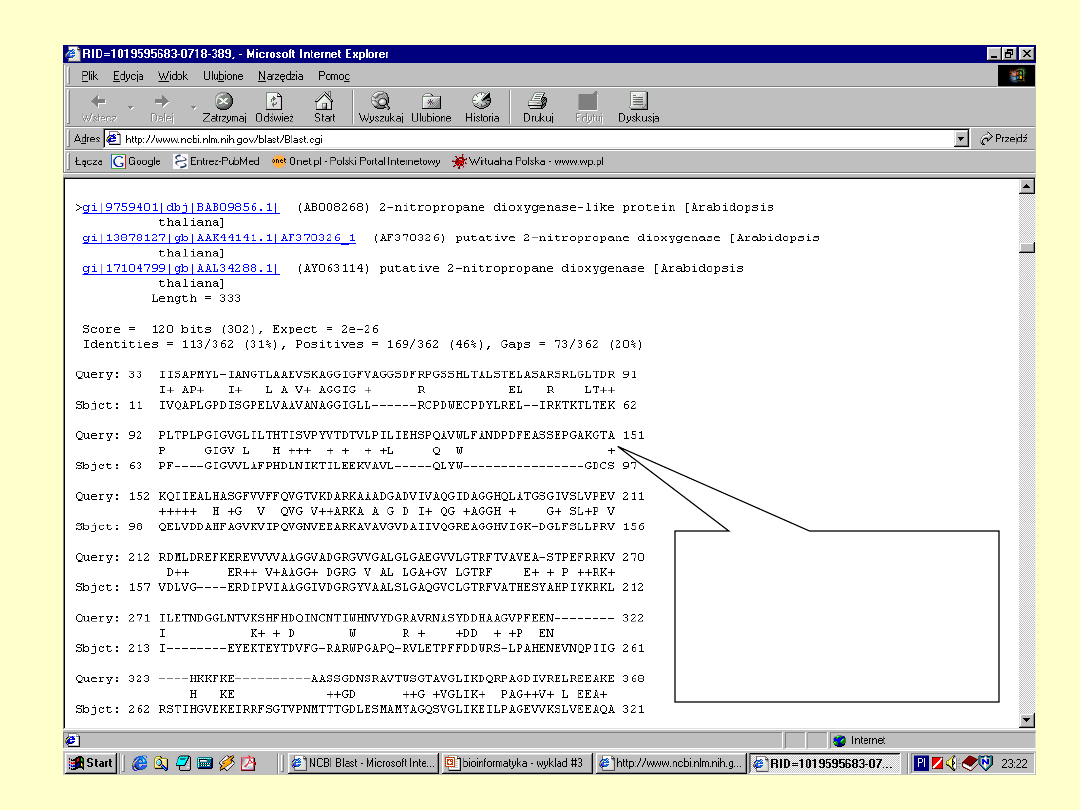
+ - określa parę
aminokwasów
mających dodatnią
wartość substutucji
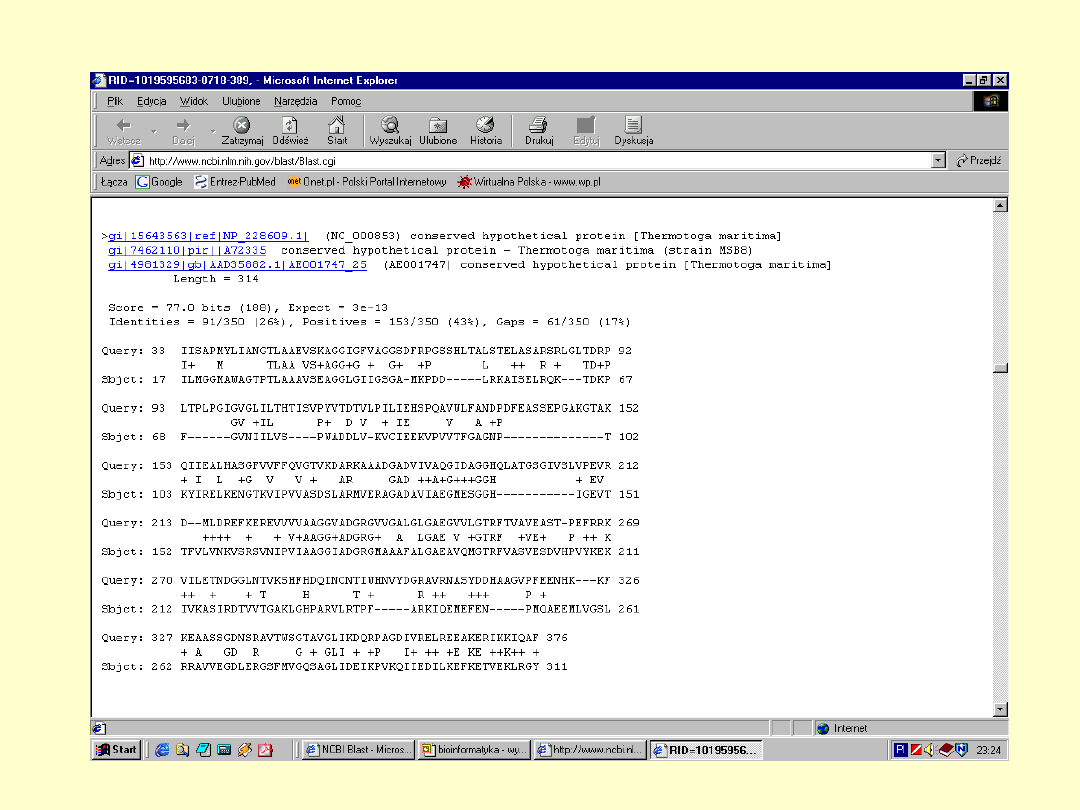
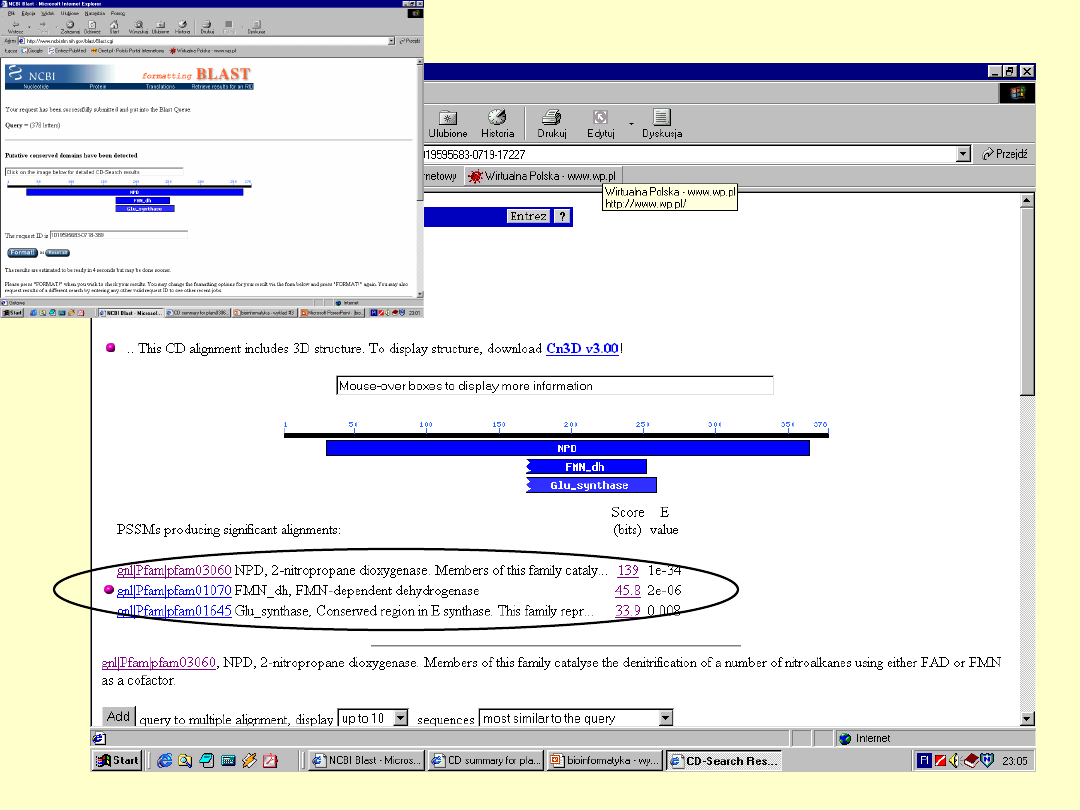
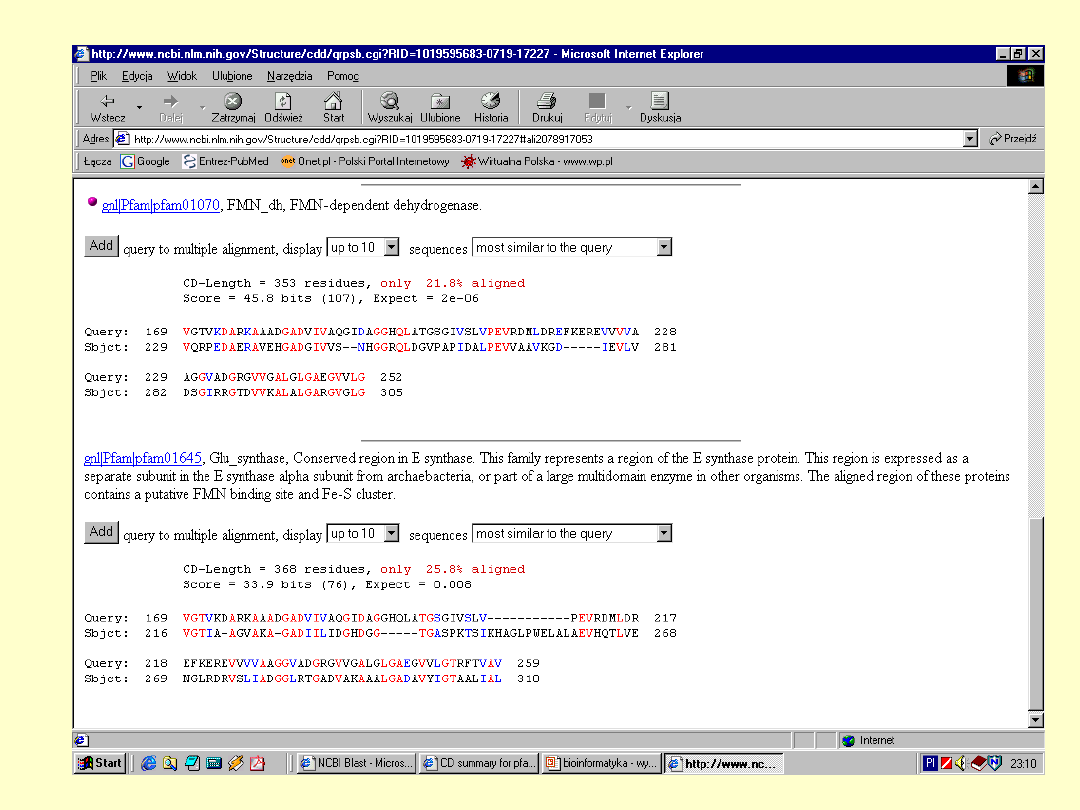
Dokładna
informacja o
białku
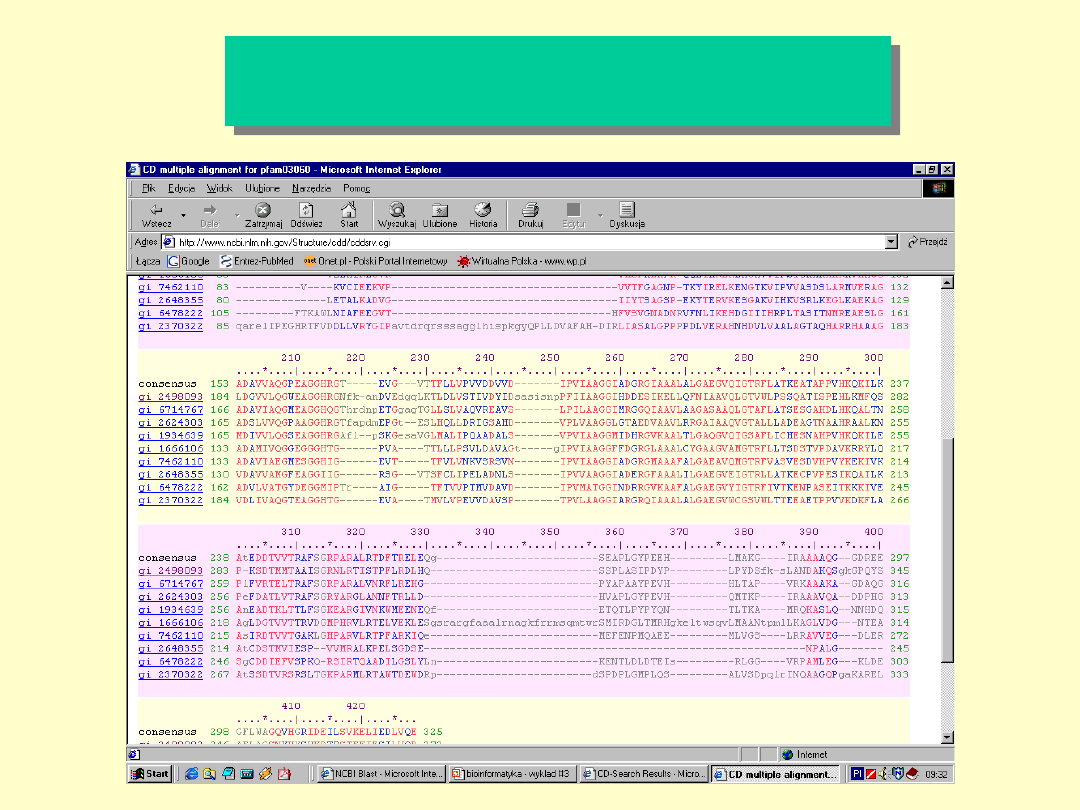
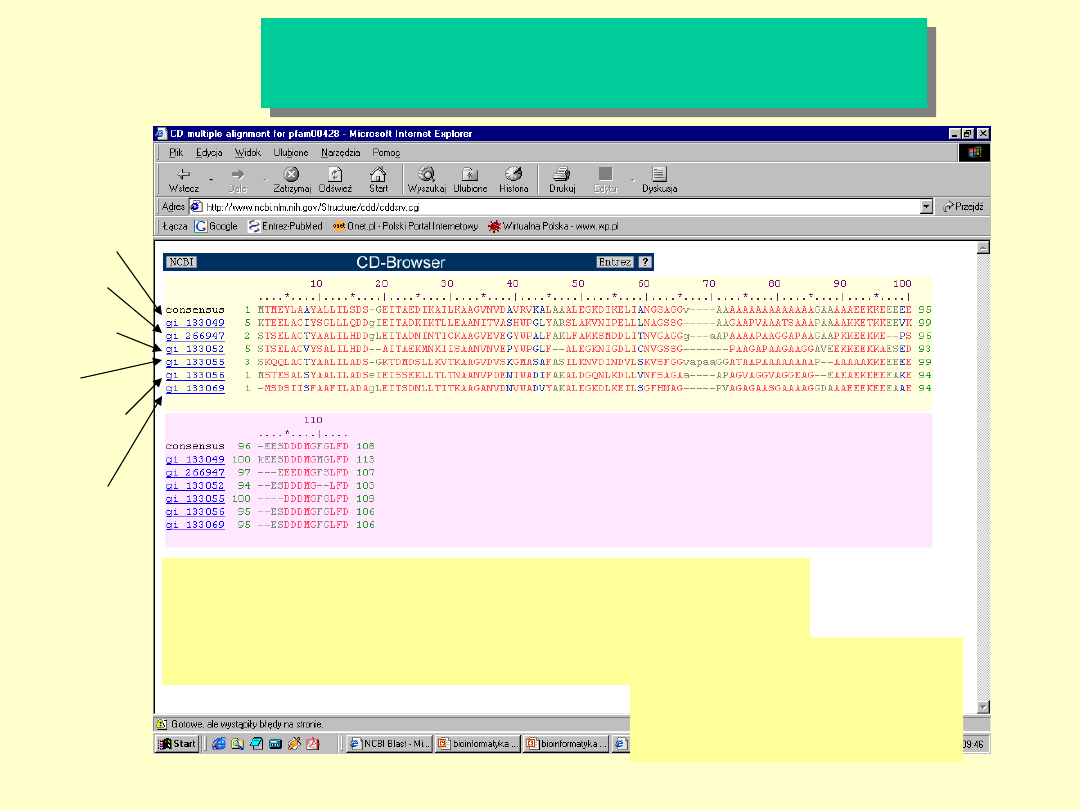
Multiple Sequence
Alignment
Multiple Sequence
Alignment
Multiple Sequence
Alignment
Multiple Sequence
Alignment
MSTESALSYA ALILADSEIE ISSEKLLTLT NAANVPVENI
WADIFAKALD GQNLKDLLVN FSAGAAAPAG VAGGVAGGEA
GEAEAEKEEE EAKEESDDDM GFGLFD
Drożdże
Meduza
Człowiek
Ryba
Kukurydz
a
Muszka
Owocowa
Drożdże, Saccharomyces
cerevisiae
Kwaśne białko rybosomowe,
P1A
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
Wyszukiwarka
Podobne podstrony:
elementy bioinformatyki wyklad2
Bioinformatyka wykład 1
Bioinformatyka wykład 3
elementy bioinformatyki wyklad4
bioinformatyka wyklad #6
Bioinformatyka wykłady
bioinfoI wyklad01
elementy bioinformatyki wyklad3
bioinfoI wyklad03
bioinfoI wyklad02
Bioinformatyka wyklad #4
elementy bioinformatyki wyklad1
Bioinformatyka wykładMocx
bioinformatyka wyklad #2
Bioinformatyka wykład 5
bioinfoI wyklad04
Bioinformatyka wykład ocx
elementy bioinformatyki wyklad2
więcej podobnych podstron