Elektrochemiczne metody analizy w ochronie środowiska
Etapy procesu analitycznego

B adany
obiekt
adany
obiekt
 Pobieranie
próbki
Pobieranie
próbki
Próbka
Przygotowanie próbki

P
róbka
w postaci umożliwiającej jej analizę
Pomiar

W yniki
pomiaru
yniki
pomiaru
Interpretacja wyniku

Wynik analizy
Pobieranie próbek do analizy
Pobieranie próbek, ich opakowanie, transport oraz przechowywanie musi być przeprowadzone tak, aby zapobiec: zanieczyszczeniu próbki, utracie lotnych składników oraz reakcjom np. ze składnikami powietrza pod wpływem temperatury lub światła
Próbka musi być reprezentatywna dla danego obiektu badanego (łatwiej to osiągnąć dla układów homogenicznych, takich jak gaz lub ciecz, trudniej dla ciał stałych, które są przeważnie niehomogeniczne)
Sposoby pobierania próbek do analizy:
Próbki gazowe mogą być pobierane do specjalnych pojemników (np. worki teflonowe, pipety gazowe) lub kolektorów (próbka ulega absorpcji w różnych cieczach lub na absorbentach stałych takich jak np. węgiel aktywny, polimery porowate)
W przypadku próbek ciekłych są one często przechowywane w obniżonej temperaturze i z dodatkiem substancji zapobiegających niepożądanym reakcjom i adsorpcji na ściankach naczyń
W przypadku próbek ciała stałego należy e podać homogenizacji w sposób mechaniczny (różne typy młynków lub moździerzy)

B
 adany
obiekt
adany
obiekt

P
róbka
pierwotna

P
 róbka
laboratoryjna
róbka
laboratoryjna



Próbka analityczna próbka analityczna próbka analityczna
Przygotowanie próbek do analizy
Obejmuje różne operacje:
Rozdrabnianie, suszenie, filtracja
Przeprowadzenie próbek do roztworu – najwięcej trudności
Wydzielenie, rozdzielenie, zatężanie analitu
Maskowanie czynników zakłócających pomiar
Przeprowadzenie próbek do roztworu
Poprzez rozpuszczenie (zachodzi w wyniku pokonania energii sieci krystalicznej ciał stałych przez energię solwatacji); rozpuszczenie w przypadku próbek rzeczywistych ma ograniczone zastosowanie, ponieważ większość z nich nie rozpuszcza się ani w rozpuszczalnikach polarnych ani niepolarnych, bądź też rozpuszcza się w stopniu ograniczonym
Poprzez mineralizację (zachodzi w wyniku reakcji chemicznych)
Mineralizacja to rozkład matrycy organicznej do prostych związków nieorganicznych, substancje organiczne ulegają utlenieniu i ulotnieniu w wyniku przebiegu prostych reakcji chemicznych:
Corg →CO2 Horg→H2O Norg → N2 Sorg →SO2
Proces mineralizacji powinien spełniać następujące warunki:
Część organiczna powinna ulec utlenieniu i ulotnieniu podczas, gdy część nieorganiczna powinna ilościowo pozostać
Mineralizacja powinna przebiegać szybko
Nie powinno zachodzić zanieczyszczenie próbki
Aparatura powinna być prosta i tania
Rozróżnia się dwa sposoby mineralizacji:
Mineralizacja sucha
Mineralizacja mokra
Mineralizacja sucha polega na spaleniu próbki w piecu, w tygielku lub parownicy w temperaturze 4000 – 6000 C w czasie kliku godzin. W trakcie rozkładu powstaje popiół złożony głównie z węglanów i tlenków, które następnie roztwarza się w odpowiednim kwasie lub mieszaninie kwasów.
Zalety: bardzo łatwa w wykonaniu
Wady: możliwość strat analitu na skutek lotności (takich pierwiastków jak As, Hg, Pb, Sb, Cr, Cu), trudności w rozpuszczeniu powstałego popiołu
Przyczyny strat w czasie mineralizacji suchej:
Corg szybko przechodzi do Cpierw a ten powoli do CO2, Cpierw ma zdolności redukujące i może redukować związki metali do postaci metalicznej, a niektóre metale są lotne i opuszczają środowisko próbki
Jeżeli w próbce znajduje się chlor w postaci związków organicznych, to w czasie mineralizacji mogą tworzyć się lotne chlorki oznaczanych metali
Aby zapobiegać stratom można przeprowadzić mineralizację suchą z dodatkiem
Np. H2SO4, HNO3, Mg(NO3)2, NH4NO3, które powodują zwiększenie przejścia Cpierw do CO2
Dodatek może mieć również za zadanie zwiększenie stabilności pierwiastka oznaczanego np. przy oznaczaniu baru dodajemy do próbki CaO, tworzą się wtedy borany, które są nielotne
Mineralizacja mokra polega na rozkładzie matrycy organicznej odczynnikami utleniającymi, głównie stężonymi kwasami: HNO3, H2SO4, HClO4 w różnych kombinacjach lub 30% H2O2. Do próbek zawierających krzemionkę dodaje się HF.
W przypadku stosowania HClO4 istnieje niebezpieczeństwo wybuchu i dlatego dodaje się go w końcowym etapie rozkładu próbki zawierającej HNO3 lub do próbki wprowadza się dodatkowo kilka mg NH4VO3.
Wybrane procedury mineralizacji mokrej
Mineralizacja w układzie otwartym
Do próbki umieszczonej w kolbce wprowadza się HNO3 i H2SO4 i całość ogrzewa
Wada: nie można stosować dla pierwiastków lotnych
Mineralizacja w układzie zamkniętym
Do próbki umieszczonej w kolbce wprowadza się HNO3 i H2SO4 i całość ogrzewa, pary kwasów odparowują (głównie HNO3), ulegają skropleniu w chłodnicy i na początku wracają do kolby, pod koniec mineralizacji układ zamykamy i pary kwasów ulegające skropleniu w chłodnicy są zbierane w zapasowym zbiorniku; po odparowaniu kwasów temperatura w kolbie rośnie powyżej temperatury wrzenia kwasu HNO3, wtedy po kropli wkraplamy HNO3 (zebrany w zapasowym zbiorniku) do próbki z H2SO4 o bardzo dużej temperaturze, co przyspiesza reakcję mineralizacji.
Zalety: nie ulatniają się pierwiastki lotne, reakcja mineralizacji przebiega szybciej (ale i tak trwa kilka godzin)
Mineralizacja w układzie zamkniętym – ciśnieniowym
Polega na reakcji analizowanej próbki z kwasami mineralnymi (najczęściej HNO3 lub HNO3 + H2O2) w podwyższonej temperaturze (głównie dzięki zastosowaniu energii mikrofalowej) w zamkniętym naczyniu z teflonu, zwanym dawniej bombą teflonową, umieszczonym w chłodzonym wodą stalowym płaszczu. Czas mineralizacji pod ciśnieniem jest znacznie krótszy i wynosi od kilku do kilkunastu minut.
Proces mineralizacji prowadzi się w naczyniach:
Teflonowych – dopuszczalna temperatura do 1700C
Z węgla szklistego – dopuszczalna temperatura do 2200C
Kwarcowych – nie ma ograniczeń temperaturowych(ale są bardzo kruche i wtedy ciśnienie na zewnątrz naczynia musi być bliskie ciśnieniu wewnątrz naczynie)
Urządzenia do prowadzenia procesów mineralizacji posiadają specjalne systemy zabezpieczeń oraz wskaźnik i miernik temperatury i ciśnienia.
W zależności od rodzaju próbki i techniki dobiera się odpowiednie warunki prowadzenia mineralizacji: rodzaj substancji mineralizujących, program mocy mikrofal i ciśnienia.
W handlu dostępne są dwa rodzaje takich mineralizatorów pracujące przy: średnich ciśnieniach do 30 atm oraz wysokich ciśnieniach do 110 atm
Wady: wysoka cena aparatury, konieczność stosowania małych odważek próbek (zwykle do 1g)
Mineralizacja z wykorzystaniem ultradźwięków
Polega na umieszczeniu próbki wraz z naczynkiem reakcyjnym, zawierającym kwasy i ewentualnie utleniacze w łaźni ultradźwiękowej. Technika stosowana do roztwarzania stałych próbek środowiskowych.
Mineralizacja z wykorzystaniem mikrofal
Stosowana głównie w układach zautomatyzowanych. Próbka z kwasem mineralizującym za pomocą pompy przepływa przez wąż teflonowy pod ciśnieniem np. 30 atm nad generatorem mikrofal, gdzie jest ogrzewana i tam przebiega mineralizacja, a następnie wprowadzana jest do aparatury pomiarowej. Długość węża i szybkość przepływu próbki dobieramy tak by na wylocie uzyskać próbkę dostatecznie zmineralizowaną do stosowanej metody analitycznej.
Mineralizacja z wykorzystaniem promieniowania
M
 etody
analityczne
etody
analityczne
 Metody klasyczne
Metody instrumentalne
Metody klasyczne
Metody instrumentalne
(wagowe i miareczkowe)
Metody elektrochemiczne Metody spektroskopowe
Elektrochemiczne metody instrumentalne
Metody w których reakcja elektrodowa przebiega bez przyłożonego napięcia zewnętrznego (czy przy zerowym prądzie Faradaya)
Potencjometria (mierzymy potencjał elektrody)
Metody w których reakcja elektrodowa przebiega pod wpływem przyłożonego do elektrod napięcia z zewnętrznego źródła:
Metody oparte na procesie elektrolizy na mikroelektrodach (dużych elektrodach gdzie elektrolizie ulega cała próbka i aby powtórzyć pomiar musimy wziąć nową porcję próbki)
Elektrograwimetria (mierzymy masę produktów elektrolizy)
Kulometria (mierzymy ładunek niezbędny d przeprowadzenia reakcji)
Metody oparte na procesie elektrolizy na mikroelektrodach (małych elektrodach, gdzie elektrolizie ulega niewielka część próbki i pomiar możemy powtórzyć z tej samej porcji próbki)
Polarografia
Woltamperometria
Woltamperometria z zatężeniem
Metody, w których nie przebiega reakcja elektrodowa
Konduktometria (mierzymy przewodnictwo roztworu)
METODY POTENCJOMETRYCZNE
W metodach potencjometrycznych wykonuje się pomiar siły elektromotorycznej (SEM) ogniwa zbudowanego z dwóch elektrod (elektrody wskaźnikowej i elektrody porównawczej) zanurzonych w badanym roztworze.
SEM ogniwa jest to różnica potencjałów pomiędzy elektrodą wskaźnikową i porównawczą
Potencjał elektrody można wyznaczyć z równania Nernsta
Nie da się wyznaczyć bezwzględnej wartości potencjału elektrody i podajemy tylko względną wartość potencjału elektrody (potencjał elektrody wodorowej został przyjęty za równy zero i potencjały normalne innych elektrod podajemy względem potencjału elektrody wodorowej 0).
Proces elektrodowy, równanie Nernsta
Każdy metal zanurzony w roztworze elektrolitu wykazuje dążność do przechodzenia do roztworu, zaś jon tego metalu w roztworze dąży do przejścia do formy metalicznej i osadza się na elektrodzie
Me – ne↔Men+
Dążność metalu do przechodzenia do roztworu Nernst nazwał prężnością roztwórczą i podał równanie opisujące zależność między potencjałem elektrody a stężeniem jonu w roztworze
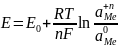 równanie Nernsta
równanie Nernsta
E0 – potencjał normalny elektrody (wartość stała) [V]
R – stała gazowa
T – temperatura bezwzględna [K]
N – ilość elektronów biorących udział w reakcji elektrodowej
F – stała
Faradaya 96500
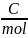
aMe+n – aktywność jonu metalu
aMe0 – jest stała = 1
Po podstawieniu stałych dla temperatury 250C i zamianie logarytmu naturalnego na dziesiętny wzór ten przyjmuje postać:
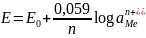
Podział elektrod ze względu na mechanizm reakcji elektrodowych:
Elektrody I rodzaju (odwracalne względem kationu lub anionu, np. elektroda srebrowa, wodorowa, miedziowa)
Elektrody II rodzaju (odwracalne względem wspólnego anionu, np. elektroda chlorosrebrowa, kalomelowa)
Elektrody III rodzaju (odwracalne względem wspólnego kationu)
Elektrody erdoks (obojętny chemicznie metal (Pt, Au), węgiel szklisty albo grafit zanurzony w roztworze zawierającym substancje w formie utlenionej jak i zredukowanej, np. elektroda chinhydronowa)
Elektrody metaliczne (np. elektroda antymonowa, bizmutowa)
Elektrody jonoselektywne inaczej membranowe (np. elektroda szklana)
Podział elektrod ze względu na funkcje jaką pełnią:
Elektrody porównawcze (odniesienia) – zachowują w warunkach pomiaru stały potencjał, praktycznie niezależny od składu badanego roztworu
Elektrody wskaźnikowe – są to elektrody, których potencjał w badanym roztworze zmienia się pod wpływem jonów na które są czułe
Elektrody I rodzaju
Elektroda srebrowa – czuła na jony Ag+
Budowa: stanowi ją drut Ag zanurzony w roztworze soli Ag+
Reakcja elektrodowa: Ag ↔ Ag+ + e
Wzór na potencjał elektrody: E = E0 + 0,059 log[Ag+]
Elektrodę tą stosuje się do:
Wyznaczania stężenia jonów Ag+
Wyznaczania PK w miareczkowaniach z zastosowaniem AgNO3
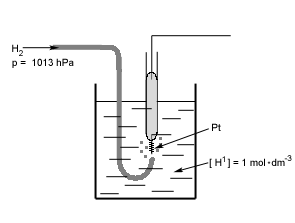
Elektroda wodorowa – czuła na jony H+
Ma małe znaczenie praktyczne, ale duże znaczenie teoretyczne
Jest wzorcem potencjału (jej potencjał w określonych warunkach przyjęty za równy 0 V i potencjały wszystkich elektrod są podawane właśnie względem elektrody wodorowej)
Budowa: stanowi ją blaszka platynowa, pokryta czernią platynową (silnie rozdrobniona Pt) zanurzona w roztworze HCl i omywana gazowym wodorem
Reakcja elektrodowa: H2↔ 2H+ + 2e
Wzór na potencjał elektrody:
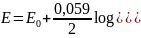
E = 0,059log[H+]
log[H+]
=
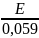
pH = -
Elektrody II rodzaju
Stanowią metale pokryte trudno rozpuszczalną solą tego metalu, znajdujące się w roztworze zawierającym anion tej soli
Elektroda chlorosrebrowa – porównawcza
Budowa: stanowi ją drut Ag pokryty AgCl zanurzony w roztworze soli o stałym stężeniu jonów Cl-
Reakcja elektrodowa: Ag – e → Ag+
Wzór na potencjał elektrody:
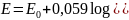
E = E0 + 0.059log[Ag+]
IR = [Ag+][Cl-]
[Ag+]
=
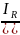
E = E0
+ 0,059log
E = E0 + 0,059logIR – 0,059log[Cl-]
E = E0’ – 0,059log[Cl-]
W przypadku, gdy aktywność jonów chlorkowych jest stała potencjał elektrody ma również wartość stałą i elektroda może być stosowana więc jako elektroda porównawcza.
Aby przygotować drut Ag pokryty AgCl:
Należy do zlewki z HCl o stężeniu około 4 mol/L zanurzyć drut Ag
Podłączyć go jako anodę, zaś jako katodę podłączyć drut Pt
Po przyłożeniu napięcia 4 V w czasie ok. 10 – 15 minut elektroda Ag pokrywa się warstwą AgCl, zaś na katodzie wydziela się wodór
Elektroda kalomelowa – porównawcza
Budowa: stanowi ją warstwa rtęci pokryta pastą rtęci i kalomelu (Hg2Cl2), nad którą znajduje się roztwór soli o stałym stężeniu jonów Cl- np. NaCl lub kCl
Reakcja elektrodowa: 2Hg ↔ Hg22+ + 2e
Wzór na potencjał elektrody:
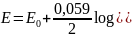
IR = [Hg22+][Cl-]2
[Hg22+]
=
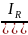
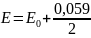 log
log
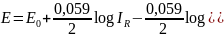

Elektrody III rodzaju
Rzadko stosowane
Elektrody redoks
Elektroda chinhydronowa
Budowa: Stanowi ją drut Pt zanurzony w nasyconym roztworze chinhydronu, czyli równomolowego produktu addycji chinonu C6H4O2 i hydrochinonu C6H4(OH)2 tworząc odwracalny układ redoks z udziałem jonów H+
Reakcja elektrodowa: C6H4(OH)2↔ C6H4O2 + 2H+ + 2e
Wzór na potencjał elektrody:
log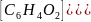
Ponieważ [C6H4O2] = [C6H4(OH)2]
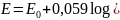
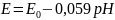
Potencjał elektrody zależy więc tylko od stężenia H+, zatem jest ona elektrodą wskaźnikową, a jej potencjał zależy od pH roztworu
Można ją stosować w zakresie pH od 1,5 do 8,5
Nie można jej stosować w obecności utleniaczy silniejszych od chinonu i reduktorów silniejszych od hydrochinonu
Elektrody metaliczne
Elektroda antymonowa
Budowa: stanowi ją pręcik z metalicznego antymonu pokryty warstwą jego tlenku, który tworzy się po zetknięciu antymonu z powietrzem
Reakcja elektrodowa: 2Sb + 3H2O ↔ Sb2O3 + 6H+ + 6e
Wzór na potencjał elektrody:
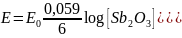
Potencjał elektrody zależy tylko od stężenia jonów H+, zatem jest ona elektrodą wskaźnikową i służy do pomiarów pH roztworów
Można ją stosować w zakresie pH od 3 do 8
Potencjał elektrody nie jest dobrze odtwarzalny, ale ze względu na wygodę jest często stosowany w przemyśle
Elektroda ta jest przydatna np. w miareczkowaniu potencjometrycznym, gdzie nie zależy nam na powtarzalnym pomiarze potencjału tylko na określeniu zmian potencjału
Elektroda bizmutowa
Budowa: jest podobna w budowie i działaniu do elektrody antymonowej.
Reakcja elektrodowa: 2Bi + 3H2O ↔ Bi2O3 + 6H+ + 6e
Wzór na potencjał elektrody:
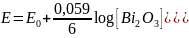
Główną jej zaletą jest to, że można ją stosować w zakresie pH od 7 do 14, czyli w silnie alkalicznym środowisku.
Elektrody jonoselektywne
Elektrody jonoselektywne )ISE) są to elektrody, których potencjał jest liniową funkcją logarytmu z aktywności (stężenia) jonu na który jest czuła dana elektroda.
Są to najpopularniejsze czujniki potencjometryczne o dużym znaczeniu praktycznym
Na dzień dzisiejszy za ich pomocą można oznaczać ok. 40 jonów oraz wiele związków organicznych
Elektrody jonoselektywne charakteryzują się dużą selektywnością i każda elektroda służy do oznaczania jednego jonu
Należy pamiętać, że nie istnieją elektrody idealnie selektywne!!!
Granica wykrywalności dla większości elektrod waha się w zakresie stężeń od 10-3 do 10-5 mol/L
Podział elektrod ISE
Ze względu na rodzaj membrany
|
ISE |
Stałomembranowe |
szklane |
do oznaczania H+ |
|
do oznaczania kationów Na+, K+, NH4+ |
|||
|
krystaliczne |
homogeniczne |
||
|
heterogeniczne |
|||
|
Ciekłomembranowe |
klasyczne |
|
|
|
polimerowe |
|||
|
Uczulane |
enzymatyczne |
||
|
gazowe |
Elektroda szklana
Pierwsza opracowana elektroda jonoselektywna do pomiaru pH
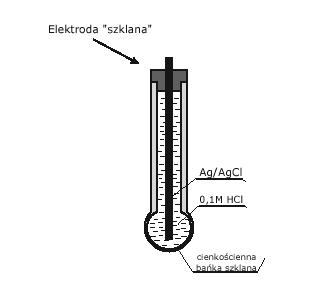
Źródłem powstawania potencjału na membranie elektrody szklanej jest żelowa struktura warstwy szkła.
Całkowity potencjał szklanej membrany jest równy różnicy potencjałów międzyfazowych, czyli potencjału na granicy próbka/zewnętrzna warstwa żelowa oraz potencjału na granicy wewnętrzna warstwa żelowa/roztwór wewnętrzny.
(rysunek)
Ponieważ aktywność H+ w obu warstwach żelowych szkła są sobie równe, a aktywność jonów H+ w buforze wewnętrznym jest utrzymywana na stałym poziomie to potencjał szklanej membrany zależy tylko od aktywności jonów H+ w roztworze próbki
E = E0 – 0,059pH
Zalety elektrod szklanych:
Wysoka stabilność potencjału
Niewrażliwość na obecność substancji utleniających i redukujących
Szeroki zakres liniowości ok. pH 1 -10 (1 – 12w przypadku szkła litowego), w tym zakresie ph jest liniową funkcja potencjału
Duży zakres temperatury nawet do 1300
Długi czas życia
Wady:
Przy wysokich wartościach pH jony Na+ wbudowują się w membranę elektrody powodując zmianę potencjału membrany szklanej w kierunku ujemnym (błąd sodowy)
Duże rozmiary
Mała odporność mechaniczna
W zależności od składu materiału membrany szklanej można otrzymać również elektrody odwracalne względem: K+, Li+, NH4+
Elektrody ze stałą membraną krystaliczną
Homogeniczne (monokryształ lub sprasowany materiał polikrystaliczny z rozdrobnionych kryształów soli trudno rozpuszczalnej)
Heterogeniczne (materiał krystaliczny zawieszony w obojętnej matrycy polimerowej (np. guma arabska))
Przykłady krystalicznych homogenicznych elektrod jonoselektywnych
-
Oznaczany jon
Skład chemiczny membrany
Zakres pomiarowy pax
F-
LaF3 [Eu3+] monokryształ
0 – 6
CN-
AgI + Ag2S
2 – 6
Ag+
Ag2S
0 – 7
Cu2+
CuS + Ag2S
0 – 8
pax - ujemny logarytm z aktywności danego jonu
Zalety stałych elektrod homogenicznych:
Wysoka selektywność, która jest spowodowana tym, że w procesie przewodzenia prądu mogą brać udział tylko jony których ładunek, promień i struktura odpowiadają jonom sieci krystalicznej membrany
Duża odporność mechaniczna
Wady stałych elektrod homogenicznych:
Trudność otrzymania membrany
Membrany z czasem ulegają procesowi starzenia i dlatego wymagają okresowo polerowania np. drobnym papierem ściernym
Pomiar mogą zakłócić jony reagujące z materiałem membrany (czyli jony tworzące osady trudniej rozpuszczalne, czynniki kompleksujące)
Elektrody stałe heterogeniczne:
Łatwe w otrzymaniu, ale mniej trwałe
Obecnie rzadko stosowane
Elektrody ciekło membranowe
Klasyczne elektrody z ciekła membraną (wykonane są z materiału hydrofobowego nasyconego roztworem wymieniacza jonowego) – obecnie wychodzą z użycia
Elektrody z membraną polimerową zwane także elektrodami pseudociekłymi – obecnie często stosowane
Typowa membrana polimerowa zawiera:
- polichlorek winylu (PCW) jako matryca
- plastyfikator
- jonofor jako substancja aktywna
Elektrody uczulane
Zbudowane są z części receptorowej i klasycznej elektrody jonoselektywnej, która pełni funkcję przetwornika informacji fizykochemicznych lub biochemicznych dostarczanych przez część receptorową
Elektrody uczulane możemy podzielić na:
- elektrody gazowe
- elektrody enzymatyczne (biosensory)
Elektrody gazowe zbudowane są z elektrody jonoselektywnej oraz hydrofobowej membrany, przez którą dyfundują cząsteczki oznaczanego gazu do wewnętrznego roztworu elektrolitu, powodując zmianę równowagi jonowej.
Przykłady elektrod gazowych:
-
Budowa elektrody
Oznaczany gaz
Wewnętrzna elektroda jonoselektywna
Roztwór wewnętrzny
ph szklana
NaHCO3
NaHSO3
NH4Cl
CO2
SO2
NH3
Np. elektroda gazowa do oznaczania CO2
CO2 + H2O↔ HCO3- + H+
Jest to reakcja odwracalna zatem zgodnie z prawem działania mas wraz ze zmianą stężenia CO2 zmienia się stężenie jonów H+, które oznacza sięga pomocą elektrody szklanej.
Elektrody enzymatyczne służą do oznaczania związków organicznych, wykorzystując fakt, że w wyniku ich reakcji hydrolizy przy katalitycznym rozdziale enzymu powstają proste jony nieorganiczne, których obecność można rejestrować za pomocą elektrod jonoselektywnych.
Przykłady elektrod jonoselektywnych
-
Budowa elektrody
Oznaczany związek organiczny
Wewnętrzna elektroda jonoselektywna
Enzym
pH szklana
Oksydaza glukozy
Glukoza
Ureaza
Mocznik
Elektroda czuła na NH+
Ureaza
Mocznik
Do oznaczania mocznika
CO(NH2)2
+ H2O
+ 2H+
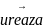 CO2
+ 2NH4+
CO2
+ 2NH4+
Wytworzone w wyniku reakcji mocznika jony amonowe powodują zmianę potencjału elektrody szklanej czułej na NH4+
Charakterystyka analityczna elektrod jonoselektywnych, czyli parametry istotne do celów analitycznych to:
Zakres pomiarowy
Nachylenie charakterystyki
Granica wykrywalności
Selektywność elektrody
Optymalny zakres pH
Stabilność
1. Zakres pomiarowy
Krzywa charakterystyki elektrody jonoselektywnej czyli wykres zależności potencjału elektrody od wykładnika z aktywności oznaczanego jonu.
(wykres) granica wykrywalności: T – wielkość teoretyczna
P – wielkość praktyczna
Zakres pomiarowy to nernstowski zakres odpowiedzi czyli zakres stężeń dla którego charakterystyka jest liniowa
2. Nachylenie charakterystyki to zmiana potencjału elektrody jaka odpowiada zmianie stężenia o jeden rząd wielkości (w zakresie liniowości krzywej kalibracyjnej)
Z
równania Nernsta nachylenie charakterystyki =
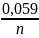 mV
mV
3. Granica wykrywalności (ang. Limit detection) jest to stężenie jonu głównego, przy którym potencjał elektrody różni się o 18/n Mv od wartości wynikającej z liniowego przebiegu charakterystyki elektrody
4. Selektywność elektrody
Idealna elektroda to elektroda której potencjał zależy tylko od stężenia jonów oznaczanych, a jej potencjał można opisać wzorem Nernsta. W praktyce potencjał elektrody zależy również od stężenia jonów przeszkadzających. Wpływ jonów przeszkadzających charakteryzuje współczynnik selektywności
Współczynnik selektywności dla jonu oznaczanego A i jonu przeszkadzającego B można wyznaczyć z następującego wzoru:
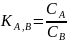
Gdzie CB jest takim stężeniem jonu przeszkadzającego, które daje taką samą odpowiedź elektrody jak stężenie jonu oznaczanego CA
K nie jest wielkością ściśle stałą dla danej elektrody i zależy od:
- siły jonowej roztworu
- stężenia jonu oznaczanego
Współczynnik selektywności K jest miarą selektywności elektrody, tzn. im większa jest jego wartość tym mniej selektywna (gorsza) jest dana elektroda, jeżeli:
KA,B << 1 to elektroda jest selektywna na jon A
KA,B nieznacznie < 1 to elektroda jest selektywna na jon A, ale na potencjał wpływa też jon przeszkadzający
KA,B = 1 to elektroda nie wykazuje selektywności
KA,B > 1 to elektroda jest selektywna na jon B
Potencjał elektrody opisuje wzór Nikolskiego:
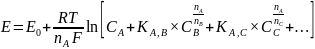
5. Optymalny zakres pH
Na potencjał większości elektrod wpływa stężenie jonów H+, ponieważ:
Mogą one brać udział w reakcji elektrodowej
Mogą wpływać na postać jonu obecnego w roztworze
Mogą wpływać na strukturę materiału elektrody
Zwykle istnieje zakres pH, w którym potencjał elektrody nie zależy od pH i jest optymalny zakres prowadzenia pomiarów.
6. Stabilność
Elektroda jest stabilna, gdy w tym samym roztworze, w tych samych warunkach elektroda ma niezmienny potencjał w czasie.
Metody potencjometryczne możemy podzielić na:
Potencjometrię bezpośrednią
Miareczkowanie potencjometryczne
W potencjometrii bezpośredniej układ elektrod złożony z elektrody wskaźnikowej i elektrody porównawczej wykorzystujemy do bezpośredniego oznaczania stężenia badanego jonu, na który jest czuła dana elektroda wskaźnikowa. Do potencjometrii bezpośredniej należą między innymi pomiary z zastosowaniem elektrod jonoselektywnych oraz pomiar pH.
Pomiary możemy prowadzić:
Metodą krzywej wzorcowej (metoda ta daje dobre wyniki, jeżeli skład roztworów wzorcowych użytych do wyznaczenia krzywej kalibracyjnej jest zbliżony do składu roztworu badanego oraz elektroda wykazuje stabilność w czasie
Metodą dodatku wzorca (jest to metoda najdokładniejsza)
W miareczkowaniu potencjometrycznym układ elektrod wykorzystujemy do wyznaczenia punktu końcowego miareczkowania. Miareczkowanie potencjometryczne jest szczególnie przydatne gdy miareczkowane roztwory są mętne, barwne i trudno wyznaczyć PK miareczkowania metodami wizualnymi.
Miareczkowanie potencjometryczne wykorzystywane jest w:
Alkacymetrii (elektroda szklana)
Kompleksometrii (elektroda czuła na jon oznaczany)
Red oksymetrii (elektroda platynowa)
Miareczkowaniu strąceniowym (elektroda srebrowa)
Sposoby prowadzenia miareczkowania potencjometrycznego:
Klasyczne
Do punktu zerowego
Różnicowe
Z dwumetalicznym układem elektrod
1. Miareczkowanie klasyczne – polega na pomiarze SEM ogniwa zbudowanego z elektrody wskaźnikowej i porównawczej zanurzonych w badanym roztworze, odczynnik (titrant) dodajemy porcjami i po każdym dodatku mierzymy potencjał, metoda dokładna, ale pracochłonna.
Sposoby wyznaczania PK w miareczkowaniu klasycznym:
Metoda środkowej
Metoda pierwszej pochodnej
Metoda drugiej pochodnej
2. Miareczkowanie do punktu zerowego – układ takiego ogniwa zbudowany jest z dwóch identycznych elektrod wskaźnikowych: jednej zanurzonej w roztworze badanym, drugiej w roztworze porównawczym, który ma taki skład jaki będzie miał roztwór badany w PK. Titrant dodajemy do roztworu badanego aż galwanometr nie wykazuje przepływu prądy (E1 = E2), czyli stężenia obu roztworów są równe.
3. Miareczkowanie różnicowe – polega na pomiarze SEM ogniwa zbudowanego z dwóch identycznych elektrod wskaźnikowych zanurzonych do dwóch naczynek zawierających ten sam roztwór badany. Do obydwóch naczynek dodajemy titranta więcej następne dodawane porcje do obu naczynek są jednakowe. Czyli zmierzony potencjał ogniwa jest potencjałem przypadającym na jednostkę objętości , otrzymujemy od razu wykres „pierwszej pochodnej”. PK odpowiada największej różnicy potencjałów pomiędzy elektrodami.
4. Miareczkowanie z dwumetalicznym układem elektrod – tą metodę stosujemy w miareczkowaniu redoks, układ składa się z elektrody wolframowej (W), której potencjał w środowisku utleniającym jest stały i platynowej (Pt) jako elektrody wskaźnikowej. Krzywa miareczkowania w tym układzie wygląda jak w miareczkowaniu klasycznym
Metody oparte na procesie elektrolizy na mikroelektrodach
Elektroliza to zespół przemian fizykochemicznych przebiegających na elektrodach zanurzonych w roztworze elektrolitu wymuszonych przepływem prądu stałego. Przemiany te obejmują:
Właściwe reakcje elektrochemiczne związane z przenoszeniem ładunku
Reakcje chemiczne przebiegające w pobliżu elektrody
Transport depolaryzatora do i od powierzchni elektrody
Elektrolizę wykorzystuje się do oznaczania pierwiastków, które mogą wydzielić się na elektrodzie w postaci elementarnej lub w postaci tlenku.
Ile było oznaczanej substancji w próbce możemy wyznaczyć na podstawie:
Różnicy mas elektrody przed i po elektrolizie (elektrograwimetria)
Ilości ładunku zużytego do całkowitego wydzielenia substancji na elektrodzie (kulometria)
W metodach tych z jednej porcji próbki można przeprowadzić tylko jeden pomiar!!!
Elektrolizę można prowadzić w układzie:
Dwuelektrodowym (elektroliza klasyczna)
Trójelektrodowym (elektroliza z kontrolowanym potencjałem elektrody)
Elektrody wykonane są najczęściej z platyny:
- katoda ma kształt walca z siatki platynowej
- anodę stanowi spirala z drutu platynowego
Elektroliza klasyczna stosowana jest gdy w roztworze znajduje się tylko jedna substancja mogąca wydzielić się na elektrodzie. W przypadku gdy tych substancji jest więcej możemy zastosować analizę klasyczną, ale do roztworu należy dodać tzw. buforu potencjału, np. oznaczane Cu2+ w obecności Pb2+ z zastosowaniem NO3- jako buforu potencjału.
W takim przypadku po wydzieleniu się miedzi na elektrodzie redukcji ulegają jony NO3-, co zapobiega wydzielaniu się ołowiu, a redukcji NO3- nie towarzyszy wydzielanie się substancji stałych na elektrodzie.
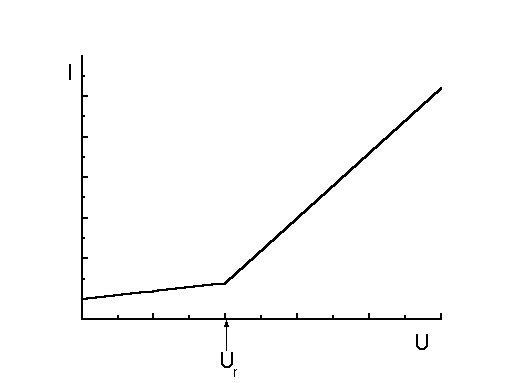
UR = EA – EK + iR + η
EA – potencjał anody
EK – potencjał katody, ulega zmianie podczas elektrolizy
η – nadpotencjał (jest zjawiskiem niekorzystnym w czasie elektrolizy)
Główne przyczyny powstawania nadpotencjału to:
Zbyt wolna dyfuzja jonów z głębi roztworu do powierzchni elektrody (aby temu zapobiec należy roztwór mieszać)
Powolny przebieg reakcji na elektrodach, zwłaszcza gdy na elektrodzie wydziela się gaz
Wartość nadpotencjału zależy od:
Rodzaju elektrody
Powierzchni elektrody (gładka czy porowata)
Rodzaju jonów (uwodnione czy skompleksowane)
Gęstości prądu (η rośnie wraz ze wzrostem gęstości prądu)
Temperatury (η maleje wraz ze wzrostem temperatury)
Elektroliza z kontrolowanym potencjałem elektrody
Prowadzi się w układzie trójelektrodowym
Jest to najczęściej stosowana metoda elektrolizy i wykorzystuje się ją do:
Oznaczania metali w stopach
Rozdzielania metali
Celów preparatywnych, tj. do syntezy związków organicznych i nieorganicznych, można w ten sposób uzyskać pośrednie produkty utleniania lub redukcji
Do wyznaczania mechanizmów procesów elektrodowych, można np. wyznaczyć ile elektronów bierze udział w reakcji
Do oczyszczania odczynników w wyniku długotrwałej elektrolizy
Optymalizacja warunków prowadzenia elektrolizy:
Dobór potencjału elektrody
Dobór postaci wydzielonego osadu
Dobór pozostałych warunków elektrolizy
Dobór potencjału elektrody
Aby wydzielić na elektrodzie tylko substancję A należy tak dobrać potencjał prowadzenia elektrolizy aby nie przekroczyć potencjału rozkładowego substancji B, Należy uwzględnić fakt, że podczas elektrolizy zmienia się potencjał katody.
(wykres)
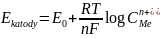
Ponieważ stężenie jonów metalu maleje w czasie prowadzenia elektrolizy zmienia się potencjał katody.
Aby wydzielić jedną substancję obok drugiej musi być odpowiednia różnica między ich potencjałami rozkładowymi!!!
Przykład:
Czy można wydzielić 99,9% Cu2+ w obecności Pb2+ jeżeli różnica między ich potencjałami rozkładowymi przed rozpoczęciem elektrolizy wynosi 100 mV?
ΔE = Ekońcowy - Epoczątkowy
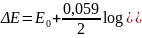
Czyli gdy różnica między potencjałami rozkładowymi Cu2+ i Pb2+ wynosi 100 mV można wydzielić 99,9% miedzi, ponieważ potencjał katody zmieni się tylko o 88 mv.
Dobór postaci wydzielonego osadu
Wydzielony osad musi dobrze przylegać do elektrody aby w czasie płukania, suszenia i ważenia nie kruszył się, co mogłoby prowadzić do ubytku osadu
Osad powinien być gładki i błyszczący:
- w tym celu do elektrolitu dodaje się tzw. czynników blaskotwórczych np. mocznik przy oznaczaniu Cu2+
- należy unikać jednoczesnego wydzielania się H2, bo w takim przypadku powstaje osad porowaty
- gęstość prądu nie może być zbyt dużą bo powstają wtedy osady gąbczaste
Części pracującej elektrody nie wolno dotykać palcami!
Dobór pozostałych warunków elektrolizy
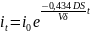
it – natężenie prądu po czasie t
i0 – natężenie prądu na początku elektrolizy
Elektroliza przebiega tym szybciej im większe jest natężenie prądu, czyli im większy jest 0,434DS/Vδ
D – współczynnik dyfuzji powinien być jak największy i dlatego roztwór należy ogrzać i intensywnie mieszać
S – powierzchnia elektrody powinna być jak największa i dlatego stosujemy elektrody siatkowe
V – objętość próbki powinna być jak najmniejsza
δ – grubość warstwy dyfuzyjnej powinna być jak najmniejsza i dlatego roztwór należy intensywnie mieszać
Elektroliza wewnętrzna
Nie wymaga stosowania żądnej aparatury generującej prąd elektryczny
Reakcja elektrodowa wymuszona jest różnicą potencjałów między dwoma metalicznymi elektrodami (np. dla układu elektrod Pb i Cu ponieważ ECu < EPb to elektroda Cu będzie katodą i będzie na niej zachodziła redukcja, a elektroda Pb będzie anoda i Pb będzie przechodzić w Pb2+ aż do momentu kiedy ECu = EPb
Materiały elektrody dobieramy tak aby uzyskać odpowiednią różnicę potencjałów
Elektroliza wewnętrzna rzadko stosowana w wykorzystuje się ją głównie do oznaczania zanieczyszczeń roztworów metalami
Wykorzystanie elektrolizy do oczyszczania ścieków:
Utlenianie jonów cyjankowych – jony cyjankowe utleniają się do cyjaninowych, stosuje się elektrolizer z anodą tytanową z powłoką platynową lub platynowo – irydową
Utlenianie rodanków – podobnie jak utlenianie cyjanków prowadzi do powstania jonów cyjaninowych, które ulegają dalszym przemianom z utworzeniem związków nietoksycznych
Utlenianie fenolu – jest wieloetapowe, elektrolizer jak przy utlenianiu cyjanków
Utlenianie elektrochemiczne w celu oczyszczania ścieków komunalnych, ścieków zawierających organiczne związki siarki, związki fosfoorganiczne, barwniki.
KULOMETRIA
Kulometria jest metodą w której ilość oznaczanej substancji wyznacza się mierząc ładunek elektryczny konieczny do jej całkowitego wydzielenia, przy założeniu 100% wydajności prądowej.
Ilościową zależność pomiędzy masą wydzielonej (przereagowanej) substancji a przepływającym ładunkiem podał Faraday w postaci dwóch praw, które łącząc dają następującą zależność:
m = k*Q
k – jest równoważnikiem elektrochemicznym i ma wartość stałą, charakterystyczną dla danej substancji
Q – jest ładunkiem jaki przepłyną prze obwód
Ponieważ
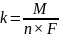 to
to
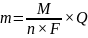
Ponieważ
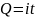 to
to
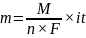
M – masa molowa
n – ilość elektronów biorących udział w reakcji
i – natężenie prądu
t – czas elektrolizy
F – stała Faradaya
A zatem mierząc wielkość ładunku można łatwo obliczyć masę wydzielonej substancji bez kalibracji metody
Aby zmierzyć ładunek w obwód musi być wbudowany kulometr
Pomiar ładunku kończymy gdy natężenie prądu osiąga wartość stałą (odpowiada ono prądowi szczątkowemu)
Kulometria jest metodą bezwzględną, czyli absolutną, w odróżnieniu od większości metod instrumentalnych, które są metodami porównawczymi wymagającymi kalibracji przy użyciu próbek wzorcowych
Kulometria jest metodą bardzo dokładną, błąd oznaczenia może wynosić nawet tylko 0,01%
Kulometria jest metodą uniwersalną, nadaje się do oznaczania zarówno makroilości i mikroilości
Kulometrię ze względu na sposób oznaczania substancji dzielimy na:
Bezpośrednią (oznaczana substancja bezpośrednia reaguje na jednej z elektrod)
Pośrednią, zwaną miareczkowaniem kulometrycznym (oznaczana substancja nie ulega reakcji elektrodowej, lecz wchodzi w reakcję z substancją wytwarzaną na jednej z elektrod w trakcie elektrolizy)
Kulometrię ze względu na sposób wykonania analizy dzielimy na:
Potencjo statyczną (elektroliza prowadzona jest przy stałym potencjale elektrody pracującej – anody lub katody), stosuje się ją zwykle do oznaczeń bezpośrednich
Amperostatyczna (elektroliza prowadzona jest przy stałym natężeniu prądu płynącego w obwodzie), stosuje się ją zwykle do oznaczeń pośrednich czyli miareczkowań
Miareczkowanie kulometryczne amperostatyczne:
W tym przypadku odczynnik którym miareczkujemy jest generowany na elektrodzie przy stałym natężeniu prądu
Metodą tą możemy przeprowadzić wszystkie miareczkowania, które można prowadzić metodą klasyczną oraz miareczkowania za pomocą odczynników nietrwałych
Nie trzeba nastawiać miana titranta
Warunki miareczkowania kulometrycznego amperostatycznego:
Reakcja elektrodowa generowana odczynnika musi przebiegać ze 100% wydajnością
Istnieje możliwość dokładnego pomiaru ładunku niezbędnego do przeprowadzenia reakcji
Istnieje możliwość wyznaczenia punktu końcowego miareczkowania
Aby spełnić warunek (1):
Elektrody muszą być rozdzielone za pomocą półprzepuszczalnej membrany lub za pomocą klucza elektrolitycznego, gdyby nie były rozdzielone to część odczynnika generowana na jednej z elektrod mogłaby ulec reakcji odwrotnej na drugiej z elektrod, w konsekwencji nie wiedzielibyśmy jaka część generowanej substancji reaguje z oznaczaną substancją
Odczynnik z którego generujemy substancję którą miareczkujemy musi być w nadmiarze aby zapobiec innym reakcjom elektrodowym
Aby spełnić warunek (2):
Warunek łatwy do spełnienia, wystarczy zmierzyć czas miareczkowania, ponieważ
Q = it gdzie i jest stałe
Aby spełnić warunek (3):
Punkt końcowy miareczkowania możemy wyznaczyć:
Za pomocą wskaźników wizualnych
Metodami instrumentalnymi np. potencjometrycznie, spektrofotometrycznie
Przykładowe reakcje generowania odczynnika (titr anta):
Na anodzie (powstaje utleniacz):
2Br- - 2e → Br2
2I- - 2e → I2
Ce3+ - e → Ce4+
Na katodzie (powstaje reduktor):
Ti4+ + e → Ti3+
Przykładowe reakcje między oznaczaną substancją a generowanym odczynnikiem (titrantem) np. Br2:
SCN- + 3Br2 + 4H2O → CN- + SO42- + 6Br- + 8H+
S 2-
- 8e → S6+
2-
- 8e → S6+
C4+ + 2e → C2+ x1
Br2 + 2e → 2Br- x3
Oznaczanie hydrazyny:
N2H4 + 2Br2 → N2 + 4HBr
N22- - 4e → N2
Br2 +2r → 2Br-
Zestaw do oznaczania jonów H+ i OH-
(rysunek)
Na anodzie utlenianie:
4 H2O ↔ 4OH- +4H+
4OH- → O2 + 2H2O + 4e
2H2O → 4H+ + O2+ 4e
Na katodzie redukcja:
2H2O ↔ 2OH- + 2H+
2H+ + 2e → H2↑
2H2O + 2e → H2↑ + 2OH-
Potencjometria kulostatyczna
Służy do oznaczeń bezpośrednich
W czasie elektrolizy utrzymuje się stały potencjał elektrody pracującej
W miarę zachodzenia reakcji natężenie prądu sukcesywnie maleje na skutek zmniejszenia się stężenia substancji, która wydziela się na elektrodzie i spadek natężenia do zera (lub do wartości prądu szczątkowego) wskazuje koniec reakcji, niepotrzebny więc jest dodatkowy detektor końca reakcji
Metody oparta na procesie elektrolizy na mikroelektrodach
Opierają się one na pomiarze prądu związanego z reakcją elektrodową przebiegającą pod wpływem przyłożonego do elektrody napięcia z zewnętrznego źródła prądu.
Polarografia obejmuje techniki z wykorzystaniem elektrody pracującej o zmieniającej się w trakcie pomiaru powierzchni, np. kapiącej elektrody rtęciowej, polarografia jest historycznie starsza niż woltamperometria
Woltamperometria obejmuje techniki z wykorzystaniem elektrody pracującej w stałej powierzchni nie zmieniającej się w czasie
Woltamperometria z zatężaniem tak jak woltamperometria, z tym że do procedury pomiarowej wprowadzony jest dodatkowy etap tak zwany etap nagromadzania.
Warunki prowadzenia pomiaru
Procesy będące podstawą pomiaru przebiegają na elektrodzie pracującej, która ma niewielką powierzchnię jest elektrodą polaryzowalną. Niewielka powierzchnia zapewnia, że mierzone prądy są małe a ilość analitu ulegającego redukcji jest tak mała, że nawet wielokrotne powtarzanie pomiaru nie powoduje zauważalnej zmiany jego stężenia
Pomiary prowadzone są w układzie trójelektrodowym składającym się z elektrody pracującej, odniesienia i pomocniczej
Dla roztworu badanego zawierającego substancję oznaczaną, czyli depolaryzator dodany musi być elektrolit podstawowy zapewniający przewodnictwo roztworu
Jeżeli elektroda pracująca polaryzowana jest napięciem ujemnym z roztworu musi być usunięty tlen, ponieważ jego obecność zakłóca pomiar
Mierzony prąd jest rejestrowany w postaci zależności od przyłożonego napięcia tworząc krzywą o kształcie fali lub piku nazywaną odpowiednio polarogramem lub woltamperogramem, potencjał elektrody pracującej jest zmieniany w trakcie pomiaru zgodnie z programem wynikającym ze stosowanej techniki
Ad. (1) warunki prowadzenia pomiaru
Elektroda pracująca:
Jest elektrodą polaryzowalną (oznacza to, że w nieobecności depolaryzatora przyjmuje potencjał zewnętrznego źródła napięcia, a w obecności depolaryzatora jej potencjał ulega odpowiednio zmianie, depolaryzatorem nazywamy substancję ulegającą reakcji elektrodowej)
W zależności od znaku przyłożonego napięcia może być katodą (potencjał ujemny) lub anodą (potencjał dodatni)
Najbardziej zbliżona do elektrody idealnie polaryzowalnej jest elektroda rtęciowa (wynika to z prawie idealnej gładkiej i czystej powierzchni rtęci, która w temperaturze pokojowej jest cieczą)
Różne konstrukcje elektrod rtęciowych (rysunki):
Kapiąca elektroda rtęciowa (KER)
Wisząca rtęciowa elektroda kroplowa (HMDE)
Elektroda rtęciowa z automatyczną generacją kropli
Błonkowa elektroda rtęciowa
KER (stosowana w polarografii)
Budowa – zbiornik rtęci połączony jest elastycznym wężykiem z kapilarą szklaną o średnicy wewnętrznej 0,02 – 0,08 mm i długości ok. 20 cm. Rtęć wypływa z kapilary grawitacyjnie tworząc doskonale powtarzalne krople. Kropla charakteryzowana jest czasem życia (1 – 6s) – czyli czasem upływającym od chwili rozpoczęcia procesu narastania do chwili oderwania się kropli pod własnym ciężarem
Zalety KER:
Doskonale odtwarzalna powierzchnia
Wraz z odrywającą się kroplą rtęci usuwane są produkty reakcji elektrodowej, co zapewnia stałe parametry elektrody
Rtęć jako metal szlachetny zachowuje się obojętnie w stosunku do większości roztworów
Możliwość pracy przy bardzo ujemnych wartościach potencjału
Wady KER:
Toksyczność
Wrażliwość na wstrząsy i zanieczyszczenia mechaniczne
Mały zakres stosowania w zakresie potencjałów dodatnich
Wisząca rtęciowa elektroda kroplowa (woltamperometria)
W tej elektrodzie rtęć wypychana jest ze zbiornika przez tłok precyzyjnie poruszany śrubą mikrometryczną. Uformowana na końcu kapilary kropla rtęci jest stabilna i może utrzymywać się prze długi okres czasu. Zwykle po przeprowadzeniu pomiaru kropla jest mechanicznie obrywana i wyciskana jest nowa kropla.
Elektroda rtęciowa z automatyczną generacją kropli, która może pracować jako:
Wisząca elektroda rtęciowa (HMDE) (woltamperometria)
Kapiąca elektroda rtęciowa (KER) (polarografia)
Błonkowa elektroda rtęciowa (MFE) (woltamperometria)
Powstaje ona przez elektrochemiczne lub mechaniczne osadzenie rtęci na powierzchni elektrody stałej – najczęściej z węgla szklistego
Elektrody te charakteryzują się korzystnym stosunkiem powierzchni do objętości umożliwiającym polepszenie czułości i rozdzielczości oznaczeń.
Wkład polskich uczonych w konstrukcję detektorów rtęciowych
Elektroda HMDE – W. Kublik z UW
Elektroda z automatyczną generacją kropli - Kowalski z AGH
Zakres polaryzacji elektrody rtęciowej w 0,1M HNO2
(wykres)
Elektroda rtęciowa posiada ograniczony zakres polaryzacji:
Katodowej (od strony potencjałów ujemnych) rozkładem elektrolitu podstawowego np. wydzieleniem wodoru
Anodowej (od strony potencjałów dodatnich) elektrochemicznym rozpuszczeniem materiału elektrody
Zakres polaryzacji zależy od składu i kwasowości elektrolitu podstawowego i przeciętnie wynosi od +0,2 do -1,5 V
Stosunkowo wąski zakres polaryzacji anodowej powoduje że elektrodę rtęciową używa się najczęściej w zakresie katodowym redukując obecne w roztworze depolaryzatory
Jeżeli chcemy prowadzić pomiary w zakresie potencjałów dodatnich to jako elektrodę pracującą wykorzystuje się:
- elektrody z metali szlachetnych np. Pt, Au, Ag
- elektrody węglowe (węgiel szklisty, grafit)
Elektrody te są to tzw. elektrody stałe
Zakres polaryzacji
Materiał Środowisko
Hg kwaśne
Ad. (2) warunki prowadzenia pomiaru
Elektroda odniesienia
Powinna być niepolaryzowana tzn. jej potencjał nie powinien ulegać zmianie w czasie przepływu prądu
W woltamperometrii są wykorzystywane głównie trzy elektrody odniesienia (chlorosrebrowa, kalomelowa, siarczanowa)
Elektroda pomocnicza
Wykonana jest zwykle z metalu szlachetnego lub węgla szklistego
Należy pamiętać że na elektrodzie pomocniczej przebiega także reakcja elektrochemiczna a jej produkty mogą zanieczyścić roztwór badany, dlatego elektroda powinna być czyszczona
Ad. (3) warunki prowadzenia pomiaru
Aby pomiar woltamperometryczny mógł być przeprowadzony do badanej próbki konieczne jest dodanie elektrolitu podstawowego
Elektrolit podstawowy (wymagania)
Powinien zapewnić przewodnictwo prądu
Wyeliminować prąd migracyjny (stężenie elektrolitu powinno być od 100 do 1000 razy większe od stężenia oznaczanego depolaryzatora)
Odpowiedni rozkład elektrolitu powinien zapewnić dobre rozdzielanie fal poszczególnych depolaryzatorów
Powinien zapewnić szeroki zakres potencjałów pracy elektrody
Nie powinien tworzyć z depolaryzatorami soli nierozpuszczalnych
Jako elektrolit podstawowy stosuje się:
Sole mocnych kwasów i mocnych zasad
Mocne kwasy
Mocne zasady
Bufory
Bufory z dodatkiem czynników kompleksujących
Stosuje się: KCl, KNO3, HCl, NaOH oraz bufory octowy…
(wykresy)
Ad. (4) warunki prowadzenia pomiaru
Roztwory mające kontakt z powietrzem zawierają rozpuszczony tlen którego stężenie wynosi ok. 10-3mol/l
Tlen obecny w roztworze przeszkadza w pomiarach polarograficznych i wolt amperometrycznych ponieważ:
Ulega redukcji według mechanizmu zależnego od kwasowości środowiska
Produkty redukcji tlenu (OH-) mogą tworzyć wodorotlenki metali których redukcja następuje przy innym potencjale niż redukcja wolnych jonów metali
Tlen obecny w roztworze może utleniać amalgamaty
Reakcje redukcji tlenu w zależności od środowiska:
W roztworach kwaśnych w pierwszym etapie powstaje ditlenek diwodoru a w drugim woda
O2 + 2H+ + 2e → H2O2
H2O2 + 2H+ + 2e → 2H2O
W środowisku obojętnym i zasadowym w obydwu etapach tworzą się jony wodorotlenowe
O2 + 2H2O +2e → H2O2 + 2OH-…
H2O2 + 2e → 2OH-
Dwustopniowa redukcja powoduje, że tlen tworzy dwie fale polarograficzne:
Pierwsza dobrze wykształcona przy potencjale ok. -0,05V
Druga bardzo szeroka przy potencjale ok. -0,9V w roztworze kwaśnym lub -1,3V w roztworze zasadowym
Wynika z tego, że dla procesów przebiegających w zakresie potencjałów dodatnich tlen nie przeszkadza i nie musi być usuwany.
Sposoby usuwania tlenu:
Sposób fizyczny polega na przepuszczeniu przez badany roztwór gazu obojętnego (N2, Ar) nie zawierającego tlenu
Sposób chemiczny polega na chemicznym związaniu rozpuszczonego tlenu. W tym celu w środowisku alkalicznym i obojętnym stosowana jest sól Na2SO3, a w środowisku kwaśnym kwas askorbinowy.
Ad. (5) warunki prowadzenia pomiaru
Rejestrowany prąd składa się z następujących prądów:
Prąd dyfuzyjny
Prąd szczątkowy
Prąd kinetyczny
Prąd katalityczny
Prąd migracyjny
Prąd pojemnościowy
Krzywa polarograficzna
(wykres)
Powyżej D – redukcja elektrodowa innego składnika
Is – prąd szczątkowy
ID – graniczny prąd dyfuzyjny
E1/2 – potencjał półfali
Ad. a
Prąd dyfuzyjny jest to prąd wynikający z redukcji lub utlenienia jonów lub cząstek depolaryzatora docierających do elektrody na drodze dyfuzji
Jest on pożądany a jego graniczna wartość jest wykorzystywana do oznaczeń ilościowych
Graniczny prąd dyfuzyjny opisuje równanie Ilkovica
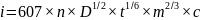 [mA]
[mA]
n – ilość elektronów biorących udział w reakcji elektrodowej
D – współczynnik dyfuzji depolaryzatora [cm2/s]
T – czas trwania kropli (s)
M – wydajność kapilary (mg/s)
C – stężenie depolaryzatora w głębi roztworu (mmol/dm3)
Staramy się przeprowadzić pomiary tak aby następujące wielkości były stałe n, D, t, m wtedy równanie przyjmuje postać
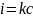
Ad. b
Prąd szczątkowy
Jest to prąd wynikający z zanieczyszczeń obecnych w roztworze
Jest to prąd niepożądany
Występuje zawsze i traktujemy go jako składnik tła
Ad. c
Prąd kinetyczny
Prąd kinetyczny: jest on związany z przejściem depolaryzatora z formy nieaktywnej w formę aktywną
Jeżeli depolaryzator znajduje się tylko w formie gotowej do reakcji elektrodowej to w takim przypadku nie mamy do czynienia z prądem kinetycznym
Jeżeli depolaryzator występuje w postaci dwóch form mamy do czynienia z prądem kinetycznym
Jeżeli na elektrodzie nie zachodzi redukcja aktywnej formy to jest stan równowagi pomiędzy formą aktywną i nieaktywną. Po przyłożeniu odpowiedniego potencjału do elektrody zaczyna zachodzić redukcja czego wynikiem jest ubytek stężenia formy aktywnej w warstwie przyelektrodowej. Ubytek ten powoduje, że forma nieaktywna zaczyna przechodzić w formę aktywną. W związku z tym wysokość fali rzeczywistej zależy od kinetyki reakcji przejścia formy nieaktywnej w aktywną a towarzyszący temu prąd to prąd kinetyczny
Wielkość prądu kinetycznego zależy od temperatury.
Np. oznaczanie aldehydu mrówkowego w wyniku jego redukcji
HCHO*H2O ↔ HCHO + H2O
Forma nieaktywna elektrodowo forma aktywna elektrodowo
(wykres)
Ad. d
Prąd katalityczny
Jest to prąd wynikający z reakcji w trakcie której depolaryzator zużywany w reakcji elektrodowej jest odtwarzany w procesie chemicznym
Jest to prąd pożądany bo powoduje wzrost czułości oznaczeń nawet 1000- krotnie
(wykres)
Reakcje przebiegające na elektrodzie będące źródłem prądu dyfuzyjnego
Fe3+ + e → Fe2+
W obecności H2O2 w warstwie przyelektrodowej przebiega dodatkowo następująca reakcja będąca źródłem prądu katalitycznego
Fe2+ + H2O2 → Fe3+ + 2OH-
Ponieważ reakcje te przebiegają cyklicznie jedna po drugiej to możliwy jest duży wzrost prądu
Ad. e
Prąd migracyjny: jest to prąd wynikający z reakcji elektrodowej depolaryzatora docierającego do elektrody pod wpływem przyciągania lub odpychania elektrostatycznego.
Transport jonów do elektrody może odbywać się:
Na drodze dyfuzji (wynikiem redukcji depolaryzatora docierającego w ten sposób jest prąd dyfuzyjny)
Pod wpływem przyciągania lub odpychania elektrostatycznego (wynikiem redukcji depolaryzatora docierającego w ten sposób jest prąd migracyjny)
Występowanie prądu migracyjnego powoduje:
Podwyższenie lub obniżenie fali (czułości), wielkość podwyższenia lub obniżenia zależy od stężenia depolaryzatora
Zakłócenia liniowej zależności prądu granicznego od stężenia
W związku z tym należy wyeliminować prąd migracyjny . Robimy to dodając elektrolit podstawowy, ponieważ wtedy na transport depolaryzatora nie ma wpływu przyciągania i odpychanie elektrostatyczne
Ad. f
Prąd pojemnościowy
Jest to prąd wynikający z doładowania elektrody do odpowiedniego potencjału
Jest niepożądany
Jest głównym składnikiem tła i dlatego wpływa decydująco na czułość oznaczeń
Prąd ten jest tym większy im większe są zmiany powierzchni elektrody, większe zmiany potencjału, więc nie występuje gdy zarówno nie ulega zmianie powierzchnia elektrody jak i nie ma zmiany potencjału
Metody polarograficzne
Polarografia stałoprądowa (klasyczna), (z ang. Direct current – dc)
Polarografia prądu maksymalnego (tastowa)
Polarografia zmiennoprądowa sinusoidalna (z ang. Alternating current – Ac)
Polarografia pulsowa różnicowa (z ang. Differentia pulse polarography – dpp)
Polarografia fali prostokątnej (kwadratowej), (z ang. Square wave –sw)
Polarografia stałoprądowa (klasyczna) dc
W metodzie tej do elektrody pracującej przykładany jest potencjał, który zmienia się liniowo w czasie i rejestrujemy w sposób ciągły zmiany natężenia prądu odpowiadające temu potencjałowi, na polarogramie otrzymujemy falę
Na prąd rejestrowany składa się zarówno prąd dyfuzyjny, jak i prąd pojemnościowy (niepożądany)
(wykresy)
Interpretacja sygnału:
E1/2 - potencjał półfali, jest on charakterystyczny dla danej substancji i wykorzystywany jest do analizy jakościowej
h=kc – wysokość fali jest proporcjonalna do stężenia i wykorzystywana jest do analizy ilościowej
(wykres)
Polarografia stałoprądowa
Charakteryzuje się słabą rozdzielczością, aby uzyskać dobrze rozdzielone fale dla dwóch depolaryzatorów różnica potencjałów ich półfal musi wynosić przynajmniej 200mV
Jest obecnie rzadko stosowana, ponieważ w metodzie tej nie ma eliminacji prądu pojemnościowego i w związku z tym granica wykrywalności jest wysoka i wynosi ok. 1*10-5mol/L
Polarografia prądu maksymalnego (tastowa)
W metodzie tej pomiar prądu przeprowadza się pod koniec czasu trwania kropli w związku z tym powierzchnia elektrody w czasie pomiaru jest stała i w ten sposób eliminowany jest częściowo prąd pojemnościowy (dla przypomnienia prąd pojemnościowy jest tym większy im większe są zmiany powierzchni elektrody)
Zmniejszenie prądu pojemnościowego, który jest głównym składnikiem tła wpływa na obniżenie granicy wykrywalności do ok. 3*10-6mol/L
Na polarogramie otrzymujemy falę a interpretacja sygnału jest taka jak w polarografii klasycznej
Polarografia zmiennoprądowa sinusoidalna (ac)
W metodzie tej na liniowo zmieniający się w czasie potencjał nakłada się potencjał sinusoidalny (płynący prąd zawiera składowe prądu stałego i składowe zmienne), ale mierzy się tylko prądy wywołane sinusoidalnymi zmianami napięcia, składowe stałe są odrzucane
Amplituda prądu sinusoidalnego wynosi od 10 d0 50 mV, częstotliwość (liczba okresów w ciągu jednej sekundy) wynosi ok. od 20 do 200 Hz
(wykresy)
Interpretacja sygnału:
Ep – potencjał piku, jest on charakterystyczny dla danej substancji i wykorzystywany jest do analizy jakościowej
h=kc – wysokość piku jest proporcjonalna do stężenia i wykorzystywana do analizy ilościowej
W metodzie tej stosuje się dwa typy przyrządów:
Polarograf ac bez detekcji fazowej, to znaczy, że rejestrujemy sumę prądu faradayowskiego związanego z reakcją elektrodową oraz prądu pojemnościowego, granica wykrywalności 10-5mol/L
Polarograf ac z detekcją fazową, która umożliwia oddzielenie zarejestrowania prądu faradayowskiego i pojemnościowego, co pozwala:
Na odrzucenie składowej pojemnościowej i zarejestrowanie tylko prądy faradayowskiego i dlatego można obniżyć granicę wykrywalności do 10-7mol/L, w ten sposób oznacza się depolaryzatory
Na odrzucenie składowej faradayowskiej i rejestrowanie tylko prądu pojemnościowego, w ten sposób oznacza się substancje powierzchniowo czynne, metoda nosi nazwę tensometrii
Zastosowanie polarografii Ac:
Obecnie praktycznie nie wykorzystuje się jej do oznaczeń depolaryzatorów
Wykorzystuje się do oznaczeń substancji powierzchniowo czynnych (tensometria)
Tensometria
(wykres)
Wady tensometrii:
Wysokość piku nie jest liniową funkcją stężenia!!
Do wykreślenia krzywej kalibracyjnej potrzebna jest duża ilość wzorców, a pomiary obarczone są dużym błędem
Pomimo tych wad metoda ta ma zastosowanie praktyczne ze względu na krótki czas analizy.
Polarografia pulsowa różnicowa (dpp)
(wykresy)
W metodzie tej na liniowo zmieniający się w czasie potencjał przykładane są pulsy potencjału, pomiaru prądu dokonujemy tuż przed pulsem i pod koniec trwania pulsu i rejestrowana jest ich różnica (w ten sposób eliminowany jest prąd pojemnościowy)
Czas trwania pulsu wynosi zwykle 50ms, amplituda 50mV lub mniej
Zasada eliminacji prądu pojemnościowego
(wykresy)
ic – prąd pojemnościowy
it – prąd faradayowski
Dzięki eliminacji prądu pojemnościowego granica wykrywalności wynosi 1*10-7mol/L
Jest to najczęściej stosowana metoda polarograficzna
Polarografia fali prostokątnej (kwadratowej) sw
(wykresy)
W metodzie tej na liniowo zmieniający się w czasie potencjał nakłada się na potencjał zmienny prostokątny (płynący prąd zawiera składowe prądu stałego i składowe zmienne)ale mierzy się tylko prąd faradayowski wywołany zamianami napięcia fali prostokątnej, składowe stałe są odrzucane
Składowa prądu pojemnościowego jest praktycznie całkowicie wyeliminowana
Amplituda prądu prostokątnego wynosi 10 – 50mV, częstotliwość wynosi ok. 100Hz
Metodę tą można stosować tylko dla reakcji odwracalnych
W metodzie tej nie jest konieczne odtlenianie roztworu (reakcja redukcji tlenu jest procesem nieodwracalnym)
Metodę tę można stosować tylko dla bardzo szybkich reakcji elektrodowych, które nadążają za bardzo szybkimi zmianami potencjału elektrody, w przypadku gdy reakcja elektrodowa jest powolna nie obserwujemy żadnego sygnału analitycznego
W metodzie tej trzeba stosować wysokie stężenie elektrolitu podstawowego
Metoda ta pozwala na oznaczanie najniższych stężeń już od 1*10-8mol/L
Woltamperometria z zatężaniem
(woltamperometria inwersyjna, analiza stripingowa)
W woltamperometrii z zatężaniem pomiar realizowany jest w dwóch etapach:
Zatężanie (nagromadzanie) na elektrodzie oznaczanego składnika
Uzyskanie sygnału (stripping), w wyniku zmiany potencjału elektrody nagromadzony depolaryzator ulega reakcji elektrodowej (redukcji, utlenieniu lub desorpcji); w etapie tym rejestrowana jest krzywa wolt amperometryczna w postaci piku
Podział metod wolt amperometrycznych ze względu na sposób zatężania:
Anodowa woltamperometria stripingowa (ASV), zatężanie w wyniku redukcji, a sygnał analityczny uzyskujemy w wyniku utlenienia nagromadzonych substancji
Katodowa woltamperometria stripingowa (CSV), zatężanie w wyniku reakcji elektrodowej prowadzącej do utworzenia trudno rozpuszczalnego związku na powierzchni elektrody, a sygnał analityczny uzyskujemy w wyniku redukcji nagromadzonych substancji
Adsorpcyjna woltamperometria stripingowa (AdSV), zatężanie w wyniku adsorpcji, a sygnał analityczny uzyskujemy w wyniku redukcji lub utlenienia nagromadzonych substancji
Anodowa woltamperometria stripingowa (ASV)
Proces zatężania realizowany jest poprzez elektrochemiczną redukcję oznaczanej substancji przy stałym potencjale najczęściej o ok. 0,2 V bardziej ujemnym niż potencjał półfali w polarografii klasycznej danej substancji, zgodnie z reakcją:
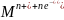
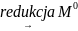
Roztwór elektroda
Po elektrolitycznym zatężeniu elektroda polaryzowana jest w kierunku anodowym i przy odpowiedniej wartości potencjału (charakterystycznego dla danego metalu) wydzielony metal przechodzi do roztworu (proces ten jest połączony z rejestracją płynącego prądu w zależności od potencjału a otrzymany wykres nazywamy woltamperogramem)

Elektroda roztwór
Anodowa woltamperometria stripingowa jest najstarszą stosowaną metodą stripingową
Za jej pomocą można oznaczać metale tworzące amalgamaty
Katodowa woltamperometria stripingowa (CSV)
Proces akumulacji oznaczanej substancji w tym przypadku polega na tworzeniu trudno rozpuszczalnych soli na powierzchni elektrody
Zazwyczaj pracującą elektrodą jest wisząca elektroda rtęciowa, chociaż w niektórych przypadkach wykorzystuje się np. elektrodę srebrną
Proces nagromadzania może przebiegać np. zgodnie ze schematem:
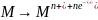
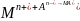
M jest materiałem elektrody
An+ jest analitem
MA jest nierozpuszczalną solą powstałą na powierzchni elektrody
Po elektrolitycznym zatężeniu elektroda polaryzowana jest w kierunku katodowym i uzyskujemy sygnał w wyniku redukcji trudno rozpuszczalnego osadu, którego będzie tyle ile było oznaczanych jonów
W ten sposób można oznaczać takie substancje jak: Cl-, Br-, I-, SCN-, S2-, Se4+ oraz niektóre związki organiczne
Np. oznaczanie Cl-
Etap nagromadzania:
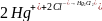
Etap uzyskania sygnału:
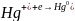
Np. oznaczanie Se4+ w obecności Cu2+
Etap nagromadzania:
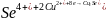
Etap uzyskania sygnału:
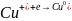
Adsorpcyjna woltamperometria stripingowa (AdSV)
W przeciwieństwie do ASV czy CSV charakteryzuje się nieelektrolityczną naturą etapu nagromadzania, zatężanie ma charakter wyłącznie adsorpcyjny i nie jest związane z przebiegiem żadnej faradayowskiej reakcji
W przypadku oznaczeń metali metodą AdSV w pierwszym etapie jony metalu reagują w roztworze z odpowiednim kompleksonem, następnie następuje adsorpcja kompleksu danego jonu z metalu z ligandem organicznym na powierzchni elektrody. W przypadku gdy ligand ma ładunek obojętny procesy te można opisać reakcjami:
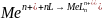
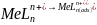
Po zakończeniu etapu nagromadzania kompleksu na powierzchni elektrody, jest ona następnie polaryzowana w kierunku katodowym lub anodowym, w zależności od tego czy podstawą krzywej wolt amperometrycznej jest reakcja redukcji czy też utleniania, w przypadku redukcji jonu metalu reakcję można zapisać równaniem:
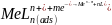
Metodą AdSV oznacza się metale, które nie tworzą amalgamatów np. klasycznym przykładem jest oznaczanie takich metali jak nikiel i kobalt w układzie z dwumetyloglioksymem (DMG) jako czynnikiem kompleksującym lub chromu z DTPA jako czynnikiem kompleksującym
Metoda AdSV jest stosowana w analizie śladowej szerokiej grupy związków organicznych, które wykazują właściwości powierzchniowo – aktywne
Podział analizy stripingowej ze względu na zastosowaną technikę uzyskania sygnału:
Technika z liniową zmianą potencjału
Technika pulsowa różnicowa
Technika fali kwadratowej
Metodą woltamperometrii z zatężaniem można oznaczać:
Wiele związków organicznych
Około 40 pierwiastków z dużą czułością (dzięki wprowadzeniu etapu zatężania) rzędu:
10-7 – 109 mol/L (najczęściej)
Do 10-14 mol/L (w przypadku wykorzystania procesów katalitycznych)
Do 10-16 mol/L (w przypadku wykorzystania enzymów)
Elektrody pracujące stosowane w woltamperometrii
Elektrody rtęciowe
Wisząca kroplowa elektroda rtęciowa (HMDE)
Filmowa elektroda rtęciowa (MFE)
Elektrody stałe:
Z węgla szklistego
Z impregnowanego grafitu
Modyfikowana
Z metali szlachetnych
Filmowa
Mikroelektrody
Elektrody stałe najczęściej mają postać dysków w osłonie z odpornego chemicznie tworzywa sztucznego. Taki kształt elektrody ułatwia mechaniczne przygotowanie powierzchni. Stosowane są elektrody dyskowe o średnicach 1 – 6mm.
Ad (1) elektrody z węgla szklistego
(rysunek)
Elektrody te są twarde i rzadko wymagają polerowania
Polerowanie ich prowadzi się najpierw na papierze ściernym a następnie Al2O3 o wielkości ziaren 0,3µm
Ad (2) elektrody z impregnowanego grafitu
Elektrody te są miękkie i często wymagają polerowania
Polerowanie ich prowadzi się tak jak dla elektrod z węgla szklistego
Ad (3) elektrody modyfikowane
Otrzymuje się nanosząc na elektrodę stałą substancje indywidualnie dobraną do oznaczania danego związku, którego zatężanie zachodzi w wyniku reakcji wymiany jonowej lub adsorpcji
Przygotowuje się elektrodę pastową z dodatkiem czynników kompleksujących (skład pasty to: sadza, olej parafinowy, czynnik kompleksujący)
Zaleta elektrod modyfikowanych:
Są mało wrażliwe na obecność substancji powierzchniowo czynnych (powszechnie obecnych w próbkach naturalnych) i dzięki temu mogą być wykorzystane do prowadzenia oznaczeń w próbkach zawierających duże stężenia tych substancji.
Ad (4) elektrody z metali szlachetnych
Są to elektrody zbudowane z drutu Ag, Au, Pt lub filmu tych metali osadzonego na drodze elektrolitycznej na innej elektrodzie stałej (najczęściej na Au)
Są rzadko stosowane
Ad (5) elektrody filmowe
Należą do nich elektroda rtęciowa, bizmutowa, ołowiowa
Otrzymuje się je przez elektrolityczne naniesienie danego metalu na elektrodę z węgla szklistego z roztworu zawierającego sól danego metalu
Ze względu na toksyczność rtęci obecnie istnieje tendencja do wyeliminowania toksycznych elektrod rtęciowych a ich miejsce w wielu procedurach zajmują nietoksyczne elektrody bizmutowe i ołowiowe
Ad (6) mikroelektrody
Są to elektrody Pt, Au lub z włókna węglowego, których średnica <30 µm
Najczęściej stosuje się je w warunkach polowych
Można je przygotować na różne sposoby, obecnie coraz częściej stosuje się mikroelektrody jednorazowego użytku
Interferencje w woltamperometrii z zatężaniem
Tworzenie się związków międzymetalicznych między wydzielonymi na elektrodzie metalami lub między wydzielonym metalem a materiałem elektrody
Nakładanie się pików
Obecność w próbce substancji organicznych o właściwościach powierzchniowo czynnych lub kompleksujących
Ad.(1)
Ponieważ pierwiastki będące w postaci związków międzymetalicznych utleniają się przy innym potencjale gdy są w wolnej postaci metalicznej na woltamperogramie rejestrowane są co najmniej dwa piki podczas ich utleniania uniemożliwiając oznaczenie takiego pierwiastka
Przykład interferencji związanych z tworzeniem się związków międzymetalicznych
(rysunki)
Sposoby eliminacji tych interferencji:
Dodatek ‘’pierwiastka trzeciego’’ (np. jonów Ga3+ w oznaczeniu Zn2+ w obecności Cu2+, w etapie nagromadzania wszystkie jony ulegają redukcji do postaci metalicznej i Ga tworzy bardzo szybko związki międzymetaliczne z Cu, a Zn jest w postaci metalicznej i daje sygnał bez interferencji
Odpowiedni dobór potencjału nagromadzania (tak aby wydzielał się na elektrodzie w postaci metalicznej tylko jeden pierwiastek)
Zastosowanie naczynka kulometryczno-woltamperometrycznego (w tym rozwiązaniu pierwiastek powodujący interferencje jest ilościowo wydzielany w części kulometrycznej, więc pierwiastek oznaczany dociera do części wolt amperometrycznej bez interferenta)
Zastosowanie wiszącej elektrody rtęciowej zamiast filmowej elektrody rtęciowej (stężenie metali wydzielonych w kropli rtęci jest dużo mniejsze niż w przypadku filmowej elektrody rtęciowej i dlatego mniejsza jest szansa tworzenia związków międzymetalicznych)
Ad. (2) Przykładem interferencji związanych z nakładaniem się pików jest np. oznaczanie Cd2+ i In3+
(rysunki)
Ad. (3) Substancje organiczne mogą:
Adsorbować się na elektrodzie częściowo lub całkowicie blokując jej powierzchnię
Mogą kompleksowa oznaczany metal, co prowadzi do zaniżenia lub zaniku sygnału od oznaczanej substancji
Sposoby eliminacji:
Usunięcie substancji organicznych przed pomiarem np.:
W wyniku naświetlenia próbki promieniowaniem UV ( substancje organiczne ulegają utlenieniu i ulotnieniu)
W wyniku przepuszczenia próbki przez kolumnę zawierającą czynnik adsorbujący substancje organiczne
Dodanie do próbki koloidalnej krzemionki na której adsorpcji ulegają substancje organiczne a krzemionka nie zakłóca pomiaru
Zastosowanie elektrod modyfikowanych, które nie są wrażliwe ma obecność substancji powierzchniowo czynnych
KONDUKTOMETYRIA polega na pomiarze przewodnictwa elektrycznego roztworu elektrolitu znajdującego się pomiędzy dwoma biernymi elektrodami. Aby zapobiec reakcjom na elektrodach w konduktometrii stosuje się prąd zmienny o częstości do 105Hz.
Przewodnictwo elektrolitu:
Zależy od stężenia elektrolitu (dla wyższych stężeń zależność nie jest liniowa)
Zależy od rodzaju elektrolitu (różne jony mają różną ruchliwość)
Jest sumą udziału wszystkich jonów
Zgodnie z prawem Ohma :
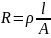
Gdzie:
R – opór przewodnika (w tym przypadku elektrolitu) [Ώ]
Ρ – opór właściwy [Ώ*cm]
l- długość przewodnika [cm]
A – pole przekroju przewodnika [cm2]
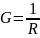 gdzie: G – przewodnictwo elektryczne (konduktancja) [S = Ώ-1]
gdzie: G – przewodnictwo elektryczne (konduktancja) [S = Ώ-1]
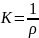 gdzie: K – przewodnictwo właściwe (Konduktywność) [S*cm-1]
gdzie: K – przewodnictwo właściwe (Konduktywność) [S*cm-1]
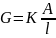 gdzie: A/l – stała naczynka konduktometrycznego, w praktyce
wyznacza się go mierząc oporność elektrolitu, którego
przewodnictwo właściwe jest znane
gdzie: A/l – stała naczynka konduktometrycznego, w praktyce
wyznacza się go mierząc oporność elektrolitu, którego
przewodnictwo właściwe jest znane
Typowy detektor konduktometryczny tworzą dwie obojętne elektrody (wykonane z platyny, złota lub grafitu) umieszczone w stałej odległości od siebie
Analizę konduktometryczną dzielimy na:
Konduktometria bezpośrednia
Konduktometria pośrednia
Miareczkowanie konduktometryczne
Konduktometria bezpośrednia polega na pomiarze bezwzględnej wartości przewodnictwa elektrolitów, nie daje selektywnej informacji o zawartości konkretnego jonu.
Konduktometria bezpośrednia jako metoda nieselektywna ma ograniczone zastosowanie głównie do:
Pomiaru przewodnictwa całkowitego (w kontroli czystości wód powierzchniowych, w kontroli procesów zmiękczania wody, niskie przewodnictwo świadczy o czystości wody)
Gdy selektywność można uzyskać w inny sposób np. konduktometryczne analizatory gazów
Konduktometria pośrednia gdy oznaczany związek reaguje ze składnikiem roztworu, można tak uzyskać selektywność pomiaru. Ma zastosowanie do oznaczania NOx po katalitycznej redukcji do amoniaku, HCl, HBr, CO2
Oznaczanie CO2 w powietrzu
(rysunek)
Przewodnictwo roztworu maleje ze wzrostem ilości pochłoniętego CO2. Oznaczenie charakteryzuje się wysoką czułością bo można oznaczyć CO2 w gazie zawierającym 0,0001% obj. CO2
Miareczkowanie konduktometryczne polega na obserwacji zmian przewodnictwa roztworu miareczkowanego, które następuje gdy do roztworu wprowadzamy jony różniące się ruchliwością od jonów obecnych w roztworze (reakcje zobojętniania, strącania, kompleksowania)
Przykład miareczkowania alkacymetrycznego
HCl + NaOH → NaCl + H2O
H+ + Cl- + Na+ + OH- → Na+ + Cl- + H2O
(rysunek)
Wykorzystanie konduktometrii w ochronie środowiska:
Oznaczanie stężeń detergentów w ściekach
Oznaczanie nawozów mineralnych w wodach irygacyjnych
Oznaczanie zawartości tlenu w wodzie (mierzy się przewodnictwo TlOH powstałego w reakcji talu z tlenem)
W analizatorach zanieczyszczeń atmosfery (np.SO2, H2SO4)
Wyszukiwarka
Podobne podstrony:
więcej podobnych podstron