
Protein Expression and Puri
Wcation 51 (2007) 1–10
www.elsevier.com/locate/yprep
1046-5928/$ - see front matter
© 2006 Elsevier Inc. All rights reserved.
doi:10.1016/j.pep.2006.06.024
Review
Strategies to maximize heterologous protein expression
in Escherichia coli with minimal cost
Wolfgang Peti
a
, Rebecca Page
b,¤
a
Brown University, Department of Molecular Pharmacology, Physiology, and Biotechnology, Box G-E3, Providence, RI 02912, USA
b
Brown University, Department of Molecular Biology, Cell Biology and Biochemistry, Box G-E4, Providence, RI 02912, USA
Received 26 April 2006, and in revised form 20 June 2006
Available online 4 July 2006
Abstract
Automation and miniaturization are key issues of high-throughput research projects in the post-genomic era. The implementation of
robotics and parallelization has enabled researchers to process large numbers of protein targets for structural studies in a short time with
reasonable cost e
Yciency. However, the cost of implementing the robotics and parallelization often prohibit their use in the traditional
academic laboratory. Fortunately, multiple groups have made signi
Wcant eVorts to minimize the cost of heterologous protein expression
for the production of protein samples in quantities suitable for high resolution structural studies. In this review, we describe recent e
Vorts
to continue to minimize the cost for the parallel processing of multiple protein targets and focus on those materials and strategies that are
highly suitable for the traditional academic laboratory.
© 2006 Elsevier Inc. All rights reserved.
Keywords: Structural biology; Heterologous protein expression; Escherichia coli; Cloning
Signi
Wcant amounts of protein, usually between 5 and
50 mg, depending on the protein size and experimental
technique used, are required for every structural biology
project
, independent of the structure elucidation
technique used, including X-ray crystallography, NMR
spectroscopy and cryo-electron microscopy. In general,
Escherichia coli is the preferred host for recombinant pro-
tein expression for structural studies
because it is
rather easy to genetically manipulate, it is relatively inex-
pensive to culture, isotope labeling protocols for NMR
spectroscopy and selenomethionine incorporation for X-
ray crystallography are established, and expression is fast,
typically producing protein in a single day. The impor-
tance of E. coli for heterologous protein production is
perhaps best highlighted by the wide variety of commer-
cial products available for the E. coli expression system.
However, there are disadvantages to using E. coli as an
expression host. Namely, E. coli is not capable of produc-
ing eukaryotic post-translational modi
Wcations, such as
glycosylation, which can be critical for the production of
folded, active protein. Equally important, some proteins,
especially larger proteins and membrane proteins, simply
fail to express in E. coli, or express, but do so insolubly as
inclusion bodies.
To overcome some of these limitations, a large number of
commercial vectors that facilitate soluble expression and sin-
gle step puri
Wcation via the use of diVerent fusion tags have
been developed. In addition, multiple E. coli strains that
facilitate the expression of membrane proteins
, proteins
with rare codons
, proteins with disul
Wde bonds
teins that are otherwise toxic to the cell, among others, are
readily available. This variety of expression vectors and cell
lines now signi
Wcantly enhances the likelihood of designing
an E. coli protein expression protocol suitable for the pro-
duction of the substantial amounts of protein required for
structural studies
. Finally, recently introduced microex-
pression incubator shakers that require as little as 500
l of
expression medium enable researchers to screen for optimal
expression conditions rapidly with reduced costs
. Once
*
Corresponding author. Fax: +1 401 863 9653.
E-mail address:
(R. Page).

2
W. Peti, R. Page / Protein Expression and Puri
Wcation 51 (2007) 1–10
the optimal expression construct is identi
Wed, standard incu-
bator shakers or fermentors are used to produce large
amounts of recombinant overexpressed protein.
During the last few years, both the pharmaceutical indus-
try and the structural genomics community have made signi
W-
cant e
Vorts to develop automated technologies to facilitate the
identi
Wcation and production of proteins suitable for func-
. This has led to new robotic
instrumentation for all steps of the structure determination
process, including rapid cloning systems
, parallel expres-
sion and puri
Wcation technologies
, nanocrystallization
and crystal growth imaging, and robotic crystal di
Vrac-
. Within the NIH Protein Structure Ini-
tiative, implementation of these technologies has resulted in
the determination of more than 1000 new protein structures
deposited in the Protein DataBank (PDB;
). However, academic laborato-
ries that usually focus on a few biologically relevant proteins
typically do not require the high-throughput demanded by
structural genomics centers and pharmaceutical industries.
Moreover, most of these technologies, such as robotics, and
novel strategies, such as commercially available rapid cloning
systems, are cost-prohibitive for most academic laboratories.
Instead, cost-e
Vective alternatives are needed. Fortunately,
during the last 5 years multiple groups have developed and
begun distributing materials that allow for the e
Ycient, paral-
lel screening of multiple constructs with minimal cost. These
techniques and materials use standard molecular biology and
protein puri
Wcation instrumentation that can be found in
most biology and certainly structural biology laboratories.
These new alternatives include new expression vectors
new or revitalized fusion tags to facilitate soluble protein
expression
, new microexpression/solubility screening
protocols
and new macroexpression methods and
instrumentation
. In this review, we describe our recent
experiences transitioning the parallel approach for protein
expression and puri
Wcation screening commonly used in
structural genomics centers to our own academic laboratories
and report on those materials and technologies we have found
most useful for our own projects.
New approaches to construct identi
Wcation
One of the most challenging steps in any structural biol-
ogy project is predicting which protein or protein fragment
will express solubly and purify for subsequent NMR spectro-
scopic or crystallographic studies. Often, small di
Verences in
the amino acid sequence itself, or in the length of the con-
struct, can transform a protein that fails to express into one
that expresses, puri
. In spite
of multiple e
Vorts to analyze the large amounts of data gen-
erated by structural genomics consortia regarding protein
expression, puri
Wcation and crystallization
, we are still
unable to predict, based on sequence alone, which proteins
will express and purify.
As with most laboratories, combined functional and
structural information is used to guide initial attempts to
identify the optimal boundaries (starting and ending resi-
dues) of a protein target/protein domain. We have found
that the ‘Fold and Function Assignment System’ (FFAS;
Vas.ljcrf.edu/Vas-cgi/cgi/Vas.pl
)
1
is a particularly use-
ful program for identifying weak structural homologs to a
sequence of interest. FFAS uses pro
Wle-proWle sequence
alignments and fold recognition to detect remote homolo-
gies not identi
Wed using other sequence comparison meth-
ods
. We use FFAS to identify the closest structural
homologs to various protein domains of interest to facili-
tate the identi
Wcation of appropriate residue boundaries for
primer design. When high resolution structures of homolo-
gous protein/protein domains are not available, we use sec-
ondary structure prediction programs such as PsiPred
to identify which regions of the domain are most likely to
form stable secondary structural elements. We also use the
programs, PONDR
and DisEMBL (HotLoops)
predict regions which are disordered. Both PONDR and
DisEMBL are computational methods based on neural
arti
Wcial networks trained for diVerent deWnitions of disor-
der, including protein sequences not visible in electron den-
sity maps (PONDR) and loops with high B-factors,
indicative of a high intrinsic mobility in these regions (Dis-
EMBL). The results from these analyses are then used to
identify multiple residues as optimal ‘start’ and ‘end’ resi-
dues for the constructs. Typically, we select between 2 and 4
‘start’ residues and 2 and 4 ‘end’ residues and use them
combinatorially to subclone anywhere between 4 and 16
di
Verent constructs of a single domain for expression, puri-
Wcation and structural analysis.
An additional approach that has proven to be success-
ful for the production of proteins for high resolution stud-
ies is ortholog screening
. Orthologs are proteins with
the same function from di
Verent species which, due to
evolution, have small di
Verences in their amino acid
sequences. While these di
Verences do not aVect protein
function, they can have signi
Wcant eVects on the ability of
a protein to express in E. coli, purify and crystallize.
Ortholog screening for high resolution studies have been
successfully used by multiple groups to obtain protein
samples suitable for high resolution studies. For example,
within the mouse homolog group of the Joint Center for
Structural Genomics (JCSG), 14 proteins were selected
for ortholog screening that had either previously failed to
crystallize or failed to express in selenomethionine media.
Using orthologous proteins, the JCSG was able to solve
the high resolution structures of 5 of these 14 protein fam-
ilies. Similarly, the Ontario Center for Structural Genom-
ics found that including even a single ortholog of a target
protein increases the number of samples for structural
studies by a factor of two
1
Abbreviations used: MCS, multiple cloning sites; TEV, Tobacco Etch
Viral; MBP, maltose binding protein; GST, glutathione S-transferase;
IMAC, immobilized metal a
Ynity chromatography; rpm, revolutions per
minute.

W. Peti, R. Page / Protein Expression and Puri
Wcation 51 (2007) 1–10
3
Parallel cloning into new vectors with novel fusion tags to
maximize the likelihood of soluble protein expression in
E. coli
The production of stable, soluble protein is one of the
most important steps in the protein structure determination
process. The experience of others
and ourselves indi-
cate that it is often necessary to express constructs of a pro-
tein of interest in frame with di
Verent protein fusion
partners in order to identify a single construct that is
expressed and soluble in amounts suitable for structural
studies. By using a parallelized matrix approach for clon-
ing, one can complete this expression and solubility screen-
ing in an e
Ycient, timely manner.
A variety of E. coli expression vectors (pET system, pBAD
system) with multiple fusion tags (hexahistidine, maltose-
binding protein, and glutathione S-transferase) under the con-
trol of di
Verent promoters (T7, trc, and araC) are widely
available. However, in many cases, the multiple cloning sites
(MCS) of these vectors often di
Ver from one another, requir-
ing new primers to be ordered and additional PCR ampli
Wca-
tion and restriction digestion steps to be carried out for every
new construct/vector pair. Thus, such vectors are not optimal
for a parallelized matrix approach to cloning. To address this
problem, a number of companies have developed novel, rec-
ombinatorial cloning systems which use recombinase (Gate-
way Technology) instead of the more traditional sticky-end or
blunt-end cloning methods to transfer gene targets from one
vector to another. For example, in the Gateway system, genes
are cloned into an entry (donor) vector which can then be
transferred, via recombination, into a variety of destination
(expression) vectors with the appropriate att recombination
elements. Although the Gateway system provides the
Xexibil-
ity of an extensive set of destination vectors, including multi-
ple E. coli, baculovirus, and mammalian expression vectors,
these systems are proprietary and its routine use can be
expensive for academic laboratories.
Novel expression vectors
We have found the EMBL system of E. coli expression
vectors, available through the European Molecular Biology
Laboratory (EMBL) protein production facility (
www.embl.de/ExternalInfo/protein_unit/draft_frames/index.
html
), most useful for implementing a parallelized matrix
approach to cloning while minimizing the associated costs
. More than 40 di
Verent E. coli expression vectors are cur-
rently available from the EMBL protein production facility.
These vectors have di
Verent promoters (T7, pBAD), include
a variety of fusion tags to facilitate soluble expression and
puri
Wcation (
), and have a choice of intervening pro-
tease sequences for tag removal (thrombin, TEV). The major
advantage of these vectors is that they have nearly identical
MCS sites. Thus, one need only amplify and digest a gene
construct once in order to subclone it into any of the avail-
able EMBL E. coli expression vectors.
The only drawback of these vectors is that they typically
have only a single restriction site for N-terminal cloning.
This is because there are ‘stu
Ver’ sequences present between
the N-terminal cloning site (NcoI) and the C-terminal clon-
ing sites (which have between 6 and 8 di
Verent restriction
sites). Thus, if you have a NcoI site in the DNA sequence of
your “to-be-expressed” protein, then you cannot use these
vectors for cloning unless they are genetically modi
Wed to
incorporate a new restriction site. Because proteins often
have multiple NcoI sites, including proteins we are studying
in our laboratories, we modi
Wed a subset of these vectors to
include an NheI site immediately following the NcoI site,
allowing these modi
Wed vectors to be used to subclone
DNA sequences with NcoI sites. We also developed a set of
our own ‘in-house’ vectors based on the expression vector
typically used in the JCSG for expression in E. coli (pMH1;
). This vector contains a small fusion tag, Thio
6
His
6
,
which includes the
Wrst six amino acids of thioredoxin, to
facilitate expression, followed by six histidine residues, to
facilitate puri
Wcation. Expression in pMH1 is controlled by
the pBAD promoter, which is tightly regulated and pre-
vents leaky expression, allowing for uniform cell culture
growth
. However, as observed by us and others,
Wnal
expression yields of protein under control of the pBAD
promoter are often dramatically lower than those obtained
using vectors under control of the T7 promoter
. Thus,
we developed two new vectors, pETRP1 and pETRP2,
which contain the Thio
6
His
6
tag both with and without a
Table 1
Subset of expression vectors typically used for the production of soluble protein in E. coli
Plasmids used for expression in E. coli; Thio
6
: MGSDKI,
Wrst six residues of thioredoxin; His
6
: HHHHHH, hexahistidine tag; Z-tag: derivative of Staph-
ylococcus protein A binding protein; GST: glutathione S-transferase; MBP: maltose binding protein; GB1: point mutant of the Streptococcus protein G
1 domain.
a
Modi
Wed by the author to include an additional N-terminal restriction site (NheI) for N-terminal cloning.
Vector
Promoter
Residue
Fusion tag
Protease site
Residues remaining
as cloning artifacts
Origin
Source
pETRP1
T7-lac
Kan
N-Thio
6
His
6
TEV
2
pBR322
Page
pETRP2
T7-lac
Kan
N-Thio
6
His
6
None
12
pBR322
Page
pETM-30
a
T7-lac
Kan
N-His
6
GST C-His
6
TEV
4
pBR322
EMBL/Page
pETM-41
a
T7
Kan
N-His
6
MBP
TEV
4
pBR322
EMBL/Page
pETZ2-1a
T7-lac
Kan
N-His
6
Z-domain
TEV
4
pBR322
EMBL
pHIS-GB1
T7-lac
Amp
N-His
6
GB1
TEV
3
pBR322
Gardner

4
W. Peti, R. Page / Protein Expression and Puri
Wcation 51 (2007) 1–10
Tobacco Etch Viral (TEV) NIa protease site, respectively,
and are under control of the T7 promoter. To date, we have
already successfully expressed more than 20 constructs
using these RP vectors, four of which have been success-
fully used for high resolution structure determination (both
NMR spectroscopy and X-ray crystallography
).
Cloning strategy
For initial cloning and expression tests, we have found it
advantageous to clone our constructs in parallel into three
di
Verent vectors: pETRP1 (N-Thio
6
His
6
, TEV protease
site), pETM-30 (N-His
6
, glutathione S-transferase [GST]
TEV protease site), and pETM-41 (N-His
6
, maltose binding
protein [MBP], TEV protease site). Usually, one of these
three tags results in soluble expression of the construct of
interest. MBP, in particular, has been shown to not only
enhance expression but to also passively promote the fold-
ing of its fused partner, thereby signi
Wcantly facilitating the
production of soluble protein
. Because an N-termi-
nal hexahistidine sequence is present in all of the vectors
(referred to as combinatorial tagging because there are two
expression/puri
Wcation tags present in the expression vector
), an additional advantage of this system is that all of
the expressed constructs can be puri
Wed using standard
immobilized metal a
Ynity chromatography (IMAC). This
makes the expressed proteins straightforward to purify in
parallel in order to rapidly identify which constructs are
suitable for large-scale expression and puri
Wcation.
If these
Wrst-round constructs fail to produce suYcient
amounts of protein for high resolution structural studies, a
second set of vectors that contain more rarely used, but also
e
Vective, fusion tags to facilitate soluble protein expression
and puri
Wcation. These include the Z-tag, a derivative of the
Staphylococcus protein A binding protein
, the GB1-tag,
a point mutant of the Streptococcus protein G
1 domain
, and NusA, the N-utilizing substance A protein
among others. The characteristics of these and other pro-
tein fusion tags are summarized in
. Our typical
work
Xowchart is shown in
TEV protease
The EMBL pETM and our in-house vectors form the
basis of our E. coli expression. These vectors are under
the control of the T7 promoter and have a pBR322 origin
of replication. This allows us to induce protein expres-
sion using IPTG or metabolically when expression is car-
ried out using the recently developed autoinduction
media (described in ‘Microexpression screening prior to
scale-up’). In addition, most of these vectors contain a
Tobacco Etch Virus NIa (TEV) protease cleavage site
between the N-terminal fusion tag and the construct of
interest. This allows us to remove any N-terminal fusion
tag using TEV protease, which can be readily expressed
and puri
Wed in the laboratory. An expression vector for a
his-tagged mutant of TEV NIa protease (His-tagged TEV
S219V), developed by Dr. D. Waugh and colleagues
,
is available from the American Type Culture Collection
(ATCC No. MBA-145). This TEV S219V mutant is
»
100-fold more resistant to auto-inactivation than wild-
type TEV, with the added bene
Wt of slightly better
catalytic activity. Additional TEV protease expression
vectors, including MBP-TEV S219V, are available from
the non-pro
Wt distributor of biological reagents, Add-
Gene (
). Finally, a vector for intracellu-
lar processing of fusion proteins by TEV protease prior
to lysis and puri
Wcation, pRK603, can also be obtained
(D. Waugh, personal communication;
). His-tagged
TEV S219V protease can be puri
Wed following protocols
such as that found at
. This allows investigators to produce
their own protease, resulting in a substantial savings in
reagent costs. In our experience, we have found TEV can
be stored at ¡80 °C for months without a signi
Wcant
decrease in catalytic activity.
Table 2
Expression and puri
Wcation characteristics of commonly used protein fusion tags
Fusion tag
Amino
acids
Size
(kDa)
Puri
Wcation
Comment
Thio
6
His
6
12
1.5
Immobilized metal
chromatography (IMAC)
Thio
6
(
Wrst six residues of thioredoxin) facilitates expression
and His
6
facilitates puri
Wcation using IMAC chromatography
His
6
-glutathione S-transferase (GST)
243
28.1
IMAC or glutathione agarose
GST: enhanced solubility; dimerization can be an issue
His
6
-maltose binding protein (MBP)
390
43.0
IMAC or amylose resin
MBP: enhanced solubility; fusion protein not always soluble
following cleavage from MBP
His
6
-disul
Wde oxidoreductase (DsbA);
disul
Wde isomerase
228
25.4
IMAC
Disul
Wde oxidoreductase (DsbA) and disulWde isomerase
(DsbC), both of which have been shown to have positive
e
Vects on expression levels when used as a fusion partner
His
6
-N-utilizing substance A protein
(NusA)
535
59.3
IMAC
Strong solubility enhancing tag
His
6
-thioredoxin A (TrxA)
135
14.7
IMAC
Solubility enhancing tag
His
6
-Staphylocuccs protein A derived
binding protein (Z-domain)
91
10.6
IMAC; protein A–Sepharose
Solubility enhancing tag
His
6
-point mutant of Streptococcus
protein G
1 domain (GB1)
85
9.7
IMAC; IgG-resins
Highly expressed and soluble; useful for NMR screening of
GB1-fusion proteins prior to tag removal
W. Peti, R. Page / Protein Expression and Puri
Wcation 51 (2007) 1–10
5
Protein coexpression
Finally, the EMBL protein production facility provides a
second series of vectors which is based on the pBAD pro-
moter with a pUC origin of replication. Because each series
of vectors confers resistance to di
Verent antibiotics (the
pETM series confers resistance to kanamycin and the pBAD
series confers resistance to ampicillin), they can also be used
in combination with one another for protein coexpression.
Microexpression screening prior to scale-up
The parallel matrix approach to construct generation
for heterologous protein expression and puri
Wcation tri-
als can rapidly produce large numbers of constructs that
must be tested for expression and solubility, especially
when comparing alternative E. coli expression strains
and/or expression conditions. For example, if four di
Ver-
ent constructs are subcloned into three di
Verent expres-
sion vectors and screened for soluble expression at two
temperatures (37 and 18 °C), 24 di
Verent expression trials
have to be carried out. To minimize the time and expense
of initial expression tests, a number of small-scale screens
have been developed that allow one to rapidly identify
which targets express and are soluble prior to large-scale
expression. Thus, these screens are essential for making
the matrix approach to protein construct generation
e
Ycient and cost-eVective.
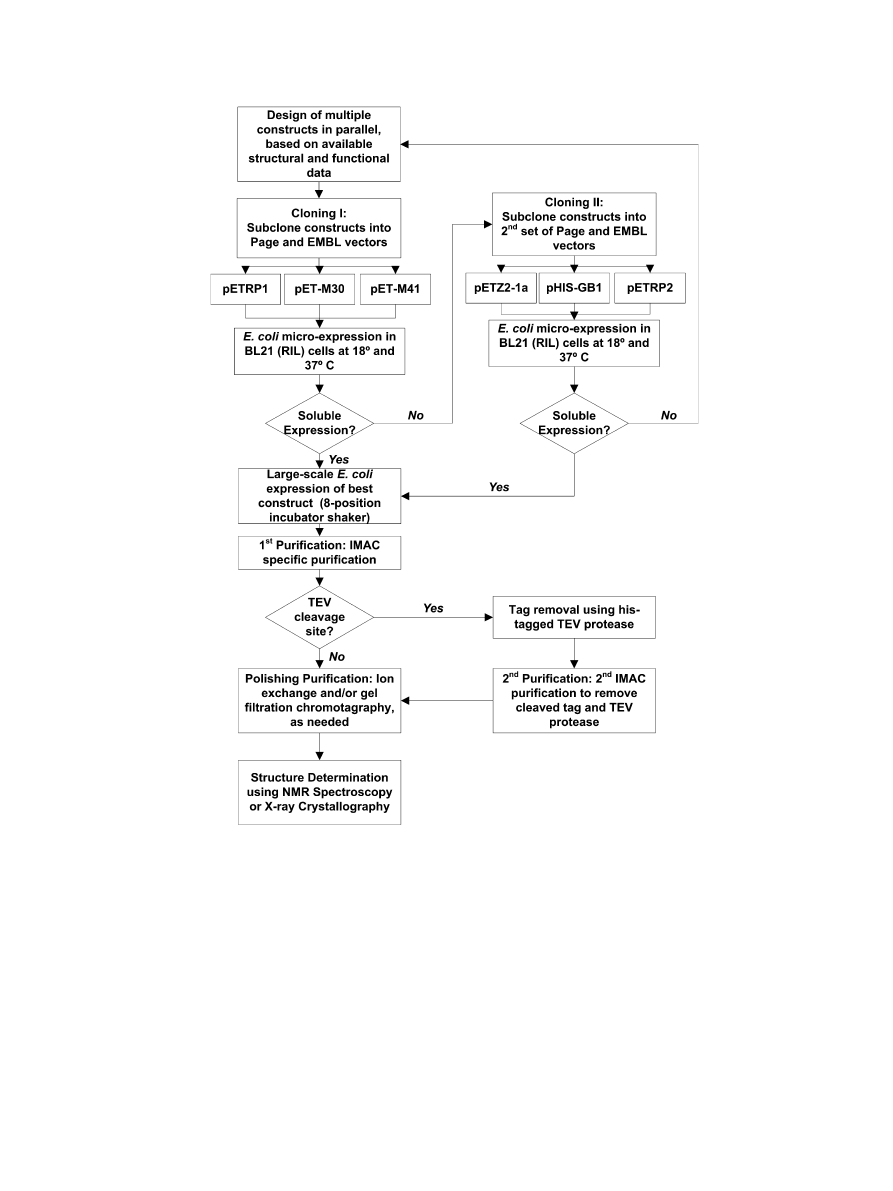
Fig. 1. Flowchart depicting the typical protein sample production strategy used by the authors.

6
W. Peti, R. Page / Protein Expression and Puri
Wcation 51 (2007) 1–10
We typically use the E. coli strain BL21 Codon-
Plus(DE3)-RIL (Stratagene) for all initial expression tests.
These cells contain a plasmid that encodes for three tRNA
genes that recognize rare codons (argU: AGA,AGG; ileY:
AUA; leuW: CUA). E. coli strains which contain additional
rare codons (such as BL21-CodonPlus(DE3)-RP/RPIL,
Stratagene; BL21-DE3-Rosetta, Novagen), are also avail-
able, when constructs fail to express in RIL cells. In addi-
tion, for tight regulation of protein expression, as is
necessary when expressing toxic proteins, we use BL21-AI
(Invitrogen) E. coli cells, which contain a chromosomal
copy of the T7 RNA polymerase under control of the arab-
inose-inducible araBAD promoter. A variety of additional
cell lines, which contain di
Verent chaperones (Chaperone
Competent Cell Lines, Takara), mutations in thioredoxin
reductase and glutathione reductase for enhanced disul
Wde
bond formation (BL21-Origami cells, Novagen), and
methionine-auxotrophs for selenomethionine incorpora-
tion for subsequent crystallographic studies (B834, Invitro-
gen), provide important alternatives.
Most microexpression screening is typically carried out
as follows. The proteins are expressed in 96-well (750
l) or
24-well deep-well (2–3 ml) blocks. The cell culture is pel-
leted by centrifugation and resuspended in chemical lysis or
sonication bu
Ver. The cells are then lysed using chemical
lysis, sonication or repeated rounds of freeze–thaw. A sam-
ple of the total lysate is removed for SDS–PAGE gel analy-
sis. The insoluble fraction of the lysate is subsequently
separated from the soluble fraction using either
Wltration or
centrifugation and a sample of the soluble fraction is used
for SDS–PAGE gel analysis. In some laboratories, the solu-
ble fraction is batch puri
Wed using small amounts of aYnity
matrices (i.e., in the EMBL Protein Production Facility,
25
l of immobilized metal aYnity chromatography,
IMAC-resin is used to purify the soluble, expressed protein
from 1.5 ml of cell culture). Finally, the expression and solu-
bility of expressed and puri
Wed proteins are determined
using SDS–PAGE electrophoresis or standard immuno-
chemistry. A number of recent reports describe such meth-
ods
and numerous commercial products, such as
ready-made chemical lysis bu
Vers including B-Per (Pierce),
Bugbuster (Novagen), and 96-well
Wltration plates (multi-
ple vendors), are available to facilitate parallelized expres-
sion screening.
Hand-in-hand with these new technologies, new instru-
mentation capable of facilitating multiple protein construct
screening has also become available
. We previously
adapted a low-cost, high-velocity incubating Glas-Col
(Glas-Col, LLC) Vertiga shaker for e
Ycient, high-through-
put E. coli microliter-scale expression screening that accu-
rately predicts protein behavior expressed in large-scale
(milliliter and liter) fermentation conditions. The apparatus
shakes cultures in three-dimensions at speeds of up to
1000 rpm, allowing small-scale (»500
L) cultures grown in
2 ml deep-well 96-well blocks to achieve optical densities
(OD
600
) of cell culture as high as 10–20. This generates su
Y-
cient material for analysis of expression, solubility and
binding to a
Ynity puriWcation matrices. Using this screen,
we showed that of 34 proteins screened, 33 consistently
expressed (or did not express) in both the small-scale (milli-
liter) and large-scale (liter) fermentation trials. More
recently, we have also shown that this Vertiga shaker can be
used to express and purify proteins in isotopically labeled
media in amounts suitable for initial structural character-
ization using microcoil NMR spectroscopy
Most recently, a novel microexpression screening proce-
dure that does not require culture growth, but instead mon-
itors soluble expression from a single colony, the colony
Wltration blot procedure (Co–Fi), has also been developed
. Rather than growing up milliliter cultures for expres-
sion screening or relying on reporter protein fusions, single
colonies are induced to express protein by transferring the
colonies to plates containing IPTG. After 6 h, the cells are
lysed by three rounds of freeze–thaw, the soluble and insol-
uble fraction separated by
Wltration and the soluble pro-
teins captured on a nitrocellulose membrane for screening
using standard immunochemicals. The Co–Fi blot, which
does not require the growth and expression small-scale cul-
tures, is ideal for screening libraries of thousands of colo-
nies using minimal time and materials.
New methods for increasing cell culture densities during
large-scale expression and improving soluble lysis
Most new expression tools have focused on fermentation
to improve E. coli cell density yields. However, fer-
mentors often require additional optimization of the
expression conditions, e.g. oxygen level, among others.
Moreover, fermentors are typically costly equipment.
Recently, new technologies and strategies have been devel-
oped that increase cell culture densities similar to those
observed with traditional fermentors, but are suitable for
traditional incubator shakers. The Vortexer incubator
shaker (Glas-Col LLC and Thomson Instrument Com-
pany) results in reproducible increases in the cell culture
densities of expressed proteins. A second option is the
BactoLift air spurge fermentor (Lofstand) in which up to
twenty 800 ml cell cultures can be grown in parallel
.
Further, the development of a new media formulation,
autoinduction media, that allows cultures to reach optical
cell densities of 20–40 OD without the need to add external
induction agents, such as IPTG
, shows highly promis-
ing possibilities for simpli
Wed parallel expression. Finally,
high volume homogenizers provide gentle E. coli lysis, max-
imizing the yield of soluble protein.
Vortexer shaker
The new Vortexer incubator shaker combines the ease of
protein expression in an incubator shaker with high
increases in cell culture densities to values typically seen in
fermentor-type setups. Multiple modi
Wcations to the regu-
lar incubator shaker setup have been made. The shaker uses
a high speed (up to 550 rpm) 3-dimensional rocking plat-

W. Peti, R. Page / Protein Expression and Puri
Wcation 51 (2007) 1–10
7
form. The orbit of Vortexer incubator shaker di
Vers from
that of traditional shakers because it rotates not only in the
X- and Y-dimensions, but also rotates in the Z-dimension.
It is temperature controlled, from 15 to 60 °C, and varies in
speed from 10 to 550 revolutions per minute (rpm). In addi-
tion, the Vortexer incubator shaker is also smaller than
more traditional incubator shakers, with dimensions of
only 28 in. (W) £ 26 in. (D) £ 22 in. (H), enabling it to
Wt on
a standard laboratory benchtop. Using the standard plat-
form, the Vortexer incubator shaker can be used to express
eight di
Verent 500–1000 ml cultures using 2.5 L Ultra-Yield
(UY; Thomson Instrument Company)
Xasks. These new
lightweight plastic UY
Xasks, which have six baZes for
increased aeration, are used for expression. Like Fernbach
Xasks, UY Xasks have a broad base 158 mm diameter, but
taper in two-stages to form a narrow neck of also 68 mm
inner diameter, and have six ba
Zes to facilitate mixing. UY
Xasks hold up to 2500 ml and are typically used to express
500–1000 ml of E. coli culture. A specially designed air-
porous seal can be used in conjunction with the UY
Xasks,
which facilitates an increase in the air-exchange compared
with aluminum foil that is usually used to prevent contami-
nation of the shaker cultures during protein expression.
We recently compared the
Wnal cell culture densities of
18 di
Verent proteins and protein domains expressed using
di
Verent media (LB, TB and autoinduction medium), diVer-
ent temperatures (18 and 37 °C), di
Verent Xasks (Fernbach
and UY) and di
Verent shakers (the vortexer incubator and
MultitronII incubator shaker, HT Infors). First, we found
that
Wnal cell culture densities were primarily dependent on
the nature of the protein expressed; i.e., certain proteins
consistently grew to higher (or lower) cell culture densities
than others, in spite of optimization of temperature, shak-
ing speed and media. For example, E. coli cultures express-
ing TEV protease reached a maximum optical cell culture
density (OD
600
) of 7.4 (rich TB medium using ultra yield
Xasks and the Vortexer incubator shaker at 18 °C). How-
ever, in most experiments, TEV cultures reached
Wnal opti-
cal densities of only 2.0 (LB medium, UY or Fernbach
Xasks, MultitronII incubator shaker, 37 °C). In contrast,
cultures expressing di
Verent proteins, such as hematopoi-
etic tyrosine phosphatase (PTN7_2)
or TM0979
were able to consistently reach much higher cell culture
densities, and in the best cases, reached OD
600
’s from 15 to
nearly 30. Second, we found that 62% of the E. coli cultures
expressed in Ultra-Yield
Xasks at 18 °C resulted in higher
Wnal cell culture densities than those expressed at the same
temperature in Fernbach
Xasks. Finally, we found 77% of
the proteins expressed in UY
Xasks at 18 °C had higher
Wnal cell culture densities when expressed in the Vortexer
shaker, while 80% of the proteins expressed in UY
Xasks at
37 °C had higher cell culture densities when expressed in the
MultitronII incubator shaker. In summary, outside of the
importance of the construct actually being expressed, we
found that the most signi
Wcant diVerences in Wnal cell cul-
ture densities depended on the media and temperature, with
the highest overall expression yields obtained for proteins
expressed at 18 °C in rich TB in the Vortexer shaker or
autoinduction medium. Both the MultitronII incubator
and Vortexer shakers are used routinely by the authors for
protein expression in E. coli.
BactoLift air spurge fermentor
While the Vortexer shaker can be used increase
Wnal cell
culture density yields, a second table top fermentor, the
BactoLift, can be used to express up to 20 di
Verent protein
samples simultaneously. The BactoLift is an air spurge fer-
mentor. Agitation is achieved by bubbling sterile air
through the culture using a
Xow-controlled air pump.
Instead of shaker
Xasks, the Bactolift attaches to standard
500 ml and 1 L centrifuge bottles. Cultures are grown
directly in the centrifuge bottles. Twelve 1-L units can be
incubated in a standard 1 £ 1.5 ft. benchtop waterbath, pro-
viding temperature regulation, and can be expanded to
accommodate 20 one-liter units. Following overnight incu-
bation, the bottles are removed from the Bactolift (which
can be repeatedly autoclaved), and are then transferred to
the centrifuge for further processing. This precludes the
need to transfer culture
Xuids from a shaker Xask, eliminat-
ing the possibility of spillage. The BactoLift was originally
developed for the production of plasmid DNA
, but has
more recently been adopted by high-throughput protein
production groups, including those at the Paci
Wc North-
west and Argonne National Laboratories
. In a compre-
hensive work comparing small-scale and large-scale
expression results, Lin and co-workers expressed 47 di
Ver-
ent proteins in 800 ml of media using the BactoLift fermen-
tor. Speci
Wcally, cell cultures were grown at 37 °C to
OD
600
’s of 0.6–0.9, induced and then allowed to express at
30 °C for 3 h, after which the bottles were removed and cen-
trifuged to pellet the cultures. They found that on average,
the
Wnal OD
600
values at harvest were 2.01 § 1.06
. These
average
Wnal cell culture densities are somewhat lower than
those of proteins expressed at 37 °C for 3 h using either the
Infors Multitron II Incubator Shaker (ATR, Laurel, MD)
or the Vortexer Shaker, which were 3.76 § 0.76 and
3.78 § 0.64, respectively. Overnight expression in a Bacto-
Lift, which would require a cooled water bath to maintain
the temperature at 18–20 °C, would likely result in higher
Wnal optical cell densities, but it is not known how they
would compare with those of the Infors Multitron II Incu-
bator Shaker or the Vortexer Shaker.
The primary advantage of the BactoLift fermentor sys-
tem is that the capacity for parallel protein expression is
high, with the ability to express 20 di
Verent proteins on a
1 L scale simultaneously. In contrast, the Infors Multitron
II Incubator and the Vortexer shaker have capacities to
express only 8–12 distinct proteins simultaneously in a
comparable 1 L scale. In addition, there is no need to trans-
fer the cultures to centrifuge tubes once expression is com-
pleted since the cultures are grown directly in the centrifuge
bottles. This is not true of the cultures grown in traditional
shakers, in which
Xasks are used for expression. Finally, it is

8
W. Peti, R. Page / Protein Expression and Puri
Wcation 51 (2007) 1–10
inexpensive, with costs of approximately $2500 for a 6-
place unit and $7500 for a 20-place unit. However, there are
drawbacks of the system. First, it is temperature-controlled
using a water bath. Thus, if expression is to be carried out
at low temperatures, such as 18 °C, to enhance the produc-
tion of soluble protein, the BactoLift fermentor must be
placed in a cold room, which is not always easily available.
Second, it requires a constant in
Xux of air via an air pump,
which occupies additional space and incurs additional rou-
tine-use costs. In general, we have found it optimal to have
at least one traditional temperature-controlled incubator
shaker, such as the Infors Multitron II Incubator, in order
to carry out protein expression on a wide variety of scales
(10 ml culture tubes; 50 ml cultures, 100 ml cultures, 1.0 L
cultures) at any temperature. Instruments like the BactoLift
fermentor or the Vortexer Incubator shaker are then pur-
chased as secondary shakers to provide additional, high-
volume expression throughput. To systematically maximize
Wnal cell culture densities, parallel fermentation systems
such as the Sixfors (HT Infors) is most useful. The Sixfors is
a system with six independent fermentor units, which can
be used to either express six di
Verent proteins or express the
same protein, but under di
Vering conditions (varying tem-
perature, pH, pO
2
, etc.) in order to identify those expression
conditions that maximize the yield of expressed, soluble
protein.
Autoinduction medium produces uniformly high cell culture
densities with strong expression
Autoinduction media can be used to express proteins
under the control of the lacUV5 promoter without the
addition of an external induction agent, such as IPTG
Instead, cultures are simply inoculated in the morning,
grown at 37 °C for 3 h and then transferred to low tempera-
ture (20 °C) for overnight growth and expression. More-
over, using this medium, cultures can repeatedly reach
Wnal
OD
600
of 20–40
. Autoinduction depends on the met-
abolic mechanisms bacteria use to regulate the use of car-
bon and energy sources present in the growth medium.
When glucose is present in the medium, the uptake of lac-
tose is blocked, thereby preventing the metabolic produc-
tion of allolactose, a natural induction agent for proteins
under control of the lacUV5 promoter. Speci
Wcally, the
presence of both glucose (0.05%) and lactose (0.2%) in
autoinduction medium enables cultures to grow to high
optical densities and once the glucose is depleted, lactose is
metabolized to allolactose leading to induction and subse-
quent expression. Because premature induction is pre-
vented by catabolite repression, autoinduction media is also
well-suited for the expression of toxic proteins. Finally,
additional formulations of autoinduction media, which
allow for speci
Wc isotopic-labeling of protein samples
required for multi-dimensional NMR spectroscopy and
enable the incorporation of selenomethionine into protein
samples required for X-ray crystallography, have also been
developed
. Because the cultures do not require the
addition of an external induction agent, the use of this
medium is particularly well suited to high-throughput envi-
ronments in which multiple proteins are expressed in paral-
lel. In particular, the chemically de
Wned autoinduction
medium suitable for isotopic-labeling with
13
C and
15
N has
been implemented for the high-throughput production of
samples suitable for structure determination using NMR
spectroscopy by the Center for Eukaryotic Structural
Genomics
. It should be noted that since the unlabeled
-lactose is also a metabolic source of carbon,
13
C and
15
N-
incorporation was typically not 100% complete, and
observed to be as low as 95%. This was determined to be
su
Ycient for NMR structural investigations.
Cell lysis
Gentle E. coli lysis following expression is essential for
successful puri
Wcation of the expressed protein. Four meth-
ods are generally used, including sonication, enzymatic lysis
using lysozyme, freeze–thaw and homogenization. Sonica-
tion is a popular technique for cell lysis. Ultrasonic probes
rapidly oscillate up and down and at a rate of 20 kHz, the
liquid sample turns into a zone of microscopic shock waves,
resulting in cell lysis by liquid shearing and cavitation. Sig-
ni
Wcant energy is released during this process, and only a
short burst of a sonicator probe can cause water to boil.
Thus, one of the primary drawbacks of sonication is that
protein samples can become overheated, resulting in pro-
tein denaturation. It is typically essential that samples be
kept on ice during the sonication process, preferably with
constant stirring to minimize heat generation near the tip of
the probe. In enzymatic lysis, lysozyme is used to digest the
peptidoglycan layer and permeabilize the outer membrane
of gram-negative bacteria. DNase is also usually added to
cleave the liberated DNA. Enzymatic lysis is a gentle,
e
Vective lysis method, but can become expensive for larger
volume cultures. An alternative to enzymatic lysis is freeze–
thaw, in which cells are resuspended in lysis bu
Ver (typically
with lysozyme and DNase1), frozen using liquid nitrogen
and then stored overnight at ¡20 or ¡80 °C or immediately
thawed at room temperature. Multiple cycles of freeze–
thaw enhance lysis. This is a gentle method, but can often
result in incomplete lysis, leading to protein loss. Notably,
certain E. coli expression cell lines are highly suited for lysis
using the freeze–thaw method. A number of cell lines have
been engineered to express T7 lysozyme, such as BL21
(DE3)pLysS cells, in order to inhibit T7 RNA polymerase
activity prior to induction thereby preventing premature
expression of the pET-derived genes. Because these cells
express lysozyme, cell lysis can be e
Yciently carried out
under milder conditions, such as freeze–thaw, without the
need to add lysozyme.
We have found high volume homogenizers, such as the
Emulsi
Xex-C3 (Avestin, Inc.) or the MicroXuidizer (Micro-
Xuidics), to be ideal for gentle cell lysis for a wide array of
culture volumes (as little as 10 ml to as many as 3 L or
more). A bene
Wt of these systems is that external enzymes,

W. Peti, R. Page / Protein Expression and Puri
Wcation 51 (2007) 1–10
9
such as lysozyme or DNase, are not required to facilitate
lysis. We use the Avestin Emulsi
Xex-C3 (“Cell cracker”) for
lysate volumes greater or equal to 10 ml. It is fast, with a
Xow-through capacity of 3 L per hour. In addition, it can
reach up to 30,000 psi and is thus suitable for lysing not
only E. coli but also yeast. Finally, it can be temperature-
controlled with an appropriate heat exchanger. We typi-
cally use three sequential passes for complete E. coli lysis.
For a 100 ml sample, the entire process, including washing
the system before and after lysis, takes about 30 min. In
between each pass, the lysate is kept on ice to minimize
transient increases in temperature. We have successfully
lysed and puri
Wed multiple proteins using this system
which, when the same cultures were lysed by sonication,
resulted in precipitated protein samples. The primary disad-
vantage of this system is its cost, »$25,000. However, con-
sidering the time and e
Vort that goes into the production of
large-scale protein samples suitable for puri
Wcation, it is
well worth the money and, because of its ease of use, is an
optimal piece of shared instrumentation for a facility or
department.
Conclusions
The production of protein samples in the amounts typi-
cally required for structural biology is often the rate-limit-
ing step of structure determination. The recent introduction
of new, inexpensive technologies and freely available clon-
ing materials have greatly facilitated the abilities of small-
to medium-sized laboratories to rapidly and cost-e
Vectively
identify protein constructs which express to high levels, are
soluble and purify readily. In particular, the EMBL catalog
of E. coli expression vectors is particularly useful because of
the large number of expression and puri
Wcation fusion tags
available and the similarity of the vectors’ multiple cloning
sites. In addition, new technologies and strategies to facili-
tate protein expression, such as the Vortexer shaker in com-
bination with Ultra-yield double-ba
Zed Xasks or the new
autoinduction medium, are resulting in signi
Wcant increases
in the
Wnal cell culture densities of expressed proteins and,
in turn, expression yields of protein in equivalent volumes
of media. Finally, new instrumentation to minimize the
time and e
Vort required for cell lysis by homogenization
have also been introduced, which greatly minimize the like-
lihood a protein sample precipitates during the lysis proce-
dure while also ensuring complete lysis.
Acknowledgments
The authors thank all the members of the Page and Peti
laboratories, especially Barbara Dancheck, Michael Hadley
and Jebecka Hudak for help with experiments, Sam Ellis
(Thompson Instrument Company) for supplies and helpful
advice and James Jacso (Glas-Col Inc.) for technical sup-
port. This work was supported by medical research grants
from the Rhode Island Foundation to R.P. and W.P. and
by a Richard B. Salomon Research Grant from Brown
University to R.P. and W.P. This work was partially sup-
ported by National Institutes of Health (NIH) Grant R01
EB003872-02.
References
[1] A.M. Edwards, C.H. Arrowsmith, D. Christendat, A. Dharamsi, J.D.
Friesen, J.F. Greenblatt, M. Vedadi, Protein production: feeding the
crystallographers and NMR spectroscopists, Nat. Struct. Biol. 7
(Suppl.) (2000) 970–972.
[2] C.W. Goulding, L.J. Perry, Protein production in Escherichia coli for
structural studies by X-ray crystallography, J. Struct. Biol. 142 (2003)
133–143.
[3] F.W. Studier, A.H. Rosenberg, J.J. Dunn, J.W. Dubendor
V, Use of T7
RNA polymerase to direct expression of cloned genes, Methods Enz-
ymol. 185 (1990) 60–89.
[4] P. Braun, J. LaBaer, High throughput protein production for func-
tional proteomics, Trends Biotechnol. 21 (2003) 383–388.
[5] B. Miroux, J.E. Walker, Over-production of proteins in Escherichia
coli: mutant hosts that allow synthesis of some membrane proteins
and globular proteins at high levels, J. Mol. Biol. 260 (1996) 289–298.
[6] U. Brinkmann, R.E. Mattes, P. Buckel, High-level expression of
recombinant genes in Escherichia coli is dependent on the availability
of the dnaY gene product, Gene 85 (1989) 109–114.
[7] W.A. Prinz, F. Aslund, A. Holmgren, J. Beckwith, The role of the thio-
redoxin and glutaredoxin pathways in reducing protein disul
Wde
bonds in the Escherichia coli cytoplasm, J. Biol. Chem. 272 (1997)
15661–15667.
[8] I. Hunt, From gene to protein: a review of new and enabling technol-
ogies for multi-parallel protein expression, Protein Expr. Purif. 40
(2005) 1–22.
[9] R. Page, K. Moy, E.C. Sims, J. Velasquez, B. McManus, C. Grittini,
T.L. Clayton, R.C. Stevens, Scalable high-throughput micro-expres-
sion device for recombinant proteins, Biotechniques 37 (2004) 364,
366, 36.
[10] W. Peti, R. Page, K. Moy, M. O’Neil-Johnson, I.A. Wilson, R.C. Ste-
vens, K. Wüthrich, Miniaturized Structural Genomics Pipeline using
Micro-expression and Microcoil NMR, J. Struct. Funct. Genomics 6
(2005) 259–267.
[11] H. Nguyen, B. Martinez, N. Oganesyan, R. Kim, An automated small-
scale protein expression and puri
Wcation screening provides beneWcial
information for protein production, J. Struct. Funct. Genomics 5
(2004) 23–27.
[12] S.A. Lesley, High-throughput proteomics: protein expression and
puri
Wcation in the postgenomic world, Protein Expr. Purif. 22 (2001)
159–164.
[13] H.E. Klock, A. White, E. Koesema, S.A. Lesley, Methods and results
for semi-automated cloning using integrated robotics, J. Struct. Funct.
Genomics 6 (2005) 89–94.
[14] W.B. Jeon, D.J. Aceti, C.A. Bingman, F.C. Vojtik, A.C. Olson, J.M.
Ellefson, J.E. McCombs, H.K. Sreenath, P.G. Blommel, K.D. Seder,
B.T. Burns, H.V. Geetha, A.C. Harms, G. Sabat, M.R. Sussman, B.G.
Fox, G.N. Phillips Jr., High-throughput puri
Wcation and quality
assurance of Arabidopsis thaliana proteins for eukaryotic structural
genomics, J. Struct. Funct. Genomics 6 (2005) 143–147.
[15] Y. Kim, I. Dementieva, M. Zhou, R. Wu, L. Lezondra, P. Quartey, G.
Joachimiak, O. Korolev, H. Li, A. Joachimiak, Automation of protein
puri
Wcation for structural genomics, J. Struct. Funct. Genomics 5
(2004) 111–118.
[16] B.D. Santarsiero, D.T. Yegian, C.C. Lee, G. Spraggon, J. Gu, D. Scheibe,
D.C. Uber, E.W. Cornell, R.A. Nordmeyer, W.F. Kolbe, J. Jin, A.L.
Jones, J.M. Jaklevic, P.G. Schutlz, R.C. Stevens, An approach to rapid
crystallization using nanodroplets, J. Appl. Cryst. 35 (2002) 278–281.
[17] G. Snell, C. Cork, R. Nordmeyer, E. Cornell, G. Meigs, D. Yegian, J.
Jaklevic, J. Jin, R.C. Stevens, T. Earnest, Automated sample mounting
and alignment system for biological crystallography at a synchrotron
source, Structure (Camb.) 12 (2004) 537–545.

10
W. Peti, R. Page / Protein Expression and Puri
Wcation 51 (2007) 1–10
[18] A.E. Cohen, P.J. Ellis, M.D. Miller, A.M. Deacon, R.P. Phizackerley,
An automated system to mount cryo-cooled protein crystals on a syn-
chrotron beamline, using compact sample cassettes and a small-scale
robot, J. Appl. Cryst. 35 (2002) 720–726.
[19] A. Dummler, A.M. Lawrence, A. de Marco, Simpli
Wed screening for
the detection of soluble fusion constructs expressed in E. coli using a
modular set of vectors, Microb. Cell Fact. 4 (2005) 34.
[20] P.B. Card, K.H. Gardner, Identi
Wcation and optimization of protein
domains for NMR studies, Methods Enzymol. 394 (2005) 3–16.
[21] S. Graslund, M. Eklund, R. Falk, M. Uhlen, P.A. Nygren, S. Stahl, A
novel a
Ynity gene fusion system allowing protein A-based recovery
of non-immunoglobulin gene products, J. Biotechnol. 99 (2002) 41–
50.
[22] R.K. Knaust, P. Nordlund, Screening for soluble expression of recom-
binant proteins in a 96-well format, Anal. Biochem. 297 (2001) 79–85.
[23] F.W. Studier, Protein production by auto-induction in high density
shaking cultures, Protein Expr. Purif. 41 (2005) 207–234.
[24] T. Mustelin, L. Tautz, R. Page, Structure of the hematopoietic tyro-
sine phosphatase (HePTP) catalytic domain: structure of a KIM
phosphatase with phosphate bound at the active site, J. Mol. Biol. 354
(2005) 150–163.
[25] A. Savchenko, A. Yee, A. Khachatryan, T. Skarina, E. Evdokimova,
M. Pavlova, A. Semesi, J. Northey, S. Beasley, N. Lan, R. Das, M.
Gerstein, C.H. Arrowmith, A.M. Edwards, Strategies for structural
proteomics of prokaryotes: quantifying the advantages of studying
orthologous proteins and of using both NMR and X-ray crystallogra-
phy approaches, Proteins 50 (2003) 392–399.
[26] J.M. Canaves, R. Page, R.C. Stevens, Protein biophysical properties
that correlate with crystallization success in Thermotoga maritima:
maximum clustering strategy for structural genomics, J. Mol. Biol.
344 (2004) 977–991.
[27] C.S. Goh, N. Lan, S.M. Douglas, B. Wu, N. Echols, A. Smith, D. Mil-
burn, G.T. Montelione, H. Zhao, M. Gerstein, Mining the structural
genomics pipeline: identi
Wcation of protein properties that aVect high-
throughput experimental analysis, J. Mol. Biol. 336 (2004) 115–130.
[28] L. Jaroszewski, L. Rychlewski, Z. Li, W. Li, A. Godzik, FFAS03: a
server for pro
Wle–proWle sequence alignments, Nucleic Acids Res. 33
(2005) W284–W288.
[29] L.J. McGu
Yn, K. Bryson, D.T. Jones, The PSIPRED protein struc-
ture prediction server, Bioinformatics 16 (2000) 404–405.
[30] K. Peng, S. Vucetic, P. Radivojac, C.J. Brown, A.K. Dunker, Z. Obra-
dovic, Optimizing long intrinsic disorder predictors with protein evo-
lutionary information, J. Bioinform. Comput. Biol. 3 (2005) 35–60.
[31] L.M. Iakoucheva, A.K. Dunker, Order, disorder, and
Xexibility: pre-
diction from protein sequence, Structure 11 (2003) 1316–1317.
[32] L.M. Guzman, D. Belin, M.J. Carson, J. Beckwith, Tight regulation,
modulation, and high-level expression by vectors containing the arab-
inose PBAD promoter, J. Bacteriol. 177 (1995) 4121–4130.
[33] T. Mustelin, L. Tautz, R. Page, The structure of the hematopoietic tyro-
sine phosphatase catalytic domain, J. Mol. Biol. 354 (2005) 150–163.
[34] M.S. Keller, W. Peti, NMR assignment of the Spinophilin PDZ
domain (493–602), J. Biomol. NMR 34 (2006).
[35] S. Nallamsetty, D.S. Waugh, Solubility-enhancing proteins MBP and
NusA play a passive role in the folding of their fusion partners, Pro-
tein Expr. Purif. 45 (2006) 175–182.
[36] R.B. Kapust, D.S. Waugh, Escherichia coli maltose-binding protein is
uncommonly e
Vective at promoting the solubility of polypeptides to
which it is fused, Protein Sci. 8 (1999) 1668–1674.
[37] D.S. Waugh, Making the most of a
Ynity tags, Trends Biotechnol. 23
(2005) 316–320.
[38] G.D. Davis, C. Elisee, D.M. Newham, R.G. Harrison, New fusion pro-
tein systems designed to give soluble expression in Escherichia coli,
Biotechnol. Bioeng. 65 (1999) 382–388.
[39] R.B. Kapust, J. Tozser, J.D. Fox, D.E. Anderson, S. Cherry, T.D.
Copeland, D.S. Waugh, Tobacco etch virus protease: mechanism of
autolysis and rational design of stable mutants with wild-type cata-
lytic pro
Wciency, Protein Eng. 14 (2001) 993–1000.
[40] R.B. Kapust, D.S. Waugh, Controlled intracellular processing of
fusion proteins by TEV protease, Protein Expr. Purif. 19 (2000) 312–
318.
[41] T. Cornvik, S.L. Dahlroth, A. Magnusdottir, M.D. Herman, R.
Knaust, M. Ekberg, P. Nordlund, Colony
Wltration blot: a new screen-
ing method for soluble protein expression in Escherichia coli, Nat.
Methods 2 (2005) 507–509.
[42] S.A. Lesley, P. Kuhn, A. Godzik, A.M. Deacon, I. Mathews, A.
Kreusch, G. Spraggon, H.E. Klock, D. McMullan, T. Shin, J. Vincent,
A. Robb, L.S. Brinen, M.D. Miller, T.M. McPhillips, M.A. Miller, D.
Scheibe, J.M. Canaves, C. Guda, L. Jaroszewski, T.L. Selby, M.A. Els-
liger, J. Wooley, S.S. Taylor, K.O. Hodgson, I.A. Wilson, P.G. Schultz,
R.C. Stevens, Structural genomics of the Thermotoga maritima prote-
ome implemented in a high-throughput structure determination pipe-
line, Proc. Natl. Acad. Sci. USA 99 (2002) 11664–11669.
[43] R.L. Alexander, R.G. Smith, Production of plasmid DNA in a simple,
inexpensive and reusable fermentor, Biotechniques 9 (1990) 358–361.
[44] C.T. Lin, P.A. Moore, D.L. Auberry, E.V. Landorf, T. Peppler, K.D.
Victry, F.R. Collart, V. Kery, Automated puri
Wcation of recombinant
proteins: combining high-throughput with high yield, Protein Expr.
Purif. (2005).
[45] W. Peti, T. Herrmann, O. Zagnitko, S.K. Grzechnik, K. Wuthrich,
NMR structure of the conserved hypothetical protein TM0979 from
Thermotoga maritima, Proteins 59 (2005) 387–390.
[46] H.K. Sreenath, C.A. Bingman, B.W. Buchan, K.D. Seder, B.T. Burns,
H.V. Geetha, W.B. Jeon, F.C. Vojtik, D.J. Aceti, R.O. Frederick, Pro-
tocols for production of selenomethionine-labeled proteins in 2-L
polyethylene terephthalate bottles using auto-induction medium, Pro-
tein Expr. Purif. 40 (2005) 256–267.
[47] R.C. Tyler, H.K. Sreenath, S. Singh, D.J. Aceti, C.A. Bingman, J.L.
Markley, B.G. Fox, Auto-induction medium for the production of [U-
15N]- and [U-13C, U-15N]-labeled proteins for NMR screening and
structure determination, Protein Expr. Purif. 40 (2005) 268–278.
Document Outline
- Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost
- New approaches to construct identification
- Parallel cloning into new vectors with novel fusion tags to maximize the likelihood of soluble protein expression in E. coli
- Microexpression screening prior to scale-up
- New methods for increasing cell culture densities during large-scale expression and improving soluble lysis
- Conclusions
- Acknowledgments
- References
Wyszukiwarka
Podobne podstrony:
Strategies for optimizing heterologous protein expression in E coli
Molecular chaperones involved in heterologous protein expression in E coli
Advanced genetic strategies for recombinant protein expression in E coli
Strategies for achieving high level expression in E coli
Rapid and efficient purification and refolding of a (His) tagged recombinant protein produced in E c
Tuning different expression parametres to achive solube recombinant proteins in E coli
Overview of bacterial expression systems for heterologous protein production from molecular and bioc
Expression of correctly folded proteins in E coli
Method for enhancing solubility of the expressed recombinant protein in E coli
Production of recombinant proteins in E coli
Protein Degradation in the Large Intestine Relevance to Colorectal Cancer
Secretory production of recombinant proteins in E coli
A Guide to the Law and Courts in the Empire
A Surgical Safety Checklist to Reduce Morbidity and Mortality in a Global Population
Natural Strategies to Kill Cancer
Peripheral clock gene expression in CS mice with
How to Change Your Life Around in 30 days
Strategie doboru partnera heteroseksualnego
Zarzadzanie strategiczne - sciaga z wykladow (1), Zarządzanie strategiczne- to dysponowanie zasobami
więcej podobnych podstron