
D
o lat osiemdziesiàtych naszego
wieku naukowcy w∏aÊciwie nie
rozumieli, skàd biorà si´ zabój-
cze w∏aÊciwoÊci komórek nowotworo-
wych – ich nie kontrolowany wzrost
i rozprzestrzenianie. Wi´kszoÊç leków
przeciwnowotworowych odkryto, dzia-
∏ajàc ró˝nymi substancjami na komórki
rakowe w hodowli i sprawdzajàc, czy
hamujà ich podzia∏y, lub te˝ wstrzyku-
jàc te substancje chorym na nowotwo-
ry zwierz´tom i obserwujàc, czy guz si´
zmniejsza. Niestety, wiele czynników,
które atakowa∏y komórki nowotworo-
we, uszkadza∏o tak˝e zdrowe tkanki –
jak szpik kostny i komórki jelita – po-
wodujàc (co zresztà nadal jest proble-
mem) przykre i czasem niebezpieczne
skutki uboczne.
Niedawno zacz´to poznawaç defekty
molekularne, decydujàce o przekszta∏-
caniu si´ komórek normalnych w no-
wotworowe [patrz: Robert A. Weinberg,
„Jak powstaje rak?”, strona 32]. Wiele
z tych zmian to mutacje w kluczowych
klasach genów, takich, które w pewnym
stopniu decydujà o podzia∏ach lub
wzroÊcie komórek. Mutacje zmieniajà
iloÊç wyprodukowanych bia∏ek kodo-
wanych przez te geny lub zaburzajà
dzia∏anie ich bia∏ek i w ten sposób za-
k∏ócajà procesy kontrolujàce podzia∏y
komórkowe.
Wiedza o zmutowanych genach
umo˝liwi farmakologom projektowanie
nowych leków, które b´dà dzia∏aç spe-
cyficznie na zmienione geny lub ich pro-
dukty. Leki te, jak si´ sàdzi, sprawià, ˝e
komórki rakowe odzyskajà swoje nor-
malne cechy, albo te˝ zabijà komórki
chore, nie uszkadzajàc nadmiernie ko-
mórek prawid∏owych. Wi´kszoÊç z tych
leków dopiero wchodzi w pierwsze sta-
dia badaƒ, wst´pne wyniki jednak dajà
nadziej´ na kontrolowanie raka na po-
ziomie molekularnym.
Defekty, które sà celem podczas tera-
pii molekularnych, wyst´pujà w trzech
klasach genów. Pierwsza klasa, tzw. on-
kogeny, stymuluje przejÊcie komórki
przez cykl komórkowy, czyli sekwen-
cj´ wydarzeƒ, w których komórka ro-
Ênie, replikuje swój DNA i si´ dzieli,
przekazujàc kompletny zestaw genów
ka˝dej komórce potomnej. Cz∏onkowie
drugiej klasy ograniczajà taki wzrost; to
tzw. geny supresory nowotworów. Ge-
ny z trzeciej klasy sterujà replikacjà i na-
prawà DNA. W przypadku wi´kszoÊci
nowotworów mutacje wyst´pujà w co
najmniej jednej z tych klas genów.
Omówimy ka˝dà z tych klas i wyja-
Ênimy zwiàzane z nimi zjawiska bioche-
miczne. Wska˝emy te˝ sposób, w jaki
lek przeciwrakowy mo˝na podawaç ko-
mórkom, oraz omówimy drog´, na ja-
kiej móg∏by on hamowaç rozwój nowo-
tworu. Na koƒcu pokrótce przedsta-
wimy perspektywy nowych metod le-
czenia. Chocia˝ dos∏ownie ka˝dy defekt
genetyczny mo˝e staç si´ punktem wyj-
Êcia do opracowania nowej terapii, sku-
pimy si´ na metodach, co do których ist-
nieje du˝a szansa, ˝e stanà si´ dost´pne
w ciàgu najbli˝szych 10 lat.
Onkogeny: aktywacja nowotworu
Onkogeny to zmutowane wersje ge-
nów sterujàcych wzrostem komórki,
zwanych czasem protoonkogenami.
Ró˝nice mi´dzy onkogenami i normal-
nymi genami mogà byç niewielkie. Cza-
sami zmutowane bia∏ko b´dàce ostatecz-
nym produktem onkogenu ró˝ni si´ od
normalnego tylko jednym aminokwa-
sem. Jednak ta jednostkowa zmiana mo-
˝e istotnie wp∏ynàç na funkcj´ bia∏ka.
116 Â
WIAT
N
AUKI
Listopad 1996
TERAPIE JUTRA
Najcz´Êciej spotykana mutacja tego
typu zachodzi w genie ras. Oko∏o
20–30% nowotworów ludzkich zawiera
nienormalny gen ras. Bia∏ko kodowane
przez ten gen zwykle zachowuje si´ jak
prze∏àcznik na szlaku przekazywania
sygna∏u nakazujàcego komórce podzia∏.
W odpowiedzi na bodêce przesy∏ane
spoza komórki bia∏ko to aktywuje
wszystkie dalsze etapy szlaku przeka-
zywania sygna∏u.
Gdy nie ma sygna∏ów z zewnàtrz,
bia∏ko Ras powinno pozostaç „wy∏àczo-
ne”. Jednak w zmutowanej wersji zacho-
wuje si´ jak prze∏àcznik w∏àczony na sta-
∏e. Ciàgle udziela komórce b∏´dnych
informacji, ka˝àc si´ jej dzieliç, kiedy ro-
biç tego nie powinna. Te obserwacje su-
gerujà, ˝e zwiàzek zdolny do zabloko-
wania dzia∏ania zmutowanego bia∏ka
Ras by∏by skutecznym lekiem przeciw-
nowotworowym. (Takie zwiàzki bloku-
jàce nazywa si´ antagonistami.) Lecz
w jaki sposób zinaktywowaç zmutowa-
ne bia∏ko Ras?
Jedno z rozwiàzaƒ nasun´∏o si´ na-
ukowcom, gdy zacz´li rozumieç, jak bia∏-
ko to powstaje. Nowo wytworzone czà-
steczki Ras sà nieczynne. Sà to prekur-
sory, które muszà przejÊç szereg mody-
fikacji biochemicznych, by staç si´ do-
jrza∏ymi, aktywnymi wersjami bia∏ka
Ras. Wówczas przyczepiajà si´ do we-
wn´trznej powierzchni b∏ony komórko-
wej, gdzie oddzia∏ujà z innymi bia∏ka-
mi i stymulujà wzrost komórek.
Modyfikacje biochemiczne zachodzà
na jednym koƒcu prekursora bia∏ka Ras,
tam gdzie enzymy dzia∏ajà na region
zwany sekwencjà CAAX. Zmiany te
przebiegajà w trzech etapach; najbardziej
istotny z nich jest pierwszy, zwany far-
nezylacjà, kiedy to do prekursora do∏à-
cza si´ 15 atomów w´gla. Reakcj´ kata-
lizuje specyficzny enzym – transferaza
farnezylu.
Jedna ze strategii blokowania aktyw-
noÊci bia∏ka Ras polega na hamowaniu
transferazy i zapobieganiu w ten sposób
modyfikacji. Naukowcy wyprodukowa-
li ju˝ kilka takich inhibitorów. W hodow-
lach komórkowych inhibitory te bloku-
jà dojrzewanie bia∏ka Ras i odwracajà
transformacj´ nowotworowà wywo∏anà
przez zmutowane geny ras. Równie za-
ch´cajàce wyniki przynios∏y testy na
zwierz´tach. Badania te wykaza∏y, ˝e in-
hibitory transferazy farnezylu zapobie-
gajà tworzeniu nowych guzów spowo-
dowanych wyst´powaniem nienormal-
nych bia∏ek Ras, a tak˝e wywo∏ujà re-
gres ju˝ istniejàcych nowotworów.
Na szcz´Êcie wyglàda na to, ˝e inhi-
bitory transferazy farnezylu sà doÊç spe-
Nowe
cele molekularne
Nowe podejÊcie w leczeniu raka
to wykorzystanie charakterystycznych
cech molekularnych komórek nowotworowych
Allen Oliff, Jackson B. Gibbs i Frank McCormick
AUDRA GERES
cyficzne. Ârodki te nie dzia∏ajà na ko-
mórki prawid∏owe czy te˝ zmienione
pod wp∏ywem innych onkogenów.
Dzi´ki specyficznoÊci inhibitorów efek-
ty uboczne ich stosowania mogà byç
minimalne. Okaza∏o si´, ˝e podawa-
nie inhibitorów transferazy farnezylu
w wysokich dawkach – takich, które
umo˝liwi∏yby wyeliminowanie ju˝ ist-
niejàcych nowotworów – w zasadzie nie
spowodowa∏o ˝adnych toksycznych
skutków w zdrowych tkankach bada-
nych zwierzàt.
Inna grupa onkogenów, które sà co-
raz bli˝sze wykorzystania w terapii prze-
ciwnowotworowej, to te, które kodujà
enzymy zwane kinazami bia∏kowymi.
(Zmutowane geny kinaz znaleziono
m.in. w przewlek∏ej bia∏aczce szpikowej,
raku piersi i raku p´cherza.) W komór-
kach normalnych kinazy bia∏kowe po-
magajà w regulacji wielu wa˝nych pro-
cesów, na przyk∏ad przesy∏aniu sygna-
∏ów mi´dzy b∏onà komórkowà a jàdrem,
inicjacji kolejnych etapów cyklu wzrostu
i podzia∏u komórki oraz w kontrolowa-
niu ró˝nych jej funkcji metabolicznych.
Kinazy bia∏kowe sterujà tymi procesa-
mi, aktywujàc inne bia∏ka w odpowie-
dzi na okreÊlone bodêce.
Kinazy mogà prowadziç do nowo-
tworów na dwa sposoby. Jednym z nich
jest nadprodukcja tych bia∏ek spowo-
dowana mutacjami w obszarach regu-
latorowych ich genów. W porównaniu
z komórkami prawid∏owymi komórki
nowotworowe wytwarzajà cz´sto nie-
zwykle du˝o takiej czy innej kinazy. Te
olbrzymie iloÊci sà przyczynà kontynu-
owania podzia∏ów komórkowych, kie-
dy nie powinny one zachodziç. Kinazà
powszechnie nadprodukowanà w tkan-
kach nowotworowych jest receptor dla
nab∏onkowego czynnika wzrostowego
(epidermal growth factor – EGF).
Kinazy mogà te˝ przyczyniaç si´ do
powstawania nowotworów, gdy majà
nieprawid∏owà budow´. Wiele komó-
rek nowotworowych zawiera kinazy
bia∏kowe, które z powodu jakiejÊ wady
strukturalnej sà zawsze w∏àczone. Prze-
prowadzajà wi´c reakcje, które stymu-
lujà niepo˝àdane podzia∏y komórek.
Przyk∏adami kinaz, które zachowujà si´
nienormalnie w niektórych nowotwo-
rach ludzkich, sà Abl, Src i kinazy za-
le˝ne od cyklin.
Inhibitor jednej czy wielu takich ki-
naz móg∏by byç oczywiÊcie skutecznym
lekiem przeciwnowotworowym. Pro-
blem polega na tym, by znaleêç substan-
cj´ zdolnà odró˝niç jednà kinaz´ od dru-
giej. Wiele z prawie tysiàca kinaz bia∏-
kowych w komórkach ssaków ma bar-
dzo podobnà budow´, szczególnie w re-
gionach czynnych biochemicznie. Tak
wi´c inhibitor jednej kinazy móg∏by za-
burzyç aktywnoÊç innych, nie spokrew-
nionych, które sà wa˝ne dla normalnych
funkcji komórkowych.
Mimo tych obaw farmakolodzy w cià-
gu ostatnich kilku lat zsyntetyzowali
i przetestowali seri´ czynników antykina-
zowych. Celem ataku wi´kszoÊci z tych
czynników sà same kinazy, ale niektóre
leki z tej grupy dzia∏ajà na poziomie ge-
netycznym (uniemo˝liwiajàc powstanie
kinaz). Na przyk∏ad przy tzw. podejÊciu
antysensowym niewielkie sztucznie wy-
tworzone fragmenty materia∏u genetycz-
nego oddzia∏ujà z mRNA komórki no-
wotworowej, nie dopuszczajàc do pro-
dukcji bia∏ek. Czàsteczki mRNA sà w za-
sadzie ruchomymi kopiami genów i fi-
zycznymi matrycami, na których komór-
ki wytwarzajà bia∏ka zakodowane w
genach [patrz: Jack S. Cohen i Michael
E. Hogan, „Nowe medykamenty gene-
tyczne”; Âwiat Nauki, luty 1995].
Zadziwiajàce jest to, ˝e inhibitory ki-
naz mogà byç doÊç selektywne. W pro-
bówce niektóre z nich wià˝à si´ z w∏aÊci-
wym celem tysiàckrotnie cz´Êciej ni˝
z nie spokrewnionymi kinazami. Bar-
dziej istotne sà wyniki uzyskane w ca-
∏ych komórkach w hodowli. Wykazujà,
˝e kilka z tych zwiàzków hamuje wzrost
komórek nowotworowych zawierajà-
cych zmutowane geny kinaz bia∏ko-
wych. Co bardziej zach´cajàce, niektóre
z nich powstrzymujà wzrost komórek
nowotworowych u zwierzàt – nasuwa
to przypuszczenie, ˝e mog∏yby dzia∏aç
tak˝e w organizmie ludzkim. Te leki da-
jà nadziej´, ˝e w ciàgu najbli˝szych kil-
ku lat wÊród Êrodków zwalczajàcych
nowotwory pojawià si´ antagoniÊci ki-
naz bia∏kowych.
Â
WIAT
N
AUKI
Listopad 1996 117
LECZENIE RAKA na poziomie molekularnym po-
lega na naprawie wadliwego DNA, wy∏àczaniu klu-
czowych bia∏ek regulujàcych wzrost i zwi´kszaniu
wra˝liwoÊci komórek nowotworowych na terapie
konwencjonalne, na przyk∏ad napromienianie.

Geny supresory nowotworów
Druga g∏ówna klasa genów odpowie-
dzialnych za powstawanie nowotwo-
rów zawiera te z nich, które normalnie
hamujà rozwój nowotworów. Rak jest
cz´sto rezultatem utraty lub niew∏aÊci-
wego funkcjonowania kluczowych bia-
∏ek regulatorowych kodowanych przez
te geny. Dwa g∏ówne bia∏ka supresoro-
we to pRB i p53.
Bia∏ko pRB (nazwane tak od „retino-
blastoma” – siatkówczaka, typu no-
wotworu, w którym po raz pierwszy
zidentyfikowano kodujàcy je gen RB)
uczestniczy w regulacji cyklu komór-
kowego. Aktywna forma tego bia∏ka ha-
muje replikacj´ DNA. W mniej wi´cej
40% ludzkich nowotworów mutacje
w genie RB unieczynniajà jego bia∏ko.
W rezultacie komórki dzielà si´ bez
przerwy.
Innà niezwykle wa˝nà czàsteczkà re-
gulatorowà jest bia∏ko p53. Cz´sto na-
zywane stra˝nikiem genomu, w normal-
nych komórkach zapobiega replikacji
uszkodzonego DNA i wywo∏uje apop-
toz´, czyli samobójstwo komórek zawie-
rajàcych nienormalny DNA. Natomiast
wadliwe czàsteczki p53 zezwalajà za-
równo na prze˝ycie komórek niosàcych
uszkodzony DNA, jak i na jego replika-
cj´. Takie komórki przekazujà ka˝dà ist-
niejàcà ju˝ mutacj´ komórkom potom-
nym, w których dzi´ki temu mogà si´
gromadziç wszystkie kolejne mutacje
potrzebne do utworzenia zagra˝ajàcych
˝yciu cz∏owieka guzów. Wydaje si´, ˝e
w wi´kszoÊci ludzkich nowotworów
gen p53 jest uszkodzony.
Jak zmierzyç si´ z wadliwymi genami
RB i p53? Rozwa˝a si´ kilka strategii.
Teoretycznie najbardziej bezpoÊrednia
polega na zastàpieniu uszkodzonego ge-
nu jego prawid∏owym odpowiednikiem.
Proces ten nazywany jest terapià geno-
wà i w doÊwiadczeniach z hodowlami
komórkowymi da∏ ju˝ zach´cajàce wy-
niki: wprowadzenie prawid∏owych ge-
nów RB lub p53 do komórek nowotwo-
118 Â
WIAT
N
AUKI
Listopad 1996
TERAPIE JUTRA
rowych hamowa∏o ich wzrost. Naukow-
cy opracowujà obecnie protoko∏y badaƒ
klinicznych. Majà nadziej´, ˝e uda im
si´ wprowadziç normalne geny p53 do
komórek nowotworowych u ludzi.
Intensywnie bada si´ ró˝ne metody
wprowadzania genów do komórek no-
wotworów. Os∏abione wirusy mog∏yby
zawieraç normalny gen i wprowadzaç
go tylko do komórek nowotworowych
[ramka obok]. To korzystajàce z wekto-
rów wirusowych podejÊcie jest jednak
wcià˝ nowe i napotyka przeszkody, z
których bynajmniej nie naj∏atwiejszà jest
atak uk∏adu odpornoÊciowego. Wirusy
mogà zostaç zabite, zanim zdo∏ajà do-
trzeç do komórek nowotworowych.
Regulowanie produktów genów
Z powodu trudnoÊci, które pi´trzà si´
przed terapià genowà, wielu onkologów
badajàcych geny supresory nowotwo-
rów przyjmuje bardziej tradycyjnà meto-
d´. Polega ona na ustaleniu ciàgu zja-
wisk b´dàcych konsekwencjà genety-
cznych defektów komórki, a nast´pnie
na opracowaniu leków zapobiegajàcych
jednemu z nich. Na przyk∏ad w zdro-
wych komórkach bia∏ko pRB blokuje ak-
tywnoÊç innego bia∏ka, czynnika trans-
krypcyjnego E2F, który w postaci nie
zwiàzanej przyspiesza syntez´ DNA.
Utrata bia∏ka pRB prowadzi wi´c do nie
kontrolowanego dzia∏ania E2F i gwa∏-
townego namna˝ania si´ komórek. Wy-
nika z tego, ˝e leki zdolne do hamowa-
nia E2F mog∏yby zatrzymaç ekspansj´
nowotworów b´dàcych wynikiem utra-
ty bia∏ka pRB.*
Obecnie trudno stwierdziç, jaki
wp∏yw mia∏by taki inhibitor na komór-
ki prawid∏owe. Ale niedawno przepro-
wadzone eksperymenty na myszach,
którym specyficznie „znokautowano”
geny E2F (a wi´c myszach pozbawio-
nych tego genu), umo˝liwiajà ju˝ mode-
lowanie ewentualnych efektów ubocz-
nych. Ekstrapolujàc wyniki na ludzi,
mo˝emy przewidzieç szkodliwe dzia∏a-
nia tych leków (i byç mo˝e znaleêç spo-
soby ich unikni´cia) na wiele lat przed
próbami klinicznymi.
Naukowcy znajà szlak biochemiczny
regulowany przez gen RB, ale o szlaku
regulowanym przez p53 niewiele wia-
domo. Nie znamy dok∏adnie molekular-
nego ciàgu zdarzeƒ wynikajàcego z utra-
ty genu p53. Dlatego te˝ w tym przy-
padku nie zidentyfikowano potencjal-
nych celów dzia∏ania leków.
Jednak dogodnà szans´ stwarza za-
dziwiajàca cecha inaktywacji bia∏ka p53.
Niektóre eksperymenty in vitro sugeru-
Wirusy przeciw nowotworom
B
yç mo˝e najskuteczniejszym sposobem dotarcia do nowotworów b´dzie u˝ycie
wirusów. W terapii genowej os∏abione wirusy dostarczajà normalne geny do wn´-
trza komórek. Potencjalnie najlepszym dostawcà terapeutycznych genów do komórek
nowotworowych jest os∏abiony adenowirus. Adenowirusy zawierajà DNA (niektóre wi-
rusy, np. retrowirusy, majà tylko RNA). JeÊli do DNA wirusa wstawi si´ terapeutycz-
ny gen, to wirus wprowadzi go do ka˝dej komórki, do której wtargnie. Nie wyrzàdzi on
komórce ˝adnej szkody, jeÊli tylko przy wstawianiu nowego genu usunie si´ z jego
DNA geny odpowiedzialne za wirulencj´.
Ponadto adenowirusy sà zdolne wybiórczo zabijaç komórki nowotworowe. Gdy ja-
kiÊ wirus wnika do normalnej komórki, to w odpowiedzi bia∏ka p53 instruujà jà, by
przesta∏a replikowaç DNA, zapobiegajàc w ten sposób tak˝e replikacji wirusa. Nato-
miast adenowirus ma bia∏ko, które ∏àczy si´ bezpoÊrednio z p53 i je unieczynnia. Wi-
rus mo˝e wtedy wykorzystaç maszyneri´ komórkowà do replikacji.
Mo˝na tak zmieniç genetycznie adenowirus, by przejà∏ kontrol´ wy∏àcznie nad ko-
mórkami rakowymi. A dok∏adniej spowodowaç, by adenowirusowe bia∏ko nie wiàza∏o
p53. Wówczas wirus nie zdo∏a unieczynniç p53 i b´dzie si´ replikowaç wy∏àcznie
w komórkach pozbawionych tego bia∏ka, czyli w wielu rodzajach komórek nowotwo-
rowych. Faktycznie, wykazano doÊwiadczalnie, ˝e takie zmodyfikowane wirusy repli-
kujà si´ skutecznie w komórkach nowotworowych i wytwarzajà identyczne wirusowe
potomstwo. Teoretycznie mog∏yby one zaka˝aç nast´pnie sàsiednie komórki nowotwo-
rowe i rozprzestrzeniç si´ w ca∏ej zrakowacia∏ej tkance. W ten sposób wszystkie ko-
mórki rakowe zosta∏yby zaka˝one i zabite.
PodejÊcia z zastosowaniem wektorów wirusowych sà jeszcze w powijakach i trzeba
przezwyci´˝yç wiele przeszkód technicznych. Najwa˝niejsza polega zapewne na ko-
niecznoÊci zainfekowania znaczàcej liczby komórek guza i na tym, by ka˝dy wprowadza-
ny gen produkowa∏ bia∏ko w iloÊci wystarczajàcej do powstrzymania podzialów komórek
nowotworowych oraz zapewnienia poprawy zdrowia pacjenta. Nale˝y si´ te˝ liczyç z wy-
stàpieniem reakcji immunologicznych na wirusowe bia∏ka – na przyk∏ad uk∏ad odporno-
Êciowy mo˝e zaatakowaç i unieszkodliwiç wirusa, zanim ten osiàgnie swój cel. Byç mo-
˝e u˝ytecznoÊç tej terapii przeciwnowotworowej b´dzie ostatecznie zale˝eç od tego,
w jakim stopniu podczas leczenia da si´ kontrolowaç odpowiedzi immunologiczne. Opra-
cowano ju˝ jeden rodzaj os∏abionego adenowirusa na potrzeby prób klinicznych, a wst´p-
ne testy na pacjentach powinny rozpoczàç si´ w ciàgu paru lat. Naukowcy badajà te˝
inne sposoby dostarczania leków, stosujàc odmienne rodzaje wirusów (np. retrowiru-
sy) oraz lipidy, które nie wywo∏ujà odpowiedzi immunologicznej.
jà, ˝e za pomocà pewnych zwiàzków
drobnoczàsteczkowych, które ∏àczà si´
ze zmutowanym bia∏kiem p53, mo˝na
mu przywróciç normalnà funkcj´. JeÊli
podobne rezultaty uda si´ uzyskaç
w komórkach nowotworowych, to pra-
wdopodobnie zaprzestanà one podzia-
∏ów, a nawet umrà, bo jednà z funkcji
normalnego bia∏ka p53 jest sk∏anianie
komórek nieprawid∏owych do samo-
zniszczenia. Opracowanie szczegó∏ów
technicznych umo˝liwiajàcych realiza-
cj´ takiego podejÊcia jest rzeczà trudnà,
ale wydaje si´, ˝e ze wzgl´du na liczb´
nowotworów zawierajàcych defektyw-
ne geny p53 mia∏oby to ogromne zna-
czenie. Prace nad mo˝liwoÊcià wyko-
rzystania tej strategii sà w toku.
Geny sprawdzajàce napraw´ DNA
Trzecia g∏ówna klasa genów mogàcych
stanowiç molekularny cel zawiera te,
które pomagajà w sprawdzaniu i utrzy-
mywaniu integralnoÊci DNA, cz´sto ule-
gajàcego uszkodzeniom w czasie normal-
nej replikacji. Bez tych mechanizmów
szansa na naprawienie uszkodzonego ge-
nu jest znikoma i wzrasta prawdopodo-
bieƒstwo przekazania defektu komór-
kom potomnym jako trwa∏ej mutacji.
W komórkach nowotworowych procesy
naprawy DNA cz´sto przebiegajà niepra-
wid∏owo. Na przyk∏ad w 10–20% ludz-
kich nowotworów jelita grubego wyst´-
pujà mutacje w genach, które zwykle
pomagajà w naprawie DNA (tj. w genach
MLH1, MSH2, PMS1 i PMS2).
W naprawie DNA biorà udzia∏ po-
Êrednio jeszcze inne geny i w rzeczywi-
stoÊci to w nich mutacje spotyka si´
znacznie cz´Êciej. Nale˝à tu geny kodu-
jàce bia∏ka punktów kontrolnych, któ-
re monitorujà przejÊcie komórki przez
cykl komórkowy i zapobiegajà rozpo-
cz´ciu nast´pnej fazy, jeÊli wczeÊniejsze
nie zakoƒczy∏y si´ poprawnie – na przy-
k∏ad DNA nie zosta∏ wiernie skopiowa-
ny. Najbardziej znane bia∏ka punktu
kontrolnego to ATM i znowu uniwer-
salne p53. Komórkom nowotworowym
bez normalnych genów ATM lub p53
brakuje tych mechanizmów sprawdza-
jàcych; uszkodzony DNA jest wi´c
„przepychany” przez proces replikacji,
co powoduje wzrost cz´stoÊci mutacji
losowych w komórkach potomnych.
Podobnie jak w przypadku zmuto-
wanych genów supresorów terapi´ ge-
nowà mo˝na by stosowaç w celu zastà-
pienia tych brakujàcych lub uszko-
dzonych genów, które kodujà bia∏ka
zwiàzane z naprawà DNA lub pokrew-
ne. Bardziej radykalne podejÊcie pole-
ga∏oby na pozwoleniu niektórym gu-
zom na ciàg∏e mutacje. Komórki
nowotworowe, które zwi´kszajà ich
tempo, p∏acà za to wysokà cen´ – wiele
mutacji jest letalnych i prowadzi do
Êmierci komórek potomnych. Guz no-
wotworowy mo˝e jednak znieÊç takie
straty, jeÊli jakieÊ nabyte mutacje zwi´k-
szà szanse prze˝ycia przynajmniej cz´-
Êci jego potomstwa. Gdy jednak muta-
cji jest zbyt wiele, ˝adne potomne ko-
mórki guza nie zdo∏ajà prze˝yç.
Â
WIAT
N
AUKI
Listopad 1996 119
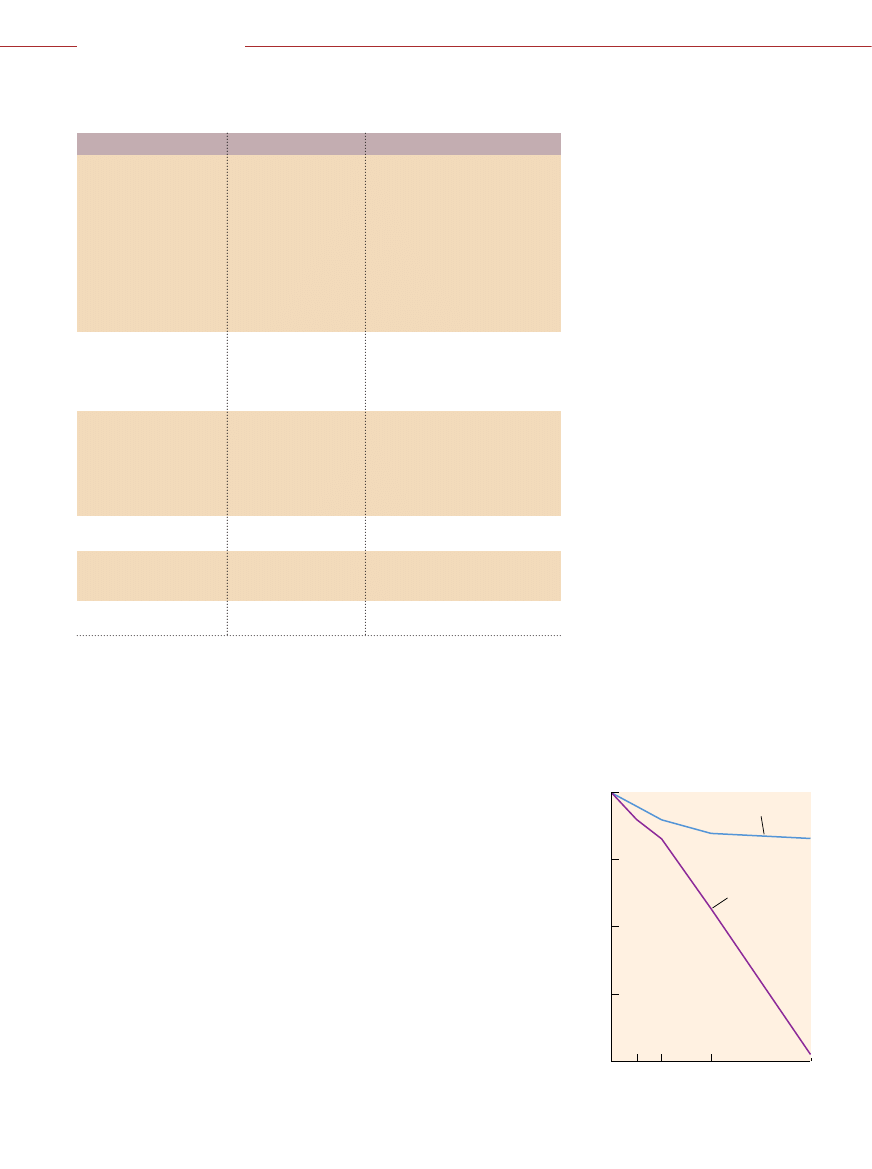
ZWIÑZEK Z LUDZKIMI
NOWOTWORAMI
CYKL KOMÓRKOWY
Zwi´kszona iloÊç
w 20% raków piersi
Aktywowane przez mutacje
w 20–30% nowotworów
Hamowanie dojrzewania
bia∏ka Ras
Aktywowane przez translokacje
chromosomowe w chronicznej
bia∏aczce szpikowej
Hamowanie kinazy
lub blokowanie syntezy
za pomocà antysensownego
kwasu nukleinowego
Aktywowane przez mutacje
w 2–5% nowotworów
Hamowanie enzymów, które
dzia∏ajà na dalszych etapach
wa˝nych szlaków
Zmutowane
lub nieobecne
w 40% nowotworów
Przywrócenie za pomocà
terapii genowej lub blokowanie
bia∏ka E2F aktywowanego
przez utrat´ pRB
Przywrócenie za pomocà
terapii genowej lub zabijania
komórek adenowirusami
Zmutowane
lub nieobecne
w 50% nowotworów
Blokowanie przeciwcia∏em
lub hamowanie funkcji
biochemicznej receptora
PODEJÂCIE
TERAPEUTYCZNE
ZEGAR
KOMÓRKOWY
JÑDRO
CYTOPLAZMA
KOMÓRKA
SSACZA
Bia∏ko
pRB
Bia∏ko
p53
Kinaza
Src
Kinaza
Abl
Bia∏ko
Ras
Receptor
czynnika
wzrostu
Komórka
si´ dzieli
Komórka si´ powi´ksza
Komórka
replikuje
DNA
Komórka
przygotowuje si´
do podzia∏u
Komórka
odpoczywa
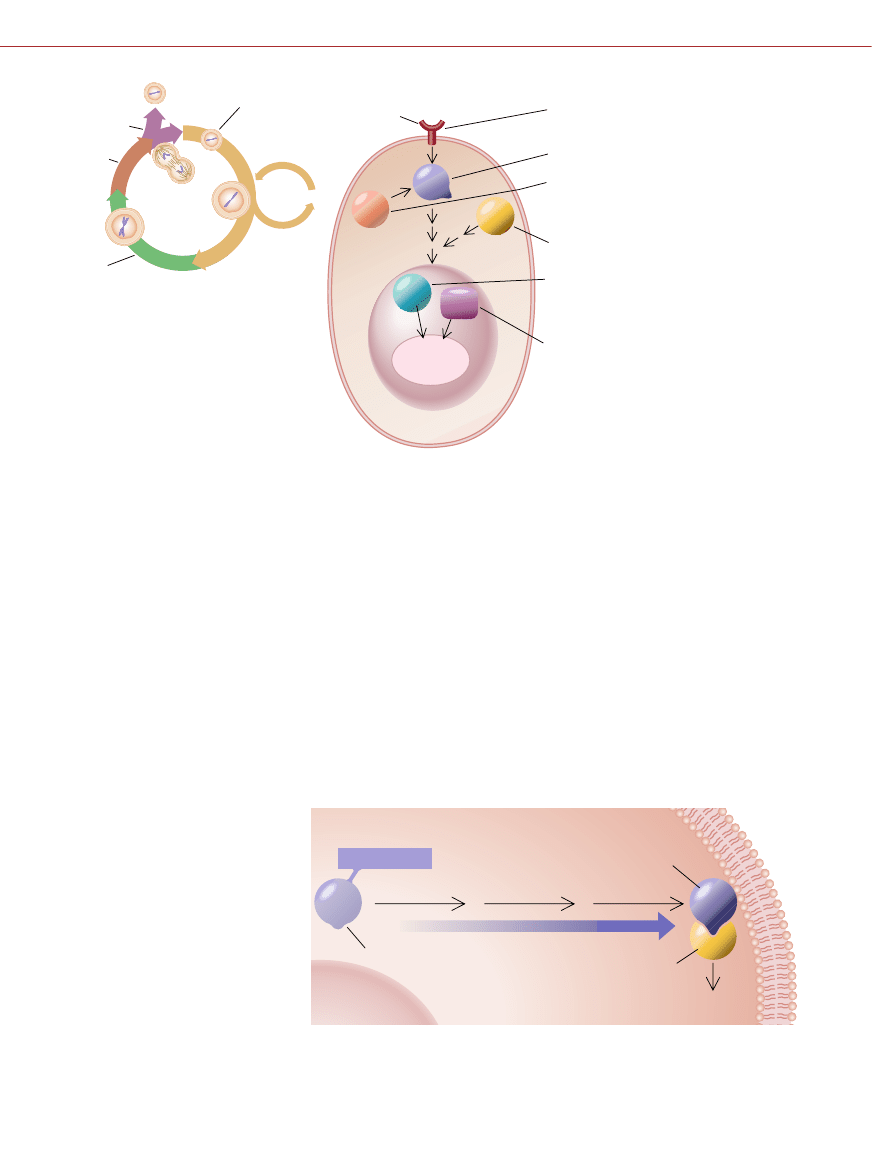
SZLAK PRZEKAZYWANIA SYGNA¸U
w komórce ssaczej
(z prawej) sk∏ada si´ z
wielu elementów, które – jeÊli ich iloÊç lub
budowa sà zmienione – mogà doprowadziç
do powstania nowotworu. Nale˝à do nich
receptory czynników wzrostu, bia∏ka Ras
i kinazy, które wspierajà ich dzia∏anie, ta-
kie jak Abl i Src. Zaburzenia pRB i p53 mo-
gà tak˝e powodowaç rozwój nowotworu.
Zmiany te przyczyniajà si´ do rozregulowa-
nia cyklu komórkowego
(u góry).
FARNEZYLACJA
PROTEOLIZA
DOJRZEWANIE
STYMULACJA
WZROSTU KOMÓRKI
BIA¸KO KOMÓRKOWE
DOJRZA¸E BIA¸KO RAS
NIECZYNNE
BIA¸KO RAS
METYLACJA
SEKWENCJA CAAX
BIA¸KO RAS poczàtkowo jest nieczynnym prekursorem. Dojrzewanie zachodzi w trzech
etapach i dotyczy sekwencji CAAX. Po modyfikacji Ras oddzia∏uje z innymi bia∏kami
i stymuluje wzrost komórki. Leki hamujàce farnezylacj´ i dzi´ki temu zapobiegajàce ak-
tywacji bia∏ka Ras mog∏yby powstrzymywaç podzia∏y komórek nowotworowych.
IAN WORPOLE
IAN WORPOLE
Jednym ze sposobów sk∏aniania ko-
mórek rakowych do wytwarzania nie-
zdolnych do prze˝ycia komórek potom-
nych jest hamowanie kilku mecha-
nizmów punktów kontrolnych naraz.
Zwyk∏e komórki dro˝d˝y poddane dzia-
∏aniu promieniowania rentgenowskie-
go umierajà dopiero po otrzymaniu wy-
sokiej dawki. JeÊli jednak mutacji ulegnie
jeden z genów punktu kontrolnego,
dro˝d˝e sà bardziej wra˝liwe na pro-
mieniowanie. Gdy jednoczeÊnie zmutu-
jà dwa takie geny lub wi´cej, komórki
stajà si´ nadwra˝liwe i zabijajà je nawet
niskie dawki.
Wiedzàc o tych zale˝noÊciach, onko-
lodzy opracowujà testy przesiewowe,
sprawdzajàce ró˝ne substancje w celu
odnalezienia zwiàzków hamujàcych
bia∏ka punktów kontrolnych. Leki mo-
g∏yby na przyk∏ad dzia∏aç na komórki
ze znanym defektem w takim w∏aÊnie
genie (powiedzmy, w zmutowanym ge-
nie p53). Majàc wiele podobnych defek-
tów, komórki rakowe powinny ∏atwo
obumieraç lub przynajmniej byç bar-
dziej podatne na inne sposoby terapii.
Kilka zwiàzków przetestowanych w ho-
dowlach komórkowych wydaje si´ obie-
cujàcych, choç badania kliniczne roz-
pocznà si´ nie wczeÊniej ni˝ na poczàtku
przysz∏ego wieku.
Oprócz atakowania genów czy bia∏ek
zaanga˝owanych we wzrost komórki
terapie molekularne mogà byç tak˝e
skierowane na inne wa˝ne czàsteczki;
jest szansa, ˝e niektóre z tych metod b´-
dà dost´pne w ciàgu 4 lat. Na przyk∏ad
wiele ró˝nych bia∏ek odpowiada za
utrzymywanie komórek w jednym miej-
scu w organizmie; opierajàc si´ na tym,
naukowcy odkryli w∏aÊciwoÊci lecznicze
inhibitorów proteaz, zdolnych zapobie-
gaç przerzutom, czyli rozprzestrzenia-
120 Â
WIAT
N
AUKI
Listopad 1996
TERAPIE JUTRA
niu si´ komórek nowotworowych [patrz:
Erkki Ruoslahti, „Jak rozsiewa si´ rak?”,
strona 42]. Inne leki pos∏u˝à do unie-
czynnienia telomerazy, enzymu, który
odbudowuje koƒce replikujàcych chro-
mosomów i w ten sposób umo˝liwia nie-
ÊmiertelnoÊç komórkom rakowym w
warunkach, w których pozosta∏e obu-
mierajà. Zwiàzki takie jak TNP-470 mo-
g∏yby zapobiegaç powstawaniu nowych
naczyƒ krwionoÊnych (angiogenezie)
[patrz: Judah Folkman, „Atak na uk∏ad
krwionoÊny guza”, strona 122].
Choç wyznaczenie celów wymienio-
nych tutaj leków jest jednym z najbar-
dziej ekscytujàcych osiàgni´ç biologii
nowotworów w ciàgu ostatnich 10 lat,
musimy ostudziç nadmierne nadzieje na
szybkie wprowadzenie tych Êrodków do
praktyki leczniczej. Nowe metody mu-
szà przezwyci´˝yç te same przeszkody,
które sta∏y przed standardowymi che-
mioterapiami. Przysz∏e leki nie tylko po-
winny zlokalizowaç swoje nowotworo-
we cele, ale te˝ znaleêç sposób, by dostaç
si´ do nich w iloÊciach zapewniajàcych
skutecznoÊç. W przypadku guzów litych
bariery sà wyjàtkowo trudne do poko-
nania, niewiele krwi dop∏ywa do ich
wn´trza, a ponadto niektóre specyfiki
mogà mieç powa˝ne trudnoÊci z wydo-
staniem si´ z naczyƒ krwionoÊnych od-
˝ywiajàcych guzy [patrz: Rakesh K. Jain,
„Bariery utrudniajàce wnikanie leków
do guzów litych”; Âwiat Nauki, wrzesieƒ
1994.] No i oczywiÊcie sà jeszcze proble-
my zwiàzane z toksycznoÊcià, efektami
ubocznymi i zjawiskiem opornoÊci ko-
mórek nowotworu na lek.
Dzi´ki wykorzystaniu najnowszych
metod nauk farmaceutycznych mo˝li-
we jest szybsze odkrywanie i opracowy-
wanie leków. Techniki te obejmujà re-
kombinacj´ DNA umo˝liwiajàcà pro-
Leczenie nowotworów
na poziomie molekularnym
Cecha nowotworu
Cele molekularne
Leki
Aktywacja onkogenu
Bia∏ka Ras
Inhibitory transferazy farnezylu:
prowadzàca do nadmiernej
L-744, 832, SCH 44342, BZA-5B
aktywnoÊci bia∏ka Ras
Kinazy: Abl,
Inhibitory kinaz tyrozynowych:
lub kinazy
receptor EGF, Erb-B2,
tyrfostyny (RG 13 022),
Src
lawendustyny (AG 957),
chinazoliny (PD 153 035)
Inhibitory antysensowne
Kinazy: PKC-
a,
Inhibitory kinaz serynowo-
Raf, cyklinozale˝ne
-treoninowych: olomucyna,
staurosporyna, butyrolakton
Inhibitory antysensowne
Utrata genów
Geny: APC, AT,
Terapia genowa przywracajàca
supresorów nowotworów
DCC
, RB, p53
normalnà funkcj´ genów
supresorów nowotworów
Czynniki antysensowne blokujàce
syntez´ E2F
Nieprawid∏owe
Enzymy naprawy
Terapia genowa przywracajàca
mechanizmy naprawy DNA
b∏´dnie sparowanych
normalnà aktywnoÊç
zasad w DNA:
enzymatycznà
MSH2, MLH1,
PMS1, PMS2
Inhibitory punktów kontroli
wzmagajàce podatnoÊç
na Êrodki uszkadzajàce DNA
Brak starzenia si´
Telomeraza
Inhibitory telomerazy
komórek nowotworowych
Angiogeneza
Czynniki wzrostu:
TNP-470; suramina
FGF, VEGF
Receptory integryny
AntagoniÊci
a
n
b
3
,
a
n
b
5
,
Przerzuty
Metaloproteazy
Inhibitory proteaz
Kolagenazy Inhibitory
kolagenaz
DAWKA (KILORADY)
PRZE˚YWALNOÂå (PROCENTY)
0
1
2
4
8
0.01
0.1
1
10
100
DRO˚D˚E NORMALNE
DRO˚D˚E
Z DEFEKTEM
PUNKTU
KONTROLNEGO
DEFEKT PUNKTU KONTROLNEGO DNA
zwi´ksza wra˝liwoÊç komórek dro˝d˝y na
promieniowanie. Dawka 8 kiloradów pozo-
stawia przy ˝yciu wiele zdrowych komórek
dro˝d˝y, ale wr´cz wymiata te, które nie po-
trafià w∏aÊciwie sprawdziç naprawy DNA.
To odkrycie Êwiadczy, ˝e zniszczenie punk-
tu kontroli DNA w komórkach nowotwo-
rowych mog∏oby uwra˝liwiaç je na terapie
konwencjonalne.
JANA BRENING
IAN WORPOLE
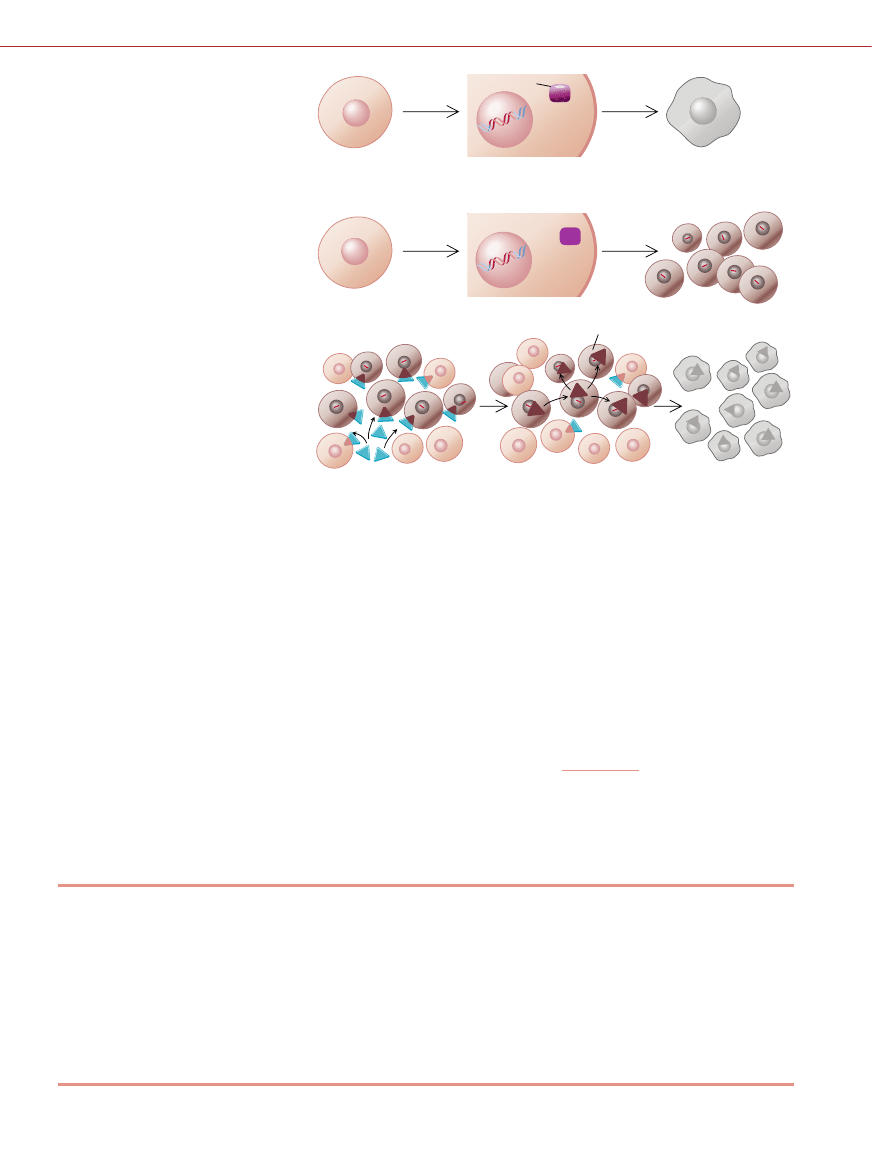
dukcj´ zwiàzków, wykorzystanie gene-
tycznie zmienionych zwierzàt jako
uk∏adów modelowych, zautomatyzo-
wane przesiewowe badania zwiàzków
na du˝à skal´, kombinatoryczne techni-
ki chemiczne oraz projektowanie leków
wspomagane komputerowo. Nawet
przy zastosowaniu takich metod wi´k-
szoÊç leków przeciwrakowych stanie si´
dost´pna po co najmniej 10 latach, liczàc
od wyznaczenia celu molekularnego do
opracowania nowego specyfiku i uzy-
skania pozwolenia na jego u˝ywanie u
ludzi.
Najpierw potrzeba dwóch do trzech
lat badaƒ molekularnych, genetycznych
i cytologicznych, by stwierdziç, ˝e dany
cel molekularny jest rzeczywiÊcie istot-
ny w rozwoju nowotworu u cz∏owieka.
Potem do dwóch lat zajmujà biochemicz-
ne badania przesiewowe, zmierzajàce
do znalezienia obiecujàcych zwiàzków.
Po odkryciu dobrego kandydata na lek
chemicy medyczni modyfikujà go, aby
uzyskaç jego optymalnà moc, specyficz-
noÊç i w∏aÊciwoÊci farmakologiczne. Te
wysi∏ki na ogó∏ trwajà kolejne trzy do
pi´ciu lat i wymagajà syntezy setek do
kilku tysi´cy pokrewnych zwiàzków.
Gdy lek znajdzie si´ ju˝ w klinice, tra-
dycyjna trzyfazowa ocena jego bezpie-
czeƒstwa, skutecznoÊci i w∏aÊciwego
dawkowania zajmuje jeszcze nast´pne
trzy do pi´ciu lat.
Ta d∏ugotrwa∏a droga prowadzàca do
odkrycia i opracowania leku to niewe-
so∏a perspektywa zarówno dla naukow-
ców zajmujàcych si´ badaniami podsta-
wowymi, jak i dla onkologów klinicz-
nych. Jednak kilka leków specyficznie
ingerujàcych w molekularne mechani-
zmy raka zbli˝a si´ ju˝ do jej koƒca.
W tym roku wesz∏y do prób klinicznych
leki antysensowne, które hamujà kina-
zy bia∏kowe. Próby kliniczne inhibito-
rów transferazy farnezylu i kilku innych
inhibitorów kinaz powinny rozpoczàç
si´ w ciàgu najbli˝szych dwóch do czte-
rech lat. Terapia genowa natomiast po-
legajàca na zastàpieniu zmutowanych
Â
WIAT
N
AUKI
Listopad 1996 121
genów przez ich prawid∏owe odpowied-
niki nie jest mo˝liwa przed up∏ywem co
najmniej 10 lat.
Oprócz precyzji dorównujàcej wiàzce
lasera podejÊcie ukierunkowane na kon-
kretne czàsteczki mo˝e mieç inne zale-
ty. Z nie znanych jeszcze przyczyn ko-
mórki rakowe z wieloma defektami mo-
lekularnymi reagujà nawet wtedy, gdy
atakuje si´ tylko jeden z nich. Niewy-
kluczone wi´c, ˝e pacjent nie b´dzie mu-
sia∏ braç kilku leków jednoczeÊnie, aby
leczenie odnios∏o skutek.
Choç przeszkody sà trudne do poko-
nania, wszystko wskazuje na to, ˝e te-
rapie nowotworowe nowej generacji b´-
dà bardziej skuteczne i mniej szkodli-
we ni˝ obecne. Dzi´ki wyznaczeniu tak
wielu molekularnych celów ataku prze-
ciwnowotworowego szansa, ˝e nowe le-
ki oka˝à si´ bronià o ra˝àcej sile dzia∏a-
nia, wydaje si´ ca∏kiem realna.
T∏umaczy∏a
Ewa Bartnik
* Ostatnie prace sugerujà, ˝e uczynienie z E2F celu
terapii przeciwnowotworowych mo˝e nie byç re-
alne. Myszy pozbawione genu E2F-1 (w sumie ist-
nieje pi´ç ró˝nych genów dla czynnika E2F) wbrew
oczekiwaniom zapadajà na ró˝ne typy nowotwo-
rów, tak wi´c E2F-1 jest obecnie uwa˝any zarazem
za onkogen i za gen supresor nowotworów, w za-
le˝noÊci od konkretnego przypadku (przyp. t∏um.).
KOMÓRKA
KOMÓRKI GUZA
OBUMIERAJÑ
NORMALNE p53
AKTYWNE p53
USZKODZONY DNA
p53 POWODUJE
SAMOZAG¸AD¢ KOMÓRKI
KOMÓRKA
WIRUS ZAKA˚A KOMÓRKI
WIRUS NAMNA˚A SI¢ TYLKO
W KOMÓRKACH NOWOTWOROWYCH
NIENORMALNE p53
U˚YCIE WEKTORA WIRUSOWEGO
NIEAKTYWNE p53
KOMÓRKI GUZA
USZKODZONY DNA
KOMÓRKI GUZA
SI¢ DZIELÑ
BIA¸KO p53 sk∏ania komórk´ do samobójstwa, gdy jej DNA jest uszkodzony na przy-
k∏ad przez leki lub promieniowanie. Jednak jego nienormalna wersja cz´sto nie jest zdol-
na zapobiec podzia∏owi komórki z wadliwym DNA. Jednym ze sposobów leczenia raka mo-
˝e byç zastosowanie wirusów tak zmienionych genetycznie, aby namna˝a∏y si´ wy∏àcznie
w komórkach z nienormalnym bia∏kiem p53. Wirus rozprzestrzenia∏by si´ wi´c tylko w ko-
mórkach nowotworowych, co prowadzi∏oby do ich Êmierci.
Informacje o autorach
ALLEN OLIFF, JACKSON B. GIBBS i FRANK McCORMICK sà farmakologami,
którzy szukajà Êrodków przeciw nowotworom na poziomie molekularnym. Oliff
otrzyma∏ stopieƒ doktora nauk medycznych w Albert Einstein College of Medi-
cine w Bronx (stan Nowy Jork), jest dyrektorem ds. badaƒ nad rakiem w Merck
Research Laboratories w West Point (Pensylwania). By∏ cz∏onkiem kilku komite-
tów naukowych i doradczych, m.in. w National Cancer Institute. Gibbs jest naczel-
nym dyrektorem ds. badaƒ nad rakiem w firmie Merck i profesorem w Univer-
sity of Pennsylvania School of Medicine. Otrzyma∏ doktorat w University of
Virginia; jest cz∏onkiem licznych komitetów redakcyjnych i doradczych. McCor-
mick za∏o˝y∏ Onyx Pharmaceuticals w Richmond (Kalifornia) w 1992 roku i jest
w tej firmie szefem ds. naukowych. Uzyska∏ stopieƒ doktorski w University of
Cambridge i by∏ wiceprezesem ds. badaƒ naukowych w Cetus Corporation i Chi-
ron Corporation.
Literatura uzupe∏niajàca
MOLECULAR THEMES IN ONCOGENESIS
. J. Michael Bishop, Cell,
vol. 64, nr 2, ss. 235-248, 25 I 1991.
PROTEINS REGULATING RAS AND ITS RELATIVES
. Mark S. Bo-
guski i Frank McCormick, Nature, vol. 366, ss. 643-654,
16 XII 1993.
TUMOR SUPPRESSOR GENES
. P. W. Hinds i R. A. Weinberg,
Current Opinion in Genetics and Development, vol. 4, nr 1,
ss. 135-141, II/1994.
PHARMACEUTICAL RESEARCH IN MOLECULAR ONCOLOGY
. J. B.
Gibbs i A. Oliff, Cell, vol. 79, nr 2, ss. 193-198, 21 X 1994.
CELL CYCLE CONTROL AND CANCER.
Leland H. Hartwell i
Michael B. Kasten, Science, vol. 266, ss. 1821-1828,
16 XII 1994.
IAN WORPOLE
Wyszukiwarka
Podobne podstrony:
więcej podobnych podstron