
Encyclopedia of Polymer Sceince and Technology
Copyright c
2005 John Wiley & Sons, Inc. All rights reserved.
IONIC LIQUIDS, POLYMERIZATION IN
Introduction
The first room temperature ionic liquid (RTIL), [EtNH
3
][NO
3
] (mp 12
◦
C), was
reported in 1914 (1), and since then a great deal of research effort has been ex-
erted to exploit possible applications of these compounds. Initially, RTILs were
used mainly as electrolytes in batteries or for metal electrodeposition; however,
nowadays they are finding an ever increasing range of applications.
Ionic liquids may be viewed as molten salts and are liquids containing only
cations and anions at ambient or near ambient (
<100
◦
C) temperature. They ex-
hibit a relatively wide electrochemical window, good electronic and ionic conduc-
tivities, a broad range of room temperature liquid compositions, and may have
negligible vapor pressure, and excellent chemical, thermal, air and moisture sta-
bility (2–6). They are composed of weakly coordinating anions, for example, BF
4
−
and PF
6
−
and, hence, are highly polar yet noncoordinating solvents. Most im-
idazolium and pyridinium ionic liquids have polarities similar to those of small
molecule alcohols (7,8). Their hydrophilicity/lipophilicity is tunable by varying the
combination of cations and anions. Thus, RTILs have been referred to as “designer
solvents” (9), and millions of different RTILs could potentially be synthesized and
optimized for specific applications. Depending on their structures, RTILs are able
to dissolve a variety of organic, inorganic, and organometallic compounds. Their
ease of handling, low vapor pressures, and potential for recycling make them
promising potentially environmentally benign reaction media to replace volatile
molecular solvents in both the chemical industry and in academic research.
Furthermore, RTILs have limited miscibility with some common organic sol-
vents but high compatibility with transition metals. As a consequence, a biphasic
or phase-separable (organic/RTIL biphasic systems) catalysis concept can be de-
veloped in which a homogeneous catalyst is immobilized in one liquid phase (RTIL)
and the reactants and/or products reside largely in another liquid phase (organic)
(10), thus enabling easy product and catalyst separation with the retention of the
transition metal catalyst in an ionic liquid phase. Various common organic reac-
tions employing metal catalysts have been conducted by employing ionic liquids as
alternative reaction media, including Diels-Alder reactions (11), Friedel-Crafts re-
actions (12), hydrogenations (13), hydroformylations (14), alkylations (15), dimer-
izations (16), Heck reactions (17), Suzuki couplings (18), Sonogashira couplings
(19), ring-closing olefin metathesis reactions (20), alcohol oxidations (21), and nu-
cleophilic substitutions (22).
1
2
IONIC LIQUIDS, POLYMERIZATION IN
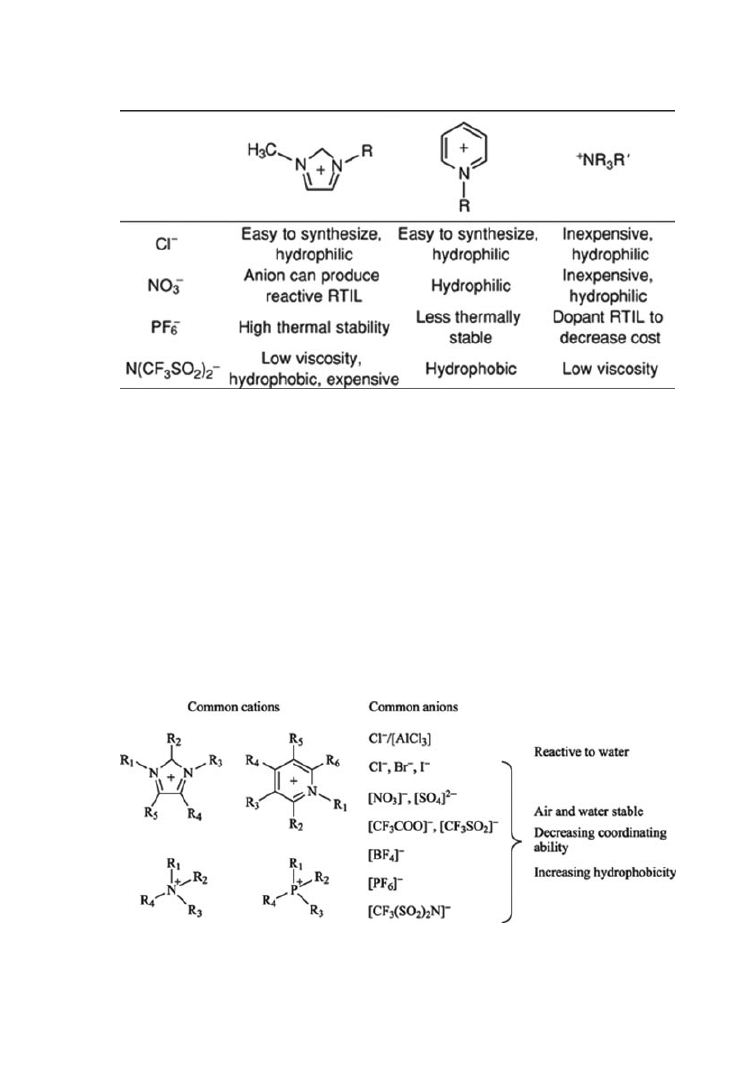
Fig. 1.
Matrix of RTIL cations and anions.
Some typical RTILs and how structure affects properties are shown in Fig-
ure 1. Representative common cations and anions used to prepare RTILs are
shown in Figure 2. RTILs are commercially available from ACROS Organic (Fisher
Scientific, UK), Chemada Fine Chemicals (Israel), Covalent Associates (Boston,
Mass.), C-TRI (Korea), CYTEC (Canada), Sigma-Aldrich (US), BASF (US), Merck
KGaA (Germany), QUILL (UK), Sachem (Austin, Tex.), Scionix (UK), and Solvent-
Innovation (Germany).
Ionic Liquids for Polymerization
One of the earliest examples of polymerization in RTILs was reported in 1990 in a
Lewis acidic ionic liquid: AlCl
3
-1-ethyl-3-methylimidazolium chloride [EMIM]Cl
Fig. 2.
Representative common cations and anions used to prepare room temperature
ionic liquids.

IONIC LIQUIDS, POLYMERIZATION IN
3
(23). It was found that the combination of TiCl
4
and AlEthylCl
2
in AlCl
3
/[EMIM]Cl
can be catalytically active for ethylene polymerization, although the yield of
polyethylene (PE) was low. This work opened a new route for making polymers
using RTILs as alternative reaction media. Later, replacing TiCl
4
with Cp
2
TiCl
2
was shown to give higher yields of PE using the same RTIL (24). More examples of
related work on using ionic liquids for polymerization or oligomerization of olefins
can be found in the literature and in patents (25,26). It was believed that the ac-
tive sites are cationic alkyl–metal complex generated by the interaction of added
transition metal catalyst with alkylaluminum moieties present in the ionic liq-
uids via halogen abstraction and alkylation. The electrochemical polymerization
of benzene in various ionic liquids has been described as a method for synthesis of
poly(p-phenylene) (27). In these studies, ionic liquids were used mainly as conve-
nient electrolytes. Carlin and Osteryoung produced a new electroactive material
by electrochemical oxidation of triphenylsilyl chloride (Ph
3
SiCl) in the acidic ionic
liquid (AlCl
3
-[EMIM]Cl) (28). The film formed exhibited reversible redox behav-
ior and was electronically conducting in the oxidized state by incorporation of the
cation of the ionic liquid into the film. However, most of the RTILs mentioned
above are the 1st generation ionic liquids, which are haloaluminate-based ionic
liquids and thus easily undergo hydrolysis to form undesirable HCl by-products.
Free Radical Polymerization
Free radical polymerization is extensively used for the synthesis of a variety of
polymeric materials due to its versatility, synthetic ease, and compatibility with
a wide variety of functional groups, which is also coupled with its tolerance to
water and protic media. However, conventional free radical polymerization has a
significant drawback, which is related to the reactivity of the propagating free rad-
ical chain ends and their propensity to undergo a variety of termination and chain
transfer reactions. The materials obtained are therefore polydisperse with limited
control over molecular weight and architecture. Ionic polymerizations (anionic or
cationic) were for many years the only “living” polymerization techniques avail-
able that achieved efficient control over the structure and architecture of vinyl
polymers. Although these techniques can generate polymers with low polydisper-
sity, controlled molecular weight, and control over chain ends and macromolecular
architectures, they are not suitable for the polymerization and copolymerization
of many vinylic monomers. This limitation is due to the incompatibility of the
growing polymer chain ends (carbanion or carbocation) with numerous functional
groups and certain monomer families. In addition, stringent reaction conditions,
including the use of ultrapure reagents and the need for total or near-total exclu-
sion of water and oxygen, limit the widespread commercial use of these techniques.
These challenges spurred polymer chemists to develop new concepts which allow
a living or controlled polymerization using special free radical polymerization
techniques. As a consequence, nitroxide-mediated polymerization (NMP), atom
transfer radical polymerization (ATRP), and radical addition-fragmentation and
transfer (RAFT) were developed. The general strategy behind these processes is
the reversible termination of the growing polymeric chain with a mediating rad-
ical to reduce the overall concentration of the propagating radical chain ends.

4
IONIC LIQUIDS, POLYMERIZATION IN
In the absence of other reactions leading to initiation of new polymer chains (ie,
no reaction of the mediating radical with the vinylic monomer), the concentra-
tion of reactive chain ends is extremely low, minimizing irreversible termination
reactions, such as combination or disproportionation. All chains would thus be
initiated only from the desired initiating species, and growth should occur in a
living fashion, allowing a high degree of control over the entire polymerization
process with well-defined polymers being obtained (29).
Although polymerization in moisture-sensitive ionic liquids has certain ad-
vantages when compared to traditional solvents, little polymerization work was
done in RTILs until the introduction of the 2nd generation of air, moisture, and
thermally stable, neutral ionic liquids, composed of dialkylimidazolium cations
and PF
6
or BF
4
anions. In spite of the fact that these ionic liquids are anticipated to
be potentially useful solvents for ionic polymerization due to their highly polar na-
ture, the first report of polymerization in the new nonhygroscopic ionic liquids was
based on a free radical polymerization process—atom transfer radical polymeriza-
tion (ATRP). Carmichael and co-workers (30) observed first-order kinetic behavior
over a wide range of reaction temperatures using ethyl-2-bromoisobutyrate ini-
tiator and CuBr/N-propyl-2-pyridylmethanimine as the catalyst pair in a 50/50
(V/V) MMA/[BMIM]PF
6
system. The reactions are faster as compared to those
in common solvents, while the polydispersities (M
w
/M
n
, where M
w
is weight-
average molecular weight and M
n
is number-average molecular weight) are nar-
row: 1.30–1.43. By extraction with toluene, the polymer can be separated from
the solution while copper catalyst remains in the ionic liquid phase, which fa-
cilitates catalyst re-use by adding fresh monomer. The use of the RTIL medium
also eliminated the need for postpurification to eliminate toxic copper salts, which
entails passing the polymer solution through purification columns. Carmichael
and co-workers attributed this polymerization behavior to the increased polarity
of ionic liquids, because a similar increase in the rate has been observed with
other polar/co-ordinating solvents. In a recent communication (31), Hong and co-
workers reported large increases in both the rate of polymerization and the molec-
ular weights that were obtained in the polymerizations of methyl methacrylate
(MMA) in [BMIM]PF
6
as compared to identical polymerizations carried out in con-
ventional organic solvents. These dramatic increases were believed to be at least in
part because of the high viscosity of the polymerization media. Thus, a “diffusion-
controlled termination” mechanism was proposed to explain the diminished chain
terminations in such a viscous system, which would account for a simultaneous in-
crease in both rate of polymerization and degree of polymerization. Very recently,
Benton and Brazel (32) reported similar behavior, high molecular weights and
rapid polymerization rates, for MMA in [BMIM]PF
6
. Harrison and co-workers
(33) used a pulse laser polymerization (PLP) technique to polymerize MMA in
[BMIM]PF
6
, which is the IUPAC-recommended standard procedure for measur-
ing the rate constant of propagation (k
p
). They found that both the propagation
and termination rates were significantly affected by [BMIM]PF
6
. They attributed
the increase of the propagation rate to the increased polarity of the ionic liquid so-
lution, which lowers the activation energy of propagation through charge-transfer
interactions. The termination rate is decreased simply because of the increased
viscosity of the polymerization medium. Both an increase of k
p
and decrease of
k
t
contribute a roughly ten-fold increase of overall rate of polymerization. Quite

IONIC LIQUIDS, POLYMERIZATION IN
5
recently, Cheng and co-workers (34) reported free radical polymerization of acry-
lonitrile in [BMIM]BF
4
using AIBN as initiator. These workers also note that ionic
liquids are excellent media for achieving high molecular weight polymers. They
attribute this result to low chain transfer constants for RTILs and the ability of
these solvents to stabilize the growing radical chain ends.
Clearly most of the early studies on polymerization in RTILs used
[BMIM]PF
6
as the medium. This was largely due to the commercial availability
and easy synthesis of this material. However, [BMIM]PF
6
can readily hydrolyze
to release toxic HF as a by-product, and there is an ever increasing focus on the
use of more benign RTILs in polymer systems. Zhang and co-workers (35) mea-
sured the polarity and viscosity of over a dozen RTILs based upon a wide range
of cations and anions, and attempted to correlate these physical data with the
low conversion conventional free radical polymerization behavior of styrene and
MMA in these RTILs. This study revealed no obvious trends between polarity
or viscosity of RTILs and polymerization rate or molecular weight of the formed
polymer. At around the same time, Strehmel and co-workers (36) reported that
the higher the viscosity of the RTIL, the higher the yields and molecular weights
of the polymers. However, unlike in the work by Zhang and co-workers, these
workers carried their polymerizations to high (near quantitative) conversions,
where the Trommsdorff effect strongly impacts polymerization behavior. Clearly,
the higher viscosities will affect polymerization behavior in this regime and make
it difficult to ascertain polymerization behavior in the non-Trommsdorf regime.
The RTILs employed in this latter study were composed of cations and anions of
similar structures except the alkyl length and positions on the cation, while either
cations or anions are structurally different in the former case. In our opinion, it is
still too early to make a conclusion regarding the effects of polarity and viscosity
of the RTIL on polymerization behavior. It would be very interesting to repeat the
experiments of Strehmel and co-workers (36) but keep conversions low. The polar-
ity of molecular solvents is a complex of many interactions including: H-bonding,
π-interaction, or van der Waals forces. In RTILs, the situation is even more com-
plicated since both cations and anions may have their own distinct interactions.
Zhang and co-workers used solvatochromic dye (Nile red) to measure the polari-
ties of RTILs and the results had similar trend as those in Reference (37). Since
Nile red is a positively solvatochromic, it preferentially interacts with anions of
RTILs. The apparent polarities deduced may be not reflect the real values, thus
further experiments need to be carried out to address this problem. In addition,
the polymers synthesized in RTILs all have similar glass transition temperatures
and microstructures as compared to those obtained in benzene or in bulk, based
upon thermal analysis and
13
C NMR experiments (38).
Biedro ´
n and co-workers (39) reported heterogeneous ATRP in [BMIM]PF
6
.
Alkyl acrylates (methyl, butyl, hexyl, and dodecyl) are either soluble, partly sol-
uble, or completely insoluble in this RTIL depending on the length of the alkyl
substituent. For the heterogeneous systems, the alkyl acrylate formed an upper
monomer phase while the CuBr/pentamethyldiethylenetriamine (PMDETA) cat-
alyst remained in the lower RTIL phase. Methyl acrylate (MA) and poly(methyl
acrylate) (PMA) are miscible with [BMIM]PF
6
and form a homogeneous polymer-
ization system, therefore, all reactions proceed in one phase. For the three other
acrylates, the growing macromolecular chains react with the monomer at the

6
IONIC LIQUIDS, POLYMERIZATION IN
interface but reside predominantly in the monomer phase. In all cases, the reac-
tions were living radical polymerizations. The advantages of heterogeneous ATRP
are easy separation of polyacrylate from the RTIL phase after reaction, with less
copper catalyst contamination and reduction of side reactions due to the absence
of catalyst in the upper monomer phase, as compared to bulk ATRP. It was found
that using a chiral RTIL (1-(R-(+)-2
-methylbutyl)-3-methylimidazolium hexaflu-
orophosphate) could impact the stereoregularity of polymer produced by ATRP
(40).
Sarbu and co-workers (41) used different catalyst systems (iron or copper
halides) to successfully carry out ATRP in a range of 1-butyl-3-methylimidazolium
ionic liquids. In iron-mediated ATRP, no additional ligand was required to achieve
a controlled polymerization of MMA although both initiation rates and rates of re-
action were low. Systems without organic ligands were effective only in a phospho-
nate ionic liquid for copper-mediated ATRP of MMA, while a ligand was required
in ionic liquids with halide or carbonate anions. ATRP in ionic liquids proceeds
with low initiation efficiency. This has been attributed to the high concentration
of the catalyst in the RTIL phase, into which the small initiators can easily diffuse
and thus generate high concentration of free radicals. The free radical can undergo
irreversible termination and cause the low initiation efficiency. However, initiator
efficiency can be improved by employing macroinitiators, which have little ten-
dency to diffuse into the RTIL phase. In addition, the catalyst can be regenerated
after removal of polymer and unreacted monomer.
Reversible atom transfer radical polymerization of MMA in [BMIM]PF
6
,
[BMIM]BF
4
, and [DMIM]BF
4
(1-dodecyl-3-methylimidazolium tetrafluoroborate)
has been described (42). In [BMIM]PF
6
, the reverse ATRP of MMA was achieved
with 2,2
-azobisisobutyronitrile (AIBN)/CuCl
2
/2,2
-bipyridine initiation system, in
which the system is homogeneous throughout the reaction. Due to a cage effect
with ionic liquids molecules, the termination of the primary radicals through de-
composition of AIBN might occur before they can initiate polymerization, which
accounts for the low initiation efficiency of AIBN. The Cu catalyst is also soluble
in the ionic liquid. As a consequence of both factors, less catalyst is needed to ef-
fectively mediate the polymerization process in ionic liquids than in other reverse
ATRPs. The ionic liquids and catalyst can be recovered and reused.
Zhang and co-workers (43) used both a bimolecular initiation system (ben-
zoyl peroxide (BPO) + 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO)) and a uni-
versal initiator system (2,2,5-trimethyl-3-(1-phenylethoxy)-4-phenyl-3-azahexane
(TMPPAH)) for the attempted nitroxide-mediated polymerization of MMA and
St. Polymers were produced but polymerizations were not living/controlled for ei-
ther monomer. Possible reasons are the low diffusion rates of mediating radicals
and slow degradation of free TEMPO at elevated temperatures in the presence of
[BMIM]PF
6
. Ryan and co-workers (44) demonstrated a controlled/living nitroxide-
mediated polymerization of MA in 50% V/V of [HMIM]PF
6
initiated by the initia-
tor pair AIBN + 4-oxo-2,2,6,6-tetramethyl-1-piperidinyl-N-oxyl (4-oxo-TEMPO) at
140
◦
C–155
◦
C. The reaction rates under these conditions were greater than for sim-
ilar reactions conducted in anisole. In both of these studies, self-polymerization
(thermal polymerization) was observed because of the high temperatures used.
Other kinds of living free radical polymerization, such as RAFT and charge trans-
fer polymerization, have also been reported in RTILs (45).
IONIC LIQUIDS, POLYMERIZATION IN
7
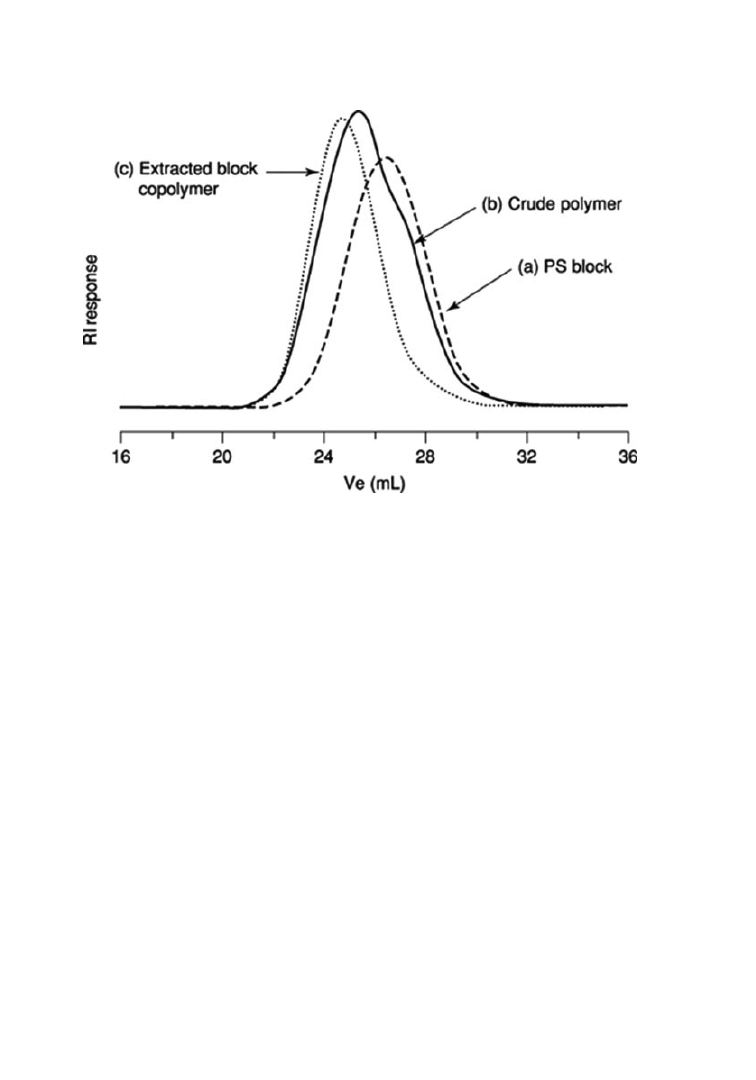
Fig. 3.
SEC traces of (a) PS block from BPO in [BMIM]PF6, (b) PS-b-PMMA before ex-
traction, and (c) PS-b-PMMA after extracting with cyclohexane and acetonitrile.
Combined with CuBr/PMDETA catalyst, dendritic polyarylether 2-
bromoisobutyrates (Gn-Br, n=1–3) can be used as effective macroinitiators for
ATRP of N-hexylmaleimide and styrene in [BMIM]PF
6
at lower temperature than
in anisole. The afforded polymers had well-defined molecular weights and low
polydispersities (1.18
< PDI < 1.36) (46). The two monomers showed a stronger
tendency to form alternating structures in RTILs as compared to polymerizations
conducted in anisole.
Limited solubility of some monomers and polymers in ionic liquids presents
opportunities to make diblock copolymers by sequential addition polymerization.
Zhang and co-workers (47) demonstrated the formation of PSt-b-PMMA by se-
quential addition in [BMIM]PF
6
through conventional free radical polymeriza-
tion using benzoyl peroxide (BPO) as initiator. St was polymerized first and the
polymer gradually precipitated out when the conversion reached around 50% due
to the insolubility of PSt in [BMIM]PF
6
. The chain coils wrapped the macrorad-
icals inside resulting in prolonged lifetimes because of diminished termination.
Unreacted St monomer was pumped out on a high vacuum line. After adding the
second monomer (MMA), diblock copolymers were formed at room temperature
although the re-initiation was not 100% (see Figure 3). Reversal of the polymeriza-
tion sequence only afforded homopolymer, PMMA. Kubisa and co-workers used a
similar strategy to produce PBA-b-PMA and PMA-b-PBA block copolymers (PBA
is poly(butyl acrylate) by different sequences of addition of monomers using the
ATRP method in the same ionic liquid (48). They found that when conversion of

8
IONIC LIQUIDS, POLYMERIZATION IN
MA, used as first monomer, exceeded 70%, a significant percentage of dead PMA
chains contaminated the final diblock copolymer. Conversely, when MA was added
to the solution of PBA in [BMIM]PF
6
, “clean” diblock was formed, essentially free
of homopolymer, and BA can be polymerized to complete conversion. Ma and co-
workers (42,49) also described successful synthesis of block copolymers, where
St was polymerized by chlorine-end-capped PMMA as macroinitiator through re-
verse ATRP in [BMIM]PF
6
, [BMIM]BF
4
, and [C
12
MIM]BF
4
.
The effect of solvents on reactivity ratios and sequence length distributions
for free radical polymerization has been extensively studied. The calculated re-
activity ratios of St and MMA (r
St
= 0.381 ± 0.02 and r
MMA
= 0.464 ± 0.02) in
[BMIM]PF
6
by nonlinear method (CONTOUR computer program (50)) are signif-
icantly different from those (r
St
= 0.54 ± 0.04 and r
MMA
= 0.50 ± 0.04) in benzene
at 60
◦
C (51). The “boot-strap” model (52), polarity of the solvents, interaction be-
tween solvent and monomers (eg solvent–monomer complex), viscosity and system
heterogeneity all possibly contribute to the different reactivity ratios in RTILs and
in benzene.
[BMIM]PF
6
was found to be an efficient plasticizer for PMMA, prepared
by in situ radical polymerization in this ionic liquid (53). The polymers have
physical characteristics comparable with those containing traditional plasticiz-
ers (phthalates, adipates, and trimellitates) and retain greater thermal stability.
RTILs are normally used as inert solvents for polymerization. However, Mays
and co-workers discovered that polymerization of MMA can be carried out at am-
bient temperature in the ionic liquid trihexyl-tetradecyl-phosphonium bis(2,4,4-
trimethylpentyl)phosphinate ([H
3
TDP] [(PM
3
)
2
P]) via a redox-initiated polymer-
ization mechanism (54). The cation of [H
3
TDP][(PM
3
)
2
P] apparently reacts with
BPO in a redox reaction, with the cation as reductant and BPO as oxidant. The
generated radical efficiently initiated polymerization at room temperature to pro-
duce PMMA with high yield and high molecular weight.
Ionic Polymerization
Living anionic polymerization usually gives narrower molecular weight distribu-
tion polymers as compared to those obtained by living free radical polymeriza-
tions. These polymerizations can proceed to complete conversion with far fewer
side reactions such as termination and chain transfer. The ability to introduce
terminal functional groups by using selective termination agents is an additional
advantage of anionic polymerization. On the other hand, the stringent purification
requirements for living anionic polymerization put some limits on the commercial
synthesis of polymers using this method. Even though RTILs are in some ways
ideal potential solvents for ionic polymerizations, because the RTIL can possibly
help to stabilize the growing carbanions or carbocations due to their ionic nature,
there are no reports in the literature on anionic polymerization in ionic liquids.
This is primarily because it is very difficult to adequately purify ionic liquids to
the standards required for anionic polymerizations. Our preliminary data (not yet
published) indicate that termination occurs due to impurities in the RTIL. Vija-
yaraghavan and co-workers (55) reported the first case of cationic polymerization
of styrene in N-butyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)amide

IONIC LIQUIDS, POLYMERIZATION IN
9
([P
14
][Tf
2
N]). The cationic initiator used was a Brønsted acid: bis(oxalato)boric
acid (HBOB). The polymerization proceeded in a living manner but the reaction
temperature was high (60
◦
C). PSt produced had low molecular weights (1300–
1700) and moderate polydispersities (1.3–1.5). The author suggested that RTILs
might promote the dissociation of the acid that can efficiently initiate polymer-
ization. Biedron and Kubisa (56) conducted cationic polymerization of styrene in
[BMIM]PF
6
with a 1-phenetyl chloride/TiCl
4
initiating system. Polymers were
obtained but chain transfer was significant, resulting in a lack of control over
molecular weight and molecular weight distribution.
Ring-Opening Polymerization
Ring-opening metathesis polymerization of norbornene using cationic al-
lenylidene
precatalys,
[(p-cymene)RuCl(Pcy
3
)( C C CPh
2
)][OTf],
in
a
biphasic
medium,
(1-butyl-2,3-dimethylimidazolium
hexafluorophosphate
([BDMIM]PF
6
)/toluene), was carried out (57). The catalyst was in the ionic liquid
phase, and the upper toluene phase dissolved the polymer that was formed. Both
the ionic liquid and catalyst could be reused up to six times without significant
loss of catalytic activity and with quantitative yields of polymer. After reloading
of catalyst, the catalyst system can still be re-used up to five times. The enhanced
recycling capabilities as compared to other alkylidene catalysts were attributed to
the ionic character of the cationic allenylidene complex, while the other systems
gradually lost their catalytic abilities within three successive runs.
Biedro ´
n and co-workers (58) presented the results of the cationic ring open-
ing polymerization of 3-ethyl-3-hydroxymethyloxetane in the most common hy-
drophilic ionic liquid, [BMIM]BF
4
, using the BF
3
-Et
2
O initiation system. The ad-
vantage of carrying out this reaction in ionic liquids over solution or bulk poly-
merizations is the high reaction temperature that can be used (up to 180
◦
C be-
cause intermolecular hydrogen bonding that leads to the formation of aggregates
is reduced at the elevated temperature). The intramolecular H-bonding, facilitat-
ing intramolecular chain transfer, is not significantly affected. The multihydroxyl,
branched structure was preserved, and the molecular weights of the polymers were
in the same range as those made in organic solvents or in bulk. On the contrary, the
application of ionic liquids can also help to reduce the reaction temperature. At
higher temperatures, side reactions may be significant. Polycarbonate made by
ring opening polymerization of ethylene carbonate experiences decarboxylation
when the reaction is carried out at 180–200
◦
C (59). In acidic ionic liquids such as
[BMIM]Cl-AlCl
3
or [BMIM]Cl-SnCl
2
, the reaction can take place at temperatures
of 100–120
◦
C depending on the ionic liquid used. The drawback of these new re-
action systems is that decarboxylation was still not negligible, and low levels of
ethylene carbonate polymers were formed.
Enzyme-Catalyzed Polymerization
Candida Antarctica lipase (lipase CA) catalyzed formation of biodegradable
polyesters in [BMIM]PF
6
and [BMIM]BF
4
has been reported (60). Ring-opening

10
IONIC LIQUIDS, POLYMERIZATION IN
polymerization of
ε-caprolactone and polycondensation of dicarboxylic acid di-
esters with 1,4-butanediol were examined. Higher molecular weight products and
improved conversions suggest the potential for green polymer chemistry by the
combination of nontoxic enzyme catalyst, mild reaction conditions with potentially
environmentally benign solvents.
Condensation Polymerization
Polyimides and polyamides are important materials due to their attractive ther-
mal, mechanical, and electrical properties. Usually, the synthesis of high molec-
ular weight polyimides in organic solvents requires high temperatures and the
presence of an acidic catalyst. Vygodskii and co-workers (61) studied the conden-
sation polymerization behavior in RTILs composed of different anions and cations.
High molecular weight polyimides and polyamides were obtained using RTILs as
novel reaction media, without addition of catalyst.
Transition-Metal Catalyzed Polymerization
Poly(phenyl-acetylene) (PPA) is a conjugated polymer with interesting photo-
conductivity, photoluminescence, nonlinear optical, and membrane properties. It
can be obtained by different polymerization methods including radical, cationic,
metathesis catalyst or Ziegler–Natta polymerizations. High molecular weight PPA
can also be obtained by Rh(I) catalyzed phenylacetylene polymerization in ionic
liquids such as n-butylpyridinim tetrafluoroborate ([bupy]BF
4
) or [BMIM]PF
4
(62). The catalyst used was either (diene)Rh(acac) or [(diene)RhCl
2
]
2
, and the
cocatalyst used was triethylamine. The polymer was separated from the ionic
liquid using either extraction with toluene or filtration by adding methanol into
ionic liquids to form a suspension of PPA in the solvent mixture methanol/RTIL.
Extraction usually facilitates catalyst recycling, but cannot permit the maximum
product recovery. While more polymers can be recovered through the filtration
method, the Rh(I) complex solution after filtration shows no catalytic activity. The
molecular weights ranged from 55,000 to 200,000.
Rogers
and
co-workers
(63)
used
1-hexylpyridinium
bis(trifluoromethanesulfonyl) imide ([C
6
Pyr][NTf
2
])/methanol as a solvent
pair for palladium-catalyzed alternating copolymerization of styrene and carbon
monoxide, in which the palladium catalyst was LPd(OAc)
2
(L=2,2
-bipyridine
and 1,10-phenanthroline). [C
6
Pyr][NTf
2
] and catalyst can be recycled, and the
yields and molecular weights were higher as compared to those obtained when
the polymerization was carried out in methanol alone. Furthermore, the catalyst
stability and propagation rate were improved due to the inhibited chain transfer
and catalyst decomposition in [C
6
Pyr][NTf
2
].
Electrochemical Polymerization
Electrochemical polymerization is a major method to synthesize conducting poly-
mer for potential application in energy storage devices, electrochromic devices,

IONIC LIQUIDS, POLYMERIZATION IN
11
and light-emitting diodes. This process shows some advantages over chemical
synthesis including faster reactions, simple procedures, generation of the poly-
mer in the doped state, and easy control of the film thickness. Naudin and co-
workers (64) have used ionic liquids for the electrochemical polymerization of
poly(3-(4-fluorophenyl)thiophene) (PFPT). The electrochemical behavior was sim-
ilar to that in common nonaqueous electrolyte. However, X-ray photoluminescence
spectroscopy revealed the presence of some ionic liquid residue in the formed film.
Other kinds of conjugated polymers such as polypyrrole, polyaniline, and unsub-
stituted polythiophene were also synthesized in ionic liquid (65).
Other Applications of Ionic Liquids in Polymers Systems
Polyaniline nanoparticles with diameters from about 30–80 nm can be fabri-
cated by interfacial polymerization at the interface between aqueous media and
RTILs (66). Electronic devices such as electrochemical mechanic actuators have
improved cycle life and larger generated strain using [BMIM]PF
6
, [BMIM]BF
4
, or
[BMIM][NTf
2
] as electrolytes as compared to traditional organic or aqueous sol-
vents, because ionic liquids have high ion conductivity, large electrochemical win-
dows, and fast ion mobility (67). Single-ion conductive membrane material, made
out of polymerizable ionic liquids by incorporating some ethylene oxide spacer
cross-linkers, has high ionic conductivity (1.37
×10
− 4
S cm
− 1
), and the film formed
is transparent and free standing (68). In order to overcome migration of ions of
RTILs along with potential gradient with target ions in membrane application,
poly(VdF-co-HFP) containing zwitterionic ionic liquids was reported as a new poly-
mer gel electrolyte which only transports target ions (69). RTILs have been used
as porogens in cross-linked polymer systems formed by free radical polymerization
of cross-linking monomers (70). RTILs are also effective solvents for regenerating
cellulose via the aid of microwave irradiation (71). Forming hydrogen-bonding be-
tween hydroxyl functions on the polymer chains and anions of RTILs such as Cl
−
,
Br
−
, and SCN
−
, which are hydrogen bond acceptors, can break the intermolecu-
lar hydrogen bonding between linear glucose polymer chains. A recent review on
application of ionic liquids in polymerization is available in the literature (72).
Conclusions
Room temperature ionic liquids have proved over recent years to be useful and
unique reaction media for a variety of chemical reactions. Some potential benefits
of working with RTILs as solvents for polymerization are wide and varied (Table
1):
Faster reaction rates and better selectivities are often observed in RTILs as
compared to those in common organic solvents. In addition to these advantages,
the nonvolatility, nonflammability, and recycling potential also help RTILs be-
come a preferred alternative reaction medium to meet environmental and other
requirements. Although polymerization in 1st generation RTILs was not very at-
tractive when compared to similar reactions carried out in organic solvents, the
2nd generation RTILs are showing much greater promise. This is evidenced by

12
IONIC LIQUIDS, POLYMERIZATION IN
Table 1. Potential Benefits of Polymerization in RTILs
• Ability to rapidly generate high molecular weight polymers with low residual monomer
• Simplified methods to synthesize complex molecular architectures such as block- and
graft copolymers, and copolymers with new monomer sequences, as compared with
more complicated techniques such as anionic, cationic and living radical polymerization
• Ability to design reactions to be carried out at much higher temperatures, as RTIL
volatility is much lower and thermal stability is much higher than traditional solvent
• Improvement in polymer physical properties, such as tensile strength, elastic modulus
and impact strength, based on the reaction mechanism in RTILs
• Ability to work with biological components in a nondenaturing environment during
reactions that incorporate enzymes, proteins, and other biologically active agents into
polymer networks.
the increasing number of papers published in this area every year, and interest-
ing and important discoveries. When conventional free radical polymerizations
are carried out in RTILs, there is an increase in the propagation rate due to the
high polarity of the RTIL and a decrease in the termination rate because of the
high viscosity of the RTILs, and thus the overall reaction rate is greatly increased.
For living/controlled polymerization such as ATRP, the reactions in RTILs are still
controllable with faster reaction rates and easy separation of catalysts and/or lig-
ands from the resulting polymers. Several other polymerization processes have
been conducted in RTILs, with the notable exception of anionic polymerization,
which needs careful work in order to find the right RTILs, purification techniques,
and polymerization conditions.
BIBLIOGRAPHY
1. P. Walden, Bull. Acad. Imper. Sci. (St. Petersburg) 1800 (1914).
2. C. L. Hussey, Pure Appl. Chem. 60, 1763–1772 (1988).
3. K. R. Seddon, in G. Mamantov and R. Marassi, eds., Molten Salt Chemistry, Reidel
Publishing Co., Dordrecht, The Netherlands, 1987, p. 365.
4. J. S. Wilkes, J. A. Levisky, R. A. Wilson, and C. L. Hussey, Inorg. Chem. 21, 1263–1264
(1982).
5. C. L. Hussey, in G. Mamantov and C. Mamantov, eds., Advances in Molten Salts Chem-
istry, Vol. 5, Elsevier Science Publishing Co., New York, 1983, pp. 185–230.
6. (a) M. K. Dieter, C. J. Dymek Jr., N. E. Heimer, J. W. Rovang, and J. S. Wilkes, J. Am.
Chem. Soc. 110, 2722 (1988);
(b) J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc.,
Chem. Commun. 965–967 (1992); (c) H. L. Ngo, K. LeCompte, L. Hargens, and A. B.
McEwen, Thermochim. Acta 357–358, 97–102 (2000).
7. S. N. V. K. Aki, J. F. Brennecke, and A. Samanta, Chem. Commun. 413 (2001).
8. H. Zhang, K. Hong, and J. W. Mays, in Abstract of Papers, ACS 227th National Meeting,
Anaheim, CA, USA, 2004.
9. M. Freemantle, Chem. Eng. News 32 (Mar. 30, 1998); 37 (May 15, 2000); 21 (Jan. 1,
2001); 26 (May 3, 2004).
10. B. Cornils and W. A. Herrmann, in B. Cornils and W. A. Herrmann, eds., Applied
Homogeneous Catalysis with Organometallic Compounds, Weinheim, New York, 1996,
Chap. 4.1, p. 1167.

IONIC LIQUIDS, POLYMERIZATION IN
13
11. T. Fischer, A. Sethi, T. Welton, and J. Woolf, Tetrahedron Lett. 40, 793 (1999).
12. A. Stark, B. L. MacLean, and R. D. Singer, J. Chem. Soc., Dalton Trans. 63 (1999).
13. Y. Chauvin, L. Mussmann, and H. Olivier, Angew. Chem. Int. Ed. 34, 2698 (1995).
14. C. P. Mehnert, R. A. Cook, N. C. Dispenziere, and M. Afeworki, J. Am. Chem. Soc. 124,
12932 (2002).
15. M. Badri, J. J. Brunet, and R. Perron, Tetrahedron Lett. 33, 4435 (1992).
16. (a) J. E. L. Dullius, P. A. Z. Suarez, S. Einloft, R. F. de Souza, J. Dupont, J. Fischer, and
A. De Cian, Organometallics 17, 815 (1988); (b) S. M. P. Silva, A. Z. Suarez, R. F. de
Souza, and J. Dupont, Polym. Bull. 40, 401 (1998).
17. D. E. Kaufmann, M. Nouroozian, and H. Henze, Syn. Lett. 1091 (1996).
18. (a) C. J. Mathews, P. J. Smith, and T. Welton, Chem. Commun. 1249 (2000);
(b) R.
Rajagopal, D. V. Jarikote, and K. V. Srinivasan, Chem. Commun. 616 (2002);
(c)
J. McNulty, A. Capretta, J. Wilson, J. Dyck, G. Adjabeng, and A. Robertson, Chem.
Commun. 1986 (2002).
19. (a) S. B. Park, H. Alper, Chem. Commun. 1306 (2004); (b) T. Fukuyama, M. Shinmen,
S. Nishitani, M. Sato, and I. Ryu, Org. Lett. 4, 1691 (2002).
20. (a) Q. Yao and Y. Zhang, Angew. Chem. Int. Ed. 42, 3395 (2003);
(b) N. Audic, H.
Clavier, M. Mauduit, J. C. Guillemin, J. Am. Chem. Soc. 125, 9248 (2003).
21. I. A. Ansari and R. Gree, Org. Lett. 4, 1507 (2002).
22. D. Kim and D. Chi, Angew. Chem. Int. Ed. 43, 483 (2004).
23. R. T. Carlin, R. A. Osteryoung, J. S. Wilkes, and J. Rovang, Inorg. Chem. 29, 3003
(1990).
24. R. T. Carlin and J. S. Wilkes, J. Mol. Catal. 63, 125 (1990).
25. (a) M. F. Pinheiro, R. S. Mauler, and R. F. de Souza, Macromol. Rapid Commun. 22,
425 (2001);
(b) P. Wasserscheid, C. M. Gordon, C. Hilgers, M. J. Muldoon, and I. R.
Dunkin, Chem. Commun. 1186 (2001); (c) O. Stenzel, R. Br ¨
ull, U. M. Wahner, R. D.
Sanderson, and H. G. Raubenheimer, J. Mol. Catal. A: Chem. 192, 217 (2003).
26. (a) EP 558187 (1993), P. W. Ambler, P. K. G. Hodgson, and N. J. Stewart; (b) WO
9521871 (1995), A. A. K. Abdul-Sada, P. W. Ambler, P. K. G. Hodgson, K. R. Seddon,
and N. J. Stewart; (c) EP 791643 (1997), P. M. Atkins, M. R. Smith, and B. Ellis; (d)
WO 0032658 (2000), V. Murphy.
27. (a) V. M. Kobryanskii and S. A. Arnautov, J. Chem. Soc., Chem. Commun. 9, 727 (1992);
(b) V. M. Kobryanskii and S. A. Arnautov, Makromol. Chem. 193, 455 (1992); (c) D. C.
Trivedi, J. Chem. Soc., Chem. Commun. 544 (1989); (d) S. A. Arnautov, Synth. Met.
84, 295 (1997); (e) L. M. Goldenberg, R. A. Osteryoung, Synth. Met. 64, 63 (1994).
28. R. T. Carlin and R. A. Osteryoung, J Electrochem. Soc. 141, 1709 (1994).
29. (a) C. J. Hawker, A. W. Bosman, and E. Harth, Chem. Rev. 101, 3661 (2001);
(b) M.
Kamigaito, T. Ando, and M. Sawamoto, Chem. Rev. 101, 3689 (2001);
(c) K. Maty-
jaszewski and J. Xia, Chem. Rev. 101, 2921 (2001).
30. A. J. Carmicheal, D. M. Haddleton, S. A. F. Bon, and K. R. Seddon, Chem. Commun.
1237 (2000).
31. (a) K. Hong, H. Zhang, J. W. Mays, A. N. Visser, C. S. Brazel, J. D. Holbrey, W. M.
Reichert, and R. D. Rogers, Chem. Commun. 1368 (2002);
32. (b) M. G. Benton and C. S. Brazel, Polym. Int. 53, 1113 (2004).
33. (a) S. Harrison, S. R. MacKenzie, and D. M. Haddleton, Chem. Commun. 2850 (2002);
(b) S. Harrison, S. R. MacKenzie, and D. M. Haddleton, Macromolecule 36, 5072 (2003).
34. A. L. Cheng, Y. Zhang, T. Zhao, and H. Wang, Macromol. Symp. 216, 9 (2004).
35. H. Zhang, K. Hong, and J. W. Mays, in R. Rogers and C. S. Brazel, eds., Ionic Liquids
in Polymer System, ACS Symposium Series, Anaheim CA, Spring, 2004, in press.
36. V. Strehmel, H. Kraudelt, H. Wetzel, E. G¨ornitz, and A. Laschewsky, in R. Rogers and
C. S. Brazel, eds., Ionic Liquids in Polymer System, ACS Symposium Series, Anaheim
CA, Spring, 2004, in press.

14
IONIC LIQUIDS, POLYMERIZATION IN
37. (a) J. F. Deye, T. A. Berger, and A. G. Anderson, Anal. Chem. 62, 615 (1990);
(b) A.
J. Carmicheal and K. R. Seddon, J. Phys. Org. Chem. 13, 591 (2000); (c) S. V. Dzyuba
and R. A. Bartsch, Tetrahedron Lett. 43, 4657 (2002).
38. H. Zhang, L. Bu, M. Li, K. Hong, A. E. Visser, R. D. Rogers, and J. W. Mays, in R. D.
Rogers and K. R. Seddon, eds., Ionic Liquids: Industrial Application for Green Chem-
istry, ACS Symposium, 2001.
39. T. Biedro ´
n and P. Kubisa, Macromol. Rapid Commun. 22, 1237 (2001).
40. T. Biedro ´
n and P. Kubisa, Polym. Int. 52, 1584 (2003).
41. T. Sarbu and K. Matyjaszewski, Macromol. Chem. Phys. 202, 3379 (2001).
42. (a) H. Ma, X. Wan, X. Chen, and Q. Zhou, J. Polym. Sci.. Part A: Polym. Chem. 41, 143
(2003); (b) H. Ma, X. Wan, X. Chen, and Q. Zhou, Polymer 44, 5311 (2003).
43. H. Zhang, K. Hong, and J. W. Mays, Polym. Bull. 52, 9 (2004).
44. J. Ryan, F. Aldabbagh, P. B. Zetterlund, and B. Yamada, Macromol. Rapid Commun.
25, 930 (2004).
45. (a) S. Perrier, T. P. Davis, A. J. Carmichael, and D. M. Haddleton, Chem Commun. 2226
(2002); (b) S. Perrier, T. P. Davis, A. J. Carmichael, and D. M. Haddleton, Euro. Polym.
J. 39, 417 (2003); (c) R. Vijayaraghavan and D. R. MacFarlane, Aust. J. Chem. 57, 129
(2004).
46. Y. Zhao, J. Zhang, J. Jiang, C. Chen, and F. Xi, J. Polym. Sci., Part A: Polym. Chem. 40,
3360 (2002).
47. H. Zhang, K. Hong, and J. W. Mays, Macromolecules 35, 5738 (2002).
48. T. Biedro ´
n and P. Kubisa, J Polym. Sci., Part A: Polym. Chem. 40, 2799 (2002).
49. (a) H. Ma, X. Wan, X. Chen, and Q. Zhou, J Polym. Sci., Part A: Polym. Chem. 41, 143
(2003); (b) H. Ma, X. Wan, X. Chen, and Q. Zhou, Polymer 44, 5311 (2003).
50. A. M. van Herk, J. Chem. Educ. 72, 138 (1995).
51. H. Zhang, K. Hong, M. Jablonsky, and J. W. Mays, Chem Commun. 1356 (2003).
52. H. Harwood, J. Macromol. Sci. Macromol Symp. 10/11, 331 (1987).
53. M. P. Scott, C. S. Brazel, M. G. Benton, J. W. Mays, J. D. Holbrey, and R. D. Rogers,
Chem. Commun. 1370 (2002).
54. H. Zhang and J. W. Mays, manuscript in preparation (2005).
55. R. Vijayaraghavan, and D. R. MacFarlane, Chem. Commun. 700 (2004).
56. T. Biedro ´
n and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem. 42, 3230 (2004).
57. S. Csihony, C. Fischmeister, C. Bruneau, I. T. Horv ´ath, and P. H. Dixneuf, New J. Chem.
26, 1667 (2002).
58. T. Biedro ´
n, M. Bednarek, and P. Kubisa, Macromol. Rapid Commun. 25, 878 (2004).
59. J. Kadokawa, Y. Iwasaki, and H. Tagaya, Macromol. Rapid Commun. 23, 757 (2002).
60. H. Uyama, T. Takamoto, and S. Kobayashi, Polym. J. 34, 94 (2002).
61. Y. S. Vygodskii, E. I. Lozinskaya, and A. S. Shaplov, Macromol. Rapid Commun. 23,
676 (2002).
62. P. Mastrorilli, C. F. Nobile, V. Gallo, G. P. Suranna, and G. Farinola, J. Mol. Catal. A:
Chem. 184, 73 (2002).
63. M. A. Klingshirn, G. A. Broker, J. D. Holbrey, K. H. Shaughnessy, and R. D. Rogers,
Chem Commun. 1394 (2002).
64. E. Naudin, H. A. Ho, S. Branchaud, L. Breau, D. B´elanger, J. Phys. Chem. B 106, 10585
(2002).
65. (a) K. Sekiguchi, M. Atobe, and T. Fuchigami, J. Electroanal. Chem. 557, 1 (2003); (b)
J. M. Pringle, J. Efthimiadis, P. C. Howlett, J. Efthimiadis, D. R. MacFarlane, A. B.
Chaplin, S. B. Hall, D. L. Officer, G. G. Wallace, and M. Forsyth, Polymer 45, 1447
(2004); (c) J. H. Shi, C. H. Yang, G. Y. Gao, and Y. F. Li, Chin. J. Chem. Phys. 17, 503
(2004).
66. H. Gao, T. Jiang, B. Han, Y. Wang, J. Du, Z. Liu, and J. Zhang, Polymer 45, 3017
(2004).

IONIC LIQUIDS, POLYMERIZATION IN
15
67. (a) J. Ding, D. Zhou, G. Spinks, G. Wallace, S. Forsyth, M. Forsyth, and D. MacFarlane,
Chem. Mater. 15, 2392 (2003); (b) W. Lu, A. G. Fadeev, B. Qi, E. Smela, B. R. Mattes,
J. Ding, G. Spinks, J. Mazurkiewics, D. Zhou, G. G. Wallace, D. R. MacFarlane, S. A.
Forsyth, and M. Forsyth, Science 297, 983 (2002).
68. S. Washiro, M. Yoshizawa, H. Nakajima, and H. Ohno, Polymer 45, 1577 (2004).
69. H. Ohno, M. Yoshizawa, and W. Ogihara, Electrochim. Acta. 48, 2079 (2003).
70. P. Snedden, A. I. Cooper, K. Scott, and N. Winterton, Macromolecule 36, 4549 (2003).
71. R. P. Swatloski, S. K. Spear, J. D. Holbrey, and R. D. Rogers, J. Am. Chem. Soc. 124,
4974 (2002).
72. P. Kubisa, Prog. Polym. Sci. 29, 3 (2004).
H
ONGWEI
Z
HANG
University of Tennessee
J
IMMY
W. M
AYS
Oak Ridge National Laboratory
Wyszukiwarka
Podobne podstrony:
Ionic Liquids, Polymerization in
Ionic liquids as solvents for polymerization processes Progress and challenges Progress in Polymer
Ionic liquids solvent propert Journal of Physical Organic Che
Density and viscosity of several pure and water saturated ionic liquids Green Chemistry
Ionic liquids perspectives for organic and catalytic rea~E90
04 Laws of Microactuators and Potential Applications of Electroactive Polymers in MEMS
Degradable Polymers and Plastics in Landfill Sites
Polymer supported catalysis in synthetic organic chemistry
Composition and Distribution of Extracellular Polymeric Substances in Aerobic Flocs and Granular Slu
GLOSSARY OF BASIC TERMS IN POLYMER SCIENCE
Liquidity In Forex Markets
Liquid Crystalline Polymers, Main Chain
Method Development in High Performance Liquid Chromatography
więcej podobnych podstron