
Journal of Neuro-Oncology 51: 231–243, 2001.
© 2001 Kluwer Academic Publishers. Printed in the Netherlands.
The role of p53 in human cancer
David Malkin
Division of Hematology/Oncology, Department of Pediatrics, The Hospital for Sick Children, University of
Toronto, Ontario, Canada
Key words: p53, tumor suppressor gene, apoptosis, growth arrest, DNA repair
Summary
In the two decades since its original discovery, p53 has found a singularly prominent place in our understanding
of human cancer. Although the biochemistry of p53 has been worked out in some detail, our knowledge of the
biologic consequences of p53 dysfunction is still quite rudimentary. Over the next several years, it will be important
to determine how best to harness the complex properties of p53’s ability to induce cellular growth arrest and cell
death to generate novel, effective approaches to cancer therapy. Furthermore, a clearer appreciation of the direct
interaction of epigenetic factors with p53 will lead to development of strategies to inhibit tumour initiation and
progression. The next decade promises to offer exciting opportunities to apply our vast knowledge of this intriguing
tumor suppressor to clinical advantage.
With over 13,000 published manuscripts to its name,
the p53 tumor suppressor gene and its correspond-
ing protein have become the most intensely studied
molecules in cancer research since their original dis-
covery in 1979 [1,2]. p53 has received the monikers
‘guardian of the genome’ and ‘gatekeeper of the cell’
for its role in preventing the accumulation of genetic
alterations through the regulation of critical check-
points in response to distinct exogenous stresses. The
p53 protein is a transcription factor that is stabilized and
activated in response to a number of stimuli including
exposure of cells to DNA damaging agents, hypoxia,
growth factors, or activated oncogenes. Activation of
p53 allows it to function as a tumor suppressor through
a number of growth controlling endpoints. The most
widely studied downstream effects of p53 activation
include growth arrest and apoptosis, although senes-
cence, differentiation, and anti-angiogenesis have also
been implicated in p53 activation. This review will
discuss the biology and biochemistry of p53 and the
molecular pathway that it controls in the context of
human neoplasia. The reader is also referred to numer-
ous extensive review articles and web sites that focus
on specific aspects of this intriguing gene [3–18]. The
eventual elucidation of the physiology of p53 will be
critical to our understanding of the complex nature of
human cancer.
p53 tumor suppressor gene structure
Alterations of the p53 tumour suppressor gene or
its encoded protein are the most frequently observed
somatic genetic events in human cancer [18]. Germline
p53 alterations are found in the majority of fami-
lies with Li–Fraumeni syndrome (LFS), an autoso-
mal dominantly inherited disorder characterized by
the occurrence of early-onset breast cancer, sarcomas
and other neoplasms (see below) [19–22]. The human
p53 gene, located on chromosome 17p13, encodes
a 53 kDa nuclear phosphoprotein. The nucleotide
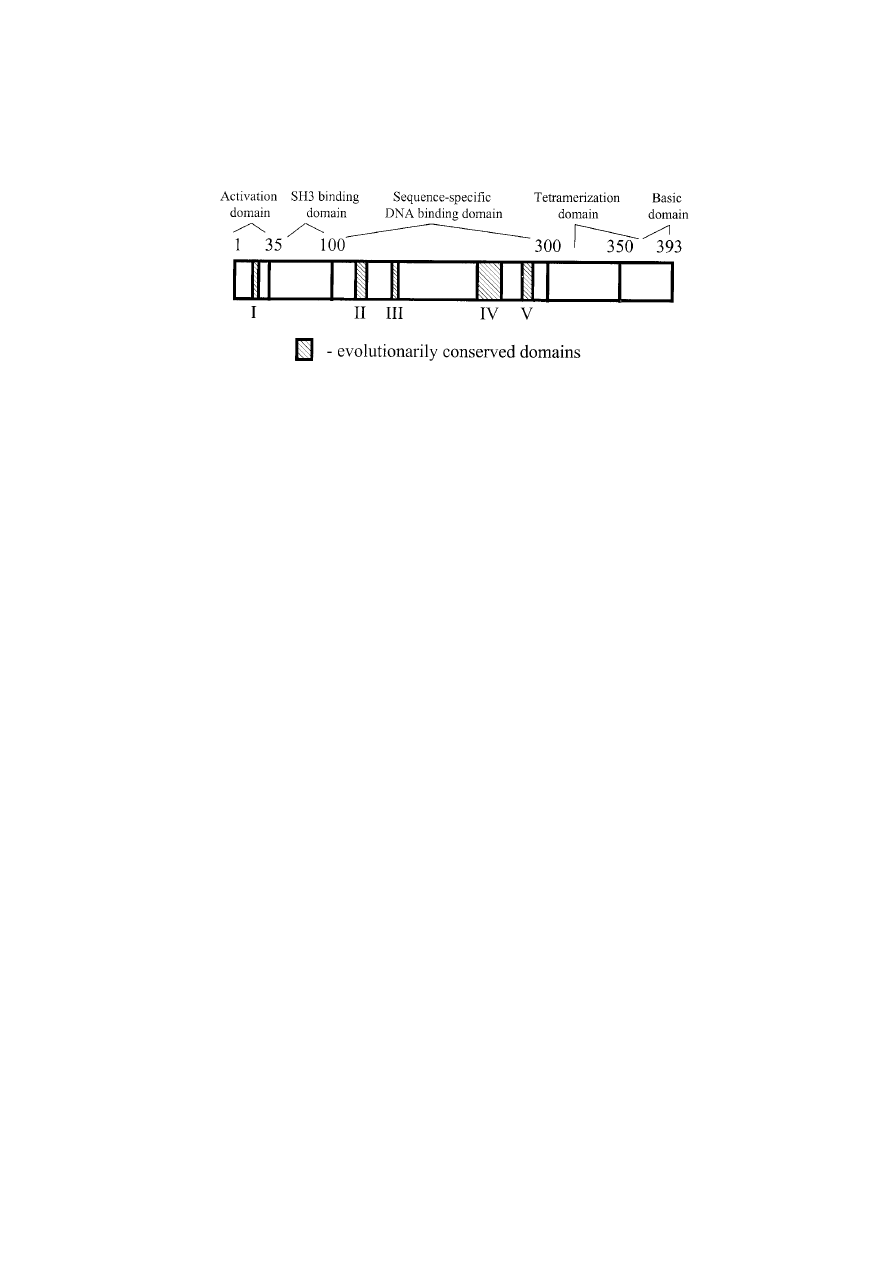
sequence predicts 393 amino acids that encode five
conserved domains [23] that are essential for nor-
mal p53 growth-suppressing functions and encom-
pass the most frequently mutated codons [24,25].
Through cross-species comparisons of the amino
acid sequences of p53 proteins, these conserved
domains have been shown to encompass residues
13–23, 117–142, 171–181, 234–250, and 270–286 [17]
(Figure 1).
The human p53 protein has been divided structurally
and functionally into four domains. The first 42 amino
acids at the N-terminus constitute the transcriptional
activation domain. This region interacts with the basal
transcriptional machinery of a cell in positively regulat-
ing expression of other growth regulatory genes [26].
232
Figure 1. Basic structure of the p53 protein. It encodes 393 amino acids. Five highly conserved domains (I–V) are represented by the
hatched boxes, and confer distinct functions as described in the text. Interactions of other molecules with p53 are critical to the regulation
of p53 function.
p53 transcriptional activation is negatively regulated by
the adenovirus E1B-55 kDa protein, the large T antigen
of SV40 polyomavirus and the E6 protein of human
papillomavirus, and the human Mdm2 protein, all of
which bind to the N-terminus of p53 [27].
The sequence-specific DNA binding domain of p53
is found between amino acids 102 and 292, and rec-
ognizes and binds to consensus target sequences. The
DNA-binding domain harbors four of the five highly
conserved regions, and it is also within this domain that
80–90% of p53 mutations have been identified [28].
The p53 protein forms a tetramer in solution; the
C-terminus of the protein is responsible for the for-
mation of this structure. The oligomerization domain
is contained within amino acids 323–356. Adjacent to
the oligomerization domain is a region (amino acids
363–393) referred to as a transcriptional regulatory
domain that regulates sequence specific DNA binding
and a DNA damage recognition domain [29]. Three
nuclear localization signals are scattered throughout
the C-terminal region of p53 [30]. Disruption of any
of these regions commonly leads to inactivation of p53
function and malignant transformation of a cell.
p53 function
In normal mammalian cells, p53 is expressed at
extremely low levels because the protein is rapidly
degraded following synthesis, exhibiting a half-life of
20–30 min [31]. p53 is targeted for degradation by a
proteosome complex following ubiquitination. Mdm2
participates in the regulation of the stability of p53 by
helping to mediate this degradation. Mdm2 can interact
with p53 in undamaged cells and target it for ubiquitin-
mediated degradation [32]. Mdm2 also binds to the
p53 protein and inhibits the ability of p53 to act as a
transcription factor. Mdm2 binds to the N-terminus of
p53, within the transactivation domain where p53, as a
transcription factor, contacts other components of the
basal transcriptional machinery. The binding of Mdm2
inhibits normal function of this region of p53, reduc-
ing the ability of p53 to activate gene expression [33].
The promoter of the mdm2 gene contains a p53 bind-
ing site and is transcribed in a p53-dependent manner
[34]. This has led to a model in which p53 up-regulates
the Mdm2 protein, therefore providing a negative reg-
ulatory feedback loop for p53 activity. The control that
Mdm2 exerts over p53 is essential for normal develop-
ment. This has been demonstrated by embryonic lethal-
ity in Mdm2-deficient mice that can be rescued by the
simultaneous deletion of p53 [35] (see Mouse Models
below).
Numerous cellular functions have been attributed to
p53 and have been extensively reviewed in the litera-
ture [17,36]. p53 acts either as a DNA-binding depen-
dent transcriptional activator or as a DNA-binding
independent repressor of several downstream targets
involved in cell growth, differentiation and prolifera-
tion. DNA damaging agents such as
γ -radiation and
certain chemotherapeutic drugs, including adriamycin
and cisplatin, cause rapid nuclear accumulation of
wild-type p53. Hypoxia, changes in pH, nucleo-
side pool depletion, antioxidants, cold shock, heat
shock, and other stresses induce p53 accumulation
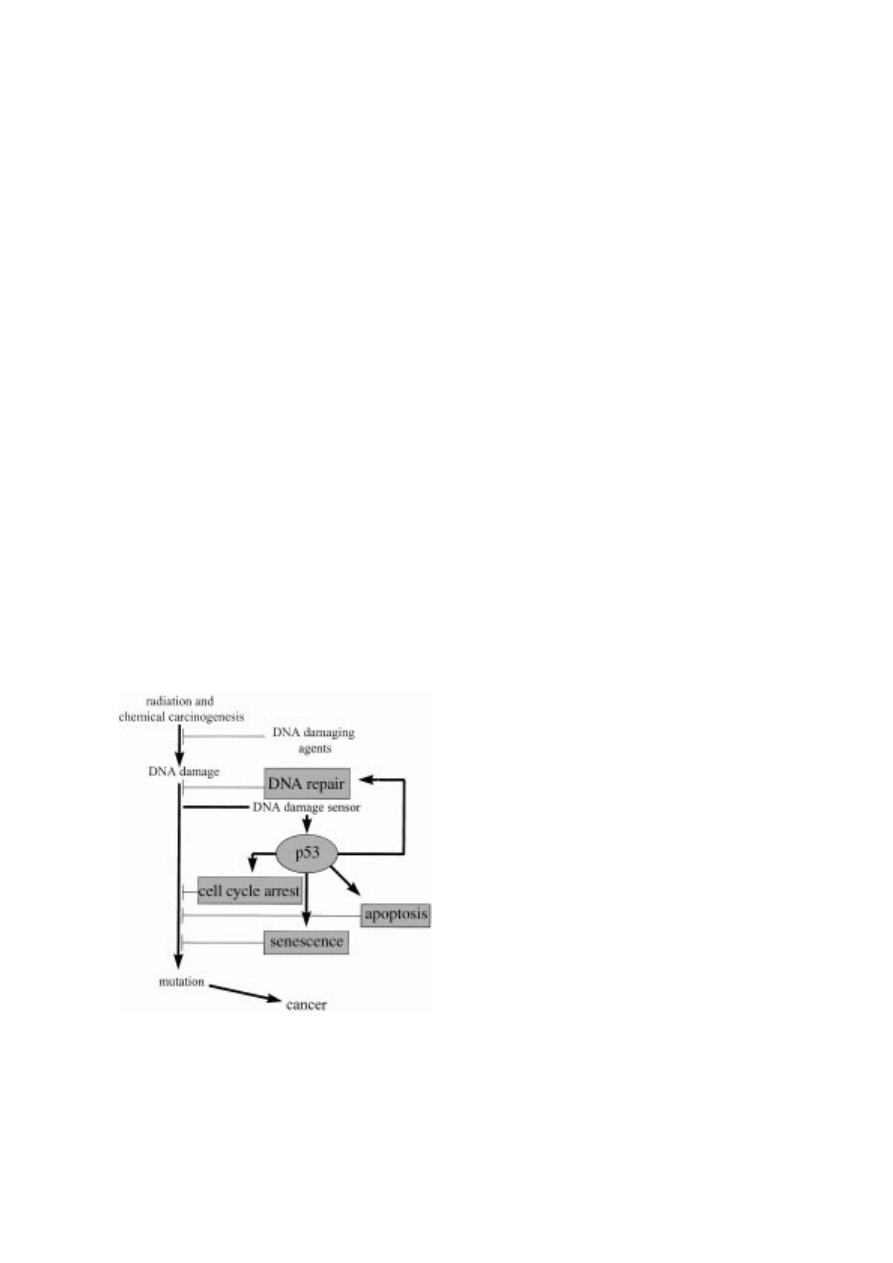
(Figure 2). Whatever the upstream stimulus, activation
of p53 can induce cell cycle arrest at G
1
/S or G
2
/M
checkpoints.
p53 structural analyses [37–40] demonstrate several
conserved amino acid residues that form actual contacts
with the DNA helix. This cooperative binding greatly
facilitates interactions with p53 response elements.

233
Figure 2. Numerous factors activate p53 to initiate a biochemical cascade that ultimately leads to cell growth arrest and/or apoptosis.
Both DNA damaging and non-DNA damaging agents can induce p53 (see text for details). The mechanisms of many of these modes of
p53 induction are poorly understand and the focus of intense investigation.
Abrogation of p53 function most frequently occurs
through inactivating mutations of amino acid residues
that alter p53’s structural or functional integrity leading
to genomic instability that increases the degree of can-
cer susceptibility. Coincident mutations in p53 and the
retinoblastoma susceptibility gene (RB1) cooperate in
the transformation of certain cell types in mice [41,42].
p53 inactivation also occurs via protein–protein inter-
actions (e.g. Mdm2-p53 or WT1-p53), intracellular
co-expression of viral oncoproteins (e.g. SV40-Tag or
HPV-E6), or nuclear exclusion of p53 protein by short-
lived anchor proteins [43,44].
The change in p53 levels result for the most part from
post-translational modifications of the p53 polypep-
tide [45]. Following DNA damage, p53 is stabi-
lized and activated. The events that occur upstream
of p53 have only recently begun to be elucidated.
Some of the advances in the field of p53 activation
have been extensively reviewed elsewhere [17,36].
Phosphorylation of p53 in response to DNA dam-
age appears to be one important mechanism by which
its activation is modulated. p53 is phosphorylated at
several serine residues within its amino- and carboxy-
terminal domains, and many of these phosphorylations
are inducible in the presence of DNA damage. For
example, phosphorylation of serines 15, 20, 37, and
392, among others, is induced by various forms of
DNA damage including XRT and UV (ultraviolet) light
[46]. Many protein kinases, some of which have been
shown to be involved in the detection, signaling and/or
repair of DNA damage, can phosphorylate p53 in vitro
and/or in vivo including ATM kinase, ATM-related
kinase (ATR), checkpoint kinases (Chk1 and Chk2),
DNA-dependent protein kinase (DNA-PK), and casein
kinase II (ckII). Phosphorylation of either serine 15 or
20 can reduce the ability of Mdm2 to negatively reg-
ulate p53. Thus, p53 stability in response to certain
DNA damaging agents is believed to occur, at least
in part, by phosphorylation of p53 at serine 15 and/or
20, thereby disrupting the Mdm2/p53 complex, and
increasing the half-life and transcriptional activation
properties of p53.
p53 activation may also occur through acetyla-
tion. For example, CBP/p300, a protein related to the
retinoblastoma susceptibility protein RB1, is able to
acetylate p53 at lysines 373 and 382 in vitro, and the
acetylation of these residues has been found to activate
p53 sequence-specific DNA binding [47].
234
p53 and apoptosis
p53 can also respond to cellular stress by signal-
ing a complex cellular physiologic pathway to induce
programmed cell death. Programmed cell death, also
known as apoptosis, is a process of cell suicide that
occurs through characteristic morphologic changes.
These include cell shrinkage, nuclear condensation,
DNA fragmentation, and plasma membrane blebbing
[48]. These morphologic changes result from the activ-
ity of intracellular cysteine proteases called caspases
[49]. Although the mechanisms by which p53 ini-
tiates apoptosis remain to be fully elucidated, sev-
eral transcriptional targets of p53 have been identified
that mechanistically link p53 to caspase activation and
apoptosis (Figure 3).
Apoptosis and growth arrest are conferred via
p53-mediated
transcriptional
activation
of
the
cyclin-dependent kinase (Cdk) inhibitor p21
CIP1
, or as
a component of a spindle checkpoint [50] that ensures
maintenance of diploidy during mitosis [51]. p21
CIP1
in turn induces cell cycle arrest through its ability to
bind to several G
1
cyclins, cyclin dependent kinases
and PCNA [52], thereby blocking DNA replication.
In addition, p53 can induce apoptosis through tran-
scriptional activation of death genes such as Bax, a
pro-apoptotic member of the Bcl-2 protein family,
Figure 3. Fundamental pathway for p53 function. The biochem-
ical interactions responsible for these properties are discussed
more fully in the text.
as well as by transcription-independent mechanisms
[53–63]. Cell cycle arrest and apoptosis appear to be
differentially regulated functions, with uncoupling
between the two dependent on the degree of DNA
damage, presence of growth factors, cell type and
specific p53 mutant forms. Even within the same
cell type, the cellular environment can dictate life or
death. For example, DNA damage causes lympho-
cytes to undergo cell cycle arrest in the presence of
interleukin-3, but in its absence, the same DNA dam-
age causes p53-dependent apoptosis [64]. The deletion
of p21 can cause cells that would otherwise undergo
p53-dependent cell cycle arrest to undergo apoptosis
instead, underscoring the major role of genetic back-
ground in determining these cellular responses [65].
The first p53 target gene identified to encode a can-
didate effector of p53-mediated apoptosis was bax, a
pro-apoptotic protein that is a member of the Bcl-2
family of proteins. The ratio of Bax : Bcl-2 appears to
be important in determining whether cells live or die.
Essentially Bax activation induces apoptosis whereas
Bcl-2 expression is associated with cell survival. Bax
and Bcl-2 control apoptosis at the level of mitochon-
drial cytochrome c release; Bax promotes its release
whereas Bcl-2 blocks the release of cytochrome c
from the mitochondria [66]. Once released from the
mitochondria, cytosolic cytochrome c (in concert with
APAF1) appears to medidate the activation of initia-
tor caspase 9, which triggers a caspase cascade lead-
ing to apoptosis [67]. The relative contribution of Bax
to p53-mediated apoptosis appears to be cell type
dependent. Thus, Bax is not required for radiation
induced, p53-dependent apoptosis in thymocytes [68],
but Bax-deficient fibroblasts appear to be compromised
in DNA-damage induced apoptosis [69].
Cell surface death receptors can also transmit rapid
apoptotic signals initiated by the binding of death lig-
ands. Transcription of the death receptor Fas is induced
by p53 through a p53-response element. This induc-
tion has been shown to contribute to cell death by
chemotherapeutic agents, but as in the case of Bax, the
role of Fas transactivation in p53-mediated apoptosis
appears to be cell type and signal-dependent. Follow-
ing DNA damage, p53 can also stimulate the expression
of another death receptor, KILLER/DR5 [70]. When
Fas or KILLER/DR5 binds to the extracellular signal-
ing molecules Fas ligand or TRAIL respectively, they
initiate signaling cascades that result in the activation
of caspases, leading to apoptosis.
Other genes that are induced in response to p53
may also be involved in apoptosis. IGF-BP3 binds
235
insulin-like growth factor-I and prevents it from initi-
ating anti-apoptotic signals [71]. The overexpression
of PAG608, a protein that localizes to the nucleolus,
leads to morphologic changes characteristic of apopto-
sis [72]. Finally, a group of PIGs (p53-induced genes)
were recently identified that appear to increase cellular
oxidation. When oxidation was blocked, p53-mediated
apoptosis was inhibited, suggesting that p53 may acti-
vate apoptosis via cellular oxidation [73].
Inherited p53 mutations, the Li–Fraumeni
syndrome and its variant phenotypes
In 1969, an inherited cancer predisposition syndrome
was reported by Li and Fraumeni on the basis of char-
acterization of four families in which at least two cases
of sarcoma occurred in early life [19,20]. Other can-
cers noted at an increased frequency in these fami-
lies included premenopausal breast cancer, leukemia
and other sarcomas. Based on prospective analysis of
these and other families, the ‘classic’ syndrome was
subsequently defined as a proband with sarcoma diag-
nosed under age 45 years, with a first-degree relative
with any cancer under 45 years, plus another first or
second-degree relative with either any cancer under 45
years or a sarcoma at any age [74,75]. In addition to
sarcomas and premenopausal breast cancer, an excess
of brain tumors, leukemias, and adrenocortical carci-
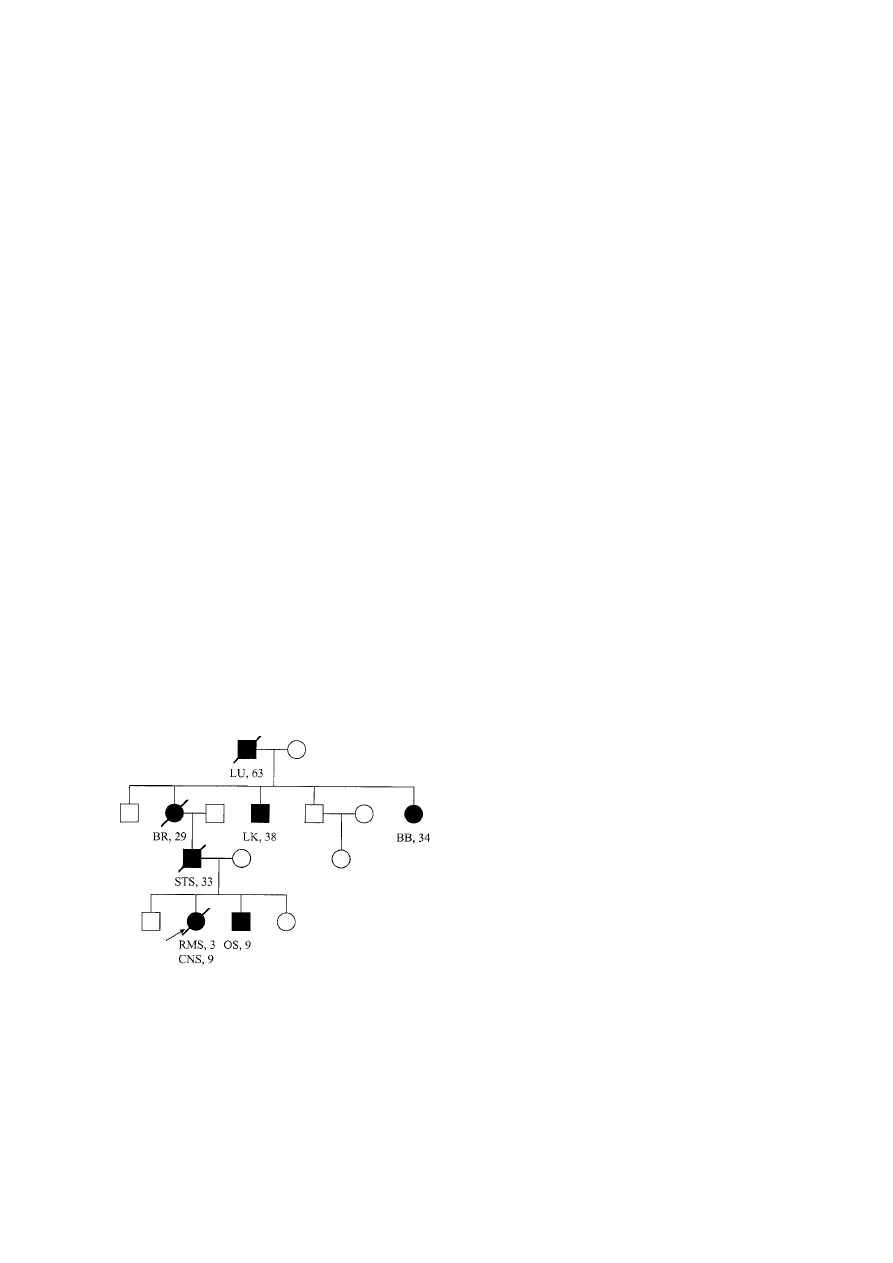
nomas were noted [75] (Figure 4). As more families
Figure 4. ‘Classic’ Li–Fraumeni syndrome pedigree. Circles –
females; Squares – males; Shaded – diagnosed with cancer;
slash – deceased; LU – lung cancer; BR – breast cancer; LK –
leukemia; BB – bilateral breast cancer; STS – soft tissue sarcoma;
RMS – rhabdomyosarcoma; OS – osteosarcoma; CNS – brain
tumor.
have been ascertained, the list of possible or probable
component tumors has been expanded to include gas-
tric cancer, lymphoma, and possibly early onset lung
cancer, choroid plexus carcinoma and colorectal cancer
[76–78]. Birch and colleagues described several fam-
ilies that did not conform to the criteria of the classic
LFS, and they termed these LFS-Like (LFS-L) [79].
The LFS-L families were defined on the basis of a
proband with any childhood cancer or sarcoma, brain
tumor or adrenocortical carcinoma diagnosed under 45
years of age with one first- or second-degree relative
with a typical LFS cancer diagnosed at any age, plus
a first- or second-degree relative in the same parental
lineage with any cancer diagnosed under the age of 60
years. In addition to the wide spectrum of tumor types
observed in LFS, Hisada and colleagues have shown
that gene carriers are at significant risk of developing
multiple synchronous or metachronous non-therapy
induced neoplasms [80]. In particular, the overall rel-
ative risk of occurrence of a second cancer was 5.3
(95% CI
= 2.8–7.8), with a cumulative probability of
second cancer occurrence of 57%. Given the high mor-
tality rate for affected members of LFS families, it was
not possible to obtain DNA from extended pedigrees
to carry out linkage analysis.
In 1990, Malkin and colleagues took a candidate
gene approach to determine the underlying genetic
lesion in LFS [81]. Based on earlier observations that
somatic mutations of the p53 tumor suppressor gene
were observed in greater than 50% of sporadic human
cancers [82], and that p53 transgenic mice carrying
mutant p53 alleles developed a wide spectrum of malig-
nancies [83], these investigators elected to examine
this gene in constitutional DNA of LFS kindreds.
Although heterozygous point mutations were initially
detected in 5 of 5 families, numerous subsequent stud-
ies by these and other investigators have shown that
only 60% to 80% of ‘classic’ LFS families harbor
detectable germline p53 mutations [85–87], while the
majority of LFS-L families do not have detectable p53
mutations in the coding regions of the gene [79,88].
A number of possible explanations have been pro-
posed to explain the lack of p53 alterations in some
‘classic’ LFS families and in most LFS-L kindreds.
Lesions within introns or the regulatory regions of
the gene have recently been suggested, although their
functional significance is unclear [89]. The types and
position of germ line p53 mutations closely reflect
those observed in sporadic tumors, and the majority
of reported mutations are missense mutations within
the conserved domains of the gene. Although it was

236
originally observed that mutations in LFS occurred in
a tight cluster within exon 7 [81], subsequent studies
have confirmed that in fact, mutations occur through-
out the gene – though primarily confined to highly con-
served regions.
Several groups have examined the role of other tumor
suppressor genes associated with cancer syndromes
associated with occurrence of multiple tumors. To date,
these studies have been non-informative for germline
alterations of PTEN, p16
INK4a
, and p19
Arf
. Other genes
involved in p53-mediated cellular growth regulatory
pathways, either effectors, targets or binding partners
of p53, have been postulated to be involved in tumor
predisposition in LFS and LFS-L families with no
detectable germline p53 alterations. Although muta-
tions of downstream targets of p53 have yet to be iden-
tified in these families, a recent intriguing observation
of heterozygous germline mutations in the checkpoint
kinase hCHK2 in one LFS family and one LFS-L fam-
ily suggests an alternative mechanism for functional
p53 inactivation in LFS [90]. This gene is the human
homolog of the yeast Cds1 and Rad53 G2 checkpoint
kinases that are involved in preventing cellular entry
into mitosis in response to DNA damage [91]. Although
the checkpoint kinase pathway genes may be impli-
cated in cancer predisposition in this setting, the rele-
vance of these observations has recently been brought
into question with the detection of functionally neu-
tral polymorphisms in homologous fragments of genes
related to hCHK2 in LFS families lacking germline p53
mutations [92]. The search then continues for expla-
nations of the genetic predisposition to cancer in these
families.
p53-deficient mice were generated by Donehower
and colleagues in 1992, and subsequently by other
groups [93–95] (see below). These mice have a strik-
ing propensity to develop a wide spectrum of cancer at
extremely early age (
<9 months), with a relative preva-
lence of lymphomas. Interestingly, p53-heterozygous
mice, harboring one wild-type and one deficient allele,
also have a high incidence of cancer, although the
tumors develop at a much slower rate [96]. Further-
more, in a pattern similar to the human LFS, these mice
have a higher incidence of sarcoma development. Mul-
tiple primary tumors occur as well, again mimicking
the human LFS phenotype. Although p53 behaves as a
classic tumor suppressor gene, less than 50% of tumors
from p53-heterozygous mice and LFS patients have
evidence of loss of heterozygosity [96,97]. It remains
unclear in these patients how the retained wild-type p53
allele is functionally inactivated en route to malignant
transformation of the cell.
A number of studies have analysed groups of patients
with tumors characteristic of LFS, yet lacking char-
acteristic family histories of cancer, for germline
p53 mutations. Such mutations have been identi-
fied in approximately 50% to 80% of children with
adrenocortical carcinoma [98,99], 10% of children
with osteosarcoma [100], and 10% of children with
rhabdomyosarcoma [101,102]. The age of onset of
tumors in the latter group of patients is strikingly lower
(average age approximately 22 months) than in RMS
patients with intact germline p53 [101]. These obser-
vations suggest a possible difference in the biologic
nature of malignant transformation of cells where p53
is altered as an early in contrast to a late event. One third
of children with sarcomas plus either multiple primary
tumors, or a family history of cancer have germline p53
mutations. However, although breast cancer is a princi-
pal component of LFS, only 1% to 2% of women with
familial, early-onset, or bilateral breast cancer harbor
germline p53 mutations [103,104].
Presymptomatic molecular testing for p53 germline
mutations in members of Li–Fraumeni kindreds has
been met with significant controversy. Because of
the variable expressivity, the diverse tumor spectrum,
and lack of clear clinical surveillance, preventative
or treatment recommendations, it is unclear how to
manage the detection of a p53 mutant carrier. Fur-
thermore, the concept of predictive genetic testing
of a child for a disease which may (or may not)
occur in young adulthood poses significant chal-
lenges to our perception of the ethics of disclosure
of genetic test results, where the potential benefi-
ciary of these results may wish to uphold the right
to ‘not know.’ In an attempt to address these issues,
guidelines for testing have been established by both
the American Society of Human Genetics in a state-
ment and the American Society of Clinical Oncology
[105,106]. These guidelines form a useful founda-
tion on which to build practical testing parameters as
better defined genotype : phenotype correlations are
generated.
More than 80% of p53 mutations (somatic or
germline) are missense and occur in the core DNA-
binding domain. Residues 175, 245, 248, 273 and
282 are the most frequently altered codons. Recently,
rare mutations in the nuclear localization signal
and tetramerization domain have been documented
and functionally characterized. Alterations that would

237
affect splicing events and lead to disruptions of the gene
product have also been reported.
Given this clinical heterogeneity, it is important to
establish how different p53 mutations predispose to the
formation of specific tumours. In vitro analysis of func-
tional activity of p53 alterations [107,108] reveals that
not all are associated with inhibition of growth arrest,
apoptosis, transcriptional activation, or an increased
cancer risk. In humans, the limited organ or target cell
specificity of p53 mutations [109] may be due to vary-
ing genetic backgrounds, acquisition of unique subse-
quent gene alterations in target tissues, or the influence
of epigenetic or environmental factors. Because it is
difficult to study in vivo factors that may influence p53
function in humans, the development of a mouse carry-
ing specific p53 mutations on a homogeneous genetic
background offers significant advantages. A model that
reflects the human p53 mutant genotype may provide a
formidable tool with which to study the role of natural
somatic and germline p53 mutations in carcinogenesis,
and could lead to the development of novel treatment
strategies.
Tumour development in mice with
germline p53 alterations
Transgenic animals carrying distinct deregulated onco-
genes develop tumours that appear to be limited to cer-
tain cell types. To better study p53 in vivo, mice have
been created that either lack functional p53 [93] or
express dominant-negative mutant alleles that inhibit
wt-p53 function [83].
p53 transgenic mice carry transgenes that encode for
proteins differing from wt-p53 either by an
193
Arg
>
Pro or
135
Ala
> Val substitution [83]. The transgenes
are under transcriptional control of the endogenous pro-
moter and the mice also carry two wt-p53 alleles. The
transgene is expressed in a wide range of tissue, yet
tumours (primarily osteosarcomas, lymphomas, and
lung adenocarcinomas) occur in only 20% of the mice,
suggesting intrinsic tissue-specific differences.
Homologous recombination has also been used in
mouse embryonic stem (ES) cells to derive ‘null’
p53 alleles [93–95]. Two separate models result from
the replacement of exons 2–6 with a neo
r
cassette
[94,95]. The third comprises insertion of a polII
promoter-driven neo
r
cassette into exon 5, as well as
a deletion of 350 nucleotides of intron 4 and 106
nucleotides of exon 5 [93]. None of the p53
−/−
mice
express detectable intact or truncated mRNA or pro-
tein. The null p53 allele has been established in the
germline of chimeric mice with either a mixed or inbred
(C57BL/6
× 129/Sv) [93,95], pure 129/Sv, or 129/O1a
backgrounds. In all mice, spontaneous development
of different tumour types, predominantly lymphomas
and sarcomas, occurred in
>75% before 6 months of
age. Multiple tumours were noted in
∼30% of tumour-
bearing p53
−/−
mice. Tumour formation, primarily sar-
comas, is delayed in heterozygotes [96]; however, 50%
develop tumors by 18 months of age. The spectrum of
tumours in p53
+/−
mice resembles the LFS phenotype
more closely than the spectrum in p53
−/−
mice.
The tumourigenic activity of the
135
Val transgene is
influenced by the presence or absence of wt-p53. Mice
carrying the transgene were crossed with p53
−/−
mice
[110]. The transgene accelerated tumor formation in
p53
+/−
, but not p53
−/−
mice, suggesting that this loss-
of-function mutation had a dominant-negative effect
with respect to tumour incidence and cell growth rates.
Although the tumour spectrum was similar in trans-
genic and p53
−/−
mice, the transgenics showed a higher
predilection to lung adenocarcinomas. Recently, it has
been suggested that reduction of p53 dosage in p53
+/−
mice itself can promote cancer formation [96].
p53-deficient mice are more sensitive to the effects
of certain carcinogenic agents than p53-wild-type mice
and the types of cancers that develop are predicted
by the agent used. p53-deficient and p53-Tg mice
exposed to sub-lethal doses of
γ -irradiation develop
tumours, usually sarcomas, earlier than untreated ani-
mals [111,112]. This susceptibility is associated with
a 2-fold increase in accumulation of radiation-induced
dsDNA breaks compared to that seen in p53
+/+
ani-
mals. These studies confirm that p53 prevents accu-
mulation of cells sustaining radiation- or chemically
induced DNA damage. Given the important role of
p53 in cell cycle control, a p53-deficient state would
be expected to deregulate differentiation and develop-
ment and yield aberrant morphogenesis and embry-
onic lethality. In fact, the teratogen/DNA-damaging
carcinogen benzo[a]pyrene [113] and the anticonvul-
sant/teratogenic drug phenytoin [114], induce a 2-4X
increase in in utero fetal death, teratogenicity and post-
partum lethality in pregnant p53
+/−
mice, further sup-
porting the embryoprotective role of p53.
The cumulative data from these studies indicate
that loss or alterations of p53 may accelerate prior
tumour predisposition, that the rate and spectrum of
development of some cancers may be strain-dependent

238
or influenced by modifier genes, and that normal mouse
development is possible even in the absence of p53.
Studies of p53
−/−
mice have proven valuable, yet
tumourigenesis in this model reflects the complete
absence of gene function, a phenomenon that is not
generally observed in humans. Although the tumour
spectrum in current p53-altered mice is highly variable,
none spontaneously mimic the human germline pheno-
type in its predominance of sarcomas and breast cancer,
nor the high frequency of carcinomas associated with
sporadic missense p53 mutations. Even the transgenic
135
Val and
193
Pro mutations are not reported in human
cancers. Although these phenotypic differences could
be species-dependent, one could postulate that creating
specific p53 missense mutations could provide a more
realistic model of p53-dependent carcinogenesis.
p53 gene therapy
The introduction of wild-type p53-expressing plasmids
into tumor cells can induce over-expression of recom-
binant wild-type p53 protein and drive cells into either
growth arrest or apoptosis [115,116]. Numerous studies
have examined the effects of exogenous p53 gene trans-
fer using an adenoviral vector on a variety of cancer cell
lines, both in vitro and in vivo. The use of Ad5CMV-p53
gene therapy in vitro has resulted in a cytotoxic effect
in cell lines of many different cancer types that harbor
either mutant or deleted p53 [117,118]. The effect of
p53 gene therapy on ‘normal’ cells has been more con-
troversial. While a number of groups have reported that
both normal human fibroblasts and mammary epithelial
cells have been spared from the cytotoxic effects of this
treatment [119], other fibroblast strains have exhibited
a significant decrease in survival following p53 gene
therapy [120].
p53 may enhance the cytotoxicity induced by ion-
izing radiation and some chemotherapeutic agents.
Therefore, the use of p53 gene therapy in combina-
tion with these therapeutic agents has been studied
in head and neck, colon and esophageal cancer cell
lines, as well as others, with additional cytotoxicity
being consistently observed. p53 gene therapy has also
been used in cancer xenograft models in combination
with either radiation therapy or chemotherapy. Most of
the reported data demonstrate significant tumor growth
inhibition or regression following intratumoral injec-
tion of Ad5CMV-p53 combined with either radiation
or chemotherapy [121]. Due to significant cytotoxic
effects of Ad5CMV-p53 gene therapy on cancer cells
both in vitro and in vivo, clinical trials have been limited
in a number of tumor types [122]. While the clinical
benefit has been limited to date, no major side effects
have been noted, and Phase II and Phase III clinical
trials are currently in progress.
One of the major obstacles of in vivo gene therapy is
the difficulty in specifically targeting transgene expres-
sion to the tumor cells. Recently, a number of target-
ing strategies have been reported, including the use of
‘oncolytic viruses’ such as ONYX-015. This tumor tar-
geting virus is an adenovirus that is missing only the
gene encoding the E1B 55kDa protein, a protein that
binds to and inhibits p53-activated transcription, and
is essential for viral replication [123]. The rationale
behind the use of this virus is that in cells with wild-
type p53, replication of ONYX-15 would be inhibited
because p53 would remain active. However, in cells
deficient in p53, the virus would be able to replicate,
lyse the host cell, and proceed to infect and replicate
in adjacent cells also lacking wild-type p53. The virus
has had some success in clinical trials, and it may, in
fact, be more widely applicable than originally antici-
pated [124]. It appears as though the virus is active not
only in tumor cells with mutant p53, but also in tumor
cells with wild-type p53, which most likely have other
defects in the p53 pathway [125].
The p53 family
Most genes occur in families and, until recently, p53
was thought to be an exception to this pattern. However,
two new members of the p53 family have now been
identified [126–131]. p73 is a putative tumour suppres-
sor with considerable structural and functional homol-
ogy to p53. The structure of p73 resembles that of p53
in all three principle functional domains with a remark-
able 63% similarity in the DNA-binding region [126].
Like p53, p73 is able to induce apoptosis by activating
p21
WAF1
/CIP1
[132]. One difference between p53 and p73
was thought to be p53’s unique ability to be induced
in the presence of DNA damage. This, however, has
been challenged as recent evidence has demonstrated
that p73 is a target of the non-receptor tyrosine kinase
c-Abl in response to DNA damaging agents such as ion-
ising radiation and cisplatin [133,134]. p73 is mapped
to chromosome 1p36, a region frequently deleted in
a variety of human tumours [135–137]. This led to
the belief that p73 could be a tumour suppressor
that plays a role in the pathogenesis of these malig-
nancies. However, several studies have documented

239
only rare mutations in a variety of human cancers
[126,138,139].
p51, also known as Ket, p40, p63 and p73L, more
closely resembles p73 than p53. Like p73 and p53, p51
has a transactivation, DNA-binding and oligomerisa-
tion domain and is able to induce growth arrest and
apoptosis in a manner similar to p53 [129]. p51 maps
to chromosome 3q27-28, a region frequently deleted in
bladder cancers but amplified in some cervical, ovar-
ian and lung cancers [127–130]. Like p73, mutations
in p51 appear to be rare [129]
The physiologic role of p73 also remains unknown.
p73 is expressed in a variety of tissues including thy-
mus, prostate, heart, liver, skeletal muscle and pancreas
[130]. The two transcripts first discovered, p73
α and
p73
β, have been found to have DNA-binding capacity
in vitro [140] and are able to interact with themselves
and each other. Both transcripts can activate transcrip-
tion from p53-responsive promoters and induce apop-
tosis when overexpressed. Recently, it has been found
that p73 is a target of c-Abl in response to DNA damage
[133,134]. This introduces another parallel with p53.
However, there is increasing evidence that p73 serves
a different physiologic role than p53. While MDM2
binds to p73, it does not degrade it as it does p53 [141].
p73 is also immune to inactivation by viral oncopro-
teins such as the SV40 T antigen, the adenovirus E1B
55 K protein and the human papillomavirus E6 pro-
tein [139]. Knockout mice may provide the clues as to
the true function of p73. Deletion of p73 causes severe
developmental disorders in mice while p53 null mice
develop normally but have an increased risk of devel-
oping tumours [142]. It seems that p73 may in fact be
a developmental gene.
New light has been shed on the physiologic role of
p51 by the work of Celli et al. [144]. In a study of
26 families with EEC (ectrodactyly, ectodermal dys-
plasia, cleft lip) syndrome, linkage analysis had led
the authors to a region of chromosome 3q27 that con-
tained the p51 gene (denoted as p63 by the authors).
In a mutation analysis examining exons 5–14, nine of
the 26 individuals were found to harbour heterozygous
p51 mutations. Interestingly, all of the missense muta-
tions were in the DNA-binding domain of p73 and most
occurred at the exact amino acids corresponding to the
three ‘hot spot’ amino acids in p53. The mutations were
found to act in dominant manner creating a phenotype
similar to that of knockout mice with symptoms such
as sparse hair, dry skin, glandular dysplasia, oligodon-
tia and limb abnormalities. Also, it is suggested that
cleft lip in humans is the equivalent of the dysplastic
maxilla and mandible seen in the knockout mice. How-
ever, as with the knockout mice, EEC patients do not
have increased risk of developing cancer. The findings
of the authors are consistent with previous studies that
suggest that p51 is an important gene in development.
References
1. Lane DP, Crawford LV: T antigen is bound to a host protein
in SV40-transformed cells. Nature 278: 261–263, 1979
2. Linzer DI, Levine AJ: Characterization of a 54K dalton
cellular tumor antigen present in SV40-transformed cells
and uninfected embryonal carcinoma cells. Cell 17: 43–52,
1979
3. p53 in Dundee website – www.dundee.ac.uk/pathology/
p53.htm
4. Protein interactions with p53 – www.dundee.ac.uk/
pathology/p53inter.htm
5. 9th p53 Workshop – www.athens.wistar.upenn.edu/
∼p53/
6. p53 researchers – www.athens.wistar.upenn.edu/
∼p53/
dirfp.html
7. p53 database at IARC – www.iarc.fr/p53/homepage.htm
8. p53 database at Institut Curie – perso.curie.fr/Thierry.
Soussi/index.html
9. Weizmann Institute p53 site – www.bioinformatics/
weizmann.ac.il/hotmolecbase/entries/p53.htm
10. p53 structure – www.pds.med.umich.edu/users/frank/logo.
html
11. p53 database – www.biomedcomp.com/4d.acg$tsrchname?
Name
= p53
12. Apotosis sites – www.access.digex.net/
∼regulate/apolist.
html
13. OMIM p53 site – www3.sncbi.nlm.nih.gov/htbin-post/
Omim/dispmim?191170
14. OMIM
mdm2
site
–
www.ncibi.nlm.nih.gov/htbin-
post.Omim/dispmim?164785
15. Mdm2
database
–
www.infosci/coh.org/mdm2asp/
default.asp
16. Levine AJ: p53, the cellular gatekeeper for growth and
division. Cell 88: 323–331, 1997
17. Prives C, Hall PA: The p53 pathway. J Pathol 187: 112–116,
1999
18. Harris CC: Structure and function of the p53 tumor sup-
pressor gene: clues for rational cancer therapeutic strate-
gies. J Natl Cancer Inst 88: 1442–1455, 1996
19. Li FP, Fraumeni JF Jr: Soft-tissue sarcomas, breast cancer,
and other neoplasms. A familial syndrome? Ann Intern
Med 71: 747–752, 1969
20. Li FP, Fraumeni JF Jr: Rhabdomyosarcoma in children:
epidemiologic study and identification of a familial cancer
syndrome. J Natl Cancer Inst 43: 1365–1373, 1969
21. Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE,
Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA,
Friend SH: Germline p53 mutations in a familial syndrome
of breast cancer, sarcomas, and other neoplasms. Science
250: 1233–1238, 1990

240
22. Malkin D: The Li–Fraumeni syndrome. In: Vogelstein B,
Kinzler KW (eds) Genetic Basis of Human Cancer 1st Edi-
tion. McGraw-Hill, Inc. New York, pp. 393–412
23. Soussi T, Caron de Fromentel C, May P: Structural aspects
of the p53 protein in relation to gene evolution. Oncogene
5: 945–951, 1990
24. Harris CC, Hollstein M: Clinical implications of the p53
tumor suppressor gene. New Engl J Med 329: 1318–1323,
1993
25. Cariello NF, Cui L, Beroud C, Soussi T: Database and soft-
ware for the analysis of mutations in the human p53 gene.
Cancer Res 54: 4454–4459, 1994
26. Lu H, Levine AJ: Human TAFII31 protein is a transcrip-
tional coactivator of the p53 protein. Proc Natl Acad Sci
USA 92: 5154–5158, 1995
27. Lin J, Wu X, Chen J, Chang A, Levine AJ: Functions of the
p53 protein in growth regulation and tumor suppression.
Cold Spring Harb Symp Quant Biol 59: 215–223, 1994
28. May P, May E: Twenty years of p53 research: structural
and functional aspects of the p53 protein. Oncogene 18:
7621–7636, 1999
29. Wang Y, Prives C: Increased and altered DNA binding of
human p53 by S and G1/M but not G1 cyclin-dependent
kinases. Nature 376: 88–91, 1995
30. Dang CV, Lee WM: Nuclear and nucleolar targeting
sequences of c-erb-A, c-myb, N-myc, p53, HSP70, and
HIV tat proteins. J Biol Chem 264: 18019–18023, 1989
31. Kubbutat MH, Ludwig RL, Ashcroft M, Vousden KH: Reg-
ulation of Mdm2-directed degradation by the C terminus
of p53. Mol Cell Biol 18: 5690–5698, 1998
32. Varshavsky A: The ubiquitin system. Trends Biochem Sci
22: 383–387, 1997
33. Momand J, Zambetti GP, Olson DC, George D, Levine AJ:
The mdm-2 oncogene product forms a complex with the
p53 protein and inhibits p53-mediated transactivation. Cell
69: 1237–1245, 1992
34. Barak Y, Juven T, Haffner R, Oren M: mdm expression is
induced by wild-type p53 activity. EMBO J 12: 461–468,
1993
35. Jones SN, Roe AE, Donehower LA, Bradley A: Rescue of
embryonic lethality in Mdm-2 deficient mice by absence
of p53. Nature 378: 206–208, 1995
36. Ljungman M: Dial 9-1-1 for p53: mechanisms of p53 acti-
vation by cellular stress. Neoplasia 2: 208–225, 2000
37. Cho Y, Gorina S, Jeffrey PD, Pavletich NP: Crystal struc-
ture of a p53 tumor suppressor-DNA complex: understand-
ing tumorigenic mutations. Science 265: 346, 1994
38. Clore GM, Omichinski JF, Sakaguchi K, Zambrano N,
Sakamoto H, Appella E, Gronenborn AM: High-resolution
structure of the oligomerization domain of p53 by mutidi-
mensional NMR. Science 265: 386, 1994
39. Lee W, Harvey TS, Yin Y, Yau P, Litchfield D,
Arrowsmith CH: Solution structure of the tetrameric min-
imum transforming domain of p53. Nature Struct Biol 1:
877, 1994
40. Kussie PH, Gorina S, Marechal V, et al.: Structure of the
MDM2 oncoprotein bound to the p53 tumor suppressor
transactivation domain. Science 274: 948, 1996
41. Williams BO, Remington L, Albert DM, Mukai S,
Bronson RT, Jacks T: Cooperative tumorigenic effects of
germline mutations in Rb and p53. Nature Genet 7: 480,
1994
42. Harvey M, Vogel H, Lee EY, Bradley A, Donehower LA:
Mice deficient in both p53 and Rb develop tumors primarily
of endocrine origin. Cancer Res 55: 1146, 1995
43. Knippschild U, Oren M, Deppert W: Abrogation of wild-
type p53-mediated growth inhibition by nuclear exclusion.
Oncogene 12: 1755, 1996
44. Moll UM, Ostermeyer AG, Haladay AG, Windfield B,
Frazier M, Zambetti G: Cytoplasmic sequestration of wild-
type p53 protein impairs G1 checkpoint after DNA dam-
age. Mol Cell Biol 16: 1126, 1996
45. Kastan MB, Onyekwere O, Sidransky D, Vogelstein B,
Craig RW: Participation of p53 protein in the cellular
response to DNA damage. Cancer Res 51: 6304–6311,
1991
46. Lakin ND, Jackson SP: Regulation of p53 in response to
DNA damage. Oncogene 18: 7644–7655, 1999
47. Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M,
Vassilev A, Anderson CW, Appella E: DNA damage acti-
vates p53 through a phosphorylation-acetylation cascade.
Genes Dev 12: 2831–2841, 1998
48. Kerr JFR, Wyllie AH, Currie AR: Apoptosis: a basic bio-
logical phenomenon with wide-ranging implications in tis-
sue kinetics. Br J Cancer 26: 239–257, 1972
49. Salvasen GS, Dixit VM: Caspases: intracellular signaling
by proteolysis. Cell 91: 443–446, 1997
50. Cross SM, Sanchez CA, Morgan CA, Schimke MK,
Ramel S, Idzerda RL, Raskind WH, Reid BJ: A p53-
dependent mouse spindle checkpoint. Science 267: 1353,
1995
51. Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande
Woude GF: Abnormal centrosome amplification in the
absence of p53. Science 271: 1744, 1996
52. Pines J: p21 inhibits cyclin shock. Nature 369: 520, 1994
53. Vogelstein B, Kinzler KW: p53 function and dysfunction.
Cell 70: 523, 1992
54. Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T:
p53 is required for radiation-induced apoptosis in mouse
thymocytes. Nature 362: 847, 1993
55. Lowe SW, Ruley HE, Jacks T, Housman DE: p53-
dependent apoptosis modulates the cytotoxicity of anti-
cancer agents. Cell 74: 957, 1993
56. Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O,
Oren M: Wild-type p53 can inhibit oncogene-mediated
focus formation. Proc Natl Acad Sci USA 86: 8763, 1984
57. Gottlieb T, Oren M: p53 in growth and neoplasia. Biochem
Biophys Acta Rev Cancer 1287: 77, 1996
58. Fan S, Smith MK, Rivet DJ, Duba D, Zhan Q, Kohn K,
Fornace AJ, O’Connor PM: Disruption of p53 function
sensitizes breast cancer MCF-7 cells to cisplatin and pen-
toxifylline. Cancer Res 55: 1649, 1995
59. Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L,
Kimchi A, Oren M: Wild-type p53 induces apoptosis of
myeloid leukemic cells that is inhibited by interleukin-6.
Nature 352: 345, 1991

241
60. Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J:
Induction of apoptosis by wild-type p53 in a human colon
tumour-derived cell line. Proc Natl Acad Sci USA 89: 4495,
1992
61. Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC,
Hooper ML, Wyllie AH: Thymocyte apoptosis induced
by p53-dependent and independent pathways. Nature 362:
849, 1993
62. Miyashita T, Reed TC: Tumour suppressor p53 is a direct
transcriptional activator of the human bax gene: Cell 80:
293, 1995
63. Miyashita T, Krajewski S, Krajewski H, et al.: Tumour sup-
pressor p53 is a regulator of bcl-2 and bax gene expression
in vitro and in vivo. Oncogene 9: 1799, 1994
64. Canman CE, Griber T, Coutts S, Kastan MB: Growth factor
modulation of p53-mediated growth arrest versus apopto-
sis. Genes Dev 9: 600–611, 1995
65. Polyak K, Waldman T, He TC, Kinzler KW, Vogelstein B:
Genetic determinants of p53-induced apoptosis and growth
arrest. Genes Dev 10: 1945–1952, 1996
66. Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I,
Jansen B, Borner C: Bcl-2 proloongs cell survival after
Bax-induced release of cytochrome c (see comments).
Nature 391: 496–499, 1998
67. Srinivasula SM, Ahmad M, Fernandes-Alnemri T,
Alnemri ES: Autoactivation of procaspase-9 by Apaf-1-
mediated oligomerization. Mol Cell 1: 949–957, 1998
68. Knudson CM, Tung KS, Tourtelotte WG, Brown GA,
Korsmeyer SJ: Bax-deficient mice with lymphoid hyper-
plasia and male germ cell death. Science 270: 96–99, 1995
69. Schmitt CA, McCurrach ME, de Stanchina E, Wallace-
Brodeur RR, Lowe SW: NK4a/ARF mutations accelerate
lymphomagenesis and promote chemoresistance by dis-
abling p53. Genes Dev. 13: 2670–2677, 1999
70. Wu GS, Burns TF, McDonald ER, Jiang W, Meng R,
Kranz ID, Kao G, Gan DD, Zhou JY, Muschel R,
Hamilton SR, Spinner NB, Markowitz S, Wu G,
ed-Deiry WS: KILLER/DR5 is a DNA damage-inducible
p53-regulated death receptor gene. Nat Genet 17: 141–143,
1997
71. Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I,
Faha B, Seizinger BR, Kley N: Induction of the growth
inhibitor IGF-binding protein 3 by p53. Nature 377:
646–649, 1995
72. Israeli D, Tessler E, Haupt Y, Elkeles A, Wilder S,
Amson R, Telerman A, Oren M: A novel p53-inducible
gene, PAG608, encodes a nuclear zinc finger protein
whose overexpression promotes apoptosis. EMBO J 16:
4384–4392, 1997
73. Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B:
A model for p53-induced apoptosis. Nature 389: 300–305,
1997
74. Li FP, Fraumeni JF, Mulvihill JJ, Blattner WA,
Dreyfus MG, Tucker MA, Miller RW: A cancer family syn-
drome in twenty-four kindreds. Cancer Res 48: 5358–5362,
1988
75. Garber JE, Goldstein AM, Kantor AF, Dreyfus MG,
Fraumeni JF, Li FP: Follow-up study of twenty-four
families with Li–Fraumeni syndrome. Cancer Res 51:
6094–6097, 1991
76. Garber JE, Burke EM, Lavally BL, et al.: Choroid plexus
tumors in the breast cancer-sarcoma syndrome. Cancer 66:
2658–2660, 1990
77. Horio Y, Suzuki H, Ueda R, et al.: Predominantly tumor-
limited expression of a mutant allele in a Japanese
family carrying a germline p53 mutation. Oncogene 9:
1231–1235, 1994
78. Varley JM, Evans DG, Birch JM: Li–Fraumeni syndrome –
a molecular and clinical review. Br J Cancer 76: 1–14, 1997
79. Varley JM, McGown G, Thorncroft M, et al.: Germ-line
mutations of TP53 in Li–Fraumeni families: an extended
study of 39 families. Cancer Res 57: 3245–3252, 1997
80. Hisada M, Garber JE, Fung CY, Fraumeni JF, Li FP: Mul-
tiple primary cancers in families with Li–Fraumeni syn-
drome. J Natl Cancer Inst 90: 606–611, 1998
81. Malkin D, Li FP, Strong LC, Fraumeni JF, Nelson CE,
Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA,
Friend SH: Germline p53 mutations in a familial syndrome
of breast cancer, sarcomas, and other neoplasms. Science
250: 1233–1238, 1990
82. Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R,
Cleary K, Bigner SH, Davidson N, Baylin S, Devilee P,
Glover T, Collins FS, Weston A, Modali R, Harris CC,
Vogelstein B: Mutations in the p53 gene occur in diverse
human tumor types. Nature 342: 705–708, 1989
83. Lavigueur A, Maltby V, Mock D, Rossant J, Pawson T,
Bernstein A: High incidence of lung, bone and lymphoid
tumors in transgenic mice overexpressing mutant alleles of
the p53 oncogene. Mol Cell Biol 9: 3982–3991, 1989
84. Frebourg T, Barbier N, Yan YX, Garber JE, Dreyfus M,
Fraumeni JF Jr, Li FP, Friend SH: Germ-line p53 mutations
in 15 families with Li–Fraumeni syndrome. Am J Hum
Genet 56: 608–615, 1995
85. Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H:
Tumours associated with p53 germline mutations: a syn-
opsis of 91 families. Amer J Pathol 150: 1, 1997
86. Birch JM, Hartley AL, Tricker KJ, Prosser J, Condie A,
Kelsey AM, Harris M, Morris-Jones PH, Binchy A,
Crowther D, Craft AW, Eden OB, Evans DGR,
Thompson E, Mann JR, Martin J, Mitchell E, Santibanez-
Koref MF: Prevalence and diversity of constitutional
mutations in the p53 gene among 21 Li-Fraumeni families.
Cancer Res 54: 1298, 1994
87. Srivastava S, Zou Z, Pirollo K, Blattner W, Cheng EH:
Germ-line transmission of a mutated p53 gene in a cancer-
prone family with Li–Fraumeni syndrome. Nature 348:
747, 1990
88. Eeles RA: Germline mutations in the p53 gene. In: Pon-
der BAJ, Cavenee WK, Solomon E (eds) Cancer Surveys:
Genetics and Cancer: A Second Look Cold Spring Harbor
Laboratory Press 101–124, 1995
89. Barel D, Avigad S, Mor C, Fogel M, Cohen IJ, Zaizov R:
A novel germ-line mutation in the noncoding region of the
p53 gene in a Li–Fraumeni family. Cancer Genet Cytogenet
103: 1, 1998
90. Bell DW, Varley JM, Szydlo TE, et al.: Heterozygous germ
line hCHK2 mutations in Li–Fraumeni syndrome. Science
286: 2528–2531, 2000

242
91. Hirao Z, Kong YY, Matsuoka S, et al.: DNA damage-
induced activation of p53 by the checkpoint kinase Chk2.
Science 287: 1824–1827, 2000
92. www.sciencemag.org/cgi/content/full/289/5478/359a
93. Donehower LA, Harvey M, Slagle BL, et al.: Mice defi-
cient for p53 are developmentally normal but susceptible
to spontaneous tumors. Nature 356: 215–221, 1992
94. Jacks T, Remington L, Williams BO, et al.: Tumor spectrum
analysis in p53-mutant mice. Curr Biol 4: 1–7, 1994
95. Purdie CA, Harrison DJ, Peter A, et al.: Tumour incidence,
spectrum and ploidy in mice with a large deletion in the
p53 gene. Oncogene 9: 603–609, 1994
96. Venkatachalam S, Shi YP, Jones SN, et al.: Retention of
wild-type p53 in tumors from p53 heterozygous mice:
reduction of p53 dosage can promote tumor formation.
EMBO J 17: 4657–4667, 1998
97. Varley JM, Thorncroft M, McGown G, Tricker K,
Birch JM, Evans DG: A novel deletion within exon 6 of
TP53 in a family with Li–Fraumeni-like syndrome, and
LOH in a benign lesion from a mutation carrier. Cancer
Genet Cytogenet 90: 14–16, 1996
98. Wagner J, Portwine C, Rabin K, Leclerc JM, Narod SA,
Malkin D: High frequency of germline p53 mutations in
childhood adrenocortical cancer. J Natl Cancer Inst 86:
1707–1710, 1994
99. Varley JM, McGown G, Thorncroft M, et al.: Are there low-
penetrance TP53 alleles? Evidence from childhood adreno-
cortical tumors. Am J Hum Genet 65: 995–1006, 1999
100. McIntyre JF, Smith-Sorensen B, Friend SH, et al.:
Germline mutations of the p53 tumor suppressor gene in
children with osteosarcoma. J Clin Oncol 12: 925–930,
1994
101. Diller L, Sexsmith E, Gottlieb A, Li FP, Malkin D:
Germline p53 mutations are frequently detected in young
children with rhabdomyosarcoma. J Clin Invest 95:
1606–1611, 1995
102. Moutou C, Le Bihan C, Chompret A, et al.: Genetic trans-
mission of susceptibility to cancer in families of children
with soft tissue sarcomas. Cancer 78: 1483–1491, 1996
103. Borresen A-L, Andersen TI, Garber J, et al.: Screening for
germ line TP53 mutations in breast cancer patients. Cancer
Res 52: 3234–3236, 1992
104. Sidransky D, Tokino T, Helzlsouer K, et al.: Inherited
p53 gene mutations in breast cancer. Cancer Res 52:
2984–2986, 1992
105. Statement of the American Society of Clinical Oncol-
ogy: genetic testing for cancer susceptibility, Adopted on
February 20, 1996. J Clin Oncol 14: 1730–1736, 1996
106. Statement of the American Society of Human Genetics on
genetic testing for breast and ovarian cancer predisposition.
Am J Hum Genet 55: i–iv, 1994
107. Quesnel S, Verselis S, Portwine C, Garber J, White M,
Feunteun J, Malkin D, Li FP: p53 compound heterozygos-
ity in a severely affected child with Li–Fraumeni syndrome.
Oncogene 18: 3970, 1999
108. Flaman JM, Frebourg T, Moreau V, Charbonnier F,
Martin C, Chappuis P, Sappino AP, Limacher IM, Bron L,
Benhatter J: A simple p53 functional assay for screening
cell lines, blood and tumours. Proc Natl Acad Sci USA 92:
3963, 1995
109. Birch JM, Blair V, Kelsey AM, Evans DG, Harris M,
Tricker KJ, Varley JM: Cancer phenotype correlates
with constitutional TP53 genotype in families with the
Li–Fraumeni syndrome. Oncogene 17: 1061, 1998
110. Harvey M, Vogel H, Morris D, Bradley A, Bernstein A,
Donehower LA: A mutant p53 transgene accelerates tumor
development in heterozygous but not nullizygous p53-
deficient mice. Nature Genet 9: 305, 1995
111. Lee JM Bernstein A: p53 mutations increase resistance to
ionizing radiation. Proc Natl Acad Sci USA 90: 5742, 1993
112. Lee JM, Abrahamson JLA, Kandel R, Donehower L,
Bernstein A: Susceptibility to radiation-carcinogenesis and
accumulation of chromosomal breakage in p53-deficient
mice. Oncogene 9: 3731, 1994
113. Nicol CJ, Harrison ML, Laposa RR, Gimelshtein IL,
Wells PG: A teratologic suppressor role for p53 in
benzo[a]pyrene-treated transgenic p53-deficient mice.
Nature Genet 10: 181, 1995
114. Wells PG, Kim PM, Laposa RR, Nicol CJ, Parman T,
Winn LM: Oxidative damage in chemical teratogenesis.
Mutat Res 12: 65, 1997
115. Ramqvist T, Magnusson K, Wang Y, Szeley L, Klein G:
Wild-type p53 induces apoptosis in a Burkitt lymphoma
(BL) line that carries mutant p53. Oncogene 8: 1495–1500,
1993
116. Show P, Bovey R, Tardy S, Sahli R, Sordat B, Cost J: Induc-
tion of apoptosis by wild-type p53 in a human colon tumor-
derived cell line. Proc Natl Acad Sci USA 89: 4495–4499,
1992
117. Hamada K, Alemany R, Zhang WW, Hittelman WN,
Lotan R, Roth JA, Mitchell MF: Adenovirus-mediated
transfer of a wild-type p53 gene and induction of apoptosis
in cervical cancer. Cancer Res 56: 3047–3054, 1996
118. Pirollo KF, Hao Z, Rait A, Jang YJ, Fee WE, Ryan P,
Chiang Y, Chang EH: p53 mediated sensitization of squa-
mous cell carcinoma of the head and neck to radiotherapy.
Oncogene 14: 1735–1746, 1997
119. Clayman GL, Frank DK, Bruso PA, Goepfert H:
Adenovirus-mediated wild-type p53 gene transfer as a sur-
gical adjuvant in advanced head and neck cancers. Clin
Cancer Res 5: 1715–1722, 1999
120. Li JH, Lax SA, Kim J, Klamut H, Liu FF: The effect of
combining ionizing radiation and adenoviral p53 therapy
in nasopharyngeal carcinoma. Int J Radiat Oncol Biol Phys
43: 607–616, 1999
121. Badie
B,
Kramar
MH,
Lau
R,
Boothman
DA,
Economou JS, Black KL: Adenovirus-mediated p53 gene
delivery potentiates the radiation-induced growth inhi-
bition of experimental brain tumors. J Neuro-Oncol 37:
217–222, 1998
122. Schuler M, Rochlitz C, Horowitz JA, Schlegel J,
Perruchoud AP, Kommoss F, Bolliger CT, Kauczor HU,
Dalquen P, Fritz MA, Swanson S, Herrmann R, Huber C: A
phase I study of adenovirus-mediated wild-type p53 gene
transfer in patients with advanced non-small cell lung can-
cer. Hum Gene Therapy 9: 2075–2082, 1998
123. Bischoff JR, Kirn DH, Williams A, Heise C, Horn S,
Muna M, Ng L, Nye JA, Sampson-Johannes A, Fattaey A,

243
McCormick F: An adenovirus mutant that replicates selec-
tively in p53-deficient human tumor cells. Science 274:
373–376, 1996
124. Pennisi E: Training viruses to attack cancers. Science 282:
1244–1246, 1998
125. Heise C, Sampson-Johannes A, Williams A, McCormick F,
Von Hoff D, Kirn D: ONYX-015, and E1B gene-
attenuated adenovirus, causes tumor-specific cytolysis and
antitumoral efficacy that can be augmented by standard
chemotherapeutic agents. Nat Med 3: 639–645, 1997
126. Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC,
Valent A, Minty A, Chalon P, Lelias JM, Dumont X,
Ferrara P, McKeon F, Caput D: Monoallelically expressed
gene related to p53 at 1p36, a region frequently deleted in
neuroblastoma and other human cancers. Cell 90: 809–819,
1997
127. Kaelin WG: The p53 gene family. Oncogene 18:
7701–7705, 1999
128. Schmale S, Bamberger C: A novel protein with strong
homology to the tumour suppressor p53. Oncogene 15:
1363–1367, 1997
129. Osada M, Ohba M, Kawahara C, et al.: Cloning and func-
tional analysis of human p51, which structurally and func-
tionally resembles p53. Nature Med 4: 839–843, 1998
130. Senoo M, Seki N, Ohira M, et al.: A second p53 related
protein, p73L, with homology to p73. Biochem Biophys
Res Commun 248: 603–607, 1998
131. Yang A, Kaghad M, Wang Y, et al.: p63, a p53 homolog at
3q27-29, encodes multiple products with transactivating,
death inducing and dominant negative activities. Molecular
Cell 2: 305–316, 1998
132. Jost CA, Marin MC, Kaelin WG: p73 is a human p53-
related protein that can induce apoptosis. Nature 389:
191–194, 1997
133. Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG,
Levrero M, Wang JY: The tyrosine kinase c-Abl regulates
p73 in apoptotic response to cisplatin-induced DNA dam-
age. Nature 399: 806–809, 1999
134. Agami R, Blandino G, Oren M, Shaul Y: Interaction of
c-Abl and p73
α and their collaboration to induce apoptosis.
Nature 399: 809–813, 1999
135. Takada N, Ozaki T, Ichimaya S, Todo S, Nakagawara A:
Identification of a transactivation activity in the COOH-
terminal region of p73 which is impaired in the naturally
occurring mutants found in human neuroblastomas. Cancer
Res 59: 2810–2814, 1999
136. Ichimaya S, Nimua Y, Kageyama H, Takada N,
Sunahara M, Shishikura T, Nakamura Y, Sakiyama S,
Seki N, Ohira M, Kaneko Y, McKeon F, Caput D,
Nakagawara A: p73 at chromosome 1p36.3 is lost in
advanced stage neuroblastoma but its mutation is infre-
quent. Oncogene 18: 1061–1066, 1999
137. Mihara M, Nimura Y, Ichimaya S, Sakiyama S, Kajikawa S,
Adachi W, Amano J, Nakagawara A: Absence of
mutation of the p73 gene localized at chromosome
1p36.3 in hepatocellular carcinoma. Br J Cancer 79:
164–167, 1999
138. Tsujimoto T, Mochizuchi S, Iwadate Y, Namba H,
Nagai M, Kawamoto T, Sunahara M, Yamamura A,
Nakagawara A, Sakiyama S, Tagawa M: The p73 gene is
not mutated in oligodendrogliomas which frequently have
a deleted region at chromsome 1p36.3. Anticancer Res 20:
2495–2497, 2000
139. Yokomizo A, Mai M, Bostwick DG, Tindall DJ, Qian J,
Cheng L, Jenkins RB, Smith DI, Liu W: Mutation and
expression analysis of the p73 gene in prostate cancer.
Prostate 39: 94–100, 1999
140. Marin MC, Kaelin WG: p63 and p73: old members of a
new family. Biochem Biophys Acta 17: M93–M100, 2000
141. Ongkeko WM, Wang XQ, Siu WY, Lau AW, Yamashita K,
Harris AL, Cox LS, Poon RY: MDM2 and MDMX bind and
stabilize the p53-related protein p73. Curr Biol 9: 829–832,
1999
142. White E, Prives C: DNA damage enables p73. Nature 399:
734–737, 1999
143. Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B,
Smits AP, Newbury-Ecob R, Hennekam RC, Van Buggen-
hout G, van Haeringen A, Woods CG, van Essen AJ, de
Waal R, Vriend G, Haber DA, Yang A, McKeon F, Brunner
HG, van Bokhoven H: Heterozygous germline mutations
in the p53 homolog p63 are the cause of EEC syndrome.
Cell 99: 143–153, 1999.
Address
for
correspondence:
David
Malkin,
Division
of
Hematology/Oncology, Room 9402, University Wing, The Hospital
for Sick Children, 555 University Avenue, Toronto, Ontario, Canada
M5G 1X8; Tel.: (416) 813-5977; Fax.: (416) 813-5327; E-mail:
david.malkin@sickkids.on.ca
Wyszukiwarka
Podobne podstrony:
Newell, Shanks On the Role of Recognition in Decision Making
THE ROLE OF CATHARSISI IN RENAISSANCE PLAYS - Wstęp do literaturoznastwa, FILOLOGIA ANGIELSKA
The Role of Women in the Church
Newell, Shanks On the Role of Recognition in Decision Making
The Role of Vitamin A in Prevention and Corrective Treatments
The Role of Design in Establishing a Brand
the role of networks in fundamental organizatioonal change a grounded analysis
The role of BRCA1 in DNA damage response
The Role of Dreams in Religious Enculturation
The Role of Fiestas in Andean Transnational Migration
Rescher, Nicolas The Role Of Rhetoric In Rational Argumentation
Fourth Lecture Universal Corporatism The Role of Intellectuals in the Modern World
[Solomon, Greenbegr & Pyszczynski] Tales from the crypt on the role of death in life
The Role of Women in the Christian Church doc
Mencej The Role of Legend in Constructing Annual Cycle
Against Bolshevism; Georg Werthmann and the Role of Ideology in the Catholic Military Chaplaincy, 19
The Role of Language in the Creation of Identity Myths in Linguistics among the Peoples of the Forme
więcej podobnych podstron