2007/2008
Wykłady genetyka
Genetyczne choroby charakterystyczne dla grupy etnicznej:
o Amerykanie
Talasemia α i β
Niedobór dehydrogenazy glukozo-6-fosforanowej
Hemogobinopatie HbS
o Chińczycy
talasemia α
Niedobór dehydrogenazy glukozo-6-fosforanowej
o Eskimosi
Wrodzony przerost nadnerczy
o Indianie Hopi
Albinizm
o Europejczycy
mukowiscydoza
fenyloketonuria
o Finowie
biegunki chlorkowe
miażdżyca wrodzona
ceroidolipofuscynoza
o mieszkańcy basenu morza śródziemnego
talasemia β,
hemoglobinopatia HbS
o Meksykanie
cukrzyca
rozszczep wargi i podniebienia
o Irlandczycy
bezmózgowie
o Apacze
ciężki niedobór odpornorności
o Japończycy
rozszczep wargi i podniebienia
o rdzenni Amerykanie
rozszczep wargi i podniebienia
cukrzyca
o Afrykanie
Talasemia
Hemoglobinopatia HbS
Przyczyny: odmienna dieta, różne czynniki społeczne.
Jasna karnacja i kolor włosów ludności Europy Północnej wykształciły się wskutek adaptacji do klimatu, w którym niedobór światła słonecznego powoduje zmniejszenie ilości melaniny i wit. D.
Strona 1 z 16
2007/2008
W pasie okołorównikowym częstość nosicieli może wynosić 25-40 % dla hemoglobiny sierpowatej.
Istnieje niewiele genów dla jednej konkretnej populacji. Przykładem choroby uwarunkowanej genem (unikatowym w populacji) jest dysplazja mózgowo-kostno-nerkowa wyłącznie u bliźniąt w populacji Hutterita, w której występuje kojarzenie spokrewnionych osób.
Analizy DNA:
o anemia sierpowata: β-globina
o cukrzyca typu I: insulina
o choroba Taya Sachsa: β-heksozoaminidaza A
o dystorfia mięśniowa typu Duchenn'a i Beckera: dystorfina
o fenyloketonuria: hydroksylaza fenyloalaninowa
o galaktozemia: galaktotransferaza
o głuchota: postać autosomalna recesywna – koneksyna Z(lub C)6
o hemofilia A i B: czynnik VIII i IX
o mukowiscydoza: CFTR
o niedokrwistość hemolityczna: dehydrogenaza gluko-6-fosforanowa o talasemia: α i β-globina
o zespół Marfana: fibrylina 1
o obrzęk naczynioruchowy: inhibitor C1
o wrodzony zespół złożonego niedoboru odporności: deaminaza adenozynowa Różnice powstają głównie przy:
dziedziczeniu wieloskładnikowym i recesywnym
odmiennej diecie
w wynniku czynników społecznych
Selekcja naturalna- rezultat niektórych różnic genetycznych między populacjami.
o jasna karnacja i kolor włosów ludności Europy Północnej, które wykształciły się wskutek adaptacji do klimatu, w którym niedobór światła obniża zawartość melaniny.
o - niedokrwistość sierpowato – krwinkowa.
o - niedobór dehydrogenazy – 6 – fosforanowej.
o - talsemia – stały się częste w niektórych populacjach, a geny odpowiedzialne za występowanie tych chorób uodporniły te populacje na malarię
*w pasie okołorównikowym częstość nosicielstwa 20 – 40% dla hemoglobiny sierpowatej
Istnieje niewiele genów specyficznych dla jednej konkretnej populacji. Przykładem choroby uwarunkowanej genem (unikatowym w populacji) jest dysplazja mózgowo-kostno-nerkowa wyłącznie u bliźniąt w populacji Hutterita, w której występuje kojarzenie spokrewnionych osób.
Strona 2 z 16
2007/2008
Cechy uwarunkowane genetycznie:
Monogenowe (6%)
o Autosomalne
o Sprzeżone autosomalne
Typu powiązania
Typu odpychania
o Z chromosomem X
o Z chromosomem Y- holandryczne
Poligenowe (ok. 90%)
Aberacje chromosomowe (4%)
o Autosomalne
o Heterosomalne
Mutacje
Częstość występowania na 1000 ludzi
Autosomalne
o Dominujące – 20
o Recesywne- 2
Sprzężone z X – 2
Wady rozwojowe – 6
Choroby przewlekłe dorosłych – 10
Mitochondrialne – rzadkie
Mutacje somatyczne – 250
Dziedziczenie autosomalne dominujące
o otoskleroza 1:330 (340)
o hipercholesterolemia rodzinna 1:100 – 1:500
o skrzywienie palca, komptodaktylia 1:1500
o nierwiakowłókniakowatość 1:3500
o naczyniakowatość twarzy i mózgu 1:5000
o achondroplazja 1:15000 – 1:77000
o choroba Huntingtona 1:15000 ( 1:250000)
o siatkówczak oka 1:20000
o wielopalczastość 1: 25000
Strona 3 z 16
2007/2008
o zespół błękitnych białkówek 1:25000
Dziedziczenie autosomalne dominujące:
Charakteryzuje się:
zmienną ekspresją (w tej samej rodzinie może być różne nasilenie choroby/cechy)
niepełną penetracją (cecha fenotypowa może być niewidoczna mimo, że osoba jest nosicielem mutacji genowej; może występować przeskok pokoleniowy)
duże ryzyko przekazania choroby – stałe, nie zależy od liczby dzieci
mozaikowość terminalna – pewne geny występują tylko w części komórek gonad jednogo z rodziców i efektem może być urodzenie się kolejnych chorych dzieci z małżeństwa klinicznie zdrowych rodziców.
może zależeć od płci rodziców (pląsawica H. – chory ojciec; dystrofia – matka)
wzrost ryzyka z wiekiem – zespół Aperta, achondroplazji wzrasta z wiekiem ojca
stan homozygotyczności jest rzadki, jeśli występuje – ciężki przebieg, uniemożliwia prokreację, śmiertelna.
Dziedziczenie autosomalne dominujące (tylko homozygoty)
P
KK
x kk
F1
Kk Kk Kk Kk
100% cecha dominująca
F2
KK Kk Kk kk
75% cecha dominująca
Proband to osoba u której jako pierwszej wykryto chorobę (przechodzi na pokolenie).
Mozaikowatość germinalna to obecność zmutowanych genów tylko w części komórek gonad jednego z rodziców. Efektem może być urodzenie chorych dzieci z małżeństwa klinicznie zdrowych osób.
Dziedziczenie autosomalne recesywne
o mukowiscydoza 1:2500 (Murzyni 1:17000, Chińczycy 1:90000) o niedobór α1 antytrypsyny 1:4000
o fenyloketonuria 1:2000 – 1:20000
o bielactwo całkowite (albinizm) 1:10000
o alkaptonuria 1:200000
o galaktozemia 1:12000
o zespół nadnerczowo-płciowy 1:490 – 1:67000
o idiotyzm amaurotyczny 1:6000 ( 1:50000)
P
RR x rr
F1
Rr Rr Rr Rr
0%, nosicielstwo
F2
RR Rr Rr rr
25% martwe urodzenie
Strona 4 z 16
2007/2008
Każda zdrowa osoba jest nosicielem co najmniej kilku alleli recesywnych nie ujawniających się fenotypowo
oboje rodzice chorego dziecka są bezobjawowymi klinicznie heterozygotami.
Prawdopodobieństwo, że każde ze zdrowego rodzeństwa chorego dziecka jest heterozygotą wynosi 2:3.
Ryzyko urodzenia się chorego dziecka wynosi zawsze 25% i jest ono stałe i nie zależy od liczby posiadanych już dzieci.
Poziomy rozkład chorych, cześciej wśród rodzeństwa, rzadko wśród innych członków rodziny.
Liczba różnych gamet
2n
Liczba potomstwa
2n x 2n = 4n
Liczba różnych fenotypów
2n
Liczba różnych genotypów
3n
Częstość cechy dominującej w pokoleniu II
(D)=3/4
Częstość cechy recesywnej w pokoleniu II
(d)=1/4
Prawo prawdopodobieństw
Prawdopodobieństwo zjawiska złożonego równa się iloczynowi prawdopodobieństw, które występują niezależnie od siebie.
P(M, n, o, P)= P(M) x P(n) x P(o) x P(P)
Dziedziczenie dominujące sprzężone z X
o zespół Blocka i Sulzbergera 1:75000
o krzywica oporna na działanie witaminy D
o zespół Retta 1:15000 ( 1:20000)
o zespół łamliwego chromosomu X 1:1250 (1:2000)
XDXD XDXd XdXd
X d Y X D Y : hemizygoty
Objawy choroby występują bez względu na płeć.
Mężczyźni są hemizygotami i choroba ma przebieg ciężki, często letalny, przekazują gen wszystkim córkom, nigdy synom.
Częstość występowania mutacji genowej u kobiet jest 2 razy większa niż u mężczyzn Dziedziczenie recesywne sprzężone z X
o deficyt dehydrogenazy gluko-6-fosforanowej 1:3 – 1:100
o zespół FraX 1:2000
o dystorfia mięśni dziecięca postępująca typu Duchenn'a i Beckera 1:3500
Strona 5 z 16
2007/2008
o hemofilia A 1:10000 – 1:20000
o hemofilia B (choroba Christmasów) 1:30000
o daltonizm
o hyperanonemia typ II
XRXR XRXr XrXr
XRY XrY
Pewną nosicielką jest kobieta, która ma co najmniej dwóch chorych synów, chorego syna i siostrzenice lub chorego syna i brata.
Tylko mężczyźni wykazują objawy choroby, często nie dożywając wieku reprodukcyjnego.
Częstość występowania mutacji genowej u kobiet jest 2 razy większa niż u mężczyzn.
Deficyt dehydrogenazy gluko-6-fosforanowej
Niedobór chroni przed malarią,
całkowity brak enzymu jest śmiertelny.
Napadowa hemoliza krwinek czerwonych (np. po aspirynie czy sulfonamidach) po zjedzeniu roślin strączkowych np. bobu.
Dreszcze, bóle brzucha, żółtaczka.
Anemia hemolityczna
Hyperanonemia typ II
17% kobiet posiadających mutację genu na jednym z chromosomów X ujawnia lżejsze lub cięższe objawy choroby prowadzące w niektórych przypadkach do ciężkiej śpiączki hyperanonemicznej.
Choroba recesywna.
Dziedziczenie sprzężone z Y
cecha występuje tylko u osobników płci męskiej i jest przekazywana synom.
Y* - niedobry chromosom.
Dziedziczenie wieloczynnikowe:
termin wieloczynnikowy jest stosowany do opisu cech będących efektem oddziaływania kombinacji czynników genetycznych i niegenetycznych.
Jeżeli uwzględnia się tylko czynniki genetyczne to określa się to jako dziedziczenie o poligenowe
o wieloczynnikowe.
Definicja obejmuje rodzinny charakter oraz zależność od czynników środowiska.
Dziecko wysokich rodziców prawdopodobnie będzie wyższe od dziecka niższych rodziców, ale odżywianie (czynnik środowiskowy) może spowodować zmianę.
Genetyczna predyspozycja jest dziedziczona od obojga rodziców.
Czynniki środowiskowe wpływające na ukształtowanie danej cechy mogą być różne u różnych osób, czarne włosy u jednej osoby mogą być wynikiem dziedziczenia po przodkach pochodzenia azjatyckiego, a u innych pochodzenia afrykańskiego.
Cechy:
− ilościowe – także, które mogą być zmierzone, wykazują zmienność ciągłą (mogą być różne i mieszczą się pomiędzy wartościami skrajnymi)
Strona 6 z 16
2007/2008
o
inteligencja
o
ogólna liczba listewek skórynych
o
liczba erytrocytów
o
ciśnienie krwi
o
masa ciała
o
barwa skóry, oczu
− jakościowe – nieciągłe, których fenotypy występują na zasadzie wszystko albo nic.
o
schizofrenia
o
cukrzyca
o
padaczka
o wrodzone wady serca
o
rozszczep wargi i podniebienia
o
wady cewy nerwowej
Dzieci mają tendencję do osiągania średniej wartości cech swoich rodziców. Regresja do średniej.
Przeciętna wartość jest nazywana wartością międzyrodzicielską.
Każdy z rodziców przekazuje połowę cech.
Dzieci rodziców, u których występują skrajne wartości cechy mają tendencję do występowania wartości bardziej przeciętnych (określane jako regresja do średniej).
Cechy ilościowe, które u normalnych osób mają zmienność ciągłą mogą również wykazywać różne odchylenia wskutek działania silnych czynników środowiskowych, np.
po zadziałaniu promieniowania dziecko może osiągnąć niższy wzrost niż teoretycznie mogłoby mieć.
W niektórych przypadkach predyspozycja do choroby łączy się z czynnikami środowiskowymi.
Zgodnie z modelem progowym wszystkie czynniki osobnicze, genetyczne, niegenetyczne decydują o podatności danej osoby na chorobę. Podatność ta polega w populacji rozkładowi wg krzywej Gaussa. Jeżeli podatność danej osoby przekroczy wartość progową, to te osoby będą chore, pozostali są zdrowi.
(Rys z Drewy. )
Modele mieszane zakładają istnienie jednego głównego genu o silnym oddziaływaniu, który działa na tle uwarunkowań wieloczynnikowych.
Uwarunkowania mendlowskie o stosunkowo wysokiej penetracji. Modele oligogenowe zakłądają małe występowanie genów działających wspólnie, możliwe jest występowanie heterogenności.
Niektóre choroby są dziedziczone w sposób mendlowski, inne mają podłoże niegenetyczne lub wieloczynnikowe.
Charakterystyka kliniczna chorób wieloczynnikowych:
o tendencja do rodzinnego występowania, nie wykazuje jednogenowych sposobów dziedziczenia
o często występują u osób jednej płci (sprężenie odźwiernika częściej u mężczyzn, toczek u kobiet)
o ryzyko ponownego wystąpienia w rodzinie jest takie samo dla wszystkich krewnych (np.
rodzeństwo, potomstwo i rodzice probanda mają przeciętnie połowę wspólnych genów) o ryzyko powtórzenia choroby u wszystkich krewnych 1-go stopnia jest identyczne Strona 7 z 16
2007/2008
o ryzyko spada w miarę oddalania się pokrewieństwa, np. ryzyko powtórzenia rozszczepu wargi i podniebienna wynosi ok. 4% dla rodzeństwa, a tylko 0,5% dla kuzynów chorego dziecka
o ryzyko powtórzenia się choroby u krewnych 1-go stopnia jest równe pierwiastkowi kwadratowemu częstości występowania choroby w populacji ogólnej o choroby wieloczynnikowe są częstsze wsród dzieci spokrewnionych rodziców (większa ilość wspólnych genów predysponujących do wystąpienia choroby) Czynniki, które wpływają na ryzyko powtórzenia się chorób wieloczynnikowych:
Wyższe jeżeli jeden z bliskich krewnych jest chory, np. ryzyko wystąpienia rozszczepu wargi z lub bez rozszczepu podniebienia u dziecka mającego jedną osobę wśród rodzeństwa chorą – 4%, zaś gdy liczba rodzeństwa chorego to 2 osoby, to ryzyko wzrasta do 10%. Oznacza to, że małżeństwo, które ma więcej niż jedno chore dziecko ma większe predyspozycje niż małżeństwo z jednym chorym dzieckiem. Dziedziczenie to jest całkiem odmienne od jednogenowego, w którym ryzyko powtórzenia się choroby zależy od genotypu rodziców a nie od liczby chorych dzieci.
Ryzyko jest wyższe dla krewnych tych dzieci, których płeć jest rzadziej dotykana chorobą. Aby choroba ujawniła się u osób płci rzadziej dotykanej chorobą muszą one mieć większe predyspozycje, tak więc i krewni mają również większe ryzyko wystąpienia podobnej choroby, np. zwężenie odźwiernika występuje 5% częściej u mężczyzn niż u kobiet, ryzyko powtórzenia choroby u brata chorego wynosi 4% jeśli dziecko jest chłopcem, 9% jeśli to dziewczynka.
Ryzyko powtórzenia choroby u krewnych jest wyższe w wyniku ciężkiej choroby probandów, np. powtórzenie rozszczepu wargi i podniebienia wynosi 2,5% dla chorych z jednostronnym rozszczepem, a 6% z dwustronnym.
Czynniki środowiskowe mają największą możliwość spowodowania choroby u osób mających predyspozycje genetyczne. Sterowanie czynnikami środowiskowymi może być skuteczne w celu zapobiegania chorobie.
Badania bliźniąt:
Badania bliźniąt są często stosowane do odróżnienia cech wieloczynnikowych od jednogenowych lub spowodowanych czynnikami niegenetycznymi.
Bliźnięta jednojajowe (monozygotyczne) mają jednakowe geny i powinny wykazywać 100% zgodność (konkordacja) wystąpienia chorób jednogenowych.
Bliźnięta dwujajowe (dizygotyczne) powinny wykazywać 50% zgodność dla chorób dominujących jednogenowych i 25% dla recesywnych.
Choroby występujące rodzinnie nie muszą być genetyczne. Zgodność etiologiczna dla bliźniąt jedno- i dwujajowych powinna być identyczna.
Jeśli jedno z bliźniąt jednojajowych cierpi na chorobę wieloczynnikową, ryzyko powtórzenia choroby u drugiego jest mniejsze niż 100%, a często nawet niż 50%. Zgodność występowania choroby jest większa
Strona 8 z 16
2007/2008
Nazwa
jednojajowe
dwujajowe
astma oskrzelowa
47%,
24%
stopa końska
33%
7%,
reumatoidalne zapalenie
34%
4%
stawów
choroba maniakalno-
50%
5%
depresyjna
nowotwór
7%
3%
Rozszczep wargi
38%
8%
nadciśnienie
25%
7%
Choroba
Częstość w populacji
U krewnego stopnia I
Wady serca
0,5 – 1%
7%
Wrodzone zwichnięcie stawów
0,07%
3,0%
biodrowych
Atopia
2,0 – 3,0%
3-4 %
Łuszczyca
2,5%
10%
Dziedziczenie mitochondrialne:
Przykładem dziedziczenia, w którym informacja genetyczna jest przekazywana tylko od matki(dziedziczenie matczyne) jest dziedziczenie mitochondrialne. Rozwijający się zarodek obejmuje mitochondria wyłącznie od matki, gdyż stanowią one składnik cytoplazmy komórki jajowej. Do niej po zapłodnieniu plemnik wnosi tylko materiał genetyczny zawarty w jego jądrze. Od ok. 3 podziału mitochondria plemnika zanikają.
Do podstawowych cech dziedziczonych mitochondrialnie można zaliczyć:
•
specyficzność tkankową
•
nasilanie się objawów chorobowych wraz z wiekiem
W kom. jajowej znajduje się kilkaset mitochondriów ok. 100 000 cząstek mitochondrialnego DNA (u plemnika 100). Objawy kliniczne niektórych chorób mitochondrialngo ujawniają się dopiero, gdy około 85%
cząstek mtDNA zawiera zmutowane sekwencje. Tak, więc pozostaje kwestią czasu, kiesy ten stan zostaje osiągnięty.
Dziedziczenie mitochondrialne:
LHOH – zespół Lebera-Lebera, wrodzona neuropatia n. wzrokowego
MELAS – encefalopatia z kwasicą mleczanową i napadami podobnymi do udaru
CEPEO – porażenie mięsni odwodzących oka
MERFF – padaczka biochemiczna z cukrzycą i głuchotą
NARAP – neuropatia obwodowa z ataksja i barwnikowym zwyrodnieniem siatkówki Na ogół efekty mutacji mitoch. ujawniają się u osób dorosłych, u dzieci wykrywa się je rzadko. U tych małych szkrabów może występować głuchota – jeśli podaje się antybiotyki aminoglikorynowe to wówczas średni wiek utraty słuchu to 5 lat. Ta sama głuchota może pojawić się u osób , które nie przyjmowały tych antybiotyków; średni czas utraty słuchu wtedy wynosi 20 lat.
Objawy chorób mitochondrialnych w rodzinie mogą się różnić stopniem rodzicielstwa lub być odmienne.
Zależne jest to z jednej strony od % zmutowanych cząstek, a z drugiej od tego, do jakich tkanek trafia większa ilość mtDNA z defektem.
Objawy kliniczne mogą w zasadzie dotyczyć wszystkich tkanek i narządów. Wykazanie, że dana choroba dziedziczy się wyłącznie matczyne nie zawsze jest łatwe szczególnie wówczas, gdy rodziny nie są zbyt liczne albo nie można sporządzić rodowodu. Zmiany fenotypowe na ogół są stwierdzane dopiero wtedy, gdy procent zmutowanych cząstek mtDNA jest większy niż pewna wartość.
Strona 9 z 16
2007/2008
Uważa się, że musi być zmutowane 95% cząstek DNA, jeżeli zmiana dotyczy genów kodujących tRNA, u których występują tylko mutacje punktowe, a 60% cząsteczek mtDNA, jeżeli rodzajem defektu są delecje.
Stąd tez wielu członków danej rodziny może mieć zmutowane mtDNA, ale efekty mutacji będą przejmować się tylko u niektórych osób. Problem stanowi tez to, że efekty danej mutacji zależne są od tego, w której tkance dostępne jest więcej zmutowanych cząstek mtDNA.
Podczas podziału komórkowego mitochondria rozdzielają się losowo, więc na zwane brak mutacji w jednej tkance świadczy o jej nieobecności u innej. Co więcej niektóre czasteczki mtDNA mogą ulegać namnarzaniu w tkankach dzielących się (np. krew), ale nigdy w tkankach postmitotycznych (mięśnie, mózg). Znane są przypadki, w których zmutowane cząstki stwierdza się tylko w jednej tkance (np.
mięśniowej).
Poligenia:
o geny uzupełniające – komplementarne
o geny hamujące – epistatyczne
o geny sumujące się – polimeryczne
o kumulujące się – czynniki wielokrotne
Geny uzupełniające się:
np. zdolność wydzielania antygenu, np. w ślinie
A – gen antygenu A, U – gen uzupełniający
P
AAUU x aauu
F1
AaUu
F2
AU-9 Au-3 aU-3 au-1
wydzielacze : niewydzielacze = 9:7
Geny epistatyczne
np. dziedziczenie barwy upierzenia kury
C – gen barwy ciemnej, H – gen hamujący
P
CCHH x cchh
F1
CcHh
F2
CH-9 Ch-3 cH-3 ch-1
barwa ciemna : barwa jasna = 3:13
Geny polimeryczne – sumujące się:
P
AABBCC x aabbcc
F1
AaBbCc
ABC
ABc
Abc
AbC
aBC
aBc
abC
abc
ABC
6
5
4
5
5
4
4
3
ABc
5
4
3
4
4
3
3
2
Abc
4
3
2
3
3
2
2
1
AbC
5
4
3
4
4
3
3
2
suma genów dominujących
aBC
5
4
3
4
4
3
3
2
aBc
4
3
2
3
3
2
2
1
abC
4
3
2
3
3
2
2
1
abc
3
2
1
2
2
1
1
0
Strona 10 z 16
2007/2008
Gdy AaBbCc x AaBbCc to trójkąt Pascala:
0
1
1
1
1
2
1
2
1
3
1
3
3
1
4
1
4
6
4
1
5
1
5
10
10
5
1
6
1
6
15
20
15
6
1
wyższy wzrost
taki sam wzrost
niższy wzrost
najwyższe dziecko P=1:64
Grupy krwi
Poznano sekwencję 250 antygenów, które należą do 23 układów antygenowych krwinek czerwonych. Białka transportujące lub tworzące kanały w błonie komórkowej (wymiana jonowa), czy tworzące kanaliki wodne, białka adhezyjne uczestniczące w oddziaływaniu międzykomórkowym, inne są receptorami dla bakterii lub wirusów, białka enzymatyczne.
Układy antygenów
o AB0: enzym (5)
o MNS: receptor (40)
o P: receptor (1)
o Rh – transporter (52)
o Luthean – adhezja (20)
o Kell – enzym (7)
o Lewis – adhezja (19)
o Duffy – receptor (1)
o Kidd – transporter (18)
o Diego – transporter (17)
o Xg – ligand/adhezja (X)
w nawiasach!
od AB0 do Luthean – liczba antygenów
od Kell do Xg – lokalizacja w chromosomie.
Układ AB0
o 1900, układ klasyczny
o antygeny można wykryć u 6-cio tygodniowego
o przeciwciała u 6-cio miesięcznego płodu.
Geny: IA
IA1
IA2
IA3
IB
IB1
IB2
IB3
i: gen amorficzny, nie koduje żadnego enzymu
Strona 11 z 16
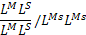
Wykłady z Genetyki Kusy
2007/2008
o dominowanie hierarchiczne: IA1 dominuje nad IA2
o kodominacja (grupy krwi AB0, Kell, MNS): IAIB
o Enzymy erytrocytarne:
o kwaśna fosfataza
o kinaza adenylowa
o białka osocza haptoglobina
o Antygeny powierzchowne komórek układ HLA
Fenotyp
Genotyp
Częstość (%)
Przeciwciała
A1
A1A1, A1A2, A1i 34
anty-B
A2
A2A2, A2i
6
anty-B
B
BB, Bi
19
anty-A
A1B
A1B
7
brak
A2B
A2B
2
brak
0
ii
32
anty-A, anty-B
•
ślina, nasienie 128-1024
•
wody płodowe 64-86 (256)
•
erytrocyty 8-31
•
łzy-2-4
•
mocz 2-4
•
Nie ma w:
o płyn mózgowo-rdzeniowy
o chrząstka
o soczewka oka
o Włosy
o zrogowaciały naskórek
Układ MNS
Fenotyp
Genotyp
Częstość (%)
MN
MN
48,3
M
MM
35,4
N
NN
16,3
geny sprzężone autosomowo (2 loci genetyczne) lub 1 para loci i allele wielokrotne Układ P
Fenotyp
Genotyp
Częstość (%)
p1
p1 p1, p1 p2, p1 pk
78,7
p2
p2 p2, p2 pk, p2 p
21,3
pk
pk pk, pk p
0,001
p
pp
0,01
Najczęściej fenotyp p1 bo dominuje – 80%
Strona 12 z 16
2007/2008
Układ Rh
(geny sprzężone autosomowo)
Fenotyp
Genotyp
Częstość (%)
Rh(-)
cde
40
Rh(+)
cDE
39,4
Cde
15
CDe
3,2
Cde
2,0
CDe
CDe
cDe
CDe
cde
cde
cde
cde
CDe
Cde
Cde
CDe
Układ Kell
allele K, k; antygen K – silny immunogen
Fenotyp
Genotyp
Częstość (%)
Kell+
KK
0,18
Kk
8,08
Kell-
kk
91,7 (Kell null)
Fenotyp kk- 92%
Układ Kidd
Jk (a+ b-)
Jka Jka
Jk (a- b+)
Jkb Jkb
Jk (a+ b+)
Jka Jkb
Jknull – brak ekspresji genu Jk, występuje gen hamujący
Układ Duffy
receptor dla Plasmodium vivax i prozapalnych cytokin Fenotyp
Genotyp
Częstość (%)
Fy (a+ b-)
Fya Fya
17
Fy (a- b+)
Fyb Fyb
34
Fy (a+ b+)
Fya Fyb
50
Fy null – a-b- nie chorują na malarię
Układ Luthean
Fenotyp
Genotyp
Lu (a+ b+)
Lua Lub
Lu (a+ b-)
Lua Lua
Lu (a- b+)
Lub Lua
Strona 13 z 16
2007/2008
Układ Lewis
Ściśle związany z układem AB0, pojawia się kilka dni po pojawieniu się pępowiny.
Fenotyp
Genotyp
Le (a+ b+)
Lea Leb
Le (a+ b-)
Lea Lea
Le (a- b+)
Leb Lea
Le null
¾ a-b- -związany z ABO w krwi pępowinowej po 10 danich życia Układy grupowe białek surowicy (dziedziczą się zgodnie z prawami Mendla) Różnią się w polu elektrycznym
Układ fosfoglukomutazy: chromosom 1, 4, 6
geny: PGM 1
2
3
1 , PGM1 , PGM1
Genotyp
Fenotyp
PGM 1
1
1 PGM1
PGM1 1-1
PGM 2
1
1 PGM1
PGM1 1-2
Układ dehydrogenazy 6-fosfoglukanianowej chromosom 1
(krwinki białe, bakterie roślin ryżu, tkanki zwierzęce)
geny: PGDA, PGDC, PGDF, PGDH, PGDR, PGDE
Fenotyp
Genotyp
I
PGDA PGDA
II
PGDA PGDC
III
PGDC PGDC
IV
PGDA PGDR
V
PGDA PGDH
Układ aminotransferazy alaninowej
(tkanki zwierzęce, bakterie)
geny: GPT1, GPT2
Fenotyp
Genotyp
Częstość (%)
GPT 1-1
GPT1 GPT1
25,2
GPT 2-1
GPT2 GPT1
50,7
GPT 2-2
GPT2 GPT2
24,1
Układ grupowy haptoglobina
(mają zdolność do wiązania wolnej hemoglobiny)
geny: Hp1:
Hp1F, Hp1S
Hp2:
Hp2FF, Hp2SS, Hp2FS
HpB, HpD, HpM, HpL
Strona 14 z 16
2007/2008
Fenotypy
Częstość (%)
Hp 1-1
13,9
Hp 2-1
45
Hp 2-2
39,2
Układ grupowy transferyn β-gloulina
geny: TfB, TfC, TfD ( TfBo-1, TfChi, TfFin )
fenotypy: Tf B-B, Tf B-C, Tf C-C, Tf B-D
układ grupowyc ceruloplazminy (α-globuliny)
geny: CpA, CpB, CpC, CpNH
fenotypy: Cp: A, AB, B, AC, BC, A-NH, B-NH
Układ kwaśnej fosfatazy
geny: PA, PB, PC
fenotypy: P: A, B, C, BA, CA, CB
Genetyka populacji
Prawo Hardyego-Weinberga: w dostatecznie licznej populacji mendlowskiej względna częstość każdego z alleli jest stała, jeżeli nie ma selekcji fenotypów, ani mutacji tych alleli.
Populacja mendlowska (panmigtyczna) to populacja której związki rozrodcze są przypadkowe i nieograniczone.
Izolacja populacji polega na braku migracji osobników i krzyżówek z osobnikami innej populacji, co uniemożliwia dopływ i odpływ genów, które charakteryzują populację.
W razie spełnienia tych warunków mówimy, że populacja znajduje się w stanie równowagi genetycznej, a względna częstość genotypów jest wyrażona równaniem: p + q = 1
p2 + 2pq + q2 = 1
p: częstość genu D
q: częstość genu d
Strona 15 z 16
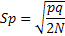
Wykłady z Genetyki Kusy
2007/2008
Czynniki wpływające na zachowanie równowagi genetycznej w populacji naturalnej:
•
mutacja
•
selekcja
•
izolacja
•
migracja
•
dobór naturalny
•
dryf genetyczny
Gdyby ich nie było to częstość genotypu byłaby taka sama.
Mutacje – oceny mogą być bezpośrednie i pośrednie.
Metoda bezpośrednia: częstość mutacji równa się pojawieniu się cechy dominującej u rodziców nie wykazujących tej cechy.
Metoda pośrednia polega na założeniu, że w populacji zachodzi równowaga pomiędzy przyrostem częstości genów spowodowanych mutacja, a zmniejszeniem się częstości zmutowanego allela w wyniku selekcji.
Mutacje w stanie homozygotycznym są zawsze szkodliwe; również niekorzystne są wszystkie nowe mutacje przy niezmienionych warunkach środowiska, zaś tempo mutacji spontanicznych wynosi ok. 10-5 / locus na jedno pokolenie zależy od środowiska. Los każdej nowej mutacji zależy od wartości przystosowawczej jej nosicieli.
Dobór naturalny wywołany działaniem czynników abiotycznych i biotycznych jest podstawowym mechanizmem mikroewolucji.
Selekcja
Jeżeli poszczególne fenotypy danej pary cech nie są równoważne pod względem śmiertelności i rozrodu, występuje dobór naturalny.
Powoduje to oddalenie od teoretycznego rozkładu cech w populacji. Dobór naturalny wywołany działaniem czynników abiotycznych i biotycznych środowiska jest podstawowym mechanizmem mikroewolucji.
Selekcja dotyczny tylko cech dziedziczących się i jest nieskuteczna w zakresie czystej linii, czyli u homozygot. Działa ona w gametogenezie, embriogenezie i rozwoju postnatalnym.
Selekc
ja cechy dominującej jest szybka, a cechy recesywnej wolna i mniej wydajna, ponieważ znaczna część genów zachowuje się w heterozygotach, które zwykle nie podlegają doborowi naturalnemu.
Współczynn
ik selekcji ujemnej jest to ułamek wskazujący, jaka część osobników o danym fenotypie ginie lub nie dochodzi do rozrodu. 0-1
Wzór na częstość fenotypu w n-pokoleniu
• fenotyp dominujący P(A)n = 2pq (1 – S)n
• fenotyp recesywny P(a)n = [q0 / (1 + nq0)]2
Dryf genetyczny
• Miarą wielkości dryfu genetycznego jest odchylenie standardowe allela ulegającego dryfowi. znak + i – oznaczają, że częstość allela może wzrastać lub maleć. Dryfowi ulegają osobnicy, nie allele!
Strona 16 z 16