Elementy katalizy
Nawet w warunkach korzystnych dla danej reakcji z termodynamicznego punktu widzenia, przebieg jej może być niekiedy bardzo powolny. Problem ten nabiera szczególnej wagi w przypadku prowadzenia procesów na skalę techniczną. Równania kinetyczne wskazują, że zwiększenie szybkości reakcji można osiągnąć zwiększając stężenia substratów lub zwiększając stałą szybkości. Możliwość zwiększania stężenia (ciśnienia) jest często ograniczona (np. ograniczona wytrzymałość aparatury, rozpuszczalność). Podwyższenie temperatury zwiększa szybkość reakcji, jednakże w przypadku reakcji egzotermicznych powoduje obniżenie stałej równowagi. Ze wzoru Arrheniusa
![]()
gdzie: A - stała całkowania (czynnik częstości, czynnik przedwykładniczy),
Ea - energia aktywacji: najmniejsza energia, jaką muszą mieć (w przeliczeniu na 1 mol) cząsteczki substratów, aby mogły wejść w daną reakcję,
![]()
(czynnik Boltzmanna) - ułamek drobin, które osiągnęły minimalną energię potrzebną, aby zaszła reakcja.
Wynika, że wzrost stałej szybkości reakcji można osiągnąć poprzez zmianę mechanizmu reakcji tak, aby obniżeniu uległa energia aktywacji lub też nastąpiło zwiększenie czynnika przedwykładniczego.
Zaobserwowano, że pewne substancje obecne w układzie mogą wpływać na szybkość reakcji pozostając w niezmienionej postaci i ilości po zakończeniu procesu. Katalizatorem nazwano substancję, która wprowadzona do układu reagującego zwiększa szybkość tej reakcji, skracając tym samym czas potrzebny do osiągnięcia stanu równowagi. Katalizatorem ujemnym (inhibitorem) określa się substancję, która zwalnia przebieg danej reakcji.
Rozważmy wpływ katalizatora na kinetykę reakcji odwracalnej
k1
A + B ↔ C + D
k2
Szybkość reakcji przebiegającej ze stałą k1 dana jest równaniem
v1 = k1 [A][B] (1)
Dla reakcji odwrotnej napiszemy
v2 = k2 [C][D] (2)

W stanie równowagi v1 = v2
Jeśli więc katalizator zwiększa n-krotnie k1, a jednocześnie nie wpływa na stałą równowagi K, to musi też n-krotnie zwiększać k2. Na przykład jony H3O+ katalizują zarówno reakcję hydrolizy estrów jak i reakcję estryfikacji.
Kataliza homogeniczna ma miejsce wtedy, kiedy katalizator stanowi jeden ze składników fazy (ciekłej lub gazowej), w której zachodzi reakcja. Przykładem może być reakcja z udziałem NO jako katalizatora
NO
SO2 + ½ O2 → SO3,
przebiegająca według mechanizmu:
NO + ½ O2 → NO2
NO2 + SO2 → SO3 + NO
O katalizie heterogenicznej mówimy wówczas, gdy katalizator stanowi odrębną fazę w układzie reagującym a reakcja przebiega na granicy faz. Najczęściej katalizator jest ciałem stałym. W przypadku koloidalnego rozdrobnienia katalizatora (np. koloidalna platyna) mamy do czynienia z katalizą, mikroheterogeniczną. Jeżeli jeden z tworzących się w reakcji produktów katalizuje daną reakcję, to proces nazywamy procesem autokatalitycznym. Rozwinięcie reakcji autokatalitycznej jest możliwe wtedy, gdy jej szybkość bez katalizatora jest różna od zera albo, gdy w układzie reakcyjnym występuje od początku reakcji katalizator.
Ogólnie można stwierdzić, że reakcje katalityczne przebiegają przez stadia pośrednie, a w końcowym etapie katalizator ulega regeneracji. Produkty przejściowe w katalizie homogenicznej to jony, rodniki, cząstki obojętne. Produktami przejściowymi w katalizie heterogenicznej są natomiast związki powierzchniowe tworzące się w wyniku chemisorpcji.
Chemisorpcja prowadzi do istotnych zmian w strukturze elektronowej cząsteczki, w wyniku, czego dochodzi do obniżenia bariery energetycznej reakcji chemicznej.
Kinetyka homogenicznych reakcji katalitycznych
Kinetykę reakcji homogenicznych możemy rozważyć na przykładzie reakcji, która bez katalizatora przebiegałaby następująco:
A + B → P
W obecności katalizatora przebiegać może według mechanizmu:
I / A + K ↔ AK (produkt przejściowy)
II AK + B → P + K
przy czym tworzenie produktu przejściowego przebiega poprzez stadium powstawania kompleksu aktywnego
A + K → [A....K] → AK
kompleks aktywny
Rozważania kinetyczne w oparciu o ten mechanizm prowadzą do wniosku, że szybkość tworzenia produktu końcowego reakcji jest proporcjonalna do stężenia katalizatora. Stwierdzenie to określa równanie Oswalda, które możemy zapisać w postaci następujących równań:
![]()
(4)
lub
![]()
(5)
gdzie x jest stężeniem produktu P, a jest stężeniem początkowym substratu, k jest stałą szybkości reakcji przebiegającej bez katalizatora, k' jest stałą szybkości reakcji katalitycznej, [K] jest stężeniem katalizatora.
Jeśli reakcja jest rzędu pierwszego, ostatnie równania można zapisać w postaci
![]()
(6)
oraz
![]()
(7)
Jeśli wyznaczymy stałe szybkości dla kilku reakcji przebiegających z udziałem katalizatora o różnych stężeniach, będziemy mogli sprawdzić, czy równanie Ostawlda jest spełnione. W tym celu należy sporządzić wykres zależności doświadczalnie wyznaczonych stałych szybkości od stężenia katalizatora:
![]()
(8)
Na podstawie tego wykresu możemy wyznaczyć stałą szybkości reakcji nie katalizowanej (jako punkt przecięcia z osią keksp ) oraz k' z nachylenia prostej.
Kinetyka heterogenicznych reakcji katalitycznych
Reakcje gazowe przy udziale stałych katalizatorów
Spośród reakcji katalitycznych, w których katalizator znajduje się w innej fazie niż reagenty, omówimy reakcje przebiegające przy udziale stałych katalizatorów. Reakcje te nazywamy niekiedy kontaktowymi. Stanowią one podstawę wielu procesów technologicznych (np. synteza amoniaku, utlenianie SO2 do SO3). Mechanizm tych reakcji jest różnorodny i bardzo złożony. Można w nim wyróżnić następujące etapy elementarne:
dyfuzja substratów do powierzchni,
adsorpcja substratów na powierzchni,
reakcja chemiczna na powierzchni,
desorpcja produktów z powierzchni,
dyfuzja produktów od powierzchni.
Są to procesy następujące po sobie, a najwolniejszy z nich będzie etapem ograniczającym szybkość całej reakcji. Procesy dyfuzji (1) i (5) są zwykle bardzo szybkie. Etapy (2) i (4) są w większości wypadków szybsze od procesu (3) i reakcja chemiczna przebiegająca na powierzchni najczęściej stanowi najwolniejsze stadium procesu katalitycznego. Warunkiem koniecznym w reakcji katalitycznej jest chemisorpcja, co najmniej jednego z substratów na powierzchni katalizatora. Chemisorpcja związana jest z reguły z wymianą elektronów między adsorbentem i adsorbowanymi substratami i utworzeniem między nimi silnych wiązań (atomowych lub jonowych). Chemisorpcji towarzyszyć może dysocjacja cząsteczek. Ze względu na duże znaczenie chemisorpcji w reakcjach kontaktowych korzystne jest możliwie duże rozwinięcie powierzchni katalizatora osiągane poprzez jego rozdrobnienie lub zapewnienie odpowiedniej porowatości.
Jeśli na katalizatorze zachodzi reakcja jednocząsteczkowa A→ produkt P, to szybkość adsorpcji substratu określa równanie
![]()
(9)
w którym p jest ciśnieniem substratu, ![]()
jest ułamkiem określającym stopień obsadzenia powierzchni katalizatora.
Szybkość desorpcji produktu można wyrazić jako
![]()
(10)
W przypadku dostatecznie szybkiego ustalania się równowagi między adsorpcją a desorpcją reagentów możemy założyć, że w stanie równowagi
![]()
(11)
Porównując prawe strony równań opisujących szybkość adsorpcji i desorpcji otrzymujemy równanie izotermy Langmuira
![]()
(12) Szybkość reakcji zachodzącej na powierzchni katalizatora zdefiniujemy jako zmianę liczby moli substratu odniesioną do jednostki czasu i jednostki powierzchni S, na której zachodzi chemisorpcja i reakcja.
![]()
(13)
[A]ads - powierzchniowe stężenie A,
![]()
- ułamek czynnej powierzchni katalizatora zajętej przez substancję A,
![]()
- sumaryczna powierzchnia ![]()
centrów aktywnych, z których każdy może związać jedną cząsteczkę A.
Rozważymy teraz dwa szczególne przypadki. Jeżeli adsorpcję produktów można zaniedbać, ciśnienie substratu jest niewielkie i współczynnik adsorpcji jest niski, to
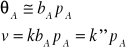
Reakcja jest reakcją pierwszego rzędu (np. rozkład HJ na Pt). Jeżeli ciśnienie A jest dostatecznie wysokie, a adsorpcja substratu silna (duża wartość ![]()
), to
![]()
W tym przypadku reakcja jest rzędu zerowego. Jeśli adsorpcja substratu A jest umiarkowanie silna, wtedy obserwuje się ułamkowy rząd reakcji. Zdarza się, że cząsteczki produktów również adsorbują się na powierzchni katalizatora. Wtedy liczba centrów aktywnych dostępnych dla cząsteczek A ulega zmniejszeniu, a mechanizm procesu staje się bardziej skomplikowany.
2

![]()
[K]
Wyszukiwarka