AMINOKWASY
Budowa aminokwasów- związki organiczne zawierające w cząsteczce przynajmniej jedną grupę aminową -NH2 i karboksylową -COOH. Są substancjami krystalicznymi, dobrze rozpuszczalnymi w wodzie.
Rozróżnia się aminokwasy:
alifatyczne i aromatyczne,
egzogenne-trzeba je dostarczyć z pożywieniem.
endogenne- wytwarzane przez organizm
białkowe i niebiałkowe
podział na podstawie właściwości fizykochemicznych lub łańcuchów bocznych R (4 grupy):
α-aminokwasy z niepolarnym łańcuchem bocznym R; właściwości hydrofobowe, rola: formowanie wiązań hydrofobowych wewn. Cząst.białka, np.: L-fenyloalanina, L-alanina, L-walina, L-leucyna, L-izoleucyna, L-prolina
α-aminokwasy zawierające niezjonizowane albo w małym stopniu ulegające proteolizie rodniki polarne; należą tu AA z grupą hydroksylową -OH (np. seryna), tiulową (cysteina), amidy AA kwaśnych; biorą udział w tworzeniu wiązań wodorowych i dwusiarczkowych w cząst. białek
AA monoamino-dikarboksylowe (kwasowe), zawierają w rodniku dodatkową grupę karboksylową, np.: kw. L-asparaginowy i kw.L-glutaminowy;
AA zasadowe (w rodniku dod.grupa zasadowa), np.: L-lizyna, L-arginina, L-histydyna.
AA kwaśne i zasadowe decydują o ładunku elektrycznym
cząst.białka.
Aminokwasy mają własności amfolitów. Ich cechą charakterystyczną jest tzw. punkt izoelektryczny.
AA białkowe- jedna grupa aminowa jest związana z tym samym atomem węgla, z którym związana jest jest grupa karboksylowa oraz atom wodoru. Ten atom węgla określa się jako atom węgla α. Wszystkie aminokwasy białkowe są α-aminokwasami.
Z atomem węgla a związane są cztery różne podstawniki a więc są to cząsteczki chiralne i mogą występować w dwóch odmianach czynnych optycznie (enancjomerach).
Konfiguracja grup związanych z atomem węgla a jest dla wszystkich aminokwasów białkowych taka sama i oznacza się ją jako L. Wszystkie aminokwasy białkowe są więc L- α-aminokwasami.
AA niebiałkowe, aminokwasy występujące w komórkach głównie roślin i mikroorganizmów, w postaci nie związanej z białkami. Są homologami, izomerami (izomeria) lub pochodnymi aminokwasów białkowych. Przykładem aminokwasu niebiałkowego u roślin jest allicyna, u zwierząt nieliczne, np. beta-alanina czy tauryna. U mikroorganizmów występują jako produkty metabolizmu i są składnikami antybiotyków (np. D-seryna, D-leucyna). Niektóre z aminokwasów niebiałkowych roślin mogą wywoływać zaburzenia u zwierząt.
Stereoizomery AA- cząsteczki chiralne występują w postaci dwóch odmian przestrzennych będących swoimi lustrzanymi odbiciami. Odmiany cząsteczek chiralnych nie są identyczne- stanowią parę izomerów nazywane są enancjomerami i należą do stereoizomerów. Enancjomery mają zdolność do skręcania płaszczyzny światła spolaryzowanego o ten sam kąt ale w przeciwną stronę. Wyjątek stanowi glicyna gdyż nie posiada asymetrycznego węgla i pozostaje obojętna optycznie
![]()
Właściwości amfoteryczne AA- AA tworzą z kwasami i zasadami sole; spowodowane są obecnością w ich cząsteczkach kwasowej gr.karboksylowej -COOH i zasadowej gr.aminowej -NH2.
W roztworach wodnych-środowisko obojętne- AA wystepują gł w postaci jonu obojnaczego.
W środowisku zasadowym jon obojnaczy wykazuje właściwości kwasowe: gr. -NH3+ reaguje z anionem OH- i z jonu obojnaczego powstaje anion AA i woda
W środowisku kwasowym wykazuje właściwości zasadwe: gr. -COO- reaguje z protonem kwasu, powstaje kation AA
![]()
Punkt izoelektryczny AA-takie pH roztworu, przy którym cząst. AA występują w formie jonu obojnaczego, którego ładunek elektryczny jest równy 0. Wartość pH obl.ze wzoru:
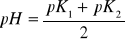
gdzie K1-stała dysocjacji gr -COO-, K2-stała dysocjacji gr -NH3+; p= -log.
Klasyfikacja AA białkowych:
Obojętne:
Alanina Ala
Asparagina Asn
Cysteina Cys
Glutamina Gln
Glicyna Gly
Izoleucyna Ile
Leucyna Leu
Metionina Met
Fenyloalanina Phe
Prolina Pro
Seryna Ser
Treonina Thr
Tryptofan Trp
Tyrozyna Tyr
Walina Val
Kwasowe - zawierają w rodniku dodatkową grupę karboksylową: (przewaga gr.kwasowych_
Kw. asparaginowy Asp
Kw. glutaminowy Glu
Zasadowe - w rodniku dod.grupa zasadowa: (przewaga gr.zasadowych)
Arginina Arg
Histydyna His
Lizyna Lys.
Właściwości białek zależą od właściwości AA składowych i struktury jaką tworzą. Wynikają one z obecności różnych grup zdolnych do jonizacji: -COOH(kwasowa), -NH2(zasadowa) oraz -OH, -SH, a także imidiazolowej, guanidynowej i reszt fosforanowych, oraz od ich ilości. Ładunek elektryczny białka również zależy od rodzaju i liczny grup zdolnych do jonizacji (a także od pH roztworu).
BIAŁKA
Polipeptydy o masie cząst. od 104 do 105, wchodzą w skład każdej kom i płynów ustrojowych; wiążą wodę niezbędną do procesów życiowych; uczestniczą w utrzymaniu stałego pH i transportowaniu związków i jonów.
Wiązanie peptydowe- w reakcji peptyzacji grupa α -NH2 jednego AA tworzy wiązanie peptydowe z grupą α-COOH drugiego AA (ten AA, którego gr.karboksylowa reaguje z gr.aminową innego AA jest pierwszy w nazwie peptydu, np.: alanyloseryna).
Wiązanie to jest sztywnym i płaskim elementem (ponieważ wiązanie N-C ma częściowo charakter wiązania podwójnego i utrudnia jego rotację).
Struktury białek:
I-rzędowa (pierwotna)-sekwencja (kolejność występowania) AA w łańcuchu polipeptydowym, utrwalana wyłącznie przez wiązania peptydowe. W strukturze tej podaje się ułożenie wszystkich silnych wiązań kowalencyjnych, m.in. disulfidowych RS-SR między dwoma resztami cysternowymi (produktem reakcji jest reszta cystyny).
II-rzędowa-sposób i stopień skręcenia/pofałdowania łańcucha polipeptydowego:
α -helisa: łańcuchy boczne znajdują się na zewnątrz , a łańcuch gł.stanowi rdzeń cylindra; stabilizowana przez wiązania wodorowe między CO i NH (wchodzące w skład wiąz.peptydowego). AA stabilizujące α-helisę: Ala, Phe, Asn, Glu, His, Met i Trp; AA destabilizujące: Val, Ile, Ser, Thr, Pro. Np.: kolagen, keratyna. Helisę kolagenu stabilizują reszty proliny i hydroksyproliny, co 3 reszta to glicyna.
β-harmonijka(kartka): wewnątrzcząst.wiąz. wodorowe; mostki wodorowe między równolegle biegnącymi w przestrzeni łańcuchami polipep; płaskie części „kartki” to sztywne wiązania peptydowe. Np.: fibroina.
III-rzędowa- powstaje w wyniku oddziaływań między łańcuchami bocznymi białka, efekt pofałdowania i zwinięcia się łańcucha polipeptydowego II-rzędowego. Stabilizowana przez wiązania wodorowe (łączy gr.boczne leżące w sąsiednich pętlach łańcucha), hydrofobowe (między niepolarnymi gr.bocznymi), jonowe (między dodatnio i ujemnie naładowanymi gr.bocznymi), kowalencyjne (mostki dwusiarczkowe; mogą łączyć fragm. jednego/różnych łańcuchów polipep.).
IV-rzędowa: określa wzajemne położenie dwóch/większej liczby łańcuchów polipeptydowych. Np.: hemoglobina (po 2 łańcuchy α i β). Identycznie zbud.łańcuch polipep. Takicj białek to protomery, podjednostki łączące się między sobą wiąz.wodorowymi, jonowymi, oddziaływaniami hydrofobowymi, czasem wiąz.dwusiarczkowymi i koorydacyjnymi.
Białke proste i złożone:
Proste-takie, kóre w wyniku hydrolizy dają tylko AA, np.: albumina surowicy krwi
Fibrylarne- zespoły rozciągniętych łańcuchów, różne wiąz.niekowalencyjne, dość trwałe, nie rozp.w H2O i solach; białka podporowe i strukturalne (skleroproteiny-u zwierzat skł.tk łącznej i strukturalnej; keratyna, kolagen, elastyna; doporne na enzymy)
Globularne- kształt eliptyczny, z pofałdowanych, zwiniętych łańcuchów o splotach utrwalonych różnego typu wiązaniami (gł.kowalenycyjnymi), łatwo rozp. w H2O; wszystkie biologicznie czynne o char.enzymów. (histony-drobnocząst.zasadowe, dużo Arg i Lys; albuminy-rozp.w H2O i roztw.soli, globuliny-źle rozp. w H2O, dobrze w soli; iałka zapasowe w liścieniach i warstwie aleuronowej ziarniaków, prolaminy-kwaśne, w skrobiowej cz.bielma; gluteiny-skrobiowa cz.bielma; 1 z podst.skł.glutenu;)
Złożone-w wyniku hydrolizy dają AA, a także inne związki, np.: węglowodany, tłuszcze, kw.nukleinowe
Fibrylarne
Globularne: fosfoproteiny-zaw.kw. fosforowy,np. kazeina, glikoproteiny-różne reszty cukrowe; wł.hormonów gonadotropowych np.erytroportyna, chromoproteiny-hemoglobina, reak.oddechowe, transport i magazynow,jonów Fe i Cu; lipoproteiny-białka+tłuszcz; nukleoproteidy-białko+kw.nukleinowy, w jądrze kom, rybosomach, biosynteza białka, dziedziczenie, gł.budulec wirusów.
Denaturacja białek- zmiany w III- i IV-rzędowej strukturze białka prowadzące do utraty aktywności biologicznej lub innej indywidualnej cechy charakterystycznej przy zachowaniu struktury I-rzędowej i wiązań kowalencyjnych. Niszczone są wiązania wodorowe, a w obecności odczynników redukujących zerwaniu ulegają wiązania disulfidowe. Odwracalna (renaturacja) lub nieodwracala. Podczas denaturacji zachodzą także zmiany rozpuszczalności i przesunięcie punktu izoelektrycznego, utrata aktyw.bioloicznej. Obserwuje się również często procesy agregacji i wytrącania, co jest związane ze zmianami stopnia hydratacji i rozpuszczalności białek. Czynniki denaturujące:
Fizyczne: temperatura, silne mieszanie, wytrząsanie, nadfiolet, promienia rentgena i jonizujące, ultradźwięki
Chemiczne: (nieznaczne nawet zmiany pH) kwasy, zasady, niektóre sole
Właściwości amfoteryczne białek- Amfoteryczny charakter białek jest wynikiem obecności i rozmieszczenia (oraz ilości) kwasowych i zasadowych grup łańcuchów bocznych. W roztworze kwaśnym białka mają ładunek dodatni w roztworze zasadowym ujemny.
Ważny fizjologicznie efekt buforowania białek zależy od równowagi:
kation białkowy
białko obojętne
anion białkowy
Punkt izoelektryczny białek-ściśle określone pH, w którym cząst.białka zawiara jednakową licznę grup dodatnich i ujemnych; wykazuje wtedy sumaryczny ładunek zerowy. W punkcie izoelektrycznym białka wykazują najmniejszą rozpuszczalność (łatwo je wytrącić z roztworu) i lepkość, oraz najsłabiej pęcznieją.
Właściwości amfipatyczne białek- białka integralne znajdujące się w błonie komórkowej mają charakter amfipatyczny. Mają zarówno regiony hydrofobowe jak i hydrofilowe i są asymetrycznie rozłożone, przechodząc na obie strony dwuwarstwy. Jest to spowodowane tym, że białka budują aminokwasy o różnej budowie łańcucha bocznego, który może mieć charakter hydrofobowy, bądź polarny. Hydrofobowe łańcuchy boczne reszt wystają w helisie na zewnątrz i mogą oddziaływać wiązaniami hydrofobowymi z łańcuchami kwasów tłuszczowych. Po każdej stronie takiej helisy znajdują się skupienia aminokwasów o obdarzonych ładunkiem (polarnych=hydrofilowych) łańcuchach bocznych, które oddziałują niekowalencyjnie z przeciwnymi ładunkami na polarnych grupach głowy lipidów błonowych.
AA o hydrofobowych łańcuchach bocznych: Gly, Ala, Val, Leu, Ile, Met, Pro, Cys, Phe, Tyr, Trp
AA o polarnych (hydrofilowych)łańcuchach bocznych: Arg, Lys, His, Asp, Glu, Ser, Thr, Asn, Gln
ENZYMY
Biokatalizatory przemian substratu w produkt; enzym wiąże się z nie zmieniając swojej udowy i z reak. Wychodzi w niezmienionej postaci; są swoiste-jeden enzym-jedna reakcja/typ reakcji.
Istota katalizy enzymatycznej-obniżenie energii aktywacji reakcji i przyspieszenie jej przebiegu.
Budowa enzymu:
KOENZYM + APOENZYM = HOLOENZYM
Koenzym-część niebiałkowa enzymu; drobnocząst.zw.org/ jony nieorganiczne/witaminy. Współdziała z apoenzymem, pomagając we właściwym rozmieszczeniu substratów w centrum aktywnym/ reagując z substratami. Ulega przekształceniu podczas reak., odtwarza się w reak.wtórnej. Jony metali (metaloenzymy) często biorą bezpośredni udział, np. w oksydoreduktazach-przenośnik e-; służą też do bezpośredniego wiąz.substratu z metaloenzymem; często stabilizują strukturę apoenzymu (wtedy są poza centrum akt.), lub pełnią obie funk.naraz: strukturalną dla apoenzymu i katalityczną. Pod względem chemicznym sa nukleotydami (składają sie z cukru(pentoza), zasady(Adenina, guanina, Tymina, cytozyna, uracyl) oraz z fosforanu (jednego lub kilku).
Działanie na przykładzie oksydoreduktazy (NAD): aldehyd 3Plicerynowy utlenia się do kw.3Pglicerynowego, a NAD ulega redukcji do NADH++H+, następnie odłącza się od enzymu I i wiąże się z enzymem II, który redukuje pirogronian do mleczanu, utleniając NADH++H+ do NAD (wraca do enz.I)
Apoenzym- część białkowa enzymu
Holoenzym- połączenie apoenzymu i koenzymu, wykazuje aktywność biologiczną.
Grupa prostetyczna- centrum aktywne enzymu. Sama posiada właściwości katalityczne, a obudowanie jej aminokwasami białka wielokrotnie przyspiesza katalizę nadając jej odpowiednią konfigurację przestrzenną i stwarzając idealne warunki do zajścia katalizowanej reakcji. Np.: pochodne hemu, obecne w hemoglobinie i mioglobinie.
Centrum aktywne, miejsce aktywne- ta część cząsteczki, która jest bezpośrednio zaangażowana w reakcji chemicznej. W białku centrum aktywne stanowią łańcuchy boczne AA, które leżą blisko siebie w cząst. choć często są od siebie bardzo oddalone w sekwencji. Częstymi AA centrum aktywnego są AA zasadowe oraz kwasowe. Aminokwasy z niepolarnym łańcuchem bocznym bardzo rzadko wchodzą w skład centrum aktywnego, ponieważ taki łańcuch boczny wchodzi w niewiele reakcji (hydrofobowe właściwości przyczyniają się do silniejszego wiąz. substratu). Ma miejsca wiązania substratu (decyduje o swoistości substratowej) i miejsce katalityczne (reakcji; AA z gr.bocznymi o szczeg. reaktywności, zwykle His, Ser, Lys). W wiązaniu substratu biorą udział siły van der Waalsa i wiąz,wodorowe.
Specyficzność reakcji enzymatycznych- działanie enzymów charakteryzuje się specyficznością - katalizuje tylko określony substrat lub określony typ reakcji chemicznej. Enzym wykazuje 3 specyficzności:
specyficzność (swoistość) substratową-a) aktywność katalityczna w stos.do substratu, który oprócz gr.reagującej ma inne gr.funkcyjne. b) dzała tylko na jeden z izomerów optycznych (s.optyczna). c) katalizuje przemiany substratów o określ.dł. łańcucha C.
regiospecyficzność- w wyniku reakcji powstaje wyłącznie jeden produkt
stereospecyficzność- wiele produktów
Klasyfikacja enzymów
Hydrolazy
Lipazy - hydroliza gr. estrowej
Nukleazy - gr. fosforanowej
Proteazy - gr. amidowej
Izomerazy
Epimerazy - izomeryzacja centrum aktywnego
Ligazy
Karboksylazy - przyłączenie CO2
Syntetazy - tworzenie nowego wiązania
Liazy
Dekarboksylazy - utrata CO2
Dehydrazy - utrata H2O
Oksydoreduktazy
Dehydrogenazy - wprowadzenie wiązania podwójnego przez oderwanie H2
Oksydazy - utlenienie
Reduktazy - redukcja
Transferazy
Kinazy - przeniesienie gr. fosforanowej
Transaminazy - przeniesienie gr. aminowej
Przebieg reakcji enzymatycznej- enzymy stanowią największą grupę tzw. biokatalizatorów, czyli katalizatorów pochodzenia biologicznego. Ich działanie polega na obniżeniu energii aktywacji poprzez zmianę mechanizmu reakcji. Nowa reakcja (z katalizatorem) może mieć większą barierę aktywacji (kataliza ujemna lub inhibicja) lub mniejszą (kataliza dodatnia) od reakcji wyjściowej.
Kinetyka reakcji enzymatycznej-
Równanie i krzywa Michaelisa-Menten:
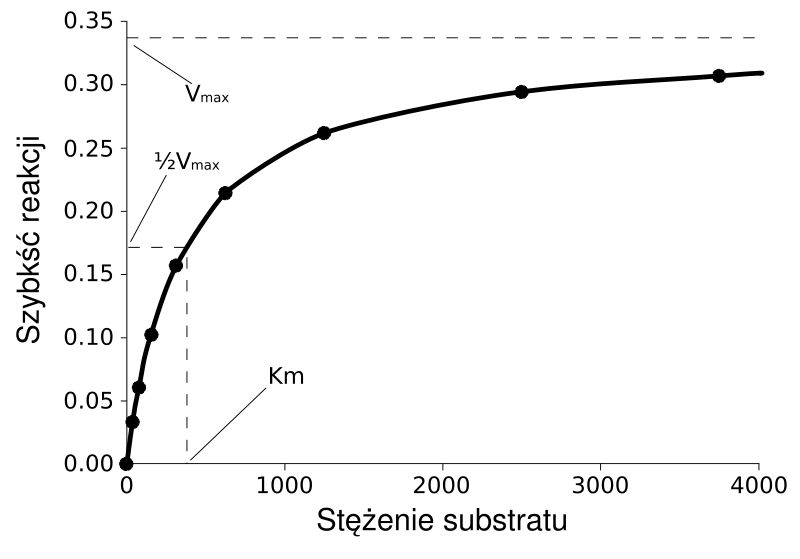
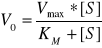
Vo-początkowa szybkość reakcji
Vmax-maksymalna szybkość reakcji
[S]-stężenie substratu
KM-stała Michaelita
Krzywa wysycenia dla reakcji enzymatycznej ukazująca zależność szybkości reakcji (V) od stężenia substratu ([S])
Stała Michaelisa (Km) - stężenie substratu, przy którym szybkość reakcji enzymatycznej jest równa połowie maksymalnej szybkości tej reakcji. Im jest ona wyższa, tym niższe powinowactwo enzymu do substratu.
KM i Vmax to wielkości charakteryzujące dany enzym
Aktywność specyficzna- określony enzym kat.: jeden typ reakcji chem. (np.przenoszenie gr.fosforanowych) lub pojedynczą reakcję jednego substratu/ zachodzącą między określonymi substratami.
Liczba obrotów - liczba moli substratu, która może przereagować w ciągu 1min z 1molem centrów aktywnych, w tem.30*C i warunkach optymal.dla enzymu.
Inhibicja niekompetycyjna- przyłączenie się inhibitora do innego miejsca enzymu niż miejsce aktywne, co powoduje zmianę konformacyjną, prowadzącą do obniżenia aktywności katalitycznej. W związku z brakiem współzawodnictwa substratu i inhibitora o miejsce aktywne, zwiększenie stężenia substratu nie może przezwyciężyć inhibicji. Inhibitor znacznie zmniejsza liczbę obrotów enzymu, nie ma wpływu na liczbę cząsteczek enzymu wiążących substrat.
Zmniejsza liczbę obrotów enzymu, nie zmniejsza liczby cząst.enzymu wiążących substrat.
Inhibicja kompetycyjna-sposób hamowania działania enzymu. Cząsteczka będąca inhibitorem ma podobną budowę do substratu hamowanego enzymu, wchodzi w centrum aktywne enzymu i blokuje je uniemożliwiając łączenie enzymu z substratem.
Zmniejsza szybkość katalizy przez zmniejszenie liczby cząst.wiążących substrat.
Regulacja allosteryczna (allosteria)-regulacja aktywności enzymu poprzez zmianę kształtu cząsteczki enzymu wywołaną przyłączeniem aktywatora lub inhibitora allosterycznego do centrum allosterycznego.
Sprzężenie zwrotne (hamowanie przez produkt)- produkt końcowy P szeregu następujących po sobie reakcji jest inhibitorem allosterycznym działającym na enzym pierwszej reakcji tego cyklu przemian
Czynniki wpływające na aktywność enzymów:
Temperatura
pH
stężenie substratu
obecność inhibitorów
ilość produktu (przy sprzężeniu zwrotnym)
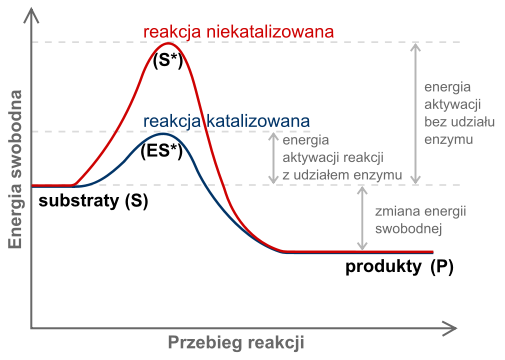
ENERGIA AKTYWACJI
Schematyczny wykres zmian energii swobodnej w czasie reakcji. By przeprowadzić substraty (S) w stan przejściowy (S*) zwykle potrzeba dużej ilości energii do pokonania progu aktywacji. Składniki reakcji w stanie przejściowym są konwertowane następnie w produkty końcowe (P). Enzymy ułatwiają tworzenie stanu przejściowego (ES*) oraz stabilizują go poprzez jego specyficzne wiązanie, zmniejszając dzięki temu ilość wymaganej energii
Wyszukiwarka