Wykład 3
Metabolizm aminokwasów i białek
Zagadnienia związane z białkami dotyczą głównie ich proteolizy i prawidłowego fałdowania. Z kolei aminokwasy traktowane są jako awaryjne źródło energii. Obok cukrów są źródłem szkieletów węglowych (pośrednio lub bezpośrednio pirogronianu) i w ten sposób trafia do metabolizmu podstawowego czyli jest źródłem energii.
Wady metaboliczne związane z białkami, dotyczą: fałdowania oraz rozkładania białek, ich biosyntezy a rzadziej rozkładu lub przyswajania aminokwasów.
Trawienie białek
Proces trawienia odbywa się przede wszystkim w początkowych odcinkach przewodu pokarmowego.
W żołądku zachodzi trawienie za pomocą pepsyny- dochodzi do denaturacji białek. Pomocne jest bardzo niskie pH (obecność jonów H+ i Cl- wydzielanych niezależnie i chroniących żołądek).
Trawienie przy pomocy enzymów proteolitycznych trzustkowych- trypsyna, chymotrypsyna, elastaza, karboksypeptydaza- są to enzymy mogące rozłożyć większość białek do oligopeptydów lub aminokwasów. Trypsyna i chymotrypsyna a następnie karboksypeptydaza trawią głównie białka globularne. Z kolei elastaza a następnie karboksypeptydaza trawią białka włókienkowe.
Otrzymane aminokwasy i oligopeptydy wchłaniane są do krwi z jelita. W czasie przechodzenia przez ściany jelit, częściowo w surowicy krwi są dotrawiane przez aminopeptydazy, dipeptydazy- tzw. unikatowe endopeptydazy (proteinazy). Otrzymujemy mieszaninę dipeptydów, tripeptydów i wolnych aminokwasów. Następnie trafiają one do surowicy krwi i potem do organów docelowych (przez wątrobę, która podobnie jak w przypadku cukrów kontroluje metabolizm aminokwasów).
Enterocyty jelita cienkiego przejmują nadzór nad dotrawianiem oligopeptydów. Jest to związane z systemem energetycznym i systemem transportu aminokwasów (każdy aminokwas ma swój transporter w poszczególnych enterocytach)
Sekrecja przy trawieniu białek związana jest w sposób bezpośredni z żołądkiem, w sposób pośredni z jelitami (gł. jelito cienkie) i w sposób nadzorczy z trzustką.
Bodźcem podstawowym do rozpoczęcia trawienia jest dostanie się do żołądka treści pokarmowej, czyli bodziec czysto mechaniczny, który oddziałuje na układ sympatyczny i parasympatyczny. Daje sygnał do żołądka, że rozpoczyna się proces trawienia. Żołądek od razu reaguje, wysyła sygnały do pozostałych części układu pokarmowego. Druga droga sygnałowa związana jest z naszą psychofizjologią jedzenia- bodziec dochodzi z receptorów czuciowych, smakowych, wzrokowych oraz nasze skojarzenia (że jemy np. cos dobrego), Sygnał taki wysyłany jest bezpośrednio do trzustki. W momencie kiedy treść pokarmowa znajduje się żołądku zaczyna on wydzielanie jonów H+ i Cl-, enzymów oraz hormonu regulacyjnego układu pokarmowego- gastrynę. Żołądek rozpoznaje etap jedzenia (w jakiej mierze jest zapełniony) na podstawie współpracy układu sympatycznego z centralnym układem nerwowym. Gastryna stymuluje zarówno autopobudzanie żołądka jak też jest sygnałem, który idzie do trzustki żeby się przygotowała na wydzielanie enzymów trawiennych, dwunastnicę itd. Regulacja wydzielania w żołądku jest sprzężeniem kilkustopniowym. Następuje wydzielanie enzymów i izoform gastryny przez 3 części żołądka: korpus, dno (odczuwa początek jedzenia), odźwiernik. Zwieracz przesuwa treść żołądka do dwunastnicy. Kiedy żołądek jest częściowo wypełniony wydzielana jest gastryna 17 i gastryna 34 oraz pepsynogen II, który jest źródłem pepsyny II- podstawowy enzym proteolityczny żołądka.
Na początku jedzenia oraz podczas dojadania między posiłkami trawienie zachodzi w dnie żołądka.
Gastryna 17 jest główną izoformą pobudzającą trzustkę.
W momencie kiedy jemy więcej pobudzona jest główna część żołądka i oprócz pepsynogenuII wydzielany jest pepsynogen I, który przekształca się (autoaktywacja) do pepsyny I- enzym bardziej odporny na pH i bardziej agresywny (mający większą ilość obrotów) jeżeli chodzi o hydrolizę. Zwiększa się ilość wydzielanych jonów H+ i Cl-. Działaja tu także czynniki tzw. autopobudzające- prawdopodobnie gastryna 34 i nieznane jeszcze izoformy gastryny.
W momencie kiedy pojawiają się pierwsze aminokwasy i oligopeptydy pobudzany jest zwieracz przesuwający treść żołądka do dwunastnicy i wtedy rozpoczyna się wydzielanie gastryny 34 i pepsynogenu II, zachodzi dotrawianie niektórych dłuższych endopeptydów.
Kiedy treść żołądka przejdzie do dwunastnicy- przemieszczają się tam głównie jony Cl- oraz proenzym pepsynogenu- część, która odcinana jest w procesie aktywacji pepsyny. Są to tzw. wydzieliny, które współpracują z gastryną w procesie wydzielania soku trzustkowego- czyli jony NaHCO3, które zobojętniają kwas oraz proenzymy enzymów trzustkowych oraz ich izoform. Aktywacja jest kaskadowa. Kaskadę tę prawdopodobnie inicjuje pepsyna ale w momencie kiedy treść staje się coraz bardziej zasadowa, następuje autoaktywacja trypsyny, chymotrypsyny, elastazy, karboksypeptydazy- one aktywują się wzajemnie. W tym momencie nastepuje już dotrawienie- pojawia się więcej wolnych aminokwasów i krótsze oligopeptydy.
Kiedy treść pokarmowa przesunie się z dwunastnicy do jelita cienkiego zaczynają dzialać wydzielane przez jelito hormony lokalne:
cholecystokinina
sekretyna,
które dodatkowo stymulują trzustkę do wydzielania kolejnych porcji enzymów proteolitycznych jeżeli jest dużo pokarmu. Poza tym stanowią sygnał dla dalszej części jelita cienkiego, że trzeba się przygotować na pobieranie i dotrawianie- sygnał przede wszystkim dla enterocytów.
W przypadku diety wysokobiałkowej oprócz centralnego układu nerwowego i szlaku pobudzenia ośrodka apetytu, stymulująco działa również acetylocholina, która jest z jednej strony hormonem współodpowiedzialnym za odczuwanie głodu a także pierwszym hormonem, który sygnalizuje, że „widzimy że jemy”.
Gastryna współdziała z histaminą. Komórki jelita cienkiego, które wydzielają somatostatynę współuczestniczą w wydzielaniu acetylocholiny.
Gastryna- autopobudza żołądek i pobudza trzustkę.
Sekretyna i cholecystokinina- autopobudzenie trzustki i pobudzenie enterocytów do procesu trawienia.
W momencie kiedy mamy porcje dipeptydów, tripeptydów i wolnych aminokwasów- wszystko jest pobierane z jelita i rozdysponowane po całym organizmie. Większość trafia do tkanek mających szybki metabolizm białkowy i mających dużo białek o krótkim okresie półtrwania czyli mięśni. Druga co do wielkości porcja trafia do wątroby, pozostałe porcje aminokwasów do innych tkanek. Najważniejsza oczywiście jest wątroba, która pełni funkcje centralną jeśli chodzi o gospodarkę aminokwasowi- nadzoruje gospodarkę, rozkład białek praktycznie we wszystkich organach, decyduje o kierowaniu aminokwasów do podstawowych szlaków metabolicznych- deaminacja oksydacyjna- wytwarzanie mocznika. Odpowiada także pośrednio za pulę aminokwasów i ewentualne wydalanie nadmiaru aminokwasów ( co jest szczególnie ważne w patologiach metabolizmu aminokwasów) przez nerki, oraz za to co się dzieje w tkankach peryferyjnych- tempo budowy białek i tempo własnego spalania aminokwasów.
Dawniej obowiązywał podział na aminokwasy niezbędne, nie-niezbędne i warunkowo niezbędne, który potem się załamał. Zapotrzebowanie na aminokwasy jest bezpośrednio związane z wiekiem organizmu:
43% aminokwasów w wieku dziecięcym można uznać za niezbędne. Jest to związane z bardzo intensywnym wzrostem w tym okresie i nawet aminokwasy, które są syntetyzowane w naszym organizmie w wątrobie muszą być suplementowane z pokarmu ponieważ wątroba nie nadąża z produkcją równą zapotrzebowaniu.
W okresie dojrzewania zapotrzebowanie na aminokwasy spada praktycznie do polowy wartości niezbędnej w okresie dzieciństwa- z tego tylko 38% można uznać za niezbędne.
W okresie dorosłym zapotrzebowanie aminokwasów ponownie dwukrotnie spada a 19% z nich to aminokwasy niezbędne
Regulacja gospodarki aminokwasowej odbywa się przede wszystkim przez wątrobę, która gospodaruje pulą 4 kwasów związanych z cyklem kwasu cytrynowego i w oparciu o nie kontroluje wszystkie aminokwasy obecne w surowicy krwi- czyli tzw. pulę aminokwasowi organizmu. W oparciu o te 4 główne kwasy (α-ketoglutaran, kw. bursztynowy, kw. fumarowy, kw. propionowy ) zachodzi synteza wszystkich aminokwasów białkowych i niebiałkowych obecnych w naszych tkankach.
Do współregulacji tych procesów służą dwa megacykle sprzężone z metabolizmem aminokwasów:
cykl biosyntezy argininy i mocznika, którego głównym zadaniem jest utrzymanie w surowicy krwi dobrego poziomu Arg (uznawana jest za główny aminokwas kontroli ogólnego metabolizmu aminokwasów) i poziom poniżej poziomu toksycznego jonów amonowych. Cykl ten przebiega następująco: jony amonowe wychwytywane są przez jelita gdzie enzym syntetaza I karbamoilofosforanu (CPS-1) i transkarbamylaza ornitynowa (OTC), które wytwarzają cytrulinę. Ona z kolei trafia do nerek gdzie jest rozkładana do Arg i wyłapywany jest mocznik, który zostaje wydalony- jest to ogólny schemat systemu awaryjnego.
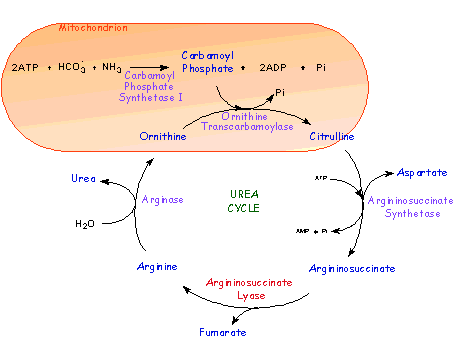
Główny system polega na wyłapywaniu jonów amonowych przez wątrobę, gdzie w pełnym cyklu mocznikowym zostaje wytworzona główna wydalana przez organizm pula mocznika- trafia z krwiobiegu do nerek i wydalana jest z moczem- jest to cykl mocznikowo- argininowy.
Drugi cykl- cykl równowagi Ala oraz Gln, która obok Arg jest drugim aminokwasem regulującym pulę aminokwasów w organizmie.
Glutamina trafia do krwiobiegu przede wszystkim z mięśni (w niewielkim stopniu z biosyntezy z wątroby) i jest rozdysponowywana do nerek gdzie rozpada się na kw. glutaminowy (Glu), następnie na glukozę, uwalniany jest mocznik, który jest wydalany. Gln jest źródłem energii dla nerek.
Podobnie jest w systemie białokrwinkowym limfocytarnym, a ewentualny nadmiar Gln trafia do jelit gdzie jest przekształcana w Ala oraz CO2 i H2O. Ala jest przerzucana do wątroby, gdzie przekształcana jest z udziałem glukoneogenezy w glukozę, a uzyskany z Ala jon amonowy przekształcany jest w mocznik, który wydalają nerki.
Jeżeli zakłócenie poziomu Gln jest poważne to znany jest szlak dodatkowy mięśni- przerzucają one do wątroby Ala żeby zrekompensować zbyt wysoki nie spalany przez organy (nerki, jelito, limfocyty) poziom Gln.
Biochemicy sportu pod koniec lat 90. usiłowali wyeliminować znaczenie aminokwasów, a badać wyłącznie równowagę ogólnych białek w organizmie, ogólnego poziomu mocznika w organizmie i równowagi pomiędzy moczem, surowicą krwi i pierwiastkami Cd. Błąd tkwił w tym, że przeceniono rolę surowicy krwi. Nie można traktować mocznika, który jest wydaliną i organizm nie jest zainteresowany jakąkolwiek równowagą między moczem a tkankami- po prostu organizm ma za zadanie jak najszybciej ten mocznik usunąć. Argumenty te spowodowały, że wycofano się z tego schematu.
Biosynteza i rozkład białek
W metabolizmie białek interesuje nas równowaga pomiędzy dwoma procesami:
biosyntezą białek
rozkładem białek
co nazywane jest poziomem obrotu białkowego.
Biosynteza białek zależy od czynników hormonalnych ogólnowzrostowych (hormony wzrostu, hormony tarczycowe) oraz od podstawowych hormonów katabolicznych i anabolicznych(insulina i glukagon) i od poziomu aminokwasów w diecie, w surowicy krwi i w wątrobie.
Rozkład białek zachodzi dwutorowo:
rozkład lizosomalny- zależny od poziomu aminokwasów i systemu insulina- glukagon (anabolizm- katabolizm)
rozkład kontrolowany białek (proteasomalny)- zależny od kortykosteroidów (hormony metabolizmu doraźnego), interleukin (precyzyjna regulacja cyklów komórkowych w poszczególnych tkankach) oraz od statusu energetycznego organizmu.
W diecie tracimy najpierw glikogen, potem białka a na samym końcu tłuszcz.
Szlak rozkładu lizosomalnego jest prawdopodobnie losowy (ostatnio sugerowane jest, że zachodzi także w sposób kontrolowany za pomocą markerów białkowych). Pobierane zostaje elementy cytoszkieletu, cytoplazmy nawet całych organelli i trawione w lizosomie.
Trawienie kontrolowane, z którego najlepiej znanym procesem jest proces rozkładu w proteasomie białek znakowanych przy czym najczęstszym znacznikiem białkowym jest ubikwityna . Znaczy ona wszystkie typy białek ponieważ posiada z jednej strony struktury hydrofobowe- spirale α- i znaczy białka z przewagą hydrofobowości na powierzchni. Z drugiej strony z kolei posiada struktury β i znaczy białka bardziej hydrofilowe na powierzchni. Znakowanie ubikwityną może zachodzić za pomocą tzw. krótkiego łańcucha- czyli 3-4 ubikwityny, lub za pomocą krótkiego łańcucha rozgałęzionego- co najmniej 5-6 ubikwityn.
Za cały mechanizm znakowania odpowiada system 3 enzymów, które wiążą z udziałem ATP najpierw ubikwitynę na zasadzie aktywacji, następnie przy pomocy enzymu pomocniczego przenoszą ubikwitynę na białko, które ma być znakowane. Wyznakowane białko trafia do proteasomu i rozkladane jest na oligopeptydy, które za pomocą odpowiednich oligopeptydaz, tripeptydaz, aminopeptydaz rozkładane są na wolne aminokwasy.
Nadal nie do końca poznano co decyduje o tym czy dane białko będzie wyznakowane i w jaki sposób. Przypuszcza się, że decyduje tutaj równowaga pomiędzy systemem znakowania ubikwityną a systemem chaperonów ( białek odpowiedzialnych za odpowiedzialne sfałdowanie innych białek). Do tej grupy należą 3 grupy białek:
chaperoniny - białka opiekuńcze, które permanentnie kontrolują prawidłowe fałdowanie białek,
białka grupy Hsp- białka szokowe zapewniające prawidłowe fałdowanie w warunkach stresowych tj. wysoka temp. czy duża kwasowość,
białka utrzymujące prawidłową modyfikacje białek np. utrzymanie prawidłowej glikozylacji, prenylacji, adenylacji.
W warunkach normalnych te dwa systemy pozostają w równowadze. Jednak w niektórych stanach może zostać ona zaburzona. Schemat ten czasem może zakłócić sytuacja gdy chaperony same zaczynają degradować białka. Poza tym stanem patologicznym jest obecność białek zagregowanych (złogi amyloidowe w chorobie Alzheimera), którymi nie interesują się ani proteasomy ani chaperony.
Okazało się również, że w różnych organizmach, a u człowieka w różnych organach oprócz ubikwityny występują inne białka, które znakują inne białka do rozkładu (np. SUMO).
Prof. Angielski przedstawił zależność, której nikt do tej pory nie obalił. Jego koncepcja głosi, że równowaga metabolizmu aminokwasowego to jedno, a równowaga dynamiczna puli białek to drugie. Podzielił białka na:
białka o długim obrocie białkowym - wolno syntetyzowane i wolno rozkładane. Równocześnie mają długi okres połtrwania sięgający nawet miesięcy (kolagen kości)
białka o szybkim obrocie białkowym- krótki okres poltrwania, np. białka metabolizmu podstawowego, które istnieją tylko kilka minut, szybko się zużywają, duża ilość obrotów enzymatycznych.
Białka o wolnym obrocie białkowym mają ogromną pulę- stanowią konstrukcję funkcjonowania złożonego organizmu. Szybkość ich syntezy i rozkładu jest mała.
Białka o szybkim obrocie białkowym maja pulę niewielką co jest rekompensowane przez bardzo szybką syntezę i bardzo szybki rozkład.
Zależność pomiędzy szybkim i wolnym obrotem może stanowić klucz do zbadania zależności między chaperonami, lizosomami i proteasomami.
Odstępstwa związane z pulą białek o wolnym i szybkim obrocie białkowym
Białka szybko odnawiające się ( szybkim obrocie białkowym):
zwiększenie puli tych białek następuje w momencie kiedy organizm musi być zmobilizowany np. trenuje. Permanentnie takie zjawisko występuje w okresie dzieciństwa kiedy organizm szybko rośnie. Charakterystyczne jest zmniejszenie się rozpadu tych białek, natomiast nie zwiększa się ich synteza.
zmniejszenie puli tych białek regulowane jest wielkością puli i zwiększeniem szybkości rozpadu. Są to stany patologiczne typu miopatie, zatrucia które powodują uwolnienie dużej ilości białek z tkanek.
W tych białkach regulacja odbywa się tylko za pomocą puli i rozkładu.
Białka wolno odnawiające się (o długim okresie połtrwania). W tym wypadku nie tylko może być naruszona pula ale i synteza i rozkład- jest to podstawowa różnica między białkami o wolnym i długim obrocie białkowym- regulacja wszystkich trzech procesów białkowych: biosynteza, pula i rozkład.
zwiększenie puli tych białek następuje gdy organizm przygotowuje się do pewnych stanów (np. u osób, które długotrwale trenują, u osób mieszkających w klimacie zmiennym i przygotowujących się do zimy, u kobiet w ciąży, u kobiet karmiących). Zwiększenie puli tych białek zachodzi z jednoczesnym zwiększeniem syntezy i rozkładu (synteza przeważa nad rozkładem), okres półtrwania tych białek trochę się skraca
zmniejszenie puli białek o długim okresie półtrwania- maleje synteza, maleje rozpad (ma przewagę nad syntezą )- pula zaczyna maleć. Taka sytuacja ma miejsce w przewlekłych stanach chorobowych (zakażenia pasożytnicze, bakteryjne) oraz w niektórych procesach naturalnych (procesy zanikowe, procesy starzenia się).
Proces patologiczny związany z pula aminokwasów i białek to głównie proces głodzenia.
Biochemicy wyróżniają 4 stany puli białkowej i aminokwasowej organizmu:
organizm odżywiany dietą wysokobiałkową- wydalane jest dużo mocznika (ciemny mocz). Jony amonowe, kw. moczowy, kreatynina i inne związki azotowe są mniej więcej w równowadze, niewiele odbiegają od osób z normalną dietą.
dieta ubogobiałkowa (wegetariańska)- w stosunku do normy obserwuje się spadek ilości mocznika nawet do połowy (mocz jest bardzo jasny). Cechą charakterystyczną jest to, że spada ilość wydalanego jonu amonowego i innych związków azotowych- kw. moczowy i kreatynina utrzymują się na poziomie normalnym ponieważ metabolizm puryn i pirymidyn i metabolizm energetyczny mięśni muszą być na rozsądnym poziomie
krótkotrwałe głodzenie- podobne cechy jak dieta wysokobiałkowa; wydalane jest bardzo dużo mocznika i bardzo dużo jonów amonowych; Poza tym obserwuje się nieznaczny ale istotny wzrost wydalania innych związków azotowych. Kwas moczowy i kreatynina są nienaruszone
długotrwałe głodzenie ( 10-40 dni)- organizm zaczyna oszczędzać mocznik ale wydala dużo jonów amonowych. W skrajnym głodzeniu pojawia się dużo kwasu moczowego i dużo kreatyniny.
Biochemicy sportowi uprościli te schematy- przedstawili nienaruszalne proporcje kreatyniny i kw. moczowego a całość zmian sprowadzili do zmian w proporcji mocznika i jonu amonowego. W warunkach normalnej diety wydalamy więcej mocznika od jonu amonowego. W głodzeniu krótko- i długotrwałym stosunek ten odwraca się- wydalane jest więcej jonu amonowego. Na tym schemacie nie są jednak uwzględnione inne wydalane związki azotowe (w niektórych przypadkach mogą być wydalane także wolne aminokwasy)
Patologie związane z metabolizmem aminokwasów i białek
Główne przemiany aminokwasów oparte na deaminacji oksydacyjnej nie mogą być zaburzone. Jakiekolwiek nieprawidłowości są letalne.
Osoby z zaburzeniami cyklu mocznikowego mogą przeżyć przy odpowiednio ustawionej diecie. Konkretnie chodzi tu o deficyt OTC (transkarbamoilazy ornitynowej). Stosuje się dietę niskobiałkową. Dochodzi do rozregulowania poziomu Arg i w sposób pochodny innych aminokwasów i niemożności wiązania odpowiednich ilości jonu amonowego- możliwość zatrucia zbyt dużym stężeniem tych jonów przy diecie wysokobiałkowej.
Zaburzenia w blokach aminokwasów nie-niezbędnych powodują niewielkie deformacje i kłopoty związane z funkcjonowaniem skóry i mięśni.
Zaburzenia aminokwasów biorących udział w konkretnych szlakach metabolicznych tworzą ważne bloki metaboliczne.
Bloki aminokwasów aromatycznych
Fenyloketonuria- związana jest ze szlakiem biosyntezy i przekształceń konkretnych aminokwasów. Zablokowanie jednego lub kilku enzymów zaangażowanych w przemianę Phe→Tyr. Jest to choroba dość powszechna i narastająca, związana ze zbyt wysokim poziomem Phe w organizmie. Następuje wydalanie fenyloketonu (objawem klinicznym jest ciemny mocz). Rozregulowana jest synteza tetrahydrobiopteryny w organizmie co pociąga za sobą kaskadę zakłóceń innych szlaków metabolicznych. Podawaniem tetrahydrobiopteryny reguluje się metabolizm chorych na fenyloketonurię.
Deficyt tyrozynazy- blok metaboliczny na etapie przekształcenia Tyr w polimery (melaniny). Albinizm właściwy- deficyt barwników w organizmie. Osoby chore podatne są na melanomę- czerniaka złośliwego skóry.
Alkaptonuria- zablokowanie rozkładu (utleniania) kw. homogentyzynowego do maleinooctanu. Brak enzymu katalizującego to przejście- oksygenazy homogentyzynianowej. Obecność czarnego pigmentu alkaptonu, który jest produktem polimeryzacji kw. homogentyzynowego. ( w stanie patologicznym odkłada się on w nerkach np. kiedy pacjent za mało pije).
Tyrozynemia (dwa typy):
rozkład pełny kw. homogentyzynowego do acetooctanu
blokuje przekształcenia acetooctanu
Ściśle związane z tkanką nerwową, bloki te prowadzą do niedorozwoju umysłowego. Blok przekształcenia tyrozyny tarczycy w trójjodotyroninę a następnie w tyroksynę co pośrednio związane jest z niedoczynnością tarczycy.
Zakłócenia rozkładu aminokwasów rozgałęzionych (Ile, Val, Leu)
Blok dehydrogenaz odpowiedzialnych za przekształcenia aminokwasów prowadzi do choroby moczu o zapachu syropu klonowego. Leczenia polega na kontroli podaży tych aminokwasów w diecie- wówczas nie ma zagrożenia długości życia ani żadnych wad rozwojowych.
Deficyt karboksylazy propionylo-CoA - enzym pośrednio uczestniczący w metabolizmie aminokwasów rozgałęzionych. Ważny w syntezie kw. tłuszczowych. Deficyt prowadzi do rozregulowania metabolizmu błon w układzie nerwowym co skutkuje niedorozwojem umysłowym.
Hiperhistydynemia- blok amoniakoliazy histydynowej, która w procesie rozkładu His jest przygotowaniem do rozerwania pierścienia. Choroba objawia się nadmiernym wydalaniem His w moczu i może prowadzić do drobnego niedorozwoju umysłowego i mięśniowego.
Hiperprolinemia, hiperhydroksyprolinemia- blok enzymatyczny powodujący przekształcenie (otworzenie pierścienia) Pro do Glu, w czym bierze udział kilka enzymów. Patologia objawia się nadmiernym wydalaniem Pro i hydroksyproliny w moczu co prowadzi do zaburzenia gospodarki tkanki łącznej- słabsze kości (kolagen)
Zakłócenia metabolizmu aminokwasów zawierających siarkę (Met, Cys)
Bloki metaboliczne:
dehydratazy serynowej- powoduje homocystynurię
dehydratazy homoserynowej- powoduje cystationurię
Jest to bezpośrednio lub pośrednio związane z białkami błonowymi (niektóre białka błonowe i transbłonowe zawierają domeny policysteinowe)- reguluje się podażą Met, Cys w diecie.
Mutacje nonsensowne czyli zamiana wstawiania aminokwasu w procesie biosyntezy białka- większość jest letalna gdyż zakłóca funkcjonowanie albo białek enzymatycznych albo strukturalnych albo transportowych.
anemia sierpowata (mutacja Hb)
stwardnienie rozsiane (regulator transmembranowy związany z prawidłowym funkcjonowaniem macierzy zewnątrzkomórkowej.
piedaldizm- mutacja związana z białkami skóry i charakterystyczne białe plamy na skórze.
Choroby związane z konkretnymi białkami i metabolizmem białek
nieprawidłowe fałdowanie białka spowodowane nieprawidłowym działaniem chaperonin, Hsp, białek prawidłowych modyfikacji potranslacyjnych- powodują syndrom marchwinowy - zakłócona fibrylna, białko włókienkowe
osteogeneza niedoskonała - powoduje nieprawidłowe zwijanie się spirali kolagenu typu I
nieprawidłowe fałdowanie α-1- antytrypsyny co powoduje: zakłócony proces krzepnięcia, transport zwrotny enzymów trzustkowych do trzustki- są to zmiany o charakterze rakowym trzustki
nieprawidłowe fałdowanie czynnika von Wildembrana( odpowiada za łączenie się w procesie krzepnięcia płytek w okaleczonych ścianach)- zaburzenia krzepnięcia krwi.
Choroba Huntingtona- nieprawidłowe fałdowanie kalmoduliny- zaburzenia w pracy synaps pobudzanych Ca (ukł. nerwowy, mięśnie)
Retinitispigmentoza- zakłócenie części białkowej rodopsyny, nieprawidłowa pigmentacja w siatkówce co kończy się różnymi stopniami niedowidzenia lub brakiem widzenia barw.
Zakłócenia proporcji izoform izoenzymów w poszczególnych tkankach
zakłócenia izoform kinazy kreatyninowej pomiędzy mięśniami szkieletowymi, m. sercowym i m. gładkimi- co prowadzi do niedorozwoju mięśnia sercowego i miopatii.
dehydrogenaza mleczanowa- jest 5 izoform, które mają bardzo konkretny wzór. W dwóch organach naruszenie porcji izoform kończy się katastrofą. W sercu- towarzyszy zmianom o char. Przedzawałowym. W wątrobie prowadzi do marskości i raka.
zasadowa fosfataza- W wątrobie zaburzenia w obrębie tego izoenzymu prowadzi do zmian zakaźnych o char. żółtaczki, marskości. Poza tym towarzyszy osteoporozie patologicznej ( młodzieńczej) oraz zmianom neoplastycznym (nowotworowym).
Obecnie w diagnostyce chorobom pasożytniczym i bakteryjnym często przypisuje się zmiany w stosunku izoform enzymów.
Serpinopatie→
→grupa chorób związana z naturalnymi białkowymi inhibitorami proteinaz serynowych- serpinami. Przykłady:
α antytrypsyna
inhibitor C1 (łączy krzepnięcie z kaskadą białka C)
antytrombina 3
inhibitor plazmminogenu
neuroserpina (odpowiada za prawidłowe krzepnięcie, lizę skrzepów, zapobieganie zatorów skrzepów)
Serpinopatie związane są z: chorobami płuc, zmianami w naczyniach krwionośnych (zmiany miażdżycowe), zmianami degeneracyjnymi, zakłóceniem angiogenezy, nieprawidłowym funkcjonowaniem płytek, demencją w oparciu o powstanie inkluzji w neuronach.
Choroby związane z nieprawidłową fosforylacją
nadmierna fosforylacja- choroby o charakterze neoplazji (nowotworu)- zakłócenie kinazy Abl, Ret
hypofosforylacje- zbyt mała fosforylacja białek- zakłócenie kinazy białkowej Ret, Kit. Zmiany objawiają się w dystrofiach ( mięśniowa, m. sercowego), agangionozach- nieprawidłowy rozwój zwojów nerwowych szczególnie w obrębie jamy brzusznej (nie trzymanie stolca)
Choroby związane z zakłóceniem glikozylacji
Chodzi tu o zakłócenie powstawania lektyn, wbudowywania glikoproteiny mannozy:
nefropatie (choroby rozwojowe nerek)
zmiany reumatoidalne
niektóre nowotwory (melanoma, nowotwory nerek)
obecność zmian glikozylacyjnych w cukrzycy
Zmiany związane z ruchem białkowym (nieprawidłowe adresowanie białek do kompartmentów komórkowych i tkankowych)
syndrom Zwellegera- w obrębie ciała noworodka nieprawidłowo są adresowane białka pomiędzy różnymi organami. Pojawiają się zmiany o charakterze zatorów żółciowych, zatorów i obumierania nerek, nienormalność w szkielecie, skórze, czaszce- dziecko się deformuje ( syndrom rozłażącego się dziecka). Dziecko umiera w przeciągu pierwszego roku.
choroby związane z kom. Odpornościowymi I-nieprawidłowy poziom N-acetyloglukozaminy co prowadzi do rozregulowania układu odpornościowego (przeżywalność jest minimalna)
zmiany gruźlicze
Zakłócenie proteasomalnego rozkładu białek
Hypercholesterolemia- choroba rodzinna zwłaszcza przy chowie wsobnym. Zakłócenie receptorów lipoproteid co prowadzi do miażdżycy i zatorów
Choroby neurodegeneracyjne- białka nie są rozkładane- gromadzą się tam gdzie nie powinny
choroba Alzheimera- złogi amyloidu tworzą się na skutek zablokowania enzymy, który powinien ten amyloid rozłożyć; złogi rozwalają komórki
choroby neurodegeneracyjne typu Creutzfelda Jakoba związane z prionami:
-ch. Creutzfelda Jakoba
-ch. Gestmana Strauszlera Szepkego- odkładanie się amyloidu w móżdżku, pierwsze objawy to zanikanie zdolności ruchu
-fatalna bezsenność rodzinna (pojawia się w wieku kilkunastu lat)
-ch. związana z ludożerstwem
Priony- przekształcenie z formy α→β ( gładkiej w szorstką), szorstkie się zlepiają tworząc złogi co rozwala tkanki nerwowe
Zakłócenie gospodarki białkowej ( syntezy i rozkładu) tarczycy→
→głównie synteza i rozkład tyroglobuliny, i w zależności czy dołączą się tu zaburzenia związane z Tyr, czy nadczynność czy niedoczynność tarczycy- występują różne jednostki chorobowe.
3
Wyszukiwarka