Ćwiczenie 2
Jakościowa analiza stałych produktów syntezy aerozolowej z wykorzystaniem spektroskopii w podczerwieni FT-IR.
Spektroskopia w podczerwieni (ang. IR - Infrared Spectroscopy) jest jedną z najczęściej stosowanych technik spektroskopowych w chemii organicznej i nieorganicznej. Polega ona na pomiarze absorpcji promieniowania podczerwonego o różnej długości fali przez badaną próbkę znajdującą się na drodze wiązki. Podstawową zaletą tej metody jest możliwość szybkiej analizy rodzaju grup funkcyjnych obecnych w próbce. Spektroskopia IR umożliwia badania substancji stałych (kryształy, proszki), cieczy i gazów.
Promieniowanie podczerwone jest częścią widma promieniowania elektromagnetycznego o długościach fal zawartych w zakresie 0,78 - 1000 μm. Od strony wyższych częstotliwości (krótszych długości fal) graniczy z promieniowaniem widzialnym (czerwień) a od strony niższych częstotliwości z promieniowaniem mikrofalowym (Rys. 1).
Rys. 1. Zakres częstotliwości promieniowania elektroamgnetycznego.
Zasada metody
W temperaturze powyżej zera absolutnego wszystkie atomy w cząsteczkach znajdują się w ciągłych ruchach wibracyjnych względem siebie. Jeśli częstotliwość tych wibracji jest równa częstotliwości promieniowania podczerwonego przechodzącego przez cząsteczkę, to cząsteczka absorbuje energię tego promieniowania. Każdy atom ma trzy stopnie swobody (x, y, z). Cząsteczka wieloatomowa złożona z n atomów posiada natomiast 3n stopni swobody.
W zależności od budowy przestrzennej cząsteczki i ilości atomów składających się na nią możemy rozróżnić następujące rodzaje drgań:
drgania walencyjne (rozciągające, Rys. 2)
symetryczne
antysymetryczne
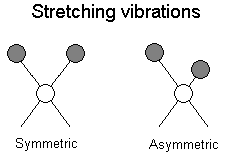
Rys. 2. Symetryczne i antysymetryczne drgania rozciągające w cząsteczce.
drgania deformacyjne (Rys. 3)
płaskie
kołyszące
nożycowe
niepłaskie
wachlarzowe
skręcające
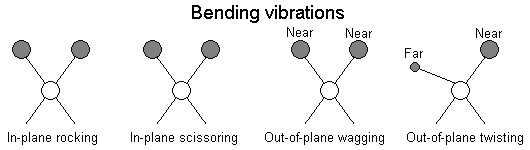
Rys. 3. Drgania deformacyjne: kolejno od lewej - kołyszące, nożycowe, wachlarzowe, skręcające.
Absorbowana energia promieniowania jest równa energii drgań występujących w cząsteczce. Powoduje to występowanie dyskretnych, skwantowanych poziomów energetycznych zawiązanych z absorpcją. Ponieważ ruchom wibracyjnym zazwyczaj towarzyszą ruchy rotacyjne (obroty), to wypadkowa energia absorbowana przez cząsteczkę nie ma postaci pojedynczej dyskretnej linii, ale tworzy tzw. pasmo absorpcyjne.
Położenia pasm absorpcyjnych zazwyczaj przedstawiane są za pomocą liczby falowej lub długości fali promieniowania przy jakiej następuje absoprcja. Liczba falowa określa liczbę fal na jednostkę długości i jest wprost proporcjonalna do częstotliwości (jak również energii) promieniowania. Jednostką liczby falowej jaką najczęściej spotyka się w badaniach IR jest [cm-1] (centymetr odwrotny). Długość fali jest natomiast odwrotnie proporcjonalna do częstotliwości (energii) i podaje się ją w [μm].
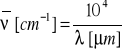
Widmo absorpcyjne jest przedstawiane w układzie współrzędnych x,y, gdzie na osi x jest liczba falowa (długość fali) promieniowania podczerwonego, a na osi y intensywność absorpcji lub procent transmitancji.
Transmitancja (T) jest wielkością określającą moc promieniowania jaka przeszła przez próbkę (I) w stosunku do mocy promieniowania padającego na próbkę (I0). Najczęściej podaje się ją w procentach (0-100%). Drugą wielkością związaną z widmami absorpcyjnymi jest absorbancja (A). Jest ona logarytmem dziesiętnym z odwrotności transmitancji.
![]()
Obszar podczerwieni wykorzystywany w badaniach podzielony jest na trzy części: bliską podczerwień (near IR), średnią podczerwień (mid IR) i daleką podczerwień (far IR). W analizie chemicznej najcześciej wykorzystywany jest obszar średniej podczerwieni (4000 - 400 cm-1) ponieważ w tym zakresie leżą pasma absorpcyjne związane z wiązaniami chemicznymi w grupach funkcyjnych najczęściej spotykanych związków chemicznych (szczególnie organicznych). Zakres średniej podczerwieni można podzielić na kilka charakterystycznych obszarów:
1000- 600 cm-1 - występują drgania deformacyjne poza płaszczyzną wiązań C-H w układach aromatycznych i alkenylowych
1500-1000 cm-1 - obszar wykorzystywany do identyfikacji (analizy jakościowej)
- drgania rozciągające wiązań pojedynczych atomów o zbliżonych masach: C-C, C-N, C-O, drgania deformacyjne różnych wiązań, drgania szkieletowe cząsteczki
2000-1500 cm-1 drgania rozciągające wiązań podwójnych C=C, C=N, N=N, N=O
2500-2000 cm-1 drgania rozciągające wiązań potrójnych C≡C , C≡N
4000-2500 cm-1 drgania rozciągające wiązań pojedynczych pomiędzy atomami znacznie różniącymi się masą np. C-H, O-H, N-H, S-H
Pasma te są schematycznie
W tabeli 1 zawarto informacje na temat położenia pasm absorpcyjnych typowych związków organicznych a Rys. 4 przedstawia przykładowe widmo IR dla etanolu.
Tabela 1. Położenie pasm absorpcyjnych dla typowych związków organicznych.
Grupa funkcyjna |
Typ |
|
Liczba falowa [cm-1] |
C-H |
sp3 (hybrydyzacja) |
R3C-H |
2850-3000 |
|
sp2 (hybrydyzacja) |
=CR-H |
3000-3250 |
|
sp (hybrydyzacja) |
C-H |
3300 |
|
aldehydy C-H |
H-(C=O)R |
2750, 2850 |
N-H |
aminy pierwszorzędowe, amidki |
RN-H2, RCON-H2 |
3300, 3340 |
|
aminy drugorzędowe, amidki |
RNR-H, RCON-HR |
3300-3500 |
|
aminy trzeciorzędowe, amidki |
RN(R3), RCONR2 |
none |
O-H |
alkohole, fenole |
wolna grupa O-H |
3620-3580 |
|
|
wiązania wodorowe |
3600-3650 |
|
kwasy karboksylowe |
R(C=O)O-H |
3500-2400 |
CN |
nitryle |
RCN |
2280-2200 |
CC |
acetylenki |
R-CC-R |
2260-2180 |
|
|
R-CC-H |
2160-2100 |
C=O |
aldehydy |
R(C=O)H |
1740-1720 |
|
ketony |
R(C=O)R |
1730-1710 |
|
estry |
R(CO2)R |
1750-1735 |
|
bezwodniki |
R(CO2CO)R |
1820, 1750 |
|
|
R(CO2)H |
1600, 1400 |
C=C |
olefiny |
R2C=CR2 |
1680-1640 |
|
|
R2C=CH2 |
1600-1675 |
|
|
R2C=C(OR)R |
1600-1630 |
-NO2 |
grupy nitrowe |
RNO2 |
1550, 1370 |
Rys. 4 Widmo IR etanolu.
Absorbancję promieniowania w zakresie podczerwieni opisuje prawo Lamberta-Beera, określające stopień pochłaniania promieniowania w zależności od długości drogi jaką pokonuje promieniowanie w badanym ośrodku (l) i stężenia molowego substancji pochłaniającej (c):
![]()
gdzie: α = 4πk/λ
k - molowy współczynnik ekscynkcji (gęstości optycznej)
λ - długość fali promieniowania
Ponieważ absorbancja jest proporcjonalna do stężenia molowego substancji pochałaniającej, to zakładając stałość drogi, długości fali i współczynnika ekstynkcji, możliwa jest analiza ilościowa. W celu wykonania takiej analizy należy wykonać pomiary absorbancji dla substancji badanej o znanym stężeniu tworząc tzw. krzywą kalibracyjną. Krzywą kalibracyjną sporządza sie dla stosunkowo niewielkich stężeń, gdyż powyżej pewnej wartości stężenia następują odstępstwa od prawa Lamberta-Beera (krzywe 2 i 3 na Rys. 5). Porównując absorbancję substancji badanej z krzywą wzorcową można określić stężenie tej substancji.
Rys. 5. Zależnośc absorbancji od stężenia.
Rodzaje spektrometrów, techniki pomiarów substancji stałych, cieczy i gazów
W badaniach metodą spektroskopii w podczerwieni wykorzystywane są dwa rodzaje spektrometrów:
Spektrometry dyspersyjne
Spektrometry fourierowskie
Spektrometr dyspersyjny składa się z trzech podstawowych elementów: źródła promieniowania, monochromatora i detektora. Źródłem promieniowania jest najczęściej włókno rozgrzane do temp. 1000 - 1800°C wytwarzające ciągłe widmo promieniowania. Monochromator jest układem w którym promieniowanie jest rozpraszane (np. z użyciem pryzmatu) a następnie wydziela z widma rozproszonego fale o ściśle określonej długości. Odbywa się to przy użyciu szczelin o regulowanej szerokości i systemu luster. Taka wiązka kierowana jest na próbkę a po jej przejściu trafia do detektora (detektor termiczny lub detektor fotonów).
Spektrometr fourierowski (FT-IR, Rys. 6) jest nowszą odmianą spektrometrów podczerwieni i wypiera on spektrometry dyspersyjne.
Rys. 6. Schemat spektrometru fourierowskiego (FTIR) z interferometrem Michelsona.
Składa się on ze źródła promieniowania (podobnego jak w spektrometrze dyspersyjnym), interferometru (zastępującego monochromator) i detektora. Najważniejszym elementem jest interferometr. Jest to najczęściej interferometr Michelsona.
Najważniejszym elementem interferometru jest ruchome lustro. Lustro to poruszając się ze stałą prędkością sinusoidalnie zmienia intensywność promieniowania padającego na próbkę. Na podstawie sygnału z detektora (po przejściu wiązki przez próbkę) tworzony jest interferogram będący zapisem natężenia sygnału interferencyjnego w funkcji czasu skanowania (ruchu) ruchomego lustra.
Operacja matematyczna (transformacja Fouriera) zamienia otrzymane widmo z postaci intensywności w funkcji czasu do postaci intensywności w funkcji częstotliwości (liczby falowej). Stąd nazwa tego typu spektrometrów - spektrometry fourierowskie.
W zależności od stanu skupienia badanej substancji stosowane są najczęściej następujące techniki pomiarowe:
Próbki stałe
pastylki w bromku potasu KBr - niewielką ilość (1-2 mg) badanej substancji stałej miesza się w moździerzu agatowym z 100-200 mg KBr (czystego i dokładnie osuszonego). Po wymieszaniu wprowadza się proszek do pastylkarki (formy do sporządzenia pastylki) którą umieszcza się pod prasą hydrauliczną. Po sprasowaniu wyjmuje się próbkę która ma postać cienkiego dysku.
metoda Nujol - do substancji reagujących z powietrzem - sporządza się pastę (w komorze suchej, atmosfera gazu obojętnego) ze sproszkowanej substancji badanej i specjalnego oleju (Nujol). Pastę umieszcza się między płytkami (wykonanymi z KBr , NaCl, itp.) i wprowadza do spektrometru.
Próbki ciekłe
niewielką ilość (kroplę) badanej cieczy umieszcza się między płytkami (KBr, NaCl) które po ściśnięciu tworzą bardzo cienką warstwę cieczy (film kapilarny) gotowy do pomiarów.
próbkę cieczy rozpuszcza się w rozpuszczalniku i umieszcza się w kuwecie cieczowej (warstwa cieczy jest znacznie grubsza niż w poprzednim przypadku) i wykonuje się pomiar. Podczas obróbki widma należy odjąć widmo czystego rozpuszczalnika od otrzymanego widma.
Próbki gazowe
do dokładnie odgazowanej (odpompowanej pod próżnią) kuwety gazowej wprowadza się (używając zaworów zamontowanych na kuwecie) próbkę analizowanego gazu. Kuwetę umieszcza się w spektrometrze i dokonuje pomiaru.
Wykonanie ćwiczenia
Analiza jakościowa analiza stałych produktów syntezy aerozolowej prowadzona jest przy użyciu spektrometru FT-IR typu Nicolet 380.
Przebieg ćwiczenia:
Przygotować próbki materiałów badanych w ilościach po ok. 0.1 g. Jeżeli materiał był wystawiony na działanie czynników atmosferycznych (nie był umieszczony w eksykatorze) należy wysuszyć go w suszarce w temp. 120 °C przez ok. 1 godzinę.
W moździerzu agatowym umieścić 1-2 mg badanej próbki i 100-200 mg dokładnie osuszonego bromku potasu KBr. Całość dokładnie wymieszać (nie ucierać!).
Umieścić mieszaninę w komorze pastylkarki upewniając się że stalowe krążki dociskowe umieszczone są prawidłowo w otworze pastylkarki.
Umieścić pastylkarkę pod prasą hydrauliczną i sprasować próbkę pod ciśnieniem 200 bar przez 1 minutę.
Wyjąć próbkę z pastylkarki i umieścić w pojemniku a następnie wprowadzić do spektrometru.
Dokonać pomiaru z zastosowaniem parametrów pracy spektrometru podanych przez Asystenta.
Zapisać i wydrukować otrzymane widma
W sprawozdaniu należy dokonać analizy jakościowej (określić jakie grupy funkcyjne/wiązania) występują w badanych proszkach. Dokonać tego na podstawie położenia pasm absorpcyjnych i baz danych widm IR
Wyszukiwarka