Laboratorium chemii i technologii radiacyjnej polimerów
Ćwiczenie 1
Dozymetria promieniowania gamma - dozymetr Frickego
Wstęp teoretyczny
Promieniowanie jonizujące to wszystkie rodzaje promieniowania, które wywołują jonizację ośrodka materialnego, tj. oderwanie przynajmniej jednego elektronu od atomu lub cząsteczki albo wybicie go ze struktury krystalicznej. Pochodzi ono z przemian jądrowych lub zostaje wytworzone w urządzeniach elektrycznych służących do przyspieszania cząstek naładowanych. Dzieli się na promieniowanie elektromagnetyczne oraz korpuskularne.
Promieniowanie elektromagnetyczne reprezentuje promieniowanie γ (monoenergetyczne), które towarzyszy przemianom jądrowym, a także promieniowanie hamowania (polienergetyczne), powstające w czasie wytracania prędkości cząstek naładowanych w polu elektrostatycznym jąder i elektronów materiału stanowiącego tarczę.
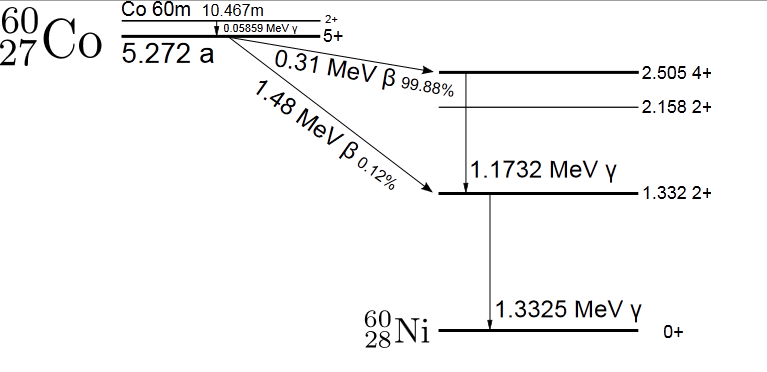
Podczas przemian jądrowych tworzą się jądra w stanie wzbudzonym, które ulegają dezaktywacji promienistej - może być to proces jednofotonowy lub zachodzący poprzez stany pośrednie (wiąże się to z emisją kilku kwantów promie-niowania γ o niższych energiach). W chemii radiacyjnej najpowszechniej stosuje się rozpad promieniotwórczy jąder 60Co jako źródło promieniowania γ. W wyniku rozpadu β- jąder izotopu
powstają trzy wzbudzone stany jąder izotopu niklu
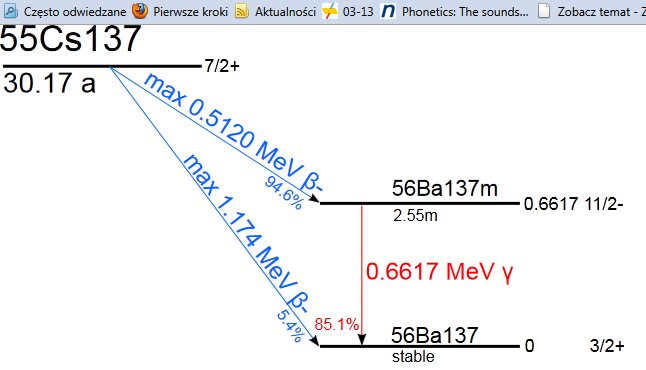
, których dezaktywa-cja pociąga za sobą emisję kwantów promieniowania γ o czterech różnych energiach, choć istotny wpływ mają jedynie kwanty o energiach 1,173 MeV i 1,333 MeV. W mniejszym stopniu wykorzystuje się rozpad promienio-twórczy cezu-137 (energia promieniowania wynosi 0,661 MeV, a okres półtrwania jest ponad pięciokrotnie dłuższy od kobaltu-60, mianowicie wynosi 30 lat wobec 5,263 lat dla 60Co).
Izotopy te wybrano ze względu na ich dostępność, odpowiednią postać fizyczną, zadowalającą szybkość rozpadu oraz dostatecznie dużą energię emitowanego promieniowania, która decyduje o zdolności penetracyjnej promieniowania.
Promieniowanie hamowania cechuje się widmem ciągłym z nałożonymi liniami charakterystycznymi dla materiału tarczy. W chemii radiacyjnej najczęściej stosuje się promieniowanie rentgenowskie (X), uzyskiwane wskutek konwersji energii kinetycznej cząstek naładowanych przyspieszanych w akceleratorach. Energia otrzymana w ten sposób zależy wprost proporcjonalnie od kwadratu liczby atomowej materiału tarczy, w której cząsteczki zostają wyhamowane, a także odwrotnie proporcjonalnie od drugiej potęgi masy cząstek oddających swoją energię, toteż konwersję energii kinetycznej przeprowadza się dla cząstek najlżejszych, czyli elektronów, hamowanych w tarczy np. wolframowej. Niestety, emisja takiego promieniowania wiąże się z wytracaniem energii przez wszystkie cząstki naładowane, w tym przez te pochodzące z rozpadu naturalnych i sztucznych izotopów promieniotwórczych.
Jako promieniowanie korpuskularne największą rolę odgrywają przyspieszone elektrony o energiach od 0,5 do 13 MeV, które uzyskuje się w różnorakich akceleratorach. Mniejszego znaczenia nabiera promieniowanie korpuskularne emitowane przez naturalne i sztuczne radionuklidy oraz cięższe cząstki naładowane przyspieszane w akceleratorach jonów, a także promieniowanie korpuskularne, towarzyszące reakcjom jądrowym, zachodzącym w reaktorach jądrowych.
Do zalet akceleratorów elektronowych należy nadawanie dowolnych, korzystnych do badań radiacyjnych energii, ustalanie natężenia wiązki, możliwość zmiany przekroju oraz kierunku strumienia, a także uzyskiwanie - prócz promieniowania ciągłego - impulsów o krótkim czasie trwania. Ta ostatnia cecha stanowi podstawę techniki radiolizy impulsowej.
Promieniowanie elektronowe można także uzyskiwać poza akceleratorami - poprzez promieniowanie β-, które zostaje wyemitowane w przemianach jądrowych naturalnych i sztucznych radionuklidów. Odznacza się ono ciągłym widmem energetycznym. Energia elektronów, własności absorpcyjne ośrodka i gęstość elektronowa materiału pochłaniającego decydują o przestrzennym rozkładzie dawki promieniowania pochłoniętego. Z praktycznego punktu widzenia należy zbadać grubość warstwy pochłaniającej promieniowanie o danej energii, w której występuje możliwie równomierny rozkład dawki zaabsorbowanej. Istotne ograniczenie stanowi niewielki zasięg promieniowania β, nawet przy maksymalnej energii (ponad 2 MeV), w fazach skondensowanych - do 4 mm dla 90Y.
W najmniejszym stopniu stosuje się strumienie cięższych jonów dodatnich jako promieniowanie korpuskularne. Promieniowanie α cechuje się znacząco większą gęstością jonizacji. Najczęściej stosowane źródło to 210Po, który w wyniku rozpadu uwalnia promieniowanie α o energii 5,3 MeV (zasięg w powietrzu: 38 mm, w wodzie: 38,9 μm). Oprócz tego było wykorzystywane również promieniowanie towarzyszące rozpadowi 226Ra czy gazowego 222Rn. W praktyce ma się do czynienia z mieszaniną promieniowań o różnych energiach, co wynika z ustalania się równowagi promieniotwórczej między głównym izotopem a produktami jego rozpadu. W badaniach stosowano także akceleratory cząstek ciężkich (protonów, deuteronów, jonów helu, jonów atomów cięższych) - uzyskiwano podówczas strumienie cząstek o bardzo wysokich energiach oraz małym natężeniu, co wywoływało przemiany jądrowe w napromienianym materiale oraz utrudniało uzyskiwanie mierzalnych stężeń produktów radiolizy.
Do podstawowych przemysłowych urządzeń radiacyjnych należą akceleratory elektronów (napromienianie materiałów, które dają się formować w postaci taśmy lub zamykać w opakowaniach o niezbyt dużej grubości warstwy i łatwych do umieszczenia na taśmie przenośnika) oraz źródła izotopowe (napromienianie dużych obiektów oraz prowadzenie syntez chemicznych w reaktorach umieszczonych w polu promienia). Jedno z przykładowych urządzeń to źródło kobaltowe, w których przenośniki skrzynkowe przemieszczają materiał do napromienienia tak, ażeby skierować skrzynki różnymi stronami do źródła. Dawkę zmienia się w zakresie od 1 do 50 J/g (od 0,1 do 5 Mrad), stosując różne prędkości przenośnika. W czasie pracy komory ładunki kobaltowe utrzymuje się przy użyciu chwytaków elektromagnetycznych w środkowym punkcie pomieszczenie do napromieniania. W razie awarii urządzenia bądź konieczności przeprowadzenia kontroli pomieszczenia napromieniań źródła opuszcza się do basenu wodnego znajdującego się w podłodze komory lub też chowa się je do pojemników ołowianych albo zasobników otoczonych odpowiednio grubą warstwą śrutu żeliwnego bądź betonu. Stropy i ściany komory napromieniań również buduje się z potężnych płyt betonowych tak, aby promieniowanie na zewnątrz nie przekraczało dopuszczalnych poziomów.
Kobalt promieniotwórczy otrzymuje się w reaktorach jądrowych w wyniku reakcji 59Co z neutronami: 59Co + n 60Co (T1/2 = 10 min) lub 59Co + n 60Co (T1/2 = 5,27 lat). Pierwszy izomer ulega w 99 % przemianie w izomer o dłuższym czasie połowicznego rozpadu. Szybkość tej reakcji zależy wprost proporcjonalnie od przekroju czynnego σ1 i gęstości strumienia neutronów. Powstający 60Co ulega dalszej przemianie, której szybkość zależy liniowo od przekroju czynnego σ2: 60Co + n 61Co. Otrzymany radionuklid produkuje się w postaci prętów lub cylindrów o różnych wymiarach i odmiennej aktywności właściwej. Ażeby uniknąć kontaminacji urządzeń radiacyjnych (kobaltowych źródeł promieniowania γ) i napromienianych materiałów, zamyka się kobalt promieniotwórczy w hermetycznych osłonach, najlepiej aluminiowych, gdyż glin nie ulega aktywacji w reaktorze bądź też daje izotopy promieniotwórcze o bardzo krótkim okresie półtrwania. Najkorzystniej uczynić to z kobaltem nieaktywnym. Do wad koszulek aluminiowych należy mała odporność chemiczna oraz mechaniczna, toteż w razie kontaktu ładunków kobaltowych z napromienianymi próbkami lub w przypadku przesuwania ich automatycznie wewnątrz źródła stosuje się koszulki stalowe, które zapobiegają uszkodzeniu osłony, a następnie rozpyleniu kobaltu. Podówczas zamyka się już aktywowany kobalt w należycie przygotowanych komorach, ponieważ żelazo aktywuje się w reaktorze do stosunkowo wolno rozpadającego się izotopu 59Fe.
W celu określenia wydajności i planowania procesów radiacyjnych potrzebna jest dokładna znajomość dawek energii dostarczanych do napromienianych materiałów. Można to uczynić poprzez obliczenia bądź drogą empiryczną. Metody kalkulacyjne wymagają dostarczenia wielu danych, jednak stanowią cenne uzupełnienie dozymetrii eksperymentalnej, gdy zachodzą różnice współczynników pochłaniania dla układu dozymetrycznego i napromienianych substancji albo kiedy należy podzielić pochłoniętą energię pomiędzy składniki mieszaniny kilku substancji lub też określić stopień nierównomierności rozkładu dawki w trakcie napromieniania przedmiotów stałych o większych wymiarach.
Do automatycznej kontroli pracy urządzeń radiacyjnych wykorzystuje się dozymetrię fizyczną, opartą na jonizacji gazu w komorach pomiarowych, na zjawisku fotoelektrycznym bądź na zmianie własności elektrycznych (np. oporności) napromienianych półprzewodników.
Dozymetria chemiczna polega na badaniu reakcji zachodzących pod wpływem promieniowania. Dobry układ dozymetryczny pozwala na pomiar dawek w możliwie szerokim zakresie od kilku krad do setek Mrad. Ponadto wydajność radiacyjna jego procesu dozymetrycznego nie powinna zależeć w znaczącym stopniu od energii czy typu promieniowania jonizującego, mocy dawki, temperatury tudzież obecności niewielkich domieszek substancji obcych. Oprócz tego jest korzystne, żeby produkty dały łatwo oznaczać się analitycznie ze sporą dokładnością, a ich ilość nie zwiększała się po przerwaniu napromieniania. Roztwory dozymetryczne winny odznaczać się trwałością na tyle dużą, żeby istniała możliwość ich wcześniejszego przygotowania, a także użycia przez dłuższy okres czasu.
Do najbardziej rozpowszechnionych ciekłych układów dozymetrycznych należy wodny roztwór soli żelaza dwuwartościowego (dozymetr Frickego lub żelazawo-żelazowy). W środowisku kwaśnym produkty radiolizy wody reagują z jonami w następujący sposób:
Roztwór zawiera rozpuszczony tlen, który wiążę całą ilość wydzielającego się wodoru atomowego w rodniki wodoronadtlenkowe HO2•. Do roztworu dodaje się również chlorek sodu, który zmniejsza czułość układu na obecność zanieczyszczeń organicznych.
Stężenie jonów żelazowych(III) oznacza się obecnie najczęściej spektrofotometrycznie (wybiera się jedną z trzech długości fal: 304, 275 lub 224 nm, dla których dokładnie wyznaczono współczynniki adsorpcji i ich zależność od temperatury). Na podstawie pomiaru adsorpcji napromienionego roztworu, znając molowy współczynnik pochłaniania ε tudzież wydajność radiacyjną utleniania jonów żelazowych(II) do żelazowych(III), oblicza się dawkę zaabsorbowaną ze wzoru:
ΔA - zmiana absorbancji (wywołana napromienieniem roztworu), k - współczynnik liczbowy, zależny od zastosowanego układu jednostek, ε - molowy współczynnik absorpcji jonów Fe2+ dla stosowanej długości fali, l - grubość warstwy roztworu w kuwecie, G(Fe3+) - wydajność jonów żelazowych(III), ρ - gęstość roztworu.
Oprócz tego stosuje się również dozymetry termoluminescencyjne i przewodnościowe, którymi bada się efekty radiacyjne w ciałach stałych, dozymetry polimerowe: folia z poli(chlorku winylu), zawierająca barwnik (np. fiolet metylowy), płytki z poli(metakrylanu metylu) - można je dołączyć bezpośrednio do materiału wprowadzanego do urządzeń radiacyjnych, przez co kontrolują stopień napromieniania każdej partii materiału.
Cel ćwiczenia
Celem ćwiczenia było poznanie działania dozymetru Frickego poprzez zmierzenie dawki promieniowania gamma pochłoniętej przez roztwór soli Fe2+ oraz wyznaczenie mocy dawki.
Wykonanie ćwiczenia
Do kolby miarowej na 50 ml odważono na wadze laboratoryjnej 0,292 g NaCl cz.d.a i 1,39 g FeSO4 • 7 H2O cz.d.a oraz odmierzono około 0,1 ml stężonego roztworu H2SO4, uzupełniono kolbę wodą destylowaną do kreski i rozpuszczono zawarte w niej sole, otrzymując roztwór A. W drugiej kolbie miarowej o pojemności 250 ml sporządzono roztwór B poprzez odmierzenie do niej 5,33 ml stężonego H2SO4 i uzupełnienie jej wodą destylowaną do kreski oraz wymieszanie otrzymanej zawartości. Do kolby miarowej o pojemności 100 ml odmierzono 2 ml roztworu A, uzupełniono roztworem B do kreski i całość wymieszano. Otrzymany w opisany wyżej roztwór dozymetryczny nasycano tlenem przez 15 minut,
a w międzyczasie 5 oznaczonych ampułek napełniono 10 ml wody destylowanej. Po natlenianiu do 6 ponumerowanych ampułek wprowadzono po 10 ml roztworu dozymetrycznego, ampułki umieszczono w koszyczku, a następnie przeprowadzono napromienianie ampułek w bombie kobaltowej, w ten sposób, że po każdych 15 minutach napromieniania wyjmowano kolejną ampułkę z roztworem dozymetrycznym, a na jej miejsce wstawiano ampułkę z wodą destylowaną. W międzyczasie włączono spektrofotometr, do pomiaru ustawiono w nim długość fali równą 304 nm, a następnie wyzerowano jego wskazania, wstawiając do komory pomiarowej dwie kuwety napełnione wodą destylowaną. Po wyjęciu napromienionych ampułek z roztworem dozymetrycznym z bomby kobaltowej zmierzono absorbancję tych roztworów oraz nienapromienionego roztworu dozymetrycznego przy ustalonej wcześniej długości fali względem wody destylowanej.
Opracowanie wyników
Tabela 1. Wyniki pomiarów absorbancji roztworu dozymetrycznego i wielkości obliczone na ich podstawie dla różnych czasów napromieniania
t [min] |
A |
ΔA |
Δc [mol/dm3] |
D [Gy] |
0 |
0,030 |
- |
- |
0 |
15 |
0,062 |
0,032 |
1,50 · 10-5 |
9,06 |
30 |
0,073 |
0,043 |
2,02 · 10-5 |
12,17 |
45 |
0,097 |
0,067 |
3,15 · 10-5 |
18,96 |
60 |
0,130 |
0,100 |
4,69 · 10-5 |
28,30 |
75 |
0,148 |
0,118 |
5,54 · 10-5 |
33,40 |
90 |
0,193 |
0,163 |
7,65 · 10-5 |
46,13 |
Przykładowe obliczenia
ΔA = A - A0, gdzie: A - absorbancja próbki po danym czasie napromieniania
A0 - absorbancja próbki nie napromienionej
ΔA = 0,062 - 0,03 = 0,032
![]()
, gdzie: ε - molowy współczynnik absorpcji; dla jonów Fe3+ w 0,8 n roztworze H2SO4 w 25ºC ε = 2130 dm3/(mol·cm)
l - grubość warstwy roztworu w kuwecie; w stosowanym spektrometrze
l = 1 cm
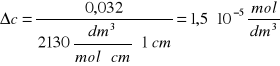
![]()
, gdzie: G(Fe3+) - wydajność radiacyjna powstawania jonów Fe3+; G(Fe3+) = 1,62 · 10-6 mol/J
ρ - gęstość roztworu dozymetrycznego; ρ = 1,024 g/cm3 =
= 1024 g/dm3
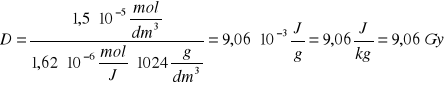
Na podstawie uzyskanych wyników sporządzono wykres dawki pochłoniętej przez roztwór od czasu napromieniania.
Metodą najmniejszych kwadratów wyznaczono moc dawki (współczynnik nachylenia prostej) wraz z błędem pomiarowym, korzystając z programu Excel.
a = 0,474175
Δa = 0,015707
Co ostatecznie, zgodnie z zasadami zaokrąglania niepewności pomiarowych, daje:
M = 0,47 ± 0,02 Gy/min
Po przeliczeniu:
M = 0,0079 ± 0,0003 Gy/s
Wnioski
Im dłuższy czas napromieniania roztworu, tym większą dawkę promieniowania pochłonął. Zależność ta jest liniowa (R2 = 0,9798). Z nachylenia prostej wyznaczyć można moc dawki. Wynosi ona 0,47 ± 0,02 Gy/min czyli 0,0079 ± 0,0003 Gy/s.
Rozrzut wyników jest najprawdopodobniej spowodowany niedokładnym sporządzeniem roztworów (niedokładnym odmierzaniem odczynników, niewystarczającym mieszaniem) czy niestarannym odmierzaniem ich ilości do analizy, a także tym, iż wskazania spektrofotometru zmieniały się w czasie i nie ulegały ustaleniu. Ponadto mieliśmy do czynienia z dawkami mniejszymi niż dolna granica stosowalności dozymetru Frickego (zakres stosowalności dozymetru Frickego zawiera się w zakresie dawek 40 - 400 Gy).
2
Wyszukiwarka