6. SPEKTROFOTOMETRYCZNE OZNACZANIE KOBALTU (II)
ZA POMOCĄ NITROZO-R-SOLI
Nitrozo-R-sól jest specyficznym odczynnikiem do spektrofotometrycznego oznaczania kobaltu w środowisku wodnym. W czasie przebiegu reakcji kompleksowania Co(II) utlenia się tlenem powietrza do Co(III):
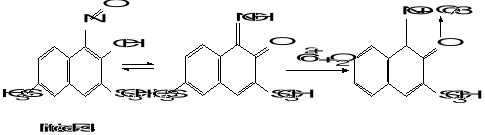
Niewielkie ilości jonów innych metali nie przeszkadzają w oznaczaniu kobaltu, ponieważ ich barwne kompleksy mogą istnieć tylko w środowisku obojętnym lub słabo kwaśnym. Silnie kwaśne środowisko wytrzymuje bez rozkładu tylko kompleks kobaltu.
Odczynniki
Roztwór wzorcowy kobaltu (II) zawierający 1 mg Co/ml.
Nitrozo-R-sól cz.d.a., 0.1% roztwór.
Bufor octanowy pH 5.6, c(CH3COOH+CH3COONa) = 1 mol/l.
Kwas solny stężony, cz.d.a.
Kwas fosforowy (V) stężony, cz.d.a.
Aparatura i sprzęt laboratoryjny
Kolby miarowe pojemności 10 ml |
7 szt. |
Zlewki pojemności 25 ml |
6 szt. |
Pipety wielomiarowe pojemności 1 ml |
3 szt. |
Pipety wielomiarowe pojemności 2 ml |
2 szt. |
Pipeta jednomiarowa pojemności 5 ml |
1 szt. |
Spektrofotometr z kuwetami 1cm.
Wykonanie ćwiczenia
1. Przygotować roboczy wzorcowy roztwór kobaltu zawierający 0.01 mg Co/ml przez rozcieńczenie 1 ml wzorca zawierającego 1 mg Co/ml do objętości 100 ml.
2. Do kolby miarowej pojemności 10 ml wprowadzić 1 ml buforu i 1 ml nitrozo-R-soli, dopełnić wodą i wymieszać. Zbadać zależność absorbancji od długości fali w zakresie od 400 do 600 nm co 10 nm względem wody jako odnośnika.
Uwaga: po zmianie długości fali, przed każdym pomiarem wyzerować przyrząd względem odnośnika.
3. Do kolby miarowej pojemności 10 ml wprowadzić 1 ml buforu, 1 ml nitrozo-R-soli, 2 ml roboczego wzorcowego roztworu kobaltu, dopełnić wodą i wymieszać. Zbadać zależność absorbancji od długości fali w zakresie od 400 do 600 nm co 10 nm. W okolicach maksimum absorbancji zagęścić odpowiednio punkty pomiarowe. Jako odnośnik stosować roztwór przygotowany w punkcie 2.
4. Na podstawie uzyskanych wyników wykonać wykres zależności absorbancji od długości fali dla nitrozo-R-soli i kompleksu kobaltu z nitrozo-R-solą. Wybrać analityczną długość fali do oznaczania kobaltu.
5. Przygotować serię wzorców do uzyskania prostej kalibracyjnej z co najmniej 5 roztworów wzorcowych. Stężenia kobaltu należy tak dobrać, aby absorbancja próbek mierzona względem ślepej próby zawarta była w zakresie od 0.1 do 1 (wykorzystać pomiar absorbancji z punktu 3).
Wariant A - w nieobecności innych jonów metali tworzących kompleksy z nitrozo-R-solą.
5.1. A. Do kolbek miarowych pojemności 10 ml wprowadzić 1 ml buforu, 2 ml nitrozo-R-soli i odpowiednią objętość roboczego wzorcowego roztworu kobaltu. Dopełnić wodą, wymieszać i po upływie 10 minut zmierzyć absorbancję. Sporządzić prostą kalibracyjną do oznaczania kobaltu.
6.A. Zmierzyć absorbancję roztworu otrzymanego do analizy po przeprowadzeniu reakcji z nitrozo-R-solą. Na podstawie otrzymanej prostej wzorcowej wyliczyć zawartość kobaltu w badanej próbce.
Wariant B - w obecności jonów metali tworzących kompleksy z nitrozo-R-solą.
5.1.B. Do zlewek pojemnosci 25 ml wprowadzić 1 ml buforu, 2 ml nitrozo-R-soli i odpowiednią objętość roboczego wzorcowego roztworu kobaltu. Zawartość zlewek ogrzać do wrzenia. Ostudzić nieco roztwory, dodać 0.5 ml stężonego kwasu ortofosforowego (V) i 0.5 ml stężonego roztworu kwasu solnego. Ogrzać roztwory do wrzenia i utrzymywać w tym stanie w ciągu minuty. Ostudzić roztwory i przenieść ilościowo do kolbek miarowych pojemności 10 ml, dopełnić wodą, wymieszać i zmierzyć absorbancję. Sporządzić prostą kalibracyjną do oznaczania kobaltu.
6B. Zmierzyć absorbancję roztworu otrzymanego do analizy po przeprowadzeniu reakcji z nitrozo-R-solą i po rozłożeniu kompleksów z innymi metalami. Na podstawie otrzymanej prostej wzorcowej wyliczyć zawartość kobaltu w badanej próbce.
11. POTENCJOMETRYCZNY POMIAR pH PRZY UŻYCIU ELEKTRODY SZKLANEJ. OCENA KWASOWOŚCI PREPARATÓW FARMACEUTYCZNYCH
Celem ćwiczenia jest przeprowadzenie potencjometrycznego pomiaru pH w ogniwie jednoprętowym złożonym z elektrody szklanej i nasyconej elektrody chlorosrebrowej. Elektroda szklana stanowi pod względem elektrochemicznym złożony układ, którego potencjał zależy od stosunku stężeń jonów wodorowych po obu stronach membrany szklanej, od potencjału elektrody wyprowadzającej oraz od niewielkiej, dochodzącej do kilkunastu miliwoltów wartości tzw. potencjału asymetrii.
Potencjał (25 °C) najczęściej stosowanej elektrody szklanej, wypełnionej roztworem kwasu solnego lub roztworem buforowym zawierającym jony chlorkowe, z wyprowadzeniem chlorosrebrnym, daje się opisać równaniem
Eszkl.= EAg/AgCl - 0.059 lg aCl- + 0.059 pHw - 0.059 pH = Eas
gdzie:
EAg/AgCl |
- potencjał normalny elektrody chlorosrebrnej, |
aCl- |
- aktywność jonów chlorkowych roztworu wypełniającego, |
pHw |
- pH roztworu wypełniającego, |
pH |
- pH roztworu badanego, |
Eas |
- potencjał asymetrii. |
Wartości EAg/AgCl, aCl-, pHw oraz Eas charakterystyczne dla danej elektrody i z wyjątkiem Eas niezmienne, mogą razem wzięte stanowić tzw. „normalny” potencjał elektrody szklanej. Wyrażenie na potencjał elektrody szklanej można zatem podać w sposób analogiczny do potencjału innych elektrod wskaźnikowych stężenia jonów wodorowych.
Eszkl. = „E0szkl.” - 0.059 pH
Jeżeli potencjał elektrody wyrażony jest względem dowolnej elektrody odniesienia, wtedy wartość jego równa sile elektromotorycznej ogniwa, złożonego z elektrody szklanej i elektrody odniesienia wynosi
E = Eg - 0.059 pH
gdzie Eg stanowi różnicę między „E0szkl.” i potencjałem elektrody odniesienia.
Z uwagi na to, że w przypadku elektrody szklanej wartość współczynnika liczbowego w równaniu (1) nie zawsze wykazuje wartość teoretyczną oraz, że potencjał asymetrii ulega zmianom w czasie, konieczna jest znajomość charakterystyki elektrody szklanej, względnie towarzyszące każdemu pomiarowi pH kalibrowanie układu pomiarowego na dwa, a w najgorszym przypadku na jeden wzorcowy roztwór buforowy.
Odczynniki
Polopiryna, asprocol, calcipiryna - preparaty farmaceutyczne
Roztwory buforowe wzorcowe od pH 1 do pH 10.
Aparatura i sprzęt laboratoryjny
Naczyńka szklane - ilość odpowiadająca przygotowanym roztworom buforowym. |
|
Zlewki pojemności 100 ml |
4 szt. |
Kolby miarowe pojemności 50 ml |
4 szt. |
Lejek szklany |
1 szt. |
Bagietka szklana |
1 szt. |
Sączki |
|
pH-metr typu N-517 MERA ELWRO.
Wykonanie ćwiczenia
1. Charakterystyka elektrody szklanej.
1.1. Zgodnie z instrukcją obsługi używanego przyrządu zmierzyć potencjał elektrody szklanej w roztworach buforowych o pH 1 - 10.
1.2. Przygotować roztwory o pH nieznanym
Tabletkę np. polopiryny rozpuścić w 30 ml destylowanej wody. Roztwór przesączyć do kolby miarowej pojemności 50 ml, przemywając sączek małymi porcjami (5 ml) trzykrotnie destylowanej wody. Roztwór w kolbie uzupełnić do kreski wodą destylowaną i wymieszać. Podobnie przygotować roztwory pozostałych preparatów farmaceutycznych. Zmierzyć w przygotowanych roztworach potencjał elektrody szklanej względem elektrody kalomelowej.
1.3. Z uzyskanych w pkt. 1.1. wartości wykreślić zależności potencjału elektrody od pH roztworu i poprowadzić między nimi prostą o najbardziej prawdopodobnym nachyleniu.
Z wykresu odczytać:
- wartość Eg,
- współczynnik nachylenia prostej do osi odciętych ,
- wartość pH, przy której elektroda szklana wykazuje potencjał równy potencjałowi elektrody chlorosrebrowej.
U w a g a : przy opracowywaniu wyników ćwiczenia można wykorzystać program komputerowy „Obliczenia”.
1.4. Posługując się wykresem E = f(pH) znaleźć wartość pH roztworów badanych.
2. Pomiar pH na skali potencjałów po wykalibrowaniu elektrod na dwa roztwory buforowe.
2.1. Na podstawie zależności potencjału elektrody od pH roztworu wybrać po dwa roztwory buforowe tak, by jeden miał niższą, a drugi wyższą od roztworu badanego wartość pH.
2.2. Wyznaczyć kolejno potencjał elektrody szklanej względem nasyconej elektrody kalomelowej w obu roztworach buforowych oraz w roztworze badanym. Obliczyć pH badanego roztworu ze wzoru
gdzie:
EB1 |
- potencjał elektrody szklanej w buforze o pH = pHB1 , |
EB2 |
- potencjał elektrody szklanej w buforze o pH = pHB2 , |
Ex |
- potencjał elektrody szklanej w buforze o pH = pHx. |
Przy czym
pHB1 < pHx < pHB2
Wyprowadzić stosowany wzór.
3. Pomiar pH na skali pH po wykalibrowaniu pH-metru na jeden roztwór buforowy.
Pomiary przeprowadzić zgodnie z instrukcjami obsługi na wszystkich oddanych do dyspozycji przyrządach stosując elektrody szklane różnego typu. Wyciągnąć wnioski z poczynionych obserwacji.
4. Pomiar pH na skali pH po wykalibrowaniu przyrządu na dwa roztwory buforowe.
Wykorzystując możliwość dostosowania czułości pH-metru typu N-517 do obniżonego nachylenia chrakterystki elektrody szklanej, przeprowadzić pomiary pH na skali pH przyrządu wykalibrowanego uprzednio na dwa roztwory buforowe zgodnie z instrukcją obsługi.
U w a g i:
- przed każdym pomiarem należy elektrody kilkakrotnie przepłukać wodą destylowaną i osuszyć (dotykając delikatnie płatkami ligniny);
- do roztworu badanego należy zanurzyć całą banieczkę elektrody szklanej;
- po skończonych pomiarach elektrody opłukać wodą destylowaną. Elektrodę szklaną przechowywać zanurzoną w wodzie destylowanej, a kalomelową w nasyconym roztworze chlorku potasu.
5. Opracowanie wyników.
5.1. Zestawić uzyskane wyniki oznaczenia pH oraz ocenić, jakie może być źródło występujących pewnych rozbieżności wyników
5.2. Podać składy użytych preparatów farmaceutycznych, pH ich roztworów i porównać je między sobą.
5.3. Na podstawie uzyskanych danych doświadczalnych (![]()
) oraz w oparciu o wzór Eg = Eºszkl. - NEK obliczyć stężenie roztworów wypełniających elektrody.
19. POTENCJOMETRYCZNE I KONDUKTOMETRYCZNE MIARECZKOWANIE MIESZANINY WODOROTLENKU SODU
I JODKU POTASU
Celem ćwiczenia jest określenie przebiegu zmian zachodzących podczas równoczesnego miareczkowania potencjometrycznego i konduktometrycznego mieszaniny jodku potasu i wodorotlenku sodu, oraz porównanie obu miareczkowań między sobą.
Miareczkowanie potencjometryczne prowadzi się w układzie elektrod srebrnej i nasyconej elektrody kalomelowej jako elektrody odniesienia. Duża różnica rozpuszczalności jodku srebra (I) i wodorotlenku srebra (I) pozwala na uzyskanie dwóch wyraźnych skoków potencjału oraz na wygodną ocenę położenia obu punktów równoważnikowych na krzywej miareczkowania potencjometrycznego.
Miareczkowanie konduktometryczne jest przykładem miareczkowania strąceniowego, w którym występują dwa czytelne punkty równoważnikowe związane z wytrącaniem najpierw jodku srebra (I) a następnie wodorotlenku srebra (I), który rozkłada się natychmiast z wydzieleniem brunatnego tlenku srebra (I).
Końcowe punkty miareczkowań ustala się graficznie na podstawie wykresów przedstawiających w pierwszym przypadku zależność potencjału elektrody srebrnej od objętości dodanego odczynnika miareczkującego w drugim zależność przewodnictwa od objętości dodanego odczynnika miareczkującego.
Odczynniki
Wodorotlenek sodu cz.d.a., roztwór o stężeniu c(NaOH) = 0.05 mol/l.
Jodek potasu cz.d.a., roztwór o stężeniu c(KI) = 0.05 mol/l.
Azotan (V) srebra (I) cz.d.a., roztwór o stężeniu c(AgNO3) = = 0.05 mol/l.
Azotan (V) potasu cz.d.a., roztwór nasycony.
Aparatura i sprzęt laboratoryjny
Biureta pojemności 25 ml z podziałką co 0.1 ml |
1 szt. |
Pipety jednomiarowe pojemności 10 ml |
2 szt. |
Pipeta jednomiarowa pojemności 25 ml |
1 szt. |
Zlewka 200-250 ml |
1 szt. |
Mieszadło magnetyczne |
|
pH-metr typ N-517 MERA ELWRO.
Konduktometr.
Wykonanie ćwiczenia
1. Do zlewki odmierzyć po 10 ml roztworów wodorotlenku sodu i jodku potasu.
2. Przygotować elektrody: elektrody platynowe opłukać wodą destylowaną, elektrodę srebrną oczyścić mechanicznie, elektrodę kalomelową opłukać wodą destylowaną i osadzić przez korek w tygielku z dnem porowatym napełnionym nasyconym roztworem azotanu (V) potasu (klucz elektrolityczny). Elektrody umieścić w zlewce z roztworem przygotowanym do miareczkowania i uzupełnić odmierzoną ilością wody destylowanej tak, aby elektrody były całkowicie zanurzone w roztworze, a wirnik mieszadła magnetycznego mógł się swobodnie obracać (rys. 1, ćwiczenie 18).
3. Wyprowadzenia elektrod połączyć z odpowiednimi zaciskami potencjometru i konduktometru. Uruchomić mieszadło magnetyczne i wykonać pierwszy pomiar SEM zestawionego ogniwa oraz przewodnictwa (zgodnie z instrukcjami obsługi używanych przyrządów).
4. Przeprowadzić miareczkowanie dozując roztwór miareczkujacy w taki sposób, by uzyskać duże zagęszczenie pomiarów na łukach krzywej oraz w najbliższym sąsiedztwie punktów równoważnikowych. W tym celu roztwór z biurety dozować należy w sposób następujący: w granicach 0 - 6 ml porcjami po 1.00 ml, 6 - 14 ml po 0.10 ml, (w najbliższym sąsiedztwie punktu równoważnikowego gwałtowną zmianę potencjału elektrody wskaźnikowej wywołać może 1 kropla roztworu); 14 - 16 ml po 0.50 ml, 16 - 24 ml po 0.10 ml, 24 - 25 ml po 0.50 ml. Uzyskane wyniki miareczkowania potencjometrycznego przedstawić w tabeli :
Objętość AgNO3 (V) [ml] |
Potencjał elektrody srebrnej (E) [mV] |
|
|
|
|
W pomiarach konduktometrycznych przewodnictwo (konduktancję) odczytywać co 0.5 ml dodawanego titranta. Dla każdej dodanej objętości roztworu miareczkującego obliczyć poprawkę.
gdzie:
V1 |
- objętość roztworu miareczkowanego, |
V2 |
- objętość odmierzonej wody, |
V3 |
- objętość dodanego odczynnika miareczkującego. |
Pomnożyć przez obliczoną poprawkę odczytane wartości konduktancji (przewodnictwa) uzyskując wartość niezależną od zmniany objętości. Otrzymane wyniki zestawić w tabeli:
Objętość roztworu miareczkującego [ml] |
|
|
Poprawiona wartość konduktancji [mS] |
|
|
|
|
5. Opracowanie wyników miareczkowania potencjometrycznego
5.1. Wykreślić krzywą miareczkowania na podstawie wyników dokładnego miareczkowania i wyznaczyć punkty końcowe miareczkowania w sposób graficzny metodą stycznych oraz metodą kół współśrodkowych Tubbsa.
5.2. Znaleźć punkty końcowe miareczkowania metodą Hostettera i Robertsa, polegającą na odczytaniu wartości pierwszej pochodnej funkcji E = f(V) jako funkcji objętości = f(V). W tym celu w kolumnie 3 tabeli wpisać wartości .
Punkt końcowy miareczkowania przypada na objętość leżącą w obszarze maksymalnej wartości . W celu znalezienia dokładnego położenia punktu końcowego wykreślić zależność = f(V) w przedziale objętości 6 - 14 ml i 16 - 24 ml dodanego roztworu miareczkującego. Warunkiem prawidłowych wyników uzyskanych na tej drodze jest dozowanie odczynnika równymi porcjami, a dokładność zależy od gęstości punktowania krzywej. Porównć ze sobą uzyskane wyniki.
5.3. Obliczyć zawartość jonów wodorotlenowych i jodkowych (w mg) w analizowanym roztworze.
6. Opracowanie wyników miareczkowania kondukto-metrycznego.
6.1. Wykonać wykres zależności konduktancji (wartość poprawiona) od objętości miareczkującego roztworu mianowanego. Wyznaczyć punkty końcowe miareczkowania. Obliczyć zawartość jonów wodorotlenowych i jodkowych (w mg) w analizowanym roztworze.
6.2. Wyjaśnić kształt krzywej miareczkowania.
6.3. Porównać między sobą wyniki miareczkowania potencjometrycznego i konduktometrycznego. Porównać te wartości z ilościami wziętymi do miareczkowania.
31. AMPEROMETRYCZNE MIARECZKOWANIE JODU W JODYNIE BEZ PRZYŁOŻONEGO NAPIĘCIA
W miareczkowaniu amperometrycznym bez przyłożonego napięcia zewnętrznego kroplowa elektroda rtęciowa przyjmuje potencjał elektrody odniesienia. Jeżeli przy tym potencjale występuje prąd graniczny substancji miareczkowanej lub miareczkującej, to można prowadzić miareczkowanie amperometryczne bez przyłożonego zewnętrznego napięcia.
Podstawą oznaczania jest reakcja tiosiarczanu z jodem:
I2 + 2S2O32-![]()
2I- + S4O62-
Substancją elektrodowoczynną jest jod.
Odczynniki
Tiosiarczan sodu cz.d.a., roztwór o stężeniu c(S2O32-) = 0.01 mol/l.
Chlorek potasu cz.d.a., roztwór o stężeniu c(KCl) = 1 mol/l.
Jod cz.d.a., roztwór o stężeniu c(I) = 0.01 mol/l.
Jodyna.
Aparatura i sprzęt laboratoryjny
Zlewka pojemności 100 ml |
3 szt. |
Pipeta jednomiarowa pojemności 1 ml |
1 szt. |
Pipety jednomiarowe pojemności 5 ml |
2 szt. |
Pipeta wielomiarowa pojemności 10 ml |
1 szt. |
Biureta pojemności 25 ml |
1 szt. |
Cylinder pojemności 50 ml |
1 szt. |
Kolbki pojemności 25 ml |
4 szt. |
Polarograf.
Elektroda platynowa (o małej powierzchni), NEK.
Klucz elektrolityczny.
Mieszadło magnetyczne z bączkiem.
Mikroamperomierz.
Wykonanie oznaczenia
1. Sprawdzenie przebiegu krzywych natężenie - napięcie:
1.1. Do 4 kolbek pojemności 25 ml odmierzyć po 10 ml chlorku potasu, a następnie dodać: do pierwszej 5 ml roztworu tiosiarczanu; do drugiej 5 ml jodu; do trzeciej 1 ml jodu i roztworu tiosiarczanu aż do odbarwienia jodu; w czwartej pozostawić elektrolit podstawowy. Dopełnić wodą do kreski. Zarejestrować krzywe natężenie - napięcie.
1.2. Wykonać polarogramy w zakresie napięć od +0.5V do -0.5V, traktując elektrodę platynową jako katodę, a NEK jako anodę.
1.3. Na podstawie otrzymanych krzywych sprawdzić, czy przy napięciu zewnętrznym U = 0 V, któryś z roztworów osiąga prąd graniczny.
2. Przeprowadzenie oznaczenia:
2.1. Do zlewki wlać 50 ml wody destylowanej, 1 ml jodyny, 10 ml roztworu chlorku potasu.
2.2. Połączyć układ według schematu:
2.3. Roztwór miareczkować roztworem tiosiarczanu porcjami po 0.5 ml, aż do uzyskania 5 punktów o stałym natężeniu prądu.
2.4. Powtórzyć miareczkowanie.
3. Opracowanie wyników
3.1. Dla każdej objętości roztworu miareczkującego obliczyć poprawkę na rozcieńczenie:
gdzie:
P |
- poprawka na rozcieńczenie, |
Vo |
- pierwotna objętość roztworu, |
V |
- objętość dodanego odczynnika miareczkującego. |
3.2. Uzyskane wyniki zestawić w tabeli:
Objętość titranta |
Odczytywane natężenie prądu |
Poprawka na rozcieńczenie |
Poprawiona wartość natężenia prądu i = iodcz· P |
|
|
|
|
3.3. Wykreślić krzywe zależności poprawionych wartości natężenienia prądu od objętości odczynnika miareczkującego.
3.4. Wyznaczyć punkty końcowe miareczkowania.
3.5. Obliczyć zawartość jodu w jodynie.
3.6. Wyjaśnić przebieg krzywej miareczkowania.
35. KULOMETRYCZNE MIARECZKOWANIE ROZTWORU HEKSACYJANOŻELAZIANU (II) POTASU ANODOWO GENEROWANYM BROMEM
Heksacyjanożelazian (II) potasu miareczkowany jest kulometrycznie anodowo wytwarzanym bromem w środowisku kwaśnym. Punkt końcowy określany jest biamperometrycznie. Układ heksacyjanożelazian (II) / heksacyjanożelazian (III) jest układem odwracalnym. Dlatego przed punktem końcowym obserwuje się wzrost a następnie zmniejszanie się prądu w obwodzie wskaźnikowym. Po całkowitym zmiareczkowaniu heksacyjanożelazianu (II) potasu w roztworze pojawia się nadmiar bromu, co powoduje powstanie kolejnego układu odwracalnego brom/bromek i wzrost natężenia prądu przepływającego przez układ wskaźnikowy.
Odczynniki
Heksacyjanożelazian (II) potasu cz.d.a. roztwór o stężeniu c(K4Fe(CN)6)=0.1 mol/l.
Roztwór reakcyjny zawierający: bromek potasu cz.d.a. o stężeniu c(KBr)=0.1 mol/l, kwas siarkowy (VI) cz.d.a. o stężeniu c(H2SO4)=0.1 mol/l.
Aparatura i sprzęt laboratoryjny
Pipeta jednomiarowa pojemności 20 ml |
1 szt. |
Mikropipeta pojemności 100 μl |
1 szt. |
Elektrochemiczny miernik uniwersalny EMU |
|
Naczynko elektrolityczne wyposażone w elektrody układu wytwarzającego i biamperometrycznego układu wskaźnikowego |
|
Wykonanie oznaczenia
1. Do części anodowej naczynka kulometrycznego wprowadzić 50 ml roztworu reakcyjnego i 100 μl roztworu heksacyjanożelazianu (II) potasu.
2. Przestrzeń katodową naczynka wypełnić roztworem reakcyjnym.
3. Uruchomić miernik zgodnie z instrukcją obsługi. Wybrać opcję "173 Miareczkowanie kulometryczne". Zainstalować elektrody zgodnie ze schematem podanym na monitorze komputera wybierając detekcję amperometryczną. Miareczkowanie wykonać mierząc "całą krzywą". Oznacza to, że generowanie odczynnika miareczkującego będzie trwało do momentu, aż przez obwód przepłynie ładunek zadeklarowany w parametrze "Maksymalna liczba kulombów".
4. Ustalić następujące parametry detekcji punktu równoważnikowego:
Napięcie przykładane do elektrod wskaźnikowych |
150 mV |
Liczba odczytów w jednym pakiecie |
20 |
5. Ustalić następujące parametry generacji odczynnika miareczkującego:
Natężenie prądu elektrolizy |
5.0 mA |
Maksymalna liczba kulombów |
1.50 C |
Doza odczynnika miareczkującego |
5.00 mC |
Równoważnik elektrochemiczny |
3.818 mg/C |
Objętość próbki |
0.1 ml |
Nazwa odczynnika miareczkującego |
brom |
Opis próbki |
Fe (II) |
6. Wcisnąć klawisz "n" co spowoduje automatyczne rozpoczęcie wykonanie pomiarów.
7. Zanim aparat wykona same miareczkowanie stabilizuje on natężenie prądu przepływającego przez elektrody wskaźnikowe. Dalsze przejście do pomiarów następuje po wciśnięciu klawisza "k" w momencie kiedy natężenia prądu nie będzie się zmieniać w czasie.
8. Przeprowadzić obliczenia (zapisując uprzednio wyniki na dysk komputera) na komputerze zgodnie z podanymi na monitorze komputera czynnościami.
9. Wydrukować krzywą biamperometryczną włączając drukarkę i naciskając klawisz "PrtScr".
10. Obliczyć dokładne stężenie roztworu heksacyjanożela-zianu (II) potasu.
11. Objaśnić przebieg krzywej miareczkowania biamperometrycznego. Zaproponować przebieg krzywej biamperometrycznej miareczkowania tiosiarczanu jodem.
39. SPRAWDZENIE TOŻSAMOŚCI I CZYSTOŚCI CAPTOPRILU METODĄ CHROMATOGRAFII CIENKOWARSTWOWEJ
Ćwiczenie ma na celu zapoznanie studenta z metodą chromatografii cienkowarstwowej (z ang. thin layer chromatography -- TLC) oraz sprawdzenie tożsamości substancji czystej i czystości leku o nazwie CAPTOPRIL.
Substancją czynną jest (S)-1-(3-merkapto-2-metylo-1-oksopropylo)- -L-prolina (nazwa zwyczajowa: kaptopryl) o wzorze strukturalnym:
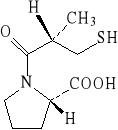
Chromatografia jest metodą rozdzielania składników jednorodnych mieszanin w wyniku różnego ich podziału między fazę ruchomą i nieruchomą układu chromatograficznego. Jeżeli fazą ruchomą jest ciecz, zaś faza nieruchoma umieszczona jest na płaszczyźnie, to mówimy o chromatografii cienkowarstwowej .
Sprawdzenie tożsamości sprowadza się do porównania wartości współczynników Rf wzorca i próbki badanej. Wykonuje się chromatogramy trzech roztworów wzorcowych. Pierwszy o stężeniu odpowiadającym stężeniu substancji w roztworze badanym, drugi pięćdziesiąt razy mniejszym i trzeci dwieście razy mniejszym. Wartość Rf plamy głównej otrzymanej z roztworu badanego powinna odpowiadać Rf plamy otrzymanej z roztworu wzorcowego pierwszego. Sprawdzenie czystości polega na porównaniu roztworów wzorcowych drugiego i trzeciego z dodatkowymi plamami roztworu badanego (jeżeli się pojawiają). Pojedyncze plamy nie powinny być większe ani intensywniejsze niż plama roztworu wzorcowego trzeciego, a suma wielkości i intensywności wszystkich dodatkowych plam nie powinna być większa od wielkości i intensywności plamy pochodzącej od roztworu drugiego. Wtedy stężenie pojedynczego zanieczyszczenia nie jest większe od 0.5% w stosunku do substancji czynnej, a suma wszystkich zanieczyszczeń 2%.
Składniki analizowanej próbki nie są barwne, należy więc przeprowadzić ich wizualizację. Chromatogramy wywołuje się najczęściej odczynnikami chemicznymi reagującymi z poszczególnymi substancjami, tworząc z nimi barwne produkty, np. pary jodu reagują z wieloma związkami tworząc plamki ich produktów o żółtym lub brązowym zabarwieniu.
Odczynniki
Toluen do HPLC.
Kwas octowy lodowaty do HPLC.
Metanol do HPLC.
Kaptopryl, preparat farmaceutyczny „CAPTOPRIL” (12.5 mg, 25 mg) "Jefa" Jelenia Góra.
Aparatura i sprzęt laboratoryjny
Kolby miarowe pojemności 10 ml |
5 szt. |
Cylinder pojemności 100 ml |
1 szt. |
Cylinder pojemności 25 ml |
1 szt. |
Pipetki kapilarne pojemności 25 μl |
5 szt. |
Mikropipeta automatyczna pojemności 50 μl |
1 szt. |
Mikropipeta automatyczna pojemności 200 μl |
1 szt. |
Zlewki pojemności 30 ml |
2 szt. |
Naczynka wagowe |
2 szt. |
Moździerz |
|
Łopatka |
|
Lejek (mały )
Sączek
Waga analityczna.
Płytka szklana pokryta żelem krzemionkowym Silica gel S60 (wymiary 10 x 20 cm)
Suszarka.
Komora jodowa.
Komora chromatograficzna.
Wykonanie ćwiczenia
1. Przygotowanie komory chromatograficznej.
- przygotowanie fazy ruchomej: odmierzyć toluen, kwas octowy lodowaty, metanol w stosunku objętościowym 75 : 25 : 1. Roztwór dobrze wymieszać,
- komorę przemyć 25 ml roztworu fazy ruchomej,
- wyciąć z bibuły pasek o wymiarach 56 x 12 cm,
- wyłożyć nią ściany komory chromatograficznej w taki sposób, aby jej końce wystawały poza nią,
- ostrożnie wlać resztę fazy ruchomej do komory chromatograficznej,
- przykryć komorę płytką szklaną zaginając na zewnątrz końce bibuły,
- pozostawić na godzinę w celu nasycenia komory (jest to czas na przygotowanie roztworów).
2. Przygotowanie roztworów wzorcowych .
ROZTWÓR WZORCOWY 1 - roztwór kaptoprylu o stężeniu 10 mg/ml: odważyć 0.10 g kaptoprylu i rozpuścić w 5 ml metanolu, przenieść do kolby miarowej pojemności 10 ml i uzupełnić metanolem do kreski.
ROZTWÓR WZORCOWY 2 - roztwór kaptoprylu o stężeniu 0.2 mg/ml: pobrać 200 μl roztworu wzorcowego 1 i przenieść go do kolbki miarowej pojemności 10 ml, a następnie uzupełnić metanolem do kreski .
ROZTWÓR WZORCOWY 3 - roztwór kaptoprylu o stężeniu 0.05 mg/ml: pobrać 50 μl roztworu wzorcowego 1 i przenieść go do kolby miarowej pojemności 10 ml, a następnie uzupełnić metanolem do kreski.
3. Przygotowanie roztworów badanych.
3.1. Roztwor badany: CAPTOPRIL 25 mg, średnia masa tabletki 0.155 g .
- odważyć ilość sproszkowanych tabletek odpowiadającą 100 mg substancji czynnej, to jest 0.618 g. Ilość tę należy wsypać do kolby miarowej pojemności 10 ml, dodać 5 ml metanolu i wytrząsać przez około 20 min. Dopełnić metanolem do kreski. Roztwór przesączyć i analizować przesącz.
3.2. Roztwor badany: CAPTOPRIL 12.5 mg, średnia masa tabletki 0.101g .
- odważyć ilość sproszkowanych tabletek odpowiadającą 100 mg substancji czynnej, to jest 0.805 g, ilość tę należy wsypać do kolby miarowej pojemności 10 ml, dodać 5 ml metanolu i wytrząsać przez około 20 min. Dopełnić metanolem do kreski. Roztwór przesączyć i analizować przesącz .
4. Przygotowanie płytki chromatograficznej:
- zaznaczyć cienko ołówkiem linię startu na wysokości 2 cm od dołu płytki,
- od lini startu odmierzyć piętnastocentymetrową drogę migracji,
- zaznaczyć punkty, na które będzie się nanosić roztwory,
- na linię startu, susząc płytkę suszarką, nanieść mikropipetką pojemności 25 μl roztwory wzorcowe i roztwór badany tak, aby plamki nie nachodziły na siebie,
- zwrócić szczególną uwagę na to, aby nie uszkodzić warstwy adsorpcyjnej,
- płytkę wysuszyć suszarką i umieścić w komorze chromatograficznej w celu rozwinięcia chromatogramu,
- obserwować czoło rozpuszczalnika; kiedy pokona ono drogę 15 cm wyjąć płytkę z komory i wysuszyć w strumieniu ciepłego powietrza.
5. Chromatogram wywołać w komorze jodowej do momentu pojawienia się widocznych plam. Po upływie tego czasu płytkę ostrożnie wyjąć z komory, otrzymane plamy, linię startu i linię końcową zakreślić ołówkiem. Wykonać kserokopię płytki.
6. Opracowanie wyników .
- wyznaczyć wartość współczynników Rf
Rf = A/B
Rf - określa stosunek drogi migracji substancji chromatografowanych (od punktu startu do środka plamki - A) do drogi przebytej przez fazę ruchomą (od lini startu do czoła fazy ruchomej - - B), gdzie B w tym przypadku wynosi 15 cm.
- zinterpretować otrzymane wyniki. Uzasadnić czy lek nadaje się do handlu?
143
9
Wyszukiwarka