Rys.1
ćwiczenie 1
IZOLACJA DNA I RNA Z KOMÓREK EUKARIOTYCZNYCH
Wiele specjalistycznych badań biologii molekularnej rozpoczyna się od izolacji genomowego DNA. Biologia molekularna zajmuje się szczegółowymi badaniami różnych elementów genomu, budową poszczególnych genów, ich funkcją a także poznawaniem mechanizmów odpowiedzialnych za regulację ich ekspresji. Ponieważ jednak obecnie także wiele innych dziedzin biologii stara się zgłębić podstawy genetyczne badanych przez siebie zjawisk, w pracach tych także zachodzi konieczność izolacji materiału genetycznego. W takich dziedzinach biologii, jak taksonomia czy antropologia, badacze coraz częściej opierają swoje wnioskowanie dotyczące pokrewieństwa pomiędzy badanymi populacjami na podstawie zmienności zapisanej w materiale genetycznym. Zmienność genetyczna jest również wykorzystywana przez medycynę sądową w takich sprawach jak ustalanie ojcostwa lub identyfikacja osób na podstawie biologicznego materiału dowodowego. Materiał genetyczny powszechnie wykorzystywany jest również w diagnostyce medycznej w celu identyfikacji u pacjentów alleli odpowiedzialnych za rozwój poszczególnych chorób. Natomiast w badaniach agrotechnicznych, genomowy DNA izolowany jest w pracach zmierzających do identyfikacji markerów molekularnych sprzężonych w określonymi właściwościami gatunków hodowlanych. Ponadto, genomowy DNA jest obiektem prac, których celem jest uzyskanie takich modyfikacji genetycznych, które polepszają właściwości produkcyjne uprawianych gatunków.
We wszystkich tych przypadkach zachodzi konieczność izolacji DNA z badanego materiału biologicznego. Zastosowana metoda izolacji powinna zapewniać uzyskanie odpowiedniej ilości DNA, charakteryzującego się wysoką czystością i niskim stopniem degradacji. Ilość preparatu uzyskanego po izolacji, w zależności od jego przeznaczenia, powinna wynosić od kilkaset nanogramów (ng) - jeżeli zamierzamy użyć go do reakcji PCR, do kilku lub kilkunastu mikrogramów (g) DNA - w przypadku testów takich jak hybrydyzacja. Uzyskanie właściwej wydajności jest szczególnie ważne w tych eksperymentach, w których dysponujemy ograniczoną ilością materiału biologicznego, np. w przypadku biopsji lub materiału kryminalistycznego.
Zwracając uwagę na czystość izolowanych kwasów nukleinowych musimy uwzględniać dwa typy zanieczyszczeń. Pierwszy z nich obejmuje wszystkie elementy komórkowe, inne niż DNA, jak również składniki buforów używanych podczas doświadczenia. Obecność tego typu zanieczyszczeń w preparacie DNA może blokować aktywność enzymów, które używamy do analizy kwasów nukleinowych w kolejnych etapach badań. Drugim typem zanieczyszczeń może być DNA pochodzący z innego źródła niż badany materiał. Ponieważ obecnie do badania DNA używamy bardzo czułe techniki, nawet niewielka ilości obcego materiału, np. DNA pochodzący od osoby wykonującej eksperyment, może mieć wpływ na wynik doświadczenia.
Izolacja RNA, podobnie jak izolacja DNA, jest również punktem wyjścia dla wielu eksperymentów. Wykonuje się ją głównie podczas badania ekspresji określonych genów, np. gdy chcemy ustalić czy dany gen ulega transkrypcji w określonej tkance, jaki jest poziom tej ekspresji i jakie czynniki warunkują ten proces. Ponadto, wyizolowany mRNA danego genu, po przepisaniu na cDNA, daje możliwość odczytania sekwencji kodującej, czyli pełnej sekwencji aminokwasowej budującej dane białko.
Pomimo, że dotychczas opracowano wiele technik izolacji DNA i RNA, większość z nich obejmuje analogiczne, przedstawione poniżej etapy.
Fizyczne zniszczenie struktury tkankowej i komórkowej materiału biologicznego, tzw. homogenizacja. Etap ten na ogół wykonuje się rozcierając materiał biologiczny po zalaniu ciekłym azotem. Kryształki lodu, tworzące się w tkankach podczas gwałtownego zamrażania, niszczą ściany i inne struktury uwalniając zawartość komórek. W przypadku zwierząt głównym utrudnieniem podczas izolacji DNA i RNA jest obecność znacznej ilości białek, które nadają komórkom zwartą strukturę. Dlatego w celu ich zniszczenia preparaty często poddawane są działaniu enzymów proteolitycznych, czyli trawiących białka, tj. proteinaza K.
Uwolnienie DNA. DNA chroniony jest w komórce w różnych strukturach błonowych, tj. jądro, mitochondria, chloroplasty a ponadto występuje w kompleksach z różnymi białkami, np. z histonami w chromatynie. W celu degradacji błon komórkowych i białek, do buforów ekstrakcyjnych dodawane są detergenty, tj. SDS, CTAB, izotiocjanian guanidyny. Detergenty te powodują rozpad struktur lipidowych oraz denaturację białek komórkowych. Zdenaturowane białka tracą swoją strukturę a tym samym zdolność do wiązania się z DNA. Ponadto detergenty inaktywują enzymy nukleolityczne, czyli takie, które po uwolnieniu ze struktur komórkowych mogłyby rozpocząć degradację DNA lub RNA, który zamierzamy wyizolować.
Usunięcie zdegradowanych elementów komórkowych. Do usuwania z preparatu składników komórkowych można stosować dwa podejścia. W starszych technikach poszczególne składniki usuwane są indywidualnie przy użyciu różnych procedur, np. białka można oddzielić od kwasów nukleinowych stosując ekstrakcję fenolem i chloroformem. Lipidy usuwamy wykonując ekstrakcję chloroformem. Polisacharydy, występujące obficie w tkanek roślinnych, usuwamy wykorzystując detergent CTAB. RNA możemy usunąć trawiąc preparat enzymem rybonukleolitycznym, np. RNazą A. W nowszych technikach, składniki komórkowe usuwane są przy wykorzystaniu złóż mających wysokie powinowactwo do kwasów nukleinowych. Złoża te upakowane są w niewielkich minikolumnach chromatograficznych. Po nałożeniu zdegradowanego ekstraktu komórkowego na kolumnę wiąże ona specyficznie DNA lub RNA, pozwalając tym samym na odsączenie innych składników.
Oddzielenie kwasów nukleinowych od związków użytych na różnych etapach izolacji, czyli detergentów, soli, inhibitorów. Składniki te, po spełnieniu swojej funkcji, muszą zostać dokładnie usunięte z preparatu, gdyż ich obecność w preparacie uniemożliwiałaby późniejsze wykorzystanie DNA lub RNA do prac wykonywanych z enzymami, np. z polimerazą używaną do reakcji PCR. Sole i detergenty usuwamy wytrącając kwasy nukleinowe przy użyciu etanolu lub izopropanolu. Resztki fenolu można usunąć podczas ekstrakcji chloroformem. W przypadku zastosowania minikolumn, związki te usuwane są razem ze składnikami komórkowymi.
Przygotowanie kwasów nukleinowych do przechowywania. DNA po oczyszczeniu przechowywany jest w postaci roztworu wodnego lub w buforach typu TE. Bufory te zawierają Tris, odpowiedzialny za utrzymanie stałego pH roztworu, a także EDTA, który wiąże jony dwuwartościowe, np. Mg2+, Ca2+, Mn2+, Zn2+. Jony te są kofaktorami wielu enzymów nukleolitycznych stanowiących zagrożenie dla przechowywanego DNA. DNA w buforach TE należy przechowywać w +4 oC lub -20oC. RNA, ze względu na bardzo dużą podatność na degradację nukleolityczną, przechowywany jest w wodzie poddanej działaniu inhibitorów rybonukleaz.
IZOLACJA DNA Z LIŚCI
Odczynniki i sprzęt:
ciekły azot
bufory (tab.1)
chloroform-alk.izoamylowy (24/1)
izopropanol
Buf. elektroforetyczny TAE
agaroza
obciążacz LB
nożyczki,
moździerz,
p robówki eppendorfa
tipsy
mikrowirówka
sprzęt do elektroforezy agarozowej
Tab.1 Bufory używane do izolacji DNA roślinnego
bufor EB (CTAB) |
bufor WB |
bufor TE10/1 |
2% CTAB |
75 % etanol |
10 mM Tris -HCl pH8,0 |
1,4M NaCl |
10mM octan amonu |
1 mM EDTA |
100mM Tris -HCl pH8,0 |
|
|
20mM EDTA |
|
|
2% PVP |
|
|
2% β-merkaptoetanol - dodać bezpośrednio przed użyciem. |
|
|
Postępowanie:
Przygotować bufor ekstrakcyjny EB (CTAB) (ang. Extraction Buffer). Odpipetować 0,5ml buforu do probówki eppendorfa i dodać do niego 10l β-merkaptoetanolu. Następnie probówki z buforem przenieść do +65oC
Pociąć liście przeznaczone do ekstrakcji i umieścić je w czystym, schłodzonym moździerzu.
Materiał zalać ciekłym azotem i rozetrzeć bardzo dokładnie na proszek.
Przenieść ok. 100mg roztartej tkanki do probówki eppendorfa.
Zalać tkankę 0,5 ml buf. EB (CTAB), przygotowanym w p.1 i ekstrahować próbę przez ok. 30 min. w temp +65oC. Podczas inkubacji kilkukrotnie delikatnie zamieszać próbę.
Dodać 1 obj. chloroformu, a następnie całość delikatnie wytrząsać przez 5 minut do uzyskania jednolitej biało-zielonej emulsji.
Preparat wirować w mikrowirówce przez 10 minut przy 12 tyś RPM.
Fazę wodną (górną), która zawiera oczyszczony DNA, przenieść do nowej probówki eppendorfa. Fazę dolną (chloroform z białkami i lipidami), pozostawiamy w probówce, którą po zamknięciu usuwamy.
Do fazy wodnej dodając 2/3 objętości izopropanolu i całość trzymać 5 min w temp. pokojowej. Zabieg ten spowoduje wytrącenie DNA do postaci zawiesinowej.
Wytrącony DNA wirować w mikrowirówce przez 10 minut przy 12 tyś. rpm.
Roztwór znad osadu usunąć a osad przepłukać buforem WB (ang. Wash Buffer).
Ponownie odwirować preparat a po usunięciu roztworu wysuszyć osad.
Wysuszony osad DNA rozpuścić w 20l buforu TE10/1 (10mM Tris, 1mM EDTA).
10l uzyskanego preparatu połączyć z 2 l buforu LB (ang. Loading Buffer), który zawiera obciążacz oraz barwnik elektroforetyczny. Całość nałożyć do kieszonek 1% żelu agarozowego. Po elektroforezie ocenić ilość i jakość uzyskanego preparatu.
IZOLACJA DNA Z WYMAZÓW
Tab.1 Bufory używane do izolacji DNA z wymazów.
bufor EB (Prot.K) |
bufor TE10/1 |
12mM Tris-HCl pH8,0 |
10 mM Tris -HCl pH8,0 |
6mM EDTA |
1 mM EDTA |
0,5% SDS |
|
Do probówki eppendorfa nałożyć 400l buforu EB (Prot.K) i 2 l Proteinazy K (10mg/ml).
Sterylną wymazówką pobrać wymaz z jamy ustnej pocierając 5-10 razy bawełnianym wacikiem wewnętrzną część policzka.
Część wymazówki z wacikiem odciąć i umieścić w probówce eppendorfa z buforem przygotowanym w p.1. Całość przenieść do bloku grzejnego. Próbę inkubować 1 godz. w temp. 55oC.
Po inkubacji wyjąć wacik, wyciskając z niego nadmiar płynu do pozostałej części homogenatu.
Próbę poddać ekstrakcji fenolowej, dodając do jednej objętości homogenatu 0,5 objętość fenolu a następnie 0,5 objętości chloroformu. Do ekstrakcji DNA używamy fenol o pH obojętnym. Uwaga - fenol jest substancją szkodliwą, dlatego tą część pracy należy wykonywać w rękawiczkach, zachowując ostrożność!
Próbę ostrożnie wytrząsać przez 5 min, a następnie wirować w mikrowirówce przez 5 min. przy obrotach 12 tyś. RPM.
Po wirowaniu zebrać warstwę wodną, czyli górną (zawierającą DNA), a następnie przenieść ją do świeżej probówki eppendorfa. Dodać jedną objętość chloroformu.
Całość ekstrahować 2 min. a następnie wirować jak w p.6.
Fazę wodną przenieść do świeżej probówki eppendorfa a następnie wytrącić DNA etanolem. Wytrącanie wykonujemy dodając do roztworu 0,2 obj. 10M octanu amonu i 2,5 obj. (w odniesieniu do całości) 96% etanolu. Po zamieszaniu próbę umieszczamy na 5 min w -20oC.
Wytrącony DNA odwirować w mikrowirówce przy obrotach 12 tyś RPM przez 10 min.
Ostrożnie usunąć supernatant a do osadu DNA dodać 100l schłodzonego 75% etanolu i całość zamieszać tak, by etanol przepłukał wnętrze probówki z osadem. Osad ponownie osadzić na dnie probówki przez wirowanie w mikrowirówce przez 5 min, przy 12 tyś. RPM.
Roztwór znad osadu ostrożnie i bardzo dokładnie usunąć pipetmanem a osad wysuszyć.
Wysuszony osad DNA rozpuścić w 20l buforu TE10/1 (10mM Tris, 1mM EDTA).
10l uzyskanego preparatu połączyć z 2 l buforu LB (ang. Loading Buffer), który zawiera obciążacz oraz barwnik elektroforetyczny. Całość nałożyć do kieszonek 1% żelu agarozowego. Po elektroforezie ocenić ilość i jakość uzyskanego preparatu.
IZOLACJA RNA Z LIŚCI
Tab.1 Bufory używane do izolacji RNA z roślin.
bufor homogenizacyjny |
Buf. elektroforetyczny 10 x MOPS |
Bufor denaturujący do RNA |
4M tiocjanian guanidyny |
0,2 M MOPS pH 7,0 |
37 % formaldehyd - 187 l |
25 mM cytrynian sodu pH 7,5 |
50 mM octan sodu |
100 % formamid - 562 l |
0,5 % sarkozyl |
10 mM EDTA |
10 x MOPS - 112 l |
0,1 M -merkaptoetanol - dodać bezpośrednio przed użyciem |
|
10 mg/ml Bromek etydyny - 10l |
100mg materiału roślinnego, roztartego w ciekłym azocie, zawiesić w 400 l buforu homogenizacyjnego. Uwaga - izotiocjanian guanidyny obecny w buforze jest substancją żrącą. Pracę z nim należy wykonywać tylko w rękawiczkach i przy zachowaniu dużej ostrożności!
Zawiesinę inkubować w ciemnym miejscu, w temp. pokojowej przez ok. 0,5 godz.
Do mieszaniny dodać 1/10 obj. 2M octanu sodu pH 4,0 (40l) i 1 obj. kwaśnego fenolu (440l). Całość wytrząsać przez 5 min.
Do próby dodać 200 l chloroformu i kontynuować homogenizację przez 5 min.
Całość wirować w mikrowirówce przez 10 min przy 12 tys. rpm.
Fazę wodną, która zawiera RNA, przenieść do nowej probówki eppendorfa i dodać do niej 1 obj. chloroformu. Całość wytrząsać przez 1 min. a następnie wirować 5 minut przy 12 tys. rpm.
Fazę wodną przenieść do nowej probówki eppendorfa. RNA wytrącić dodając 1 obj. izopropanolu.
Wytrącony RNA odwirować przy 12 tys. rpm przez 10 min w +4oC.
W celu usunięcia resztek buf. homogenizacyjnego osad RNA przemyć 100l 75% etanolu i odwirować przez 3 min. przy 12 tys. rpm.
Dokładnie usunąć resztki etanolu i osuszyć osad.
Rozpuścić RNA w 20 l H2ODEPC.
Do 5 l preparatu dodać 1 obj. buforu denaturującego do RNA. Uwaga: bufor ten zawiera bromek etydyny, który jest rakotwórczy! Całość denaturować przez 5 min w +65oC.
Schłodzić preparat w lodzie a następnie dodać 2 l buforu LB (ang. Loading Buffer) i całość nałożyć na żel denaturujący.
Nałożyć preparat na 1,5% denaturujący żel agarozowy.
Elektroforezę prowadzić w buforze 1x MOPS przy napięciu 100 V.
Po zakończeniu elektroforezy żel analizujemy w świetle UV.
Przygotowanie żelu denaturującego
Przygotowanie aparatu do elektroforezy RNA. W celu pozbycia się RNA-az aparat do elektroforezy należy umyć detergentem, przepłukać 0,1% roztworem DEPC i wytrzeć do sucha ligniną z etanolem.
Przygotowanie 1,5% żelu agarozowego (50 ml). Do 0,7g agarozy dodać do 42,5 ml wody i zagotować. Po schłodzeniu agarozy do ok. +65oC dodać 5 ml 10x st. buforu MOPS oraz 2,5 ml 37% formaldehydu a następnie całość wylać do uprzednio przygotowanego aparatu elektroforetycznego. Uwaga - wszystkie te czynności wykonać w dygestorium przy włączonym wywiewie. Opary formaldehydu są trujące.
Firmowe zestawy do oczyszczania kwasów nukleinowych
W celu ułatwienia prac związanych z badaniem kwasów nukleinowych, firmy biotechnologiczne sprzedają specjalne kolumny do izolacji i oczyszczania DNA i RNA. Systemy te na ogół oparte są zdolności złóż krzemionkowych do wiązania kwasów nukleinowych w obecności wysokich stężeń soli chaotropowych, takich jak chlorowodorek guanidyny lub izothiocjanian guanidyny. Sole chaotropowe, powodują również rozpad błon komórkowych i denaturację białek, co sprzyja oddzieleniu kwasów nukleinowych od pozostałych składników komórki. Chaotropy ang. chaotropes, czyli ,,czyniące chaos'' są to substancje, które w roztworach wodnych wprowadzają silne zaburzenia w sieci wiązań wodorowych. W oparciu o tą zasadę opracowano systemy wykorzystywane do następujących celów:
izolacja całkowitego DNA
izolacja całkowitego RNA
izolacja plazmidowego DNA,
elucja DNA z żeli agarozowych i poliakryloamidowych
oczyszczanie kwasów nukleinowych po reakcjach enzymatycznych, takich jak: PCR, restrykcja, znakowanie.
Do sprzedawanego sprzętu firmy dołączają szczegółowe opisy odpowiednich procedur. Generalna zasada wykorzystania większości systemów obejmuje następujące etapy:
wstępne przygotowanie oczyszczanego kwasu nukleinowego. W przypadku DNA plazmidowego jest to ekstrakt bakteryjny pozbawiony DNA genoforowego. W przypadku DNA całkowitego roślin lub zwierząt jest to ekstrakt białkowy uzyskany w wyniku rozbicia tkanek w buforze zawierającym stężone sole chaotropowe. Preparaty agarozy zawierające eluowany DNA powinny zostać poddane stopieniu. Mieszaniny zawierające DNA po reakcjach enzymatycznych powinny zostać wzbogacone o odpowiednie stężenie buforu chaotropowego.
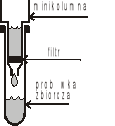
nałożenie preparatu na kolumny. W zależności od skali, w jakiej przeprowadza się oczyszczanie, mogą to być tzw. minikolumny (rys. 1), w których przepływ roztworu przez membranę zawierającą złoża krzemionkowe uzyskuje się w wyniku wirowania całej kolumny w mikrowirówce, lub większe kolumny, w których roztwór przepływa grawitacyjnie. Złoża, przez które przechodzi roztwór, wychwytują specyficzne dla nich kwasy nukleinowe, tzn. DNA lub RNA, a pozostałe składniki roztworu, stanowiące zanieczyszczenie dla badanych prób, zostają zebrane w probówce znajdującej się poniżej filtra.
płukanie preparatu. W celu dokładnego odpłukania resztek zanieczyszczeń związanych z filtrem, kolumienki przepłukuje się kilkukrotnie roztworami zawierającymi sole chaotropowe lub buforami zawierającymi etanol.
wymywanie kwasów nukleinowych z kolumny. Po usunięciu wszystkich zanieczyszczeń i osuszeniu membrany, ze złoża wymywa się oczyszczone kwasy nukleinowe, nakładając na kolumnę odpowiednią ilość wody, lub buforu TE. Zebrane z kolumny preparaty oczyszczonego DNA lub RNA, na ogół nadają się do bezpośredniego wykorzystania do dalszych prac badawczych.
Rys.1
Używanie standaryzowanych systemów do oczyszczania kwasów nukleinowych posiada następujące zalety w porównaniu do analogicznych technik, wykonywanych metodami tradycyjnymi:
czas trwania eksperymentu zostaje znacznie skrócony,
wydajność i czystość prób oczyszczonych tą drogą są wyższe,
metody te eliminują konieczność pracy z substancjami szkodliwymi, tj. fenol, chloroform,
uzyskiwane wyniki są w wysokim stopniu powtarzalne.
3
5
6