1. Podział chemii analitycznej.
Analiza jakościowa - analiza, której celem jest stwierdzenie (lub wykluczenie) obecności jakiegoś składnika w badanej próbce. Identyfikację składników próbki dokonuje się za pomocą odpowiednich odczynników analitycznych, powodujących charakterystyczną reakcję, której towarzyszy zmiana barwy roztworu, płomienia, specjalnie stopionej perły (barwne perły) albo wytrącenie osadu.
Analiza ilościowa - oznaczanie ilości wybranych składników lub ustalenie pełnego składu badanej próbki.
Chemia nieorganiczna - dział chemii zajmujący się właściwościami pierwiastków i zw. chem. (poza większością zw. węgla) oraz reakcjami chem., w których biorą udział.
Chemia organiczna - dział chemii zajmujący się właściwościami, strukturą i przemianami zw. węgla (poza niektórymi zw. tego pierwiastka, rozpatrywanymi w chemii nieorganicznej).
Analiza śladowa - wyłącznie oznaczenie domieszek (zanieczyszczeń, śladów). W tym zastosowania szczególne, np.: w technice jądrowej, w materiałach półprzewodnikowych, w analizie wody pitnej i innej.
2. Podział metod analizy ilościowej.
a) metody klasyczne - metody ilościowe, wykorzystujące pomiar masy osadu otrzymanego w toku analizy (analiza wagowa) lub objętości roztworu mianowanego zużytego w reakcji z oznaczanym składnikiem (analiza miareczkowa).
b) metody instrumentalne - obejmują metody pomiaru własności fizycznych lub fizykochemicznych badanej próbki, określające np.: własności elektryczne i elektrochemiczne, optyczne, promieniotwórczość i inne. Metody te odróżnia od metod klasycznych znacznie wyższa czułość i szybkość analizy. Stosowana aparatura pomiarowa jest na ogół bardzo kosztowna.
3. Metoda analityczna i wielkości ją charakteryzujące.
Metoda analityczna, sposób wykrywania lub oznaczania jakiejś substancji.
Wielkości charakteryzujące:
a) czułość - stosunek przyrostu mierzonego tą metodą sygnału do odpowiadającego mu przyrostu zawartości oznaczanego składnika w próbce.
b) dokładność (accuracy) - stopień zgodności wartości uzyskanej z wartością rzeczywistą.
c) precyzja (precision) - rozrzut wyników analizy uzyskanych podczas wielokrotnie przeprowadzanego oznaczania danej substancji chem. Zależy od jakości stosowanej aparatury. Liczbowo określa się odchyleniem standardowym.
d) oznaczalność - najmniejsze stężenie lub ilość składnika próbki, przy którym można oznaczyć ten składnik daną metodą.
e) wykrywalność - najmniejsze stężenie lub ilość składnika próbki, którą można wykryć daną metodą.
4. Rodzaje błędów.
a) przypadkowe - spowodowane zespołem czynników przypadkowych, działających w chwili wykonywania pomiaru. Przyczyny:
- przypadkowe zanieczyszczenie osadu lub r-ru
- przypadkowe straty
- niestabilność pracy aparatury
- zmęczenie analityka
b) systematyczne - spowodowane czynnikami działającymi w jednakowy sposób podczas wielorazowego prowadzenia oznaczenia analizy. Przyczyny:
- niewłaściwe wyskalowanie przyrządów pomiarowych
- zastosowanie niewłaściwych odczynników
- subiektywna ocena analityka
- oczekiwanie z góry jakiegoś wyniku
c) grube - znacznie odbiegają od pozostałych wyników serii. Przyczyny:
- niepoprawne wykonanie pomiaru
- używanie wadliwie działających przyrządów
- niedostateczna uwaga analityka
5. Sposoby wyrażania błędów.
a) błąd bezwzględny - wartość bezwzględna różnicy między wartością wyliczoną a rzeczywistą.
b) błąd względny - stosunek błędu bezwzględnego do wartości rzeczywistej, wyrażony w %.
6. Czynniki wpływające na wybór metody analitycznej.
a) skład próbki i wstępnie przybliżona wartość jej składnika, które należy oznaczyć:
- składniki gł. powyżej 2%
- składniki domieszkowe 2-0,01%
- składniki śladowe do 0,01%
b) decyzja o analizie pojedynczej próbki lub analizie seryjnej
c) wielkość badanej próbki
d) względy ekonomiczne
7. Rodzaje odczynników chemicznych.
- techniczne, których nie wolno stosować
- czyste (pure) - stosowane w dość prymitywnych analizach, nie wymagające dokładniejszych analiz
- chemicznie czyste (chemically pure)
- czyste do analizy (A. C. S.) - z atestem firmy produkującej, gwarantującym nieprzekraczalną zawartość określonych zanieczyszczeń. Stosuje się je z reguły do przygotowywania r-ów podstawowych zwykle mianowanych, oraz podczas wykonywania analiz bardziej odpowiedzialnych
- spektralnie czyste (spectrally pure)
- do mikroanalizy (MAR), gł. elementarnej analizy organicznej
- o specjalnej czystości
8. Podstawowe pojęcia i definicje.
a) liczność materii (n) - liczba cząstek zawartych w określonej ilości danej substancji.
b) mol - liczność materii występująca gdy liczba cząstek jest równa liczbie atomów zawartych w 12g 12C.
c) masa atomowa - średnia masa atomu pierwiastka chem. wyrażana w atom. jednostkach masy (u) lub innych jednostkach masy.
d) współczynnik równoważności - określa ilościowy udział danego reagenta w reakcji chemicznej.
e) masa cząsteczkowa - suma mas atom. pierwiastków chem. tworzących cząsteczkę, wyrażana w takich jednostkach jak masa atomowa.
9. Sposoby wyrażania stężeń.
a) stężenie molowe - liczba moli danego związku jaką zawiera 1dm3 danego roztworu ![]()
.
b) ułamek molowy - stosunek liczby moli danego związku do sumy liczby moli wszystkich składników.
c) stężenie masowe - stosunek masy wybranego związku chemicznego do objętości roztworu lub mieszaniny ![]()
.
d) ułamek masowy - stosunek masy składnika układu do masy całego układu.
e) ułamek objętościowy - stosunek objętości składnika do sumarycznej objętości wszystkich składników układu.
f) stężenie molalne - liczba moli substancji rozpuszczonej w 1kg rozpuszczalnika.
10. Prawo działania mas, stała równowagi chemicznej.
Prawo działania mas (równowagi chemicznej)
Dla każdej reakcji odwracalnej w stanie równowagi stosunek iloczynu stężeń molowych produktów do iloczynu stężeń molowych substratów podniesionych do odpowiednich potęg odpowiadających liczbom cz. biorących udział w reakcji jest równy stałej nazywanej stałą równowagi.
Stała równowagi chem.
Współczynnik opisujący stan równowagi odwracalnych reakcji chem. Stosunek iloczynu stężeń produktów do iloczynu stężeń substratów.
mA + nB ↔ pC + qD
![]()
11. Stała i stopień dysocjacji.
Stała dysocjacji jest miarą mocy słabych elektrolitów. Dla reakcji CH3COOH + H2O → H3O+ + CH3COO-, ![]()
.
Stopień dysocjacji α - stosunek liczby moli cząsteczek danego związku chem., które uległy rozpadowi na jony do łącznej liczby cząsteczek tego związku, w którym zaszło zjawisko dysocjacji elektrolitycznej. Zależy od:
- struktury związku, dla którego ten stopień jest ustalany
- rodzaju rozpuszczalnika
- obecności w roztworze innych związków zdolnych do dysocjacji
- stężenia roztworu (na ogół wzrasta w miarę rozcieńczania roztworu)
- temperatury (na ogół nieco wzrasta wraz ze wzrostem temperatury)
12. Prawo rozcieńczeń Ostwalda.
Im bardziej rozcieńczony słaby elektrolit, tym większy będzie jego stopień dysocjacji. Prawo Ostwalda nie dotyczy mocnych elektrolitów.
Wyprowadzenie wzoru dla CH3COOH
[CH3COO-] = C·α,
ponieważ ![]()
[H+] = C·α, ponieważ ![]()
[CH3COOH] = C(1 - α),
ponieważ C = Cz + Cnz
Cnz = C - Cz
Cz = α·C
Cnz = C - (α·C)
Cnz = C - (1 - α)
![]()
,
ponieważ 1 - α ≈ 1, więc:
K ![]()
α2·C => α ![]()
![]()
13. Pojęcie aktywności.
Aktywność określa zdolność jonów do wstępowania w reakcje chem. Stężenie, w którym jony wstępują w reakcje chem.
14. Współczynnik aktywności, termodynamiczna stała równowagi.
Współczynnik aktywności - stosunek aktywności danego jonu do jego stężenia w roztworze ![]()
. Jest wielkością niemianowaną. Zależy od stężenia (bardziej dla elektrolitów aniżeli nieelektrolitów) - dla roztworów nieskończenie rozcieńczonych równa się 1.
Termodynamiczna stała równowagi - stosunek iloczynu aktywności produktów dysocjacji do iloczynu aktywności substratów, podniesionych do odpowiednich potęg równych stechiometrycznym współczynnikom.
15. Dysocjacja wody, iloczyn jonowy wody.
Woda wykazuje pewne przewodnictwo elektryczne. Jest więc słabym elektrolitem, zatem ma miejsce dysocjacja elektrolityczna, zachodząca w niewielkim stopniu zgodnie z równaniem H2O ↔ H+ + OH- lub H2O + H2O ↔ H3O+ + OH-.
W temp. pokojowej stała dysocjacji:
![]()
.
Stopień dysocjacji: α = 2·10-9 - ponieważ wartość jest niewielka stężenie części niezdysocjowanej jest równe stężeniu początkowemu.
Iloczyn jonowy wody: Kwody = 1·10-14.
Stężenia H+ i OH- są sobie równe i wynoszą:
CH+ = COH- = ![]()
= 1·10-7 ![]()
.
16. Właściwości kwasów i zasad w świetle teorii Bronsteda.
Teoria protonowa, definiująca kwasy jako substancje zdolne do oddawania protonów (donor protonu), a zasady jako substancje zdolne do przyłączania protonów (akceptor protonu). Kwasy i zasady tworzą sprzężoną parę wymieniającą protony. Każdy kwas odpowiada swej zasadzie uboższej o 1 proton. Reakcja wymiany protonu między kwasem oddającym proton i zasadą przyjmującą proton - reakcja protolizy.
Woda w tym przypadku ma właściwości amfoteryczne:
a) może zachowywać się jak kwas:
NH3 + H2O ↔ NH4+ + OH-
zas1 kw1 kwas2 zas2
b) lub jak zasada:
H2O + HF ↔ H3O+ + F-
zas1 kwas1 kwas2 zas2
17. Rozpuszczalniki protonowe.
Podział rozpuszczalników protonowych:
a) protolityczne - zdolne do przyjmowania lub oddawania protonu i do autoprotolizy (możliwość przekazania protonu między własnymi cząsteczkami, np. 2H2O ↔ H3O+ + OH-, 2CH3COOH ↔ CH3COOH2+ + CH3COO-)
b) aprotyczne - niezdolne do reakcji z protonem (węglowodory aromatyczne, alifatyczne)
Podział rozpuszczalników protolitycznych:
a) protonodonorowe - łatwo oddające protony (bezwodny kwas octowy)
b) protonoakceptorowe - wykazujące tendencję do przyłączania protonów (pirydyna, ciekły amoniak)
c) amfoteryczne - z równą łatwością oddające lub przyłączające protony (woda, etanol)
18. Stałe dysocjacji sprzężonego kwasu i zasady.
Gdy w układzie kwas-zasada znajduje się cząsteczka rozpuszczalnika to np.:
H2CO3 + H2O ↔ HCO3- + H3O+ ![]()
NH3 + H2O ↔ NH4+ + OH- ![]()
KA - stała dysocjacji kwasowej, określa moc kwasu węglowego jako donora protonu dla rozpuszczalnika
KB - stała dysocjacji zasadowej, określa moc zasady jako akceptora protonu
KH2O - iloczyn jonowy wody
19. Stałe dysocjacji kwasów i zasad wieloprotonowych.
Dla kwasów (zasad) wieloprotonowych dysocjujących stopniowo gł. źródłem jonów H3O+ (OH-) jest pierwszy stopień dysocjacji. Każdemu etapowi dysocjacji odpowiada inna stała np.:
H3PO4 + H2O ↔ H2PO4- + H3O+ ![]()
H2PO4- + H2O ↔ H2PO42- + H3O+ ![]()
H2PO42- + H2O ↔ PO43- + H3O+ ![]()
20. pH, pOH
pH = -log [H+]
pOH = -log [OH-]
pH + pOH = 14
Kwasowość lub zasadowość roztworu zależy nie tylko od rodzaju rozpuszczonych kwasów i zasad, ale także od ich stężeń. W roztworach wodnych obniżenie pH zwiększa pOH i odwrotnie. Zwiększa się wtedy kwasowość lub zasadowość roztworu. W rozcieńczonych roztworach wodnych skala pH obejmuje zakres 0-14. W przypadku stężonych kwasów jeżeli st > 1mol/l to pH przyjmuje wartość ujemną, w przypadku zasad pH > 14.
21. Reakcje kwasów z zasadami.
a) mocny kwas-mocna zasada
HCl + NaOH ↔ NaCl + H2O
Powstaje sól mocnego kwasu i mocnej zasady oraz woda. Otrzymuje się roztwór obojętny (pH = pOH).
b) mocny kwas-słaba zasada
HCl + NH4OH ↔ NH4Cl + H2O
W wyniku reakcji powstaje roztwór kwaśny.
c) słaby kwas-mocna zasada
CH3COOH + NaOH ↔ CH3COONa + H2O
Powstaje sól słabego kwasu i mocnej zasady oraz woda. Otrzymuje się roztwór zasadowy.
d) słaby kwas-słaba zasada
CH3COOH + NH3 ↔ CH3COO- + NH4+
22. Roztwory buforowe - przykłady, wzory na obliczenie pH, pojemność buforowa, zastosowanie buforów.
Roztworami buforowymi nazywa się roztwory słabego kwasu i jego soli z mocną zasadą oraz słabej zasady i jej soli z mocnym kwasem, wykazujące stałe stężenie jonów wodorowych, które praktycznie nie ulega zmianie podczas rozcieńczania lub po dodaniu niewielkich ilości mocnych kwasów lub zasad. Roztworami buforowymi mogą być także roztwory soli kwasów wieloprotonowych (KH2PO4, Na2HPO4).
Roztwory buforowe zachowują podczas rozcieńczania stałe pH, ponieważ zależy ono od stosunku stężeń składników roztworu buforowego, a nie od ich bezwzględnych stężeń.
Wzory:
- dla kwasów: ![]()
- dla zasad: ![]()
Pojemność buforowa - zdolność przeciwstawiania się wpływom zmieniającym pH
![]()
dcz - zmiana stężenia mocnej zasady w roztworze
dpH - zmiana pH
β jest zawsze dodatnie, gdyż jeśli dcz > 0 to dpH > 0 (ze wzrostem stężenia zasady pH wzrasta). Jeżeli do roztworu wprowadzi się kwas, to stężenie zasady zmaleje, a więc dcz < 0, pH również zmaleje, więc dpH < 0 i β > 0.
Zastosowanie buforów:
- przy produkcji barwników, leków syntetycznych
- w procesach fermentacyjnych
- w poligrafii, przy druku w technice offsetowej
- wiele buforów jest stosowanych do kontrolowania pH gotowych produktów spożywczych, kosmetyków i leków
- jako substancje lecznicze np. do przemywania poparzonej skóry lub oczu
23. Wskaźniki kwasowo-zasadowe.
Wskaźnikami kwasowo-zasadowymi lub wskaźnikami pH nazywamy takie związki, przeważnie organiczne, które zmieniają swą barwę w określonym zakresie pH roztworu. Są stosowane do wyznaczania PK miareczkowania alkacymetrycznego i określenia pH roztworu.
Teoria Ostwalda
Wskaźniki są to słabe kwasy lub zasady organiczne, których cząsteczki niezdysocjowane mają inną barwę niż jony.
Teoria Hantzscha
Wskaźniki występują w postaci 2 lub więcej odmian tautomerycznych różniących się strukturą, barwą i pozostających w stanie równowagi. Odmianami tautomerycznymi są: pseudokwasy (związki organiczne w właściwościach związków obojętnych ale w środowisku alkalicznym mające właściwości kwasowe) i pseudozasady (w środowisku kwaśnym przechodzą w postać zasadową).
24. Reakcje utleniania i redukcji.
H2O2 jako utleniacz i reduktor:
- jako utleniacz H2O2 + 2H+ + 2e → 2H2O
np. Cr2(SO4)3 + 3H2O2 + 10KOH → 2K2CrO4 + 3K2SO4 + 8H2O
- jako reduktor H2O2 - 2e → 2H+ + O2
np. 2KMnO4 + 5H2O2 + 3H2SO4 → 2MnSO4 + K2SO4 + 8H2O + 5O2
Potencjał redoks - potencjał elektrody platynowej lub z innego metalu szlachetnego zanurzonej w roztworze zawierającym układ redoks:
![]()
25. Wzór do obliczenia stałej równowagi z wartości potencjału redoks.
Stan równowagi reakcji redoks ustala się gdy potencjały obu reakcji redoks wyrównają się:
red1 ↔ utl1 + n1e
red2 ↔ utl2 + n2e
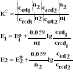
w stanie równowagi E1 = E2
![]()
26. Wpływ środowiska na reakcje redoks.
Wpływ pH
Wpływ pH uwidacznia się w przypadku, gdy w reakcji biorą udział jony H+ lub OH-, np.:
MnO4- + 8H+ + 5e → Mn2+ + 4H2O
![]()
Gdy reakcja przebiega z udziałem jonów H+ to zmniejszenie pH zwiększa potencjał.
Gdy reakcja przebiega z udziałem jonów OH- to zwiększenie pH zwiększa potencjał.
Jeżeli potencjały obu ukł. redoks są zbliżone to wtedy sytuacja jest niekorzystna gdyż reakcja jest odwracalna. Przy odpowiednim dobraniu pH reakcja może przebiegać całkowicie (zmniejszenie lub zwiększenie pH). Przykładem jest reakcja utlenienia arseninu jodem:
AsO33- + I2 + H2O ↔ AsO43- + 2I- + 2H+
![]()
![]()
AsO43- + 2H+ + 2e ↔ AsO33- + H2O
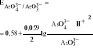
I2 + 2e → 2I-
![]()
Z powyższych wzorów wynika, że pH wywiera wpływ na potencjał układu AsO43-/ AsO33-, ponieważ we wzorze występuje stężenie jonów wodorowych. Aby zachodziła reakcja utleniania jodem należy potencjał ukł. AsO43-/ AsO33- zmniejszyć przez zwiększenie pH do 7:
![]()
Przy pH=7 nastąpiło więc duże obniżenie potencjału układu (z 0,58V do 0,17V) i w tych warunkach można przeprowadzić reakcję utlenienia AsO33- jodem.
Wpływ kompleksowania
Jony Fe3+ tworzą trwałe kompleksy fluorkowe FeF63-, dlatego dodatek fluorków do roztworu zawierającego jony Fe3+ i Fe2+ zmniejsza znacznie stężenie jonów Fe3+, co powoduje znaczne obniżenie potencjału utleniającego układu Fe3+/Fe2+. Obniżenie potencjału jest tak duże, że łatwo przebiegająca w zwykłych warunkach reakcja utleniania jonów I- jonami Fe3+ może być zahamowana.
Wpływ wytrącania osadów
Np. jodometryczne oznaczenie Cu2+:
![]()
![]()
2Cu2+ + 4I- ↔ 2CuI + I2
![]()
IrCuI = 10-12
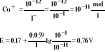
Jeżeli stężenie jodku potasu wynosi 0,1![]()
wtedy stężenie jonów [Cu+] wynosi 10-11![]()
, a więc potencjał ukł. ulega zwiększeniu.
27. Formalne potencjały redoks.
Formalny potencjał redoks (Efo) to potencjał, gdy analityczne (a nie równowagowe) stężenia postaci utlenionej i zredukowanej są równe. Rzeczywisty potencjał redoks w danych warunkach roztworu.
28. Wskaźniki redoks.
Wskaźnikami redoks są barwne substancje, które tworzą ukł. redoks, przy czym postać utleniona wskaźnika jest inaczej zabarwiona niż postać zredukowana. Postać zredukowana jest zwykle bezbarwna. Wskaźnik zmienia barwę w punkcie lub blisko punktu równoważności. Barwa wskaźnika zależy od stosunku stężeń postaci utlenionej i zredukowanej.
29. Rozdzielanie układów.
Rozdzielanie ukł. heterogenicznych:
- mechaniczne odsiewanie
- sedymentacja (osadzanie na dnie naczynia)
- dekantacja (zlewanie)
- odwirowywanie (przyspieszona sedymentacja)
- sączenie (filtrowanie)
- dializowanie (dyfundowanie cząstek przez błony przepuszczalne)
- odparowywanie składnika lotnego i suszenie substancji stałych
Rozdzielanie ukł. homogenicznych:
- przeprowadzenie składników w różne fazy
- wprowadzenie innego rozpuszczalnika
- wprowadzenie odczynnika strącającego
- metody wykorzystujące różnicę lotności substancji
- elektroliza (wew. lub zew.)
- metody strąceniowe
- metody ekstrakcyjne
- wymiana jonowa
30. Strąceniowe metody rozdzielania.
Charakterystyka osadów koloidalnych i warunki ich strącania
Posiadają nieuporządkowaną strukturę i silnie rozwiniętą powierzchnię, która może być przyczyną zanieczyszczeń. Dzielą się na serowate (AgCl) i galaretowate (Fe(OH)3), w kontakcie z rozpuszczalnikami na hydrofobowe i hydrofilowe oraz liofobowe i liofilowe. Podstawowymi procesami związanymi z osadami koloidalnymi są koagulacja i peptyzacja. Zależność między tymi procesami:
Charakterystyka osadów krystalicznych i warunki ich strącania
Posiadają uporządkowaną strukturę. Podczas rozpuszczania tworzą roztwory rzeczywiste. W zależności od warunków strącania otrzymujemy osady grubokrystaliczne (BaSO4) lub drobnokrystaliczne (MgNH4PO4).
- osad wytrącać należy z roztworów rozcieńczonych
- wytrącanie osadu powinno odbywać się w podwyższonej temp.
- powoli dodawać odczynnik strącający, ciągle mieszając
- odczynnik należy wprowadzić w odpowiednim nadmiarze
Strącanie homogeniczne
Odczynnik strącający nie jest dodawany, ale tworzy się w roztworze w wyniku reakcji chem. Takim odczynnikiem jest tioacetamid (CH3CSNH2). Rozkłada się on podczas ogrzewania i jest rozmieszczony równomiernie w całym roztworze, dlatego tworzą się zarodki krystalizacji.
31. Iloczyn rozpuszczalności.
Iloczynem rozpuszczalności trudno rozpuszczalnej substancji określa się iloczyn stężeń jonów tej substancji w roztworze pozostającym w równowadze z osadem:
AB ↔ A+ + B-
Ir = [A+][B-]
Ir posiada ściśle określoną wartość w danej temp.
Obl. rozpuszczalności (x) ze znanych wartości Ir:
![]()
32. Czynniki wpływające na rozpuszczalność osadu.
a) efekt wspólnego jonu - zmniejszenie rozpuszczalności osadu po dodaniu substancji w skład, której wchodzi jeden z jonów zw. trudno rozpuszczalnego;
b) efekt obcego jonu (efekt solny) - zwiększenie rozpuszczalności soli trudno rozpuszczalnej na skutek obecności w roztworze dużego stężenia obojętnych soli (nie mających jonów wspólnych z daną solą trudno rozpuszczalną);
c) wpływ pH - jony hydroniowe H3O+ zwiększają rozpuszczalność trudno rozpuszczalnych wodorotlenków oraz soli słabych kwasów. Dodanie do roztworu mocnego kwasu powoduje obniżenie stężenia jonów OH-, a co za tym idzie, zwiększenie stężenia jonów metalu w roztworze. Konsekwencją tego jest wzrost rozpuszczalności osadu wodorotlenku jako całości. W przypadku soli słabych kwasów, wprowadzone do roztworu jony hydroniowe wiążą aniony reszty kwasowej tworząc cząsteczki słabo zdysocjowanych kwasów. Zmniejsza się w ten sposób stężenie wolnych jonów w roztworze. Ponieważ Ir jest wartością stałą, to obniżeniu się stężenia jonów ujemnych musi towarzyszyć wzrost stężenia jonów dodatnich, czyli zwiększenie rozpuszczalności osadu;
d) kompleksowanie - dość częstym zjawiskiem jest tworzenie przez daną parę jonową nie tylko trudno rozpuszczalnych soli ale i łatwo rozpuszczalnych kompleksów. W takiej sytuacji niewielki dodatek jonu wspólnego spowoduje spadek rozpuszczalności, ale zwiększenie stężenia jonu wspólnego doprowadzi do powstawania kompleksu i w rezultacie sumarycznego zwiększenia ilości rozpuszczonego metalu, pomimo tego, że stężenie wolnego jonu metalu będzie mniejsze.
2
Wyszukiwarka