17 stycznia 2007 r.
Analiza DNA znajduje zastosowanie w wielu dziedzinach. Należą do nich przede wszystkim diagnostyka medyczna (transplantologia, diagnostyka prenatalna chorób dziedzicznych itp.), medycyna sądowa (ustalanie pokrewieństwa, badania identyfikacyjne) i kryminalistyka. Jest również niezbędna w takich dziedzinach jak: mapowanie genetyczne, antropologia, genetyka populacyjna, biologia ewolucyjna, hodowla roślin i zwierząt, systematyka itd.
Z historii badań DNA:
1902 r. odkrycie układu antygenów grupowych krwi AB0 (Landsteiner, Richter)
1958 r. opisanie układu antygenów zgodności tkankowej HLA
1970 r. początek trawienia DNA przy użyciu enzymów restrykcyjnych (Arber, Nathans, Smith)
1975 r. technika transferu i hybrydyzacji DNA (Southern)
1977 r. technika sekwencjonowania (Sanger, Maxam, Gilbert)
1978-80 r. koncepcja metody RFLP (Salomon, Bodner, Batstein)
1981 r. zsekwencjonowanie mtDNA (Anderson)
1983 r. opracowanie techniki PCR (Saiki, Mullis)
1985 r. opisanie polimorfizmu sekwencji minisatelitarnych (VNTR), sondy multilokusowe, technika „DNA fingerprinting”(Jeffreys)
1987 r. sondy jednolokusowe (Wong, Nakamura)
1991 r. opisanie polimorfizmu sekwencji mikrosatelitarnych (STR)
1993 r. rutynowe badania DNA w ponad trzydziestu krajach
2001 r. opisanie polimorfizmu pojedynczych nukleotydów (SNP)
DNA jako dowód w sądzie:
1985 r. pierwszy dowód z badań DNA w sprawie imigracyjnej
1987 r. pierwszy dowód w sprawie o morderstwo w Wielkiej Brytanii
1988 r. dowód w sprawie o morderstwo i gwałt w USA
1994 r. pierwsza formalna baza danych DNA - CODIS
2001 r. pierwszy dowód sądowy z badań DNA mitochodrialnego
Informacja genetyczna komórki. Modele dziedziczenia
Całość informacji genetycznej komórki podzielona jest pomiędzy genom jądrowy i mitochondrialny.
Genom jądrowy obejmuje 3×109 pz. Sekwencje kodujące, czyli geny, których liczba szacowana jest na ok. 30 tysięcy, stanowią tylko ok. 20% genomu. Pozostałe 80% stanowią sekwencje niekodujące.
O polimorfizmie DNA jądrowego stanowią:
tandemowe sekwencje repetytywne (20-30% wszystkich sekwencji niekodujących);
zmienność pojedynczych nukleotydów (p. niżej).
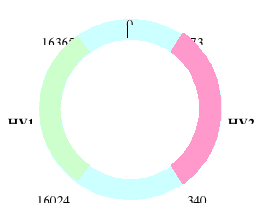
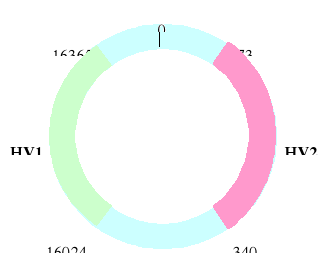
DNA mitochondrialny (mtDNA) jest kolistą cząsteczką, zawierającą jedynie 16 579 pz, w tym 37 genów. Źródłem zmienności są dwa regiony zawierające sekwencje niekodujące: HV1 i HV2, liczące w sumie 1200 pz. Region HV1, leżący pomiędzy 16024. a 16365. parą nukleotydów, zawiera 50 loci SNP. Region HV2 leży pomiędzy 73. a 340. parą nukleotydów i obejmuje 28 loci SNP. W tych dwóch regionach poszukuje się zmienności mtDNA.
Dziedziczenie informacji genetycznej zawartej w genomie jądrowym odbywa się zgodnie z prawami Mendla. Według prawa czystości gamet (I prawo Mendla) każda gameta wytwarzana przez organizm posiada tylko jeden gen z danej pary alleli. Prawo niezależnej segregacji cech (II prawo Mendla) mówi, że geny należące do dwóch różnych par alleli są rozdzielane do gamet niezależnie od siebie. Prawa te nie dotyczą DNA mitochondrialnego, które nie rekombinuje, lecz dziedziczy się wyłącznie z linii matczynej.
Wyróżniamy dwa typy zmienności dziedzicznej:
zmienność rekombinacyjna → tworzenie nowych układów istniejących już alleli na skutek różnych kombinacji cech rodzicielskich i wymiany homologicznych fragmentów DNA (tylko genom jądrowy);
zmienność mutacyjna → tworzenie nowej informacji genetycznej; nagłe, skokowe zmiany przekazywane potomstwu, powstające na skutek tranzycji, insercji, delecji lub mutacji punktowych (genom jądrowy i mitochondrialny).
Rodzaje polimorfizmu DNA:
Polimorfizm pojedynczych nukleotydów (Single Nucleotide Polymorphism, SNP).
SNP oznacza utrwalone mutacje punktowe występujące z częstością min. 1%. Polimorfizm w obrębie jednego locus jest bardzo mały, gdyż obejmuje tylko trzy możliwości (np. homozygota AA, homozygota GG, heterozygota AG). Niską zmienność rekompensuje jednak bardzo duża ilość miejsc zmiennych. Mutacja punktowa przypada średnio raz na każde 1000 pz, nie wyłączając sekwencji kodujących. Wielka liczba loci jest źródłem olbrzymiej zmienności, rozłożonej równomiernie w całym genomie, co czyni metodę SNP szczególnie wartościową w przypadku badania DNA zdegradowanego, niemożliwego do oceny innymi metodami. Przykładami SNP są m.in. polimorfizm układów grupowych krwi, lekowrażliwość, a także niektóre choroby genetycznie uwarunkowane (np. mukowiscydoza, fenyloketonuria, achondroplazja, hemofilia A, anemia sierpowatokrwinkowa itd.).
Polimorfizm DNA satelitarnego (krótkich sekwencji tandemowych).
Mianem DNA satelitarnego określa się zespoły sekwencji powtórzonych, ułożonych jedna za drugą (tandemowo). DNA satelitarny dzieli się na minisatelitarny (Variable Number of Tandem Repeats, VNTR) i mikrosatelitarny (Short Tandem Repeats, STR). Różnice między nimi przedstawia tabela:
|
DNA minisatelitarny |
DNA mikrosatelitarny |
Długość motywu repetytywnego |
9-100 pz |
2-6 pz |
Ilość tandemowych powtórzeń |
kilkadziesiąt |
kilkanaście |
Odległość między sąsiednimi loci |
500-20 000 pz |
5-500 pz |
Polimorfizm SNP a polimorfizm VNTR:
Metody wykrywania polimorfizmu DNA:
ANALIZA RESTRYKCYJNA ( = analiza długości fragmentów restrykcyjnych, Restriction Fragments Length Polymorphism, RFLP):
analiza pojedynczego locus (Single Locus Probes, SLP) → PROFIL;
jednoczesna analiza wielu loci (Multiple Locus Probes, MLP) → FINGERPRINT.
W metodzie tej wykorzystuje się trawienie DNA za pomocą enzymów restrykcyjnych, czyli enzymów bakteryjnych przecinających nić w ściśle określonym miejscu. Kolejne etapy metody to: elektroforetyczny rozdział pociętego DNA, denaturacja DNA w alkalicznym roztworze, transfer metodą Southerna na nylonową (nitrocelulozową) membranę i hybrydyzacja ze specyficznymi sondami. Wystarczy zmiana pojedynczego nukleotydu w sekwencji rozpoznawanej przez enzym, aby nie był on w stanie przeciąć DNA. Spowoduje to zmianę liczby powstałych fragmentów DNA (produktów rozkładu pierwotnej cząsteczki) oraz ich wielkości. Jedno i drugie ujawnia rozdział elektroforetyczny.
ENZYMATYCZNA AMPLIFIKACJA (reakcja łańcuchowa polimerazy, Polymerase Chain Reaction, PCR):
polimorfizm DNA mikrosatelitarnego (STR);
polimorfizm pojedynczych nukleotydów (SNP).
Reakcja łańcuchowa polimerazy jest metodą powielania łańcuchów DNA w warunkach laboratoryjnych, polegającą na sekwencji wielokrotnego podgrzewania i oziębiania próbki. Do reakcji wprowadza się matrycowy DNA, trifosforany deoksyrybonukleotydów, startery oraz termostabilną polimerazę. Na początku w wysokiej temperaturze (zwykle około 95°C) pękają wiązania wodorowe i podwójna helisa DNA rozdziela się na dwa pojedyncze łańcuchy. Obniżenie temperatury do optymalnej wartości, ściśle określonej dla danej pary starterów (45-70°C), powoduje ich przyłączenie do matrycy. Ponowne podwyższenie temperatury do około 72°C sprzyja utworzeniu kompleksu między matrycą i starterami a polimerazą DNA, co rozpoczyna syntezę nici komplementarnej do matrycy.
Wielkimi zaletami metody są jej bardzo wysoka czułość oraz wydajność. (Gdyby wydajność metody była stuprocentowa, po n cyklach reakcji z jednej cząsteczki można by uzyskać 2n cząsteczek; wydajność rzeczywista jest niewiele mniejsza).
Odmianą reakcji PCR jest tzw. PCR w czasie rzeczywistym (Real Time PCR, RT-PCR). Jest to metoda ilościowego oznaczania DNA, pozwalająca na monitorowanie reakcji w czasie, w którym ta reakcja przebiega. RT-PCR wymaga termocyklera z odpowiednim systemem biologicznym, specjalnie zaprogramowanych sond oraz dodania barwników fluorescencyjnych inkorporujących podwójną nić DNA (np. bromku etydyny). Monitorowanie zmian w stężeniu produktu następuje poprzez pomiar fluorescencji proporcjonalnej do ilości produktu w mieszaninie.
Inną odmianą jest tzw. multiplex PCR (MPCR), która polega na równoczesnej amplifikacji dwóch lub większej liczby (do 16) markerów genetycznych w jednej mieszaninie reakcyjnej. Możliwe jest to dzięki zastosowaniu kilku par starterów, determinujących amplifikację produktów różnej długości. Detekcję ułatwia oznakowanie markerów różnymi fluorochromami. Główne zalety MPCR to: obniżenie nakładu pracy, zmniejszenie kosztów, możliwość uzyskania większej ilości informacji o badanym materiale i zmniejszenie ryzyka kontaminacji.
Identyfikacja liczby powtórzeń motywu repetytywnego metodami PCR i RFLP:
TECHNIKA DOT BLOT, czyli testu kropkowego z zastosowaniem oligonukleotydu specyficznego dla danego allelu (Allele Specific Oligonucleotide, ASO).
W technice ASO wykorzystuje się oligonukleotydowe sondy komplementarne do określonego allelu danego genu (np. do genu dystrofiny zawierającego mutację powodującą dystrofię Duchenne'a). Powielony produkt reakcji PCR nanoszony jest na nylonową membranę. Następnie hybrydyzuje się go z biotynylowaną sondą, komplementarną do poszukiwanego allelu. Następnie membranę inkubuje się z enzymem peroksydazą chrzanową (horseradish peroxidase, HPR), która wiąże się z sondą poprzez kompleks biotyna-streptawidyna. Na koniec do środowiska reakcji dodaje się bezbarwny substrat, który związana peroksydaza przekształca w barwny produkt. Jeśli w powielonym fragmencie DNA nie występuje poszukiwana mutacja, sonda nie hybrydyzuje, a co za tym idzie - nie dochodzi do przyłączenia peroksydazy i zabarwienia substratu.
Technika ASO jest szczególnie użyteczna w ustalaniu nosicielstwa zmutowanego genu i szybkiej diagnostyce chorób genetycznie uwarunkowanych, takich jak fenyloketonuria, dystrofia mięśniowa, zespół łamliwego chromosomu X i in.

Na podobnej zasadzie opiera się analiza DNA przy użyciu tzw. mikromacierzy. Idea mikromacierzy wywodzi się bezpośrednio z tradycyjnych technik analizy ekspresji genów. Mikromacierz DNA (DNA microarray) jest to szklana lub plastikowa płytka z naniesionymi w regularnych pozycjach mikroskopowymi fragmentami DNA, różniącymi się od siebie sekwencją. Fragmenty te pełnią rolę sond, które wykrywają przez hybrydyzację komplementarne do siebie cząsteczki DNA lub RNA. W zależności od rodzaju i pochodzenia cząsteczek umieszczonych na szklanym podłożu wyróżnia się dwa rodzaje mikromacierzy: mikromacierze cDNA oraz mikromacierze oligonukleotydowe, zwane też „chipami DNA”. Nanosząc na mikromacierz znakowaną próbkę badanego materiału biologicznego, można w jednym eksperymencie przeanalizować ekspresję tylu genów, ile reprezentujących je sond naniesiono na podłoże (czyli nawet kilkudziesięciu tysięcy). Stało się to możliwe dzięki miniaturyzacji sond oraz zastosowaniu specjalnych barwników fluorescencyjnych. Dane zbierane są przez czytnik konfokalny, po wzbudzeniu znaczników fluorescencyjnych światłem lasera o odpowiedniej długości. Zmierzona intensywność fluorescencji, pomniejszona o wartość tła, obrazuje ilość cząsteczek, które hybrydyzowały do danego elementu mikromacierzy.
SEKWENCJONOWANIE.
Sekwencjonowanie DNA jest techniką odczytywania kolejności zasad azotowych tworzących cząsteczkę. Obecnie najczęściej używaną metodą jest metoda Sangera, oparta o syntezę DNA przez polimerazę DNA in vitro. Do reakcji dodaje się niewielką ilość trifosforanów dideoksynukleotydów (ddNTP), które są wbudowywane w DNA, ale ich dołączenie uniemożliwia dalsze wydłużanie nici. W ten sposób otrzymuje się fragmenty DNA o różnej długości zakończone specyficznym nukleotydem. Produkty reakcji rozdziela się elektroforetycznie, co powoduję ich segregację pod względem wielkości. Trifosforany dideoksynukleotydów znakowane są fluorochromami, dzięki czemu odczyt może być w pełni zautomatyzowany.
Najdoskonalszą z technik odczytu jest analiza produktów sekwencjonowania w żelach kapilarnych (kapilarna elektroforeza żelowa). Elektroforeza kapilarna (capillary eletrophoresis, CE) obejmuje rodzinę technik analitycznych, których wspólną cechą jest rozdział molekuł pod wpływem zewnętrznego pola elektrycznego, w cienkiej kapilarze o średnicy rzędu kilkudziesięciu mikrometrów. Analizator prowadzi odczyt jednocześnie w 16 kapilarach, co pozwala w ciągu godziny ocenić 600 Kpz. Ograniczeniem metody są wysokie koszty (koszt analizatora wynosi ok. 600 tys. PLN).
Najnowszą metodą analizy sekwencyjnej DNA jest pirosekwencjonowanie, zwane również sekwencjonowaniem w czasie rzeczywistym (www.pyrosequencing.com). W metodzie tej, podobnie jak w poprzedniej, na matrycy badanej nici DNA odbywa się synteza nici komplementarnej. W środowisku reakcji poza DNA i polimerazą znajdują się: siarczan 5'-fosfoadenozyny, lucyferyna oraz enzymy apyraza, ATP-sulfurylaza i lucyferaza.
Polimeraza DNA syntetyzuje łańcuch komplementarny na matrycy badanej nici DNA (powielonej metodą PCR). Do mieszaniny reakcyjnej dodawane są etapami kolejne nukleotydy. Jeśli dodany nukleotyd nie zostanie wbudowany w nowo powstającą nić, jest niszczony przez enzym apyrazę. Jeśli dodany nukleotyd zawiera komplementarną zasadę, jest wbudowywany do syntetyzowanej nici z uwolnieniem cząsteczki pirofosforanu. Uwolniony pirofosforan i siarczan 5'-fosfoadenozyny służą jako substraty dla ATP sulfurylazy, która syntetyzuje cząsteczkę ATP. Energia z ATP umożliwia katalizowaną przez lucyferazę przemianę lucyferyny do oksylucyferyny. Jest to reakcja chemiluminescencyjna. Emisja światła rejestrowana jest przez kamerę i zapisywana na wykresie reakcji w postaci piku, którego wysokość jest proporcjonalna do ilości wbudowanych nukleotydów.
Pirosekwencjonowanie jest najszybszą obecnie metodą analizy DNA. Jej wadą jest ograniczona długość nici, którą można jednorazowo poddać sekwencjonowaniu.
Analiza DNA w medycynie sądowej
Zastosowanie analizy DNA w medycynie sądowej obejmuje:
Badanie pokrewieństwa:
sprawy alimentacyjne (zazwyczaj dochodzenie ojcostwa);
badania osób zaginionych;
sprawy spadkowe;
sprawy imigracyjne (w tych krajach, w których dopuszcza się łączenie rodzin).
Badania identyfikacyjne:
ustalanie pokrewieństwa osób;
identyfikacja sprawców przestępstw;
tworzenie baz profili DNA;
identyfikacja osób w katastrofach masowych.
Cechy dobrego markera DNA
Markery DNA stosowane w medycynie sądowej powinny charakteryzować się takimi cechami jak wysoki stopień polimorfizmu, wysoka heterozygotyczność i niski stopień mutacyjności. Przydatnym pojęciem jest siła dyskryminacji (power of discrimination, PD), czyli prawdopodobieństwo, że dwie losowo wybrane osoby z populacji nie będą posiadały takich samych cech.
W badaniach identyfikacyjnych przeprowadza się analizę uzyskanego profilu DNA i szacuje prawdopodobieństwo przypadkowej zgodności. Przy badaniu 3 loci STR prawdopodobieństwo to wynosi 10-4 (1:10 tys. osób). Przy badaniu 10 loci jest to już 10-13, a przy 15 loci - 10-18 osób. Zazwyczaj bada się 5 loci minisatelitarnych = 15 STR = 50 SNP.
Dokładność analizy genetycznej sprawia, że polimorfizm DNA określa się mianem „daktyloskopii molekularnej”.
Dochodzenie spornego ojcostwa
Podstawy prawne badań dochodzenia spornego ojcostwa zawiera Kodeks Rodzinny i Opiekuńczy (Tytuł II, Dział I, Rozdział I: Pochodzenie dziecka):
Proces o ustalenie ojcostwa:
Art. 84. § 1. Sądowego ustalenia ojcostwa może żądać dziecko, jego matka oraz domniemany ojciec dziecka. Jednakże matka ani domniemany ojciec nie mogą wystąpić z takim żądaniem po śmierci dziecka lub po osiągnięciu przez nie pełnoletniości.
§ 2. Dziecko albo matka wytacza powództwo o ustalenie ojcostwa przeciwko domniemanemu ojcu, a gdy ten nie żyje - przeciwko kuratorowi ustanowionemu przez sąd opiekuńczy.
§ 3. Domniemany ojciec dziecka wytacza powództwo o ustalenie ojcostwa przeciwko dziecku i matce, a gdy matka nie żyje - przeciwko dziecku.
Art. 85. § 1. Domniemywa się, że ojcem dziecka jest ten, kto obcował z matką dziecka nie dawniej niż w trzechsetnym, a nie później niż w sto osiemdziesiątym pierwszym dniu przed urodzeniem się dziecka.
§ 2. Okoliczność, że matka w tym okresie obcowała także z innym mężczyzną, może być podstawą do obalenia domniemania tylko wtedy, gdy z okoliczności wynika, że ojcostwo innego mężczyzny jest bardziej prawdopodobne.
Art. 86. Powództwo o ustalenie lub zaprzeczenie pochodzenia dziecka oraz o unieważnienie uznania dziecka może wytoczyć także prokurator.
Proces o zaprzeczenie ojcostwa:
Art. 62. § 1. Jeżeli dziecko urodziło się w czasie trwania małżeństwa albo przed upływem trzystu dni od jego ustania lub unieważnienia, domniemywa się, że pochodzi ono od męża matki. Domniemania tego nie stosuje się, jeżeli dziecko urodziło się po upływie trzystu dni od orzeczenia separacji.
§ 2. Jeżeli dziecko urodziło się przed upływem trzystu dni od ustania lub unieważnienia małżeństwa, lecz po zawarciu przez matkę drugiego małżeństwa, domniemywa się, że pochodzi ono od drugiego męża.
§ 3. Domniemania powyższe mogą być obalone tylko na skutek powództwa o zaprzeczenie ojcostwa.
Art. 63. Mąż matki może wytoczyć powództwo o zaprzeczenie ojcostwa w ciągu sześciu miesięcy od dnia, w którym dowiedział się o urodzeniu dziecka przez żonę.
Badania przeprowadzane w celu ustalenia ojcostwa ocenia się wg parametru znanego jako siła wyłączenia (power of exclusion, PE). Jest to odsetek niesłusznie pozwanych mężczyzn, którzy zostaną wyłączeni w toku badania.
Kolejność badań w rutynowym procesie o ustalenie ojcostwa obejmuje:
Ekspertyzę Iº HLA → wyłączenie ojcostwa lub jego brak.
Ekspertyzę DNA → wyłączenie ojcostwa lub jego potwierdzenie z prawdopodobieństwem graniczącym z pewnością.
Identyfikacja płci
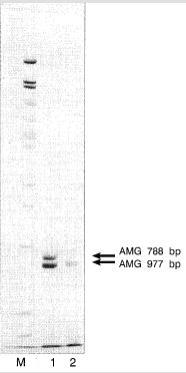
Profil genetyczny płci ustala się w oparciu metodę PCR, na podstawie analizy genu amelogeniny (amelogenin, AMG). Gen ten jest w 90 % homologiczny dla chromosomów X i Y. Różnica jest skutkiem delecji, występującej na chromosomie Y. Przy zastosowaniu takiej samej pary starterów PCR amplifikacji ulegają więc dwa odcinki DNA o różnych długościach. Krótszy, o wielkości 788 pz, jest charakterystyczny dla chromosomu Y (Y AMG). Dłuższy, o wielkości 977 pz, jest swoisty dla chromosomu X (X AMG). Stwierdzenie w elektroforezie mniejszego i większego produktu PCR świadczy o obecności w śladzie chromosomów X oraz Y i jest dowodem, że pochodzi on od mężczyzny. Natomiast wykrycie jedynie fragmentu dłuższego, swoistego dla chromosomu X, jest dowodem pochodzenia śladu od kobiety. Analiza genu AMG jest metodą bardzo czułą - wystarcza do niej obecność niewielkiej ilości chromosomalnego DNA (poniżej 400 pg).
Rysunek przedstawia rozdział elektroforetyczny genów Y-AMG i X-AMG.
Prążek 788 pz pochodzi z chromosomu Y, 977 pz z chromosomu X. Obecność dwóch prążków (1) wskazuje na pochodzenie materiału genetycznego od mężczyzny, obecność jednego prążka (2) na pochodzenie od kobiety; M - marker.
Inną metodą identyfikacji płci jest badanie markerów chromosomu Y. Są one absolutnie specyficzne dla mężczyzn (nie rekombinują). Genetyczna zmienność typu STR w wielu obszarach chromosomu Y tworzy tzw. haplotyp, przekazywany w linii męskiej z pokolenia na pokolenie. Badanie markerów chromosomu Y umożliwia:
stuprocentowo pewną identyfikację DNA mężczyzny (sprawcy 97% przestępstw!);
wykrywanie przestępstw na tle seksualnym (badanie mieszanin materiału genetycznego sprawcy i ofiary);
ustalanie pokrewieństwa w linii męskiej.
Odpowiednikiem markerów chromosomu Y u kobiet jest DNA mitochondrialny, który również nie rekombinuje i przekazywany jest wyłącznie w linii matczynej. Ocenie podlegają regiony HV1 i HV2, które bada się w kierunku zmienności typu SNP. Sekwencją referencyjną, służącą porównywaniu z badanym materiałem, jest tzw. sekwencja Andersona. Badanie markerów mtDNA jest użyteczne w takich sytuacjach jak:
ustalanie pokrewieństwa w linii żeńskiej;
badanie włosów, śladowych ilości DNA lub DNA zdegradowanego.
Art. 86 dotyczy sytuacji wyjątkowych (gwałt, kazirodztwo, obcowanie z nieletnim itp.).
Badania ludzkiego mtDNA doprowadziły do ustalenie jego „drzewa genealogicznego”, którego początków upatruje się w Afryce ok. 200 tys. lat temu. Wyniki badań sugerują, że wszyscy ludzie zamieszkujący dziś obszar ziemi są potomkami jednej kobiety (ewolucyjna teoria „Ewy mitochondrialnej”).


SNP
VNTR
Wyszukiwarka