8. Apolipoproteiny:
I. Rodzina A: związana z HDL
a) apoA-I
- aktywacja białka ABC, które umożliwia przeniesienie cholesterolu z komórki do cząstki HDL
- aktywator LCAT
- główna apolipoproteina lipoprotein HDL (70% ich białka)
- masa 28 kDa
- ligand dla cubiliny - umożliwia to jej resorpcję zwrotną w cewkach proksymalnych nefronów
- wysokie stężenie w osoczu: 100-130 mg/dl
- synteza w wątrobie i jelicie
- drastyczne obniżenie jej stężenia występuje w chorobie Tangierskiej (sam defekt dotyczy białka ABC1, jego skutkiem jest obniżone stężenie HDL i apoA-I)
b) apoA-II
- regulacja HTGL (wątrobowa lipaza triglicerydowa degradująca remnanty VLDL)
- masa 17 kDa
- stanowi 20% białek HDL
- obecna w 2/3 cząstek HDL
- synteza w wątrobie
- stężenie we krwi: ok. 30 mg/dl
c) apoA-IV
- aktywator LCAT
- masa 45 kDa
II. Rodzina B:
a) apoB48
- obecna w chylomikronach i ich remnantach
- pełni funkcję strukturalną
- masa 260 kDa
- syntezowana w jelicie
b) apoB100
- obecna w VLDL i LDL
- jest ligandem dla receptora wysokiego powinowacta apoB/E
- posiada masę 550 kDa
- syntezowana w wątrobie
c) obie apolipoproteiny są syntezowane przy wykorzystaniu mRNA z tego samego genu jako matrycy; w jelicie następuje modyfikacja posttranskrypcyjna mRNA, co powoduje, że translacja kończy się na 2153 kodonie (białko zawiera 2152 reszty aminokwasowe); w wątrobie powstaje białko złożone z aminokwasów kodowanych przez wszystkie 4563 kodony;
w jelicie mRNA genu APOB podlega redagowaniu - RNA editing, które prowadzi do zamiany reszty C w 2153 kodonie CAA na U, przez co z kodonu glutaminy powstaje kodon stop; reakcję katalizuje deaminaza cytydynowa
III. Rodzina C: związane z metabolizmem lipoprotein bogatych w triglicerydy, swobodnie przenoszone między różnymi frakcjami lipoprotein
- syntezowane w wątrobie i jelicie
- apoproteiny o najniższej masie cząsteczkowej (I - 6,5 kDa, II - 8,3 kDa, III - 8,7 kDa)
- apoC-I - aktywator LCAT
- apoC-II - aktywator LPL; jej defekt może prowadzić do chylomikronemii
- apoC-III - inhibitor LPL
- obecne w HDL, chylomikronach, VLDL
9. Apolipoproteina E:
a) występowanie:
- chylomikrony i ich remnanty
- VLDL
- IDL (remnanty VLDL)
- HDL
b) rola:
- eliminacja z krążenia produktów katabolizmu chylomikronów (receptor LRP)
- metabolizm VLDL - eliminacja IDL (ligand dla receptora apoB/E)
c) synteza:
- wątroba
- mózg (głównie przez neuroglej)
- śledziona
- nerki i płuca
d) masa:
- 34 kDa; 299 aminokwasów
e) gen:
- na 19 chromosomie
- 3 allele: ε2, ε3, ε4
f) polimorfizm genu:
- 6 fenotypów związanych z różnymi kombinacjami alleli: apoE2/2, apoE3/2, apoE3/3, apoE4/3, apoE4/4, apoE4/2
- poszczególne izoformy różnią się aminokwasami w pozycji 112 i 158
- izoforma apoE2 ma mniejszą zdolność wiązania się z receptorem apoB/E, co prowadzi do zmniejszenia katabolizmu remnantów VLDL i chylomikronów w wątrobie; towarzyszy temu wzmożony wychwyt LDL przy udziale „zwolnionych” receptorów apoB/E, co skutkuje obniżonym stężeniem cholesterolu całkowitego i LDL (homozygoty apoE2/2)
- izoforma apoE4 przyspiesza katabolizm lipoprotein wyposażonych w apoE - wykazuje wyższe powinowactwo do receptora apoB/E niż izoforma apoE3; wzmożony transport lipidów, w tym cholesterolu w VLDL, skutkuje zmniejszeniem ilości receptorów LDL w błonie hepatocytu; podwyższa się poziom LDL, które nie są wyłapywane przez wątrobę, przez co wzrasta ryzyko choroby niedokrwiennej serca
- fenotyp apoE4/4 obserwowany częściej w hiperlipoproteinemiach typu II, apoE2/2 w hiperlipoproteinemii typu III
g) drugi rodzaj polimorfizmu: związany z postrtranslacyjną glikozylacją - przyłączenie reszt kwasu sjalowego
h) izoforma apoE4 może odgrywać rolę w patomechanizmie choroby Alzheimera; apoE4 może tworzyć kompleksy z peptydem beta/A4 pochodzącym z białka prekursorowego amyloidu APP; brak alleli ε4 obniża ryzyko wystąpienia choroby
12. Kubilina i megalina:
Kubilina:
a) białko zakotwiczone w błonie komórkowej dzięki palmitylacji
b) masa ok. 470 kDa
c) jej ligandy:
- apolipoproteina A-I
- transferyna
- HDL
- kompleks IF-witamina B12
- RAP
- białka komórek Clara
Megalina:
a) transbłonowa
b) masa ok. 600 kDa
c) samotnie występuje w błonach:
- podocytów
- pneumocytów typu II
- komórek tarczycy
- komórek przytarczyc
- endometrium
- najądrzu
d) ligandy:
- kompleks transkobalamina-B12
- kompleks RBP-witamina A
- apolipoproteina H
- alfa-2-mikroglobulina
- transtyretyna
- PTH
- hormony peptydowe
- kompleks UPA-PAI-I
- Apo-B
W połączeniu z megaliną kubilina występuje w:
- jelicie cienkim
- cewce proksymalnej nefronu
- cytotrofoblaście
Ligandy wspólne dla megaliny i cubiliny:
- DBP - białko wiążące witaminę D
- lekki łańcuch Ig
- hemoglobina
- albumina
13. Receptor LDL (LDLR):
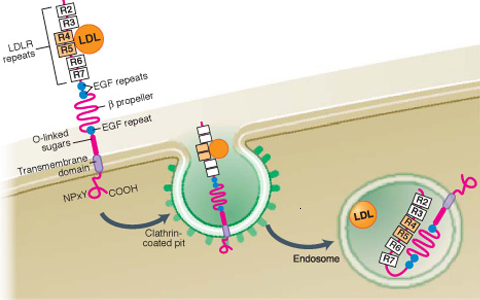
a) receptor apoB/E zwany receptorem wysokiego powinowactwa
b) ligandy:
- apoB-100
- apoE
c) aktywność hamowana przez RAP - receptor associated lipoprotein
d) funkcje:
- tkanki pozawątrobowe: pobieranie (internalizacja) LDL
- wątroba: pobieranie różnych lipoprotein wyposażonych w apoB-100
e) obecny w zagłębieniach błonowych pokrytych klatryną - zagłębienia przechodzą w pęcherzyki endocytarne
f) ekspresja genu receptora hamowana przez cholesterol
g) struktura:
- domena wiążąca ligand (przy N-końcu) - liczne wiązania dwusiarczkowe
- domena zbliżona do prekursora czynnika wzrostu EGF - EGFP
- domena bogato glikozylowana (bez znaczenia funkcjonalnego)
- domena transbłonowa
- domena cytozolowa przy C-końcu z sekwencją sygnałową
sekwencje wiążące ligand znajdują się przy N-końcu receptora: 40-aminokwasowe w 50% homologiczne powtórzenia (w liczbie 7)
domena EGFP wykazuje 30% homologii w stosunku do sekwencji EGF (3 powtórzenia); jej funkcja polega na uwalnianiu ligandu z jego połączenia z domeną wiążącą ligand
h) gen posiada 18 eksonów
i) jego mutacje powodują hiperlipoproteinemię typu II
15. Chylomikrony:
I. struktura i skład:
a) syntezowane w jelicie; trafiają do chłonki i przez przewód piersiowy do układu krwionośnego; wielkość: do 1000 nm
b) skład lipidowy:
- triglicerydy: 90-95 %
- estry cholesterolu: 2-4%
- fosfolipidy: 2-6%
- wolny cholesterol: 1%
- białka: 1%
c) odpowiedzialne w głównej mierze za transport triglicerydów egzogennych
d) skłąd białkowy:
- apoB48
- apoC-II - otrzymują je od HDL
II. metabolizm
a) otrzymują apoC-II od HDL
b) z prądem krwi trafiają do tkanki tłuszczowej żółtej i mięśni poprzecznie prążkowanych, tu poddawane są działaniu lipazy lipoproteinowej aktywowanej przez apoC-II
c) działanie LPL uwalnia wolne kwasy tłuszczowe, glicerol i apoC-II - powstaje chylomikron resztkowy (remnant chylomikronu)
d) remnanty chylomikronów są wyłapywane przez hepatocyty przy udziale odpowiednich receptorów
e) zaburzenia w metabolizmie prowadzą do hiperlipoproteinemii typu I oraz (częściowo) typu V
f) im wyższe stężenie chylomikronów tym niższe stężenie HDL
16. VLDL:
a) syntezowane przez hepatocyty
b) bogate w triglicerydy - endogenne, w przeciwieństwie do TG w chylomikronach
c) skład lipidowy:
- triglicerydy: 50-65 %
- estry cholesterolu: 8-14 %
- fosfolipidy: 12-16 %
- wolny cholesterol: 4-7 %
d) skład białkowy (białka stanowią 5-10% cząstki):
- apoB-100
- apoE
e) połączenie składników lipidowych z białkowymi następuje w aparacie Golgiego - powstaje natywny (niedojrzały) VLDL
f) metabolizm:
- w krwiobiegu natywny VLDL otrzymuje apoC-II od HDL
- dzięki aktywności CETP otrzymuje od HDL cholesterol, w zamian oddaje triglicerydy
- w naczyniach tkanki tłuszczowej i mięśni poprzecznie prążkowanych poddawany działaniu LPL
- powracają jako IDL do wątroby,
- 2/3 IDL jest wyłapywane przez receptor wysokiego powinowactwa (ligandem jest apoB-100)
- 1/3 IDL jest przez HTGL przekształcana w LDL (apoE odłącza się w tym procesie)
g) podwyższony poziom VLDL obserwowany w hiperlipoproteinemii typu IV i V
24. Modyfikacje LDL:
a) modyfikacje biłka apo B100 - skutkują jego obniżonym powinowactwem do receptora LDL
- oksydacja - pod wpływem wolnych rodników i enzymów lizosomalnych; zachodzi pod wpływem substancji uwalnianych przez komórki krwi; dotyczy zwłaszcza reszt tyrozyny i lizyny, może prowadzić do fragmentacji białka; tak zmodyfikowana apo B100 może stać się obca antygenowo
- glikacja - spada powinowactwo do receptora LDL, zwiększa się wychwyt przy udziale receptorów scavenger
- glikooksydacja
- angiotensynizacja
- tiolacja - przyłączenie homocysteiny
b) modyfikacje kwasów tłuszczowych
- utlenianie do nadtlenków kwasów tłuszczowych
c) modyfikacje cholesterolu:
- utlenianie do oksysterolu
d) modyfikacje mogą prowadzić do wytwarzania przeciwciał przeciwko cząstkom LDL
e) w ścianie naczyniowej LDL są modyfikowane przez: lipooksygenację, aktywność mieloperoksydazy, wolne rodniki takie jak: rodnik ponadtlenkowy, peroksynitryle, rodnik hydroksylowy
f) lipoproteiny LDL są zabezpieczone przed oksydacją przez znajdujące się w nich: gamma i alfa tokoferol, karotenoidy, retinoidy
g) modyfikacje prowadzą do nasilenia aterogennego wpływu LDL
Małe gęste LDL:
a) podfrakcji B lipoprotein LDL
b) wykazuje bardziej aterogenny wpływ niż podfrakcja A
c) ich obecność związana jest z insulinoopornością i nadprodukcją apo-B100
d) mechanizm zwiększenia produkcji ich komponenty białkowej w insulinooporności:
- insulina nie może hamować lipolizy w tkance tłuszczowej żółtej, co skutkuje
- zwiększoną zawartością FFA w osoczu i ich transportem do wątroby, w której
- obecne w dużej ilości FFA stymulują syntezę VLDL hamując degradację apo-B100 (która uległaby degradacji przy niskich stężeniach FFA)
- zwiększona ilość VLDL intensywnie wymienia triglicerydy na cholesterol z HDL, co w konsekwencji prowadzi do obniżenia poziomu HDL
- następuje patologiczna wymiana lipidów między VLDL a LDL, co obniża zawartość cholesterolu w cząstkach LDL; kiedy LDL zostaną poddane działaniu LPL, która uwolni z nich składniki triglicerydowe, będą posiadały mniejszą ilość cholesterolu w stosunku do podfrakcji A - tzn. będą miały obniżony stosunek zawartości cholesterolu do zawartości białka, którego będzie tyle samo - dalej jedna cząsteczka apo-B100 na jedną cząstkę LDL
e) ich aterogenność polega na:
- małym powinowactwie do receptora LDL
- długim okresie półtrwania
- intensywnym naciekaniu ściany naczyniowej
- silnym wiązaniu z glikozaminoglikanami
- przyspieszonej oksydacji
- intensywnym wychwycie przez makrofagi
18. Wykrywanie małych gęstych LDL:
a) elektroforeza w gradiencie pH
b) ultrawirowanie w gradiencie gęstości
c) NMR lipoprotein - pozwala zmierzyć stężenie samych cząstek LDL oraz ich wielkość
d) stężenie apoB-100
19. HDL:
a) wielkość: 8-13 nm
b) skład lipidowy:
- duża zawartość cholesterolu, estry: 10-20%, wolny: 5%;
- fosfolipidy stanowią 25% masy,
- triglicerydy: 7%
c) skład białkowy:
- białka stanowią ok 45% masy cząstek
- 70% białek stanowi apo AI (28 kDa) - występująca we wszystkich HDL
- 20% stanowi apo AII (17 kDa) - występuje w 2/3 cząstek HDL
- białka migrujące: apo CI, apo CII, apo CIII, apo E
Przeciwmiażdżycowe działanie HDL:
- transport zwrotny cholesterolu - usuwanie go ze ściany naczyniowej
- hamowanie adhezji monocytów
- hamowanie oksydacji LDL dzięki obecności paraoksonazy - antyoksydacyjnego enzymu wytwarzanego przez wątrobę
20. CYKL HDL:
- apo AI - niezbędny składnik, syntezowana w jelicie
- apo AI zostaje otoczone fosfolipidami i cholesterolem - proces zachodzi przy powierzchni hepatocytów, ale też w innych miejscach obfitujących w fosfolipidy i cholesterol - powstaje natywny HDL3 o kształcie dyskoidalnych
- apo AI oddziałuje z receptorem, dzięki czemu możliwy staje się transport cholesterolu z komórki do HDL za pośrednictwem białka ABC-1 (posiada 2 domeny regulowane przez ATP, jego ekspresja jest zależna od aktywacji PPAR przez tiazolidynodiony i wolne kwasy tłuszczowe); mówimy, że apo AI aktywuje ABC-1
- po odebraniu cholesterolu i jego estryfikacji przez LCAT HDL3 przechodzi w HDL2A
- HDL2A pod wpływem CETP przechodzi w HDL2B
- HDL2B poddawany jest działaniu HTGL, następuje hydroliza triglicerydów - powoduje to jego ponowne przejście w formę HDL3
21. Transport zwrotny cholesterolu:
- zachodzi dzięki cząstkom HDL odbierającym cholesterol z tkanek obwodowych; jest to możliwe dzięki białku ABC-1 komórek obwodowych, aktywowanemu przez apo AI
a) droga bezpośrednia:
- zachodzi przy udziale receptora SR B1 - scavenger receptor
- całe cząstki HDL zostają wciągnięte do wnętrza do wnętrza hepatocytu
- estry cholesterolu są hydrolizowane przez esterazę
- HDL nie zostaje całkowicie strawiony - w „okrojone” formie (większą część stanowi apo AI) zostaje wydzielony z powrotem z hepatocytu
RECEPTORY SCAVENGER
a) nie podlegają regulacji
b) ligandy:
- zmodyfikowane lipoproteiny
- polirybonukleotydy
- naturalne i zmodyfikowane polisacharydy
- endotoksyny, azbest i inne obce sunstancje
c) rozmieszczenie:
- komórki śródbłonka
- makrofagi
- komórki Browicza-Kupffera
d) należy do nich receptor SR B1
- zlokalizowany na hepatocycie
- pośredniczy w przekazywaniu cholesterolu z HDL do komórki (HDL nie podlega wówczas internalizacji)
e) receptory scavenger makrofagów z uwagi na brak regulacji odgrywają istotną rolę w miażdżycy; absorpcja nadmiernych ilości cholesterolu i zmodyfikowanych lipoprotein przez makrofagi za pośrednictwem receptorów scavenger prowadzi do powstania komórek piankowatych - foam cells, których obecność prowadzi do powstania blaszki miażdżycowej
b) droga pośrednia - jest najistotniejsza u człowieka:
- polega na wymianie lipidów między HDL a VLDL: HDL przyjmują triglicerydy przekazując część swojego cholesterolu na VLDL
- VLDL jest następnie poddawany działaniu lipazy lipoproteinowej, w wyniku czego powstają IDL - remnanty VLDL
- 2/3 remnantów VLDL internalizowane jest przez hepatocyty po związaniu przez receptor wysokiego powinowactwa
- 1/3 poddawana jest działaniu HTGL, przez co przekształca się w LDL, których 75% jest wyłapywane przez wątrobę
c) cholesterol w wątrobie:
- obniża aktywność reduktazy beta-hydroksy, beta-metylo glutarylo-S-CoA - hamuje syntezę endogennego cholesterolu
- po przekształceniu w oksysterole hamuje ekspresję receptora LDL
- jest deponowany w hepatocycie dzięki ACAT
- służy do syntezy kwasów żółciowych
22. ABCA1:
a) ATP binding cassette transporter A1
b) 2261 aminokwasów, członek nadrodziny białek ABC
c) złożone z 2 podobnie zbudowanych kowalencyjnie ze sobą związanych połówek
d) każda połówka posiada domenę wiążącą nukleotydy z dwoma konserwatywnymi motywami (Walker 1 i Walker 2) oraz domenę transbłonową i zewnątrzkomórkową (tworzącą glikozylowane pętle)
e) uczestniczy w transporcie lipidów z komórki na zewnątrz albo w mechanizmie bezpośrednim tworząc kanał, albo pełniąc rolę regulatorową (dokładny mechanizm nieznany)
f) defekt genu ABCA1 jest przyczyną choroby tangierskiej
g) C-koniec (w cytozolu) oddziałuje z syntrofiną-β2 i utrofiną
h) ABCA1 odpowiada za transport:
- fosfolipidów (przede wszystkim); głównie fosfatydylocholiny
- cholesterolu
- innych substancji - α-tokoferolu, apoE, interferonu-1β
i) mechanizm działania:
- wpływ na błonę komórkową - organizuje ją w mikrodomeny, co ułatwia przenoszenie lipidów na lipoproteiny
- bezpośrednie oddziaływanie z apoA-I
j) wzrost ekspresji genu następuje przy wysokich wewnątrzkomórkowych stężeniach cholesterolu
- powstające w komórce oksysterole tworzą kompleks z receptorami LXR i następnie aktywują ekspresję genu ABCA1
23.
1. LCAT:
a) acylotransferaza lecytyna-cholesterol
b) syntezowana w wątrobie, uwalniana do krążenia
c) estryfikuje cholesterol w HDL biorąc udział w jego zwrotnym transporcie
d) dwa typy aktywności LCAT:
- alfa - estryfikacja cholesterolu HDL
- beta - estryfikacja cholesterolu w lipoproteinach zawierających apoB; brak tego rodzaju aktywności, który może być skutkiem rozległych chorób wątroby, prowadzi do wzrostu poziomu wolnego cholesterolu
e) jego aktywatory:
- apoA-I
- apoA-IV
- apoC-I
f) aktywność LCAT ostatecznie prowadzi do zwiększenia zawartości apoE i obniżenia zawartości apoC w VLDL
g) objawy wrodzonego niedoboru LCAT:
- zwiększenie stężenia triglicerydów
- zmętnienie rogówki
- niedokrwistość
- białkomocz
- zmiany ksantomatyczne (czasam) w okolicach ścięgien, dłoni
2. LPL - lipaza lipoproteinowa śródbłonka naczyń:
a) odpowiedzialna za hydrolizę triglicerydów zawartych w chylomikronach i VLDL - przekształca je w odpowiednio: remnanty chylomikronów i IDL
b) występuje w naczyniach krwionośnych tkanek pozawątrobowych:
- mięśni szkieletowych
- tkanki tłuszczowej
- mięśnia sercowego
c) aktywatory:
- apoC-II
- heparyna
- GAG wiążące eznym do ściany naczyniowej
d) dożylne podanie heparyny powoduje uwolnienie enzymu z jego połączenia z siarczanem heparanu i nagły wzrost lipolitycznej aktywności w osoczu
e) substancje obniżające aktywność:
- apoC-III
- siarczan protaminy
f) aktywność wyższa u kobiety, obniża się z wiekiem
g) brak aktywności LPL występuje w hiperlipoproteinemii typu I
3. HTGL - wątrobowa lipaza triglicerydowa:
a) zlokalizowana na śródbłonku zatokowych naczyń wątroby
b) odpowiedzialna za katabolizm 1/3 remnantów VLDL oraz przekształcenie HDL2B w HDL3
c) substraty: triglicerydy i fosfolipidy
d) aktywowana przez:
- insulinę
- hormony tarczycy
e) do pełnej aktywności wymagane wysokie stężenie NaCl
f) aktywność wyższa u mężczyzn
4. HSL - hormon-sensitive lipase:
a) hormonozależna wewnątrzkomórkowa lipaza adipocytów (HTL)
b) uwalnia wolne kwasy tłuszczowe z adipocytów w okresie międzyposiłkowym i przy zwiększonym zapotrzebowaniu energetycznym
c) aktywność wzrasta wraz z wzrostem stężenia cAMP
d) aktywowana przez adrenalinę przez receptor beta-1-adrenergiczny
e) aktywność obniżana przez insulinę, która powoduje spadek poziomu cAMP
25. Oksysterole:
Sterole z grupami funkcyjnymi zawierającymi tlen
a) źródła:
- pożywienie
- oksydacja endogenna:
1. autooksydacja
2. swoiste monooksygenazy 7α, 20α, 22β, 23β, 25, 26, 27
3. enzymatyczna lub nieenzymatyczna peroksydacja lipidów
b) zawartość w produktach żywnościowych wzrasta wskutek obróbki technologicznej:
- obróbka termiczna: skondensowane mleko, mleko w proszku, smażone masło
- niewłaściwe przechowywanie (np. suszone żółtka)
c) oksysterole pochodzące z pożywienia przenoszone przez lipoproteiny:
- VLDL - 5α-cholestan-3β, 5α-6β-triol, 7-ketocholesterol
- LDL - 25-hydroksycholesterol
- HDL - śladowe ilości
d) powstawanie w organizmie - oksydacja endogenna:
- pod wpływem wolnych rodników - przede wszystkim rodnik hydroksylowy, w mniejszym stopniu anionorodnik ponadtlenkowy
- enzymatyczna hydroksylacja cholesterolu 7α, 20α, 22β, 23β, 25, 26, 27 lub epoksydacja w pozycji 5α-6α
- powstają też jako związki pośrednie w syntezie kwasów żółciowych i hormonów sterydowych
e) przedstawiciele (pierścienie A i B):
- 7α-hydroksycholesterol - 4-165 ug/L
- 7β-hydroksycholesterol - 0-265 ug/L
- 7-ketocholesterol - 0-373 ug/L
- 5-6-β-epoksycholesterol
- 5-6-α-epoksycholesterol
- cholestantriol
f) przedstawiciele (łańcuch boczny):
- 24-hydroksycholesterol 3-43 ug/L
- 25-hydroksycholesterol
- 26-hydroksycholesterol 30-352 ug/L
g) działanie:
- cytotoksyczne - inhibitory wzrostu komórek
- immunosupresyjne - hamowanie proliferacji i transformacji limfocytów
- hamowanie syntezy DNA oraz syntezy cholesterolu
- inhibitory kalmoduliny
- wpływ na strukturę i funkcję błony komórkowej
- hamują podziały komórek szybko proliferujących - w ten sposób ograniczają wzrost nowotworów
- toksyczne działanie dla mikroorganizmów
- mieszanina oksysteroli ma działanie aterogenne: przede wszystkim 5α-cholestan-3β oraz 5α-6β-triol
- 26-hydroksy-cholesterol jest inhibitorem reduktazy HMG-CoA
- apoptoza komórek endotelium
- hamowanie proliferacji komórek mięśni gładkich ściany naczyniowej
26. Lipoproteina (a):
a) Skład lipidowy i białkowy - jak w LDL
b) dodatkowo posiada apolipoproteinę (a) - o strukturze przypominającej precle - cringles
c) liczba domen przypominających precle uwarunkowana jest genetycznie
d) aterogenne działanie, ponieważ:
- jej wiązanie z receptorem jest utrudnione
- jej klirens jest powolny
- łatwo ulega modyfikacjom
- gromadzi się w ścianie naczyniowej
- konkuruje z plazminogenem lub t-PA - z uwagi na podobieństwo strukturalne
- stymuluje sekrecję PAI - inhibitora aktywatora plazminogenu
- obniża uwalnianie TGF-beta, który to czynnik wzrostowy hamuje wzrost SMC
e) wzrost jej stężenia występuje przy obniżonym poziomie estrogenów i w przypadkach hipoandrogenizmu
f) wartość graniczna - wartość odcięcia: 0,2 g/l (20 mg%)
g) oznaczamy jej stężenie u osób:
- po zawale serca przy prawidłowych stężeniach lipidów
- z incydentami zakrzepowymi
27. Lipoproteina x:
a) występuje w znacznej ilości we krwi pacjentów z cholestazą wątrobową
b) postać liposomu zbudowanego z podwójnej warstwy fosfolipidów (66% to lecytyna), wolnego cholesterolu (25%) i białek (5% - połowa to albuminy)
c) gęstość zbliżona do LDL
d) właściwości aterogenne nieudowodnione
28. Proaterogenny wpływ cukrzycy:
a) hiperglikemia
b) hiperinsulinemia (typ II)
c) nadciśnienie
d) dyslipidemia
e) zaburzenia krzepnięcia krwi
f) obecność advanced glication end products
g) zwiększony stres oksydacyjny
Triada lipidowa:
a) zespół zaburzeń lipidowych obecnych w cukrzycy
b) składają się na nią:
- hipertriglicerydemia
- obniżenie poziomu cholesterolu HDL
- obecność małych gęstych LDL
c) w cukrzycy nie występuje hipercholesterolemia!
d) mechanizm powstawania triady lipidowej:
- w insulinoopornych adipocytach insulina nie może hamować HSL; następuje wzrost lipolizy i zwiększone uwalnianie FFA do osocza
- w wątrobie przy dużych stężeniach insuliny pobudzona zostaje apoB-100, w wyniku czego powstaje więcej VLDL
- w cukrzycy wzrasta aktywność CETP, co prowadzi do nasilenia wymiany lipidowej między VLDL i HDL
- HDL wzbogacone w triglicerydy z VLDL są poddawane działaniu HTGL, co prowadzi do ich zagęszczenia i zmniejszenia ich masy - powstają małe gęste HDL, które przesączają się przez naczynia włosowate kłębuszków nerkowych - następuje w ten sposób utrata HDL
- CETP również przenosi triglicerydy z VLDL na LDL; wzbogacone w TG LDL są poddawane działaniu HTGL i LPL, co prowadzi do zmniejszenia ich masy - tracą uzyskane TG stając się małymi gęstymi LDL
30. Hiperlipoproteinemia typu I:
a) nazywana również hiperlipidemią swoistą, chorobą Burgera-Grutza, hiperchylomikronemią
b) spowodowana defektami genetycznymi:
- niedobór lipazy lipoproteinowej (najważniejsza przyczyna)
- defekt apoC-II - aktywatora lipazy lipoproteinowej
- obecność przeciwciał przeciwheparynowych w surowicy - obniżona aktywność LPL
c) związana z gromadzeniem się chylomikronów we krwi - chylomikronemia
d) poziom VLDL pozostaje bez zmian z powodu hamującego wpływu chylomikronów na ich syntezę
e) głównym objawem jest triglicerydemia - stężenia rzędu 10 000 mg%; cholesterol w normie lub nieznacznie podwyższony
f) chylomikrony naciekają narządy miąższowe:
- wątrobę
- trzustkę - mogą wywołać jej zapalenie aktywując zawarte w niej proenzymy enzymów proteolitycznych; zapalenie trzustki jest główną przyczyną śmierci osób dotkniętych tą hiperlipoproteinemią
- śledzionę
g) osoby nieleczone nie dożywają 20 roku życia
h) leczenie polega na podawaniu wyłącznie krótko- i średniołańcuchowych kwasów tłuszczowych; w przypadku defektu apo C-II możemy pacjentowi podać to białko
i) w teście lodówkowym osocze jest przejrzyste z kożuchem
j) w obrazie elektroforetycznym pojawia się plama chylomikronów (obecna również wtedy, gdy pacjent nie jest na czczo)
k) stosunek cholesterol całkowity/triglicerydy (Tch/TG) mniejszy od 0,2
31. Hiperlipoproteinemia typu II:
a) jest związana z defektem receptora LDL
b) może być w postaci heterozygotycznej (1/500 osób w Polsce) - liczba receptorów zmniejszona o połowę lub homozygotycznej (1/1 mln osób w Polsce) - brak receptorów
c) postać receptoronegatywna i receptorodefektywna (łagodniejsza)
d) w osoczu podwyższony poziom LDL, które przebywając we krwi przez długi okres czasu podlegają różnorodnym modyfikacjom - wzrasta ich działanie aterogenne
e) wzrost endogennej syntezy cholesterolu
f) w postaci 2a:
- hipercholesterolemia rodzinna (hipercholesterolemia pierwotna, dziedziczna)
- surowica klarowna, bez kożucha
- podwyższony poziom beta lipoprotein
- podwyższony cholesterol (całkowity i LDL), triglicerydy prawidłowe
- stosunek Tch do TG wyższy niż 1,5
g) w postaci 2b:
- surowica opalizoująca
- podwyższony poziom beta i pre-beta lipoprotein
- wzrost stężenia cholesterolu całkowitego i LDL oraz triglicerydów
- stosunek Tch do TG = 1 - 1,5
h) objawy hiperlipoproteinemii typu II:
- kępki żółte
- rąbek starczy
- żółtaki na wyprostnych częściach kończyn i w dole podkolanowym
32. Hiperlipoproteinemia typu III:
a) hiperlipoproteinemia mieszana, choroba szerokiego prążka, hiperlipoproteinemia remnantowa, dysbetalipoproteinemia
b) spowodowana nieprawidłowym fenotypem apoE - nieprawidłowy allel apoE2 obecny w układzie hetero- lub homozygotycznym zamiast apoE3:
- polimorfizm apoE ma podłoże genetyczne, dodatkowo jest związany z posttranslacyjną glikozylacją białka
- najczęściej występująca u człowieka forma to: apoE3 - najczęstszy fenotyp to apoE3/3
- w dysbetalipoproteinemii rośnie stosunek apoE2/apoE3
c) powinowactwo apoE do receptora apo B/E jest obniżone, co upośledza wyłapywanie remnantów VLDL
d) w osoczu rośnie poziom remnantów VLDL (IDL), obniża się poziom LDL
e) pod względem aterogenności remnanty VLDL porównywalne do LDL; ulegają one modyfikacjom w naczyniach, po czym naciekają ich ściany rozpoczynając proces miażdżycowy
f) odkładanie w tkankach obwodowych powoduje powstanie guzów
g) w elektroforezie brak oddzielnych prążków beta i pre-beta - występuje jeden szeroki prążek remnantów pre-beta lipoprotein - lipoprotein flotujących
h) w teście zimnej flotacji - osocze jest mętne z kożuchem
i) fenotyp apoE możemy zbadać metodą ogniskowania izoelektrycznego w gradiencie pH - umożliwia pewne rozpoznanie
33. Hiperlipoproteinemia typu IV:
a) triglicerydemia endogenna
b) podwyższone stężenie TG, prawidłowe lub nieznacznie podwyższone stężenie cholesterolu całkowitego, cholesterol HDL często obniżony; frakcja beta-lipoprotein podwyższona
c) przyczyna postaci pierwotnej nieznana, jako hiperlipoproteinemia wtórna występuje w cukrzycy i chorobach wątroby
d) wzrost syntezy i podwyższony poziom VLDL w osoczu wiąże się z:
- naciekaniem narządów miąższowych (wątroby, śledziony, trzustki)
- naciekaniem ściany naczyniowej - podwyższone ryzyko miażdżycy
- w teście lodówkowym surowica mleczna - silnie lipemiczna
- prążek pre-beta lipoprotein mocno wysycony w elektroforezie
e) stosunek Tch do TG mniejszy niż 0,9
34. Hiperlipoproteinemia typu V:
a) związana z obecnością 2 defektów:
- zwiększonej syntezy VLDL
- defektu lipazy lipoproteinowej
b) wzrost poziomu VLDL i chylomikronów - w teście zimnej flotacji osocze mleczne (mętne) z kożuchem
c) obniżona aktywność lipazy lipoproteinowej może być związana:
- z defektem apoC-II
- obecnością nieprawidłowej apoC-III będącej inhibitorem LPL
- insulinoopornością
d) patologiczny wpływ na organizm:
- naciekanie wątroby i śledziony
- naciekanie trzustki, które może wywołać jej zapalenie
- ryzyko wystąpienia miażdżycy względnie niewielkie
e) stosunek Tch do TG = 0,2 - 0,5
36. Diagnostyka lipidowa step-by-step:
1. Czy jest hiperlipidemia?
- badamy profil lipidowy wg zasad opisanych w skrypcie
2. Jaka jest przyczyna hiperlipidemii? Czy jest to hiperlipidemia pierwotna czy wtórna?
a) hiperlipoproteinemie pierwotne wykluczamy per eliminationem przeprowadzając badania mające na celu stwierdzenie, czy zaburzenia gospodarki lipidowej nie są wynikiem innych chorób:
- TSH (co 10 hiperlipidemia u kobiet jest spowodowana niedoczynnością tarczycy)
- cukrzyca
- enzymy wątrobowe
- badanie moczu
- alfa-amylaza
3. Jaki jest typ hiperlipoproteinemii?
4. Czy są nielipidowe czynniki ryzyka?
5. Jakie jest globalne ryzyko?
6. Jaki jest typ prewencji? Czy wtórna czy pierwotna?
40. Wpływ kwasów tłuszczowych na gospodarkę lipidową:
a) PUFA Omega-6 (n-6)
linolowy 18:2
oleje: słonecznikowy, sojowy,
kukurydziany, krokoszowy
arachidonowy 20:4
- spadek poziomu triglicerydów (VLDL)
- spadek HDL
- spadek LDL
b) PUFA omega 3 (patrz niżej)
Kwasy mononienasycone MUFA głównym przedstawicielem jest kwas oleinowy, dla osób które mają szczuplejszy portfel polecany jest olej rzepakowy , który nadaje się zarówno do spożycia na ciepło jak i na zimno, a dla osób z zasobnym portfelem - oliwa z oliwek.
c) kwasy mononienasycone (MUFA) cis - margaryny miękkie:
- spadek LDL
- HDL bez zmian
d) kwasy mononienasycone (MUFA) trans margaryny twarde - powstają w wyniku obróbki przemysłowej olejów, z których te kwasy są pozyskiwane, z którego jest wytwarzany tłuszcz, który następnie konsumujemy. I chodzi o to, że ten tłuszcz jest lepszy, który zawiera mniej izomeru trans, bowiem te izomery trans kwasów nienasyconych działają jak kwasy nasycone, czyli:
- wzrost LDL
- obniżenie HDL
e) kwasy nasycone Kwasy nasycone - mają działanie niekorzystne, najbardziej niekorzystne działanie ma kwas laurynowy i kwas mirystynowy, oraz palmitynowy. Zarówno te najkrótsze kaprynowy i kaprylowy, oraz długi stearynowy nie wykazują szkodliwego działania na układ lipidowy osocza. One głównie występują w produktach pochodzenia zwierzęcego, czyli zarówno w tłustym mięsie, kiełbasie zwyczajnej, bekonach, ale również występuje w olejach roślinnych palmowym i kokosowym. Na szczęście w naszej strefie geograficznej te rośliny nie rosną w celach spożywczych.
Z powyższych przyczyn, triglicerydy zaspokajające 30% potrzeb energetycznych organizmu powinny zawierać jak najwięcej kwasów MUFA cis; w tych 30%:
- 10-15% powinny stanowić tg zawierające kwasy MUFA (oczywiście cis)
- PUFA omega 6 - 6%
- PUFA omega 3 - 2% (PUFA łącznie 8%)
- kwasy nasycone - 6-7%
41. PUFA omega3 - działanie metaboliczne:
a) należą tu wielonienasycone kwasy tłuszczowe szeregu omega3:
- kwas alfa-linolenowy oleje: lniany, sojowy, rzepakowy
- kwas ikozapentaenowy - timnodonowy - oleje rybie np. tran dorszowy, oleje z makreli, ryby śledziowate i łososi
- kwas dokozapentaenowy - klupanodonowy - oleje rybie, fosfolipidy
- kwas dokozaheksaenowy - cerwonowy olej rybi, fosfolipidy
Należy tu także zmielona chrząstka rekina
b) obecne w dużych ilościach w olejach rybich
c) działanie na ścianę naczyniową:
- redukcja hiperplazji błony wewnętrznej przez hamowanie wydzielania PDGF
- hamowanie restenozy po zabiegu angioplastyki
d) wpływ na hemostazę:
- wzrost aktywności tPA
- wzrost aktywności antytrombiny III
- spadek stężenia fibrynogenu
- spadek poziomu PAI
- obniżenie lepkości krwi
e) wpływ na gospodarkę lipidową:
- spadek poziomu kwasów tłuszczowych
- spadek syntezy VLDL
- spadek syntezy apo B
- spadek syntezy kwasów tłuszczowych
- spadek LDL przy normalnych stężeniach; przy podwyższonych stężeniach (hiperlipoproteinemia) następuje wzrost frakcji LDL
- wzrost frakcji HDL
- nasilenie utleniania kwasów tłuszczowych
f) działanie antyarytmiczne
43. Pozaenergetyczne funkcje kwasów tłuszczowych:
a) utrzymanie struktury i funkcji błon komórkowych
b) hamowanie pompy sodowo-potasowej
c) hamowanie translokazy nukleotydów adeninowych
d) regulacja receptorów hormonów (T4, angiotensyny II, glikokortykoidów, erytropoetyny)
e) regulacja funkcji kanałów błonowych K, Ca, Cl
f) modulacja ekspresji genów FABP, syntetazy acylo-S-CoA, desaturazy steroilo-CoA
42. FABP
a) Fatty acid binding proteins - białka wiążące kwasy tłuszczowe
b) niskocząsteczkowe białka cytozolowe odpowiedzialne za wewnątrzkomórkowy transport kwasów tłuszczowych
c) ekspresja genów modulowana przez kwasy tłuszczowe
d) co najmniej 8 typów:
- sercowy - H (heart)
- mózgowy - B (brain)
- mielinowy - M (myelin)
- naskórkowy - E (epidermal)
- jelitowy - I (intestinal)
- wątrobowy - L (liver)
- adipocytarny - A (adipocyte)
- I-LBP - ileal lipid-binding protein - białko wiążące lipidy charakterystyczne dla jelita krętego
e) białko I-LBP wykazuje duże powinowactwo do skoniugowanych kwasów żółciowych (jelito kręte jest miejscem resorpcji kwasów żółciowych)
f) wykazują większe powinowactwo do kwasu oleinowego i arachidonowego niż palmitynowego
g) FABP preferują kwasy nienasycone (wyjątkiem jest FABP typu jelitowego)
h) powinowactwo zwiększa się wraz ze wzrostem hydrofobowego charakteru FA
i) masa: 14-15 kDa
j) decydują o prędkości pobierania FFA z osocza przez komórkę
k) transportują proliferaty peroksysomów z cytosolu do jądra, gdzie wchodzą w interakcję z PPAR
l) 1 cząsteczka białka wiąże 2 cząsteczki kwasu
44. Beta-oksydacja - przebieg, energetyka, znaczenie w metabolizmie komórki.
wewnątrz mitochondrów (matrix mitochondrialna), rozrywane są wiązania beta stąd nazwa - beta oksydacja,
dwustopniowe utlenianie, długołańcuchowych FA z wytworzeniem ATP,
odłączenie dwuwęglowego fragmentu - acetylo CoA
utlenienie w cyklu Krebsa i łańcuchu oddechowym,
z 16 węglowego kwasu tłuszczowego - 130 cz. ATP
Utlenienie długołańcuchowych FA z wytworzeniem ATP
Matrix mitochondrialna
W cytozolu AKTYWACJA kwasu tłuszczowego
Utworzenie wiązania tioestrowego z CoA
Transport przez wewnętrzną błonę mitochondrialną przy udziale ARNITYNY
Krótkie i średniołańcuchowe (do 10C) przenikają
ၢ-oksydacja - przebieg
POWTARZAJĄCE SIĘ SEKWENCJE 4 REAKCJI
Utlenienie acylo-CoA przez FAD do trans-delta2-enolilo-CoA
Dehydrogenaza acylo-CoA
Uwodnienie do 3-hydroksyacetylo-CoA
Hydrataza enoilo-CoA
Utlenienie przez NAD+ do 3-ketoacylo-CoA
Dehydrogenaza hydroksyacylo-CoA
Tioliza przez HS-CoA i powstanie acetylo-CoA i acylo-CoA skróconego o 2 węgle
beta -ketoliaza
ၢ-oksydacja - znaczenie w metaboliźmie komórki
powstanie wielu cząsteczek ATP
wytworzenie acetylo-CoA (8 moli dostarcza 12 moli ATP)
omega-oksydacja:
a) zachodzi w siateczce śródplazmatycznej
b) substratami są wolne kwasy tłuszczowe
c) polega na utlenieniu grupy CH3 (węgla omega) danego kwasu do grupy COOH
d) reakcja katalizowa przez hydroksylazy współdziałające z cytochromem P-450, NADPH + H+, i O2
e) czynnik ułatwiający - lecytyna
f) prowadzi do powstania kwasów dikarboksylowych, które trafiają do mitochondrium, gdzie są poddawane beta-oksydacji lub są wydalane z moczem
alfa-oksydacja:
a) zachodzi w wątrobie i tkance nerwowej
b) dotyczy długołańcuchowych, rozgałęzionych kwasów tłuszczowych
c) polega na:
- hydroksylacji węgla C-alfa przy udziale hydroksylazy współdziałającej z:
1. cytochromem P-450
2. NADPH + H+
3. Fe2+
4. kwasem askorbinowym
- dekarboksylacji alfa-hydroksykwasu z równoczesnym utlenieniem
45. Karnityna:
a) zmodyfikowany aminokwas
b) przenośnik reszt długołańcuchowych kwasów tłuszczowych przez wewnętrzną błonę mitochondrialną
c) synteza:
- lizyna jest modyfikowana przez przyłączenie 3 grup metylowych z S-adenozylometioniny
- grupy metylowe wiążą się z grupą ε aminową lizyny - powstaje ε-N-trimetylolizyna
- węgiel β ε-N-trimetylolizyny jest hydroksylowany przez dioksygenazę w obecności Fe2+ i O2
- β -hydroksy-ε-N-trimetylolizyna rozpada się na glicynę i na aldehyd γ-N-trimetyloaminomasłowy
- jego grupa aldehydowa jest utleniana do grupy karboksylowej przy udziale dehydrogenazy zależnej od NAD+ a węgiel β jest hydroksylowany przy udziale dioksygenazy w obecności Fe2+ i O2
- powstaje kwas β-hydroksy- γ-N-trimetyloaminomasłowy - karnityna
d) mechanizm przenoszenia reszt długołańcuchowych kwasów tłuszczowych z cytosolu do matrix mitochondrium nosi nazwę czółenka karnitynowego:
- acylotransferaza karnitynowa I katalizuje przeniesienie reszty acylowej na karnitynę - enzym związany z zewnętrzną powierzchnią wewnętrznej błony mitochondrialnej
- powstaje wysokooenergetyczne wiązanie między resztą acylową a karnityną
- acylokarnityna jest przenoszona do matrix przez translokazę
- acylotransferaza karnitynowa II związana z wewnętrzną powierzchnią wewnętrznej błony mitochondrialnej przenosi resztę acylową na HS-CoA
- malonylo-S-CoA (stężenie wysokie przy nasilonej syntezie FA) jest inhibitorem acylotransferazy karnitynowej I
e) brak acylotransferazy karnitynowej II lub niedobór karnityny powoduje niezdolność mięśni do wykorzystywania długołańcuchowych kwasów tłuszczowych jako paliwa; powoduje to:
- osłabienie i bóle mięśni
- przenikanie mioglobiny z uszkodzonych komórek mięśniowych do krwi
f) brak palmitoilotransferazy (acylotransferazy) karnitynowej I dotyczy wątroby - zmniejszenie beta-oksydacji i ketogenezy oraz hipoglikemia
g) niedobór karnityny:
- występuje zwłaszcza u noworodków (szczególnie u wcześniaków)
- niewystarczająca synteza karnityny lub jej utrata przez nerki
- niedobór również u chorych z acydurią spowodowaną kwasami organicznymi oraz po hemodializie
- występują napadowe okresy hipoglikemii wywołane upośledzeniem glukoneogenezy
- zmniejszenie beta-oksydacji i ketogenezy
- podwyższone stężenie FFA w osoczu, gromadzenie lipidów i osłabienie mięśni
h) aktywny biologicznie izomer L
i) źródła:
- z pokarmem
- w wyniku resorpcji w kanalikach nerkowych
- endogenna synteza
j) w jelicie przekształcana przez bakterie w trimetyloaminę lub gamma-butyrobetainę
46. Tłuszcze spalają się w ogniu węglowodanów:
Pojęcie dotyczy zależności między metabolizmem lipidów i węglowodanych w wątrobie.
a) wolne kwasy tłuszczowe uwalniane są z adipocytów w wyniku lipolizy
b) spalanie wolnych kwasów tłuszczowych prowadzi do wytworzenia acetylo-S-CoA
c) acetylo-S-CoA może być zużywany w cyklu Krebsa, ale tylko jeżeli dostępny jest szczawiooctan
d) szczawiooctan syntezowany jest z pirogronianu pochodząceg z glikolizy - powiązanie metabolizmu węglowodanów i lipidów
e) w okresie głodu:
- szczawiooctan zostaje zużyty do produkcji glukozy w procesie glukoneogenezy
- brakuje szczawiooctanu dla kondensacji z acetylo-S-CoA
- acetylo-S-CoA w wątrobie zużywany jest w procesie ketogenezy - tworzy się beta-hydroksymaślan, acetooctan i aceton (w wyniku nieenzymatycznej dekarboksylacji acetooctanu)
f) innymi słowy, gdy dostępność glukozy jest niewielka produkty beta-oksydacji w wątrobie nie mogą być efektywnie wykorzystywane do produkcji energii
g) zamiast do cyklu Krebsa acetylo-S-CoA z beta-oksydacji jest wykorzystywany do ketogenezy
47. Ciała ketonowe:
a) zaliczamy do nich:
- beta-hydroksymaślan
- acetooctan
- aceton
Stosunek beta-hydroksymaślan/acetooctan waha się we krwi w granicach: 1:1 - 10:1; beta-hydroksymaślan jest najobficiej występującym we krwi ciałem ketonowym
b) w warunkach prawidłowych stężenie tych substancji nie przekracza 10 mg/l (0,25 mmol/l)
c) pełnią funkcję paliwa energetycznego uwalnianego z wątroby, spalanego w tkankach obwodowych
d) powstają w procesie ketogenezy zachodzącym w mitochondriach hepatocytów
E) ketogeneza:
- jest charakterystyczna dla okresów międzytrawiennych
- jej szybkość zależy od napływu wolnych kwasów tłuszczowych
- pozwala odtwarzać mitochondrialną pulę wolnego CoA
- zapobiega powstawaniu nadmiernej ilości ATP
f) synteza ciał ketonowych:
- enzym tiolaza: 2 cząsteczki acetylo-S-CoA kondensują tworząc acetoacetylo-S-CoA;
- enzym syntaza β-hydroksy-β-metyloglutarylo-S-CoA: przyłączenie jeszcze jednej cząsteczki acetylo-S-CoA do acetoacetylo-S-CoA = powstaje β-hydroksy-β-metyloglutarylo-S-CoA
- enzym liaza β-hydroksy-β-metyloglutarylo-S-CoA: rozkład β-hydroksy-β-metyloglutarylo-S-CoA na acetooctan i acetylo-S-CoA
- acetooctan ulega spontanicznej dekarboksylacji w wyniku której powstaje aceton
- dehydrogenaza β-hydroksymaślanowa katalizuje odwracalną przemianą acetooctanu do β-hydroksymaślanu
G) wykorzystanie ciał ketonowych:
- w wątrobie nie mogą być spożytkowane jako surowiec energetyczny; acetooctan może być wykorzystany jako prekursor w syntezie cholesterolu
- w głównym szlaku utylizacji acetooctanu: aktywowany przez enzym CoA-transferazę β-ketokwasową - powstaje acetoacetylo-S-CoA, który przez tiolazę rozkładany jest do 2 cząsteczek acetylo-S-CoA
- β-hydroksymaślan jest utleniany przez dehydrogenazę β-hydroksymaślanową do acetooctanu; ten jest rozkładany przez tiokinazę do 2 cząsteczek acetylo-S-CoA
- szczególnie mięsień sercowy i kora nerki wykorzystują acetooctan jako paliwo
- mózg dzięki adaptacji może zaspokoić nawet 75% swoich potrzeb energetycznych ciałami ketonowymi
h) wzrost stężenia ciał ketonowych powyżej 12 mmol/l nie powoduje dalszego wzrostu intensywności procesu spalania ciał ketonowych - wysycenie enzymów
I) działanie patologiczne ciał ketonowych:
- acetooctan i beta-hydroksymaślan to dość silne kwasy; gromadząc się w osoczu wywołują kwasicę metaboliczną
- substancje te są wydalane z organizmu z moczem, gdzie wiążą sód i potas; przez to zubożają organizm w Na+ i K+
- skutkiem wpływu kwasicy metabolicznej wywołanej ciałami ketonowymi na OUN jest śpiączka cukrzycowa (ketonowa)
Regulacja ketogenezy:
a) dopływ wolnych kwasów tłuszczowych:
- hormony pobudzające lipolizę (m.in. adrenalina, glukagon) powodują wzrost stężenia kwasów tłuszczowych w osoczu
- zwiększone stężenie kwasów tłuszczowych prowadzi do większego natężenia procesu ketogenezy
- glikokortykoidy działają permisywnie w stosunku do lipolizy, nasilają glukoneogenezę, co zmniejsza zawartość szczawiooctanu w wątrobie, co z kolei hamuje spalanie kwasów tłuszczowych w procesie beta-oksydacji i zwiększa ketogenezę
b) palmitoilotransferaza karnitynowa I:
- hamowana przez malonylo-CoA powstający w dużych ilościach w stanie sytości w procesie karboksylacji acetylo-S-CoA
- w okresie sytości przeważa estryfikacja kwasów tłuszczowych i ich wbudowywanie do VLDL
- w okresie głodzenia wzrasta stężenie FFA - acylo-CoA hamuje w wątrobie aktywność karboksylazy acetylo-S-CoA
- stężenie malonylo-CoA spada, więcej kwasów tłuszczowych trafia do mitochondrium i ulega beta-oksydacji
- gdy stężenie FFA jest duże więcej acetylo-S-CoA wytworzonego w beta-oksydacji wykorzystywane jest w procesie ketogenezy niż w cyklu Krebsa; odbywa się to przez zmniejszenie stężenia szczawiooctanu wskutek zwiększenia stosunku NADH/NAD+ i zaburzenia równowagi szczawiooctan-jabłczan
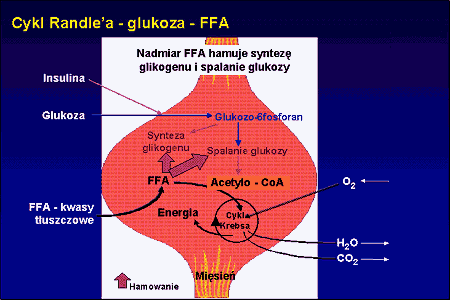
48.
Zwiększone uwalnianie FFA z tkanki tłuszczowej wisceralnej → konkurencja FFA z glukozą o możliwość przemian w cyklu Randle'a - zahamowanie przemian glukozy. Zwiększona sekrecja FFA prowadzi ponadto do: syntezy TG, sekrecji VLDL, glukoneogenezy
cykl Randle'a, zachodzi w nim synteza trójglicerydów z glukozy i glicerolu pod wpływem insuliny
50. Wpływ hormonów na gospodarkę lipidową:
a) lipolizę hamują:
- insulina
- kwas nikotynowy
- PGE1
HORMONY POBUDZAJĄCE LIPOLIZĘ:
b) lipolizę pobudzają:
- adrenalina i noradrenalina
- glukagon
- ACTH
- MSH
- GH
- TSH
- wazopresyna
- hormony tarczycy
c) metyloksantyny pobudzają lipolizę przez zahamowanie fosfodiesterazy, co skutkuje wzrostem cAMP
d) glikokortykosterydy pobudzają lipolizę przez wzrost syntezy HSL (niezależnie od cAMP)
Regulacja wydzielania glukagonu:
a) wydzielanie pobudzane przez:
- niskie stężenia glukozy
- argininę, lizynę i treoninę (działają przy niskich stężeniach glukozy)
- glikokortykosterydy i hormony przewodu pokarmowego
- adrenalinę poprzez receptory β
b) wydzielanie hamowane przez:
- podwyższone stężenia glukozy (powyżej wartości na czczo)
- kwasy tłuszczowe i ciała ketonowe
- insulinę i somatomedynę - parakrynnie
- katecholaminy poprzez receptory α
Wpływ hormonów na gospodarkę lipidową:
A. INSULINA
a) hamowanie lipolizy:
- hamowanie hormonozależnej lipazy adipocytów - wiąże się ze spadkiem cAMP
- obniża stężenie FFA i glicerolu w osoczu
b) nasilenie lipogenezy i biosyntezy acyloglicerolu
- wzrost aktywności dehydrogenazy pirogronianowej, acylotransferazy glicerolo-3-fosforanowej, karboksylazy acetylo-CoA
c) mechanizmy prowadzące do zahamowania lipolizy:
- spadek poziomu cAMP w wyniku aktywacji fosfodiesterazy
- hamowanie cyklazy adenylanowej poprzez białko Gi
- pobudzanie fosfatazy lipazy, która inaktywuje HSL
d) mechanizmy pobudzania lipogenezy:
- aktywacja lipazy lipoproteinowej śródbłonka włośniczek
- zwiększenie napływu glukozy do komórek - jest ona źródłem glicerolu i NADPH + H+ potrzebnych w lipogenezie
- stymulacja syntezy triglicerydów z glukozy
- hamowanie lipolizy
B. GLUKAGON:
a) główny hormon zwiększający uwalnianie FFA z tkanki tłuszczowej w okresach międzytrawiennych - aktywator lipolizy
b) przyspiesza beta-oksydację w wątrobie wpływając na prędkość wyłapywania przez ten narząd FFA z osocza
c) nasila proces ketogenezy:
- ogranicza szybkość syntezy cytrynianu w mitochondriach
- zmniejsza intensywność przemian cyklu Krebsa
- hamuje karboksylazę acetylo-S-CoA - hamuje syntezę kwasów tłuszczowych
- hamuje syntezę cholesterolu poprzez hamowanie fosfatazy defosforylującej reduktazę reduktazę HMG-CoA, aktywuje kinazę HMG-CoA
- wpływa na podwyższenie stężenia acetylo-S-CoA, który jest wykorzystywany w procesie ketogenezy
d) posiada receptory na hepatocytach i adipocytach:
- GR1 - aktywowany przy niskim stężeniu hormonu, oddziałuje poprzez kaskadę fosfatydyloinozytolową
- GR2 - aktywowany przy wysokich stężeniach hormonu, oddziałuje poprzez zwiększenie stężenia cAMP w komórce wskutek aktywacji cyklazy adenylanowej
C. ACTH:
a) wywiera efekt poprzez aktywację cyklazy adenylanowej
b) stymuluje syntezę i transport receptora apoB/E ułatwiając pobieranie LDL z osocza przez korę nadnercza
c) aktywuje esterazę cholesterolową
d) pobudza syntezę kortyzolu
D. KORTYZOL:
a) wpływ zaznacza się głównie w okresach międzytrawiennych
b) pobudza lipolizę w tkance tłuszczowej przez:
- nasilenie syntezy HSL
- wzrost syntezy białka potrzebnego do aktywacji HSL
- pełni funkcję przyzwalającą dla glukagonu, katecholamin i GH uwrażliwając adipocyty na lipolityczny wpływ tych hormonów
c) w wątrobie:
- przyzwalające działanie dla glukagonu
d) regulacja stosunku insulina-glukagon: hamowanie uwalniania insuliny, wzrost uwalniania glukagonu
e) hamowanie syntezy receptora insuliny
f) w rdzeniu nadnerczy pobudza syntezę katecholamin
E. KATECHOLAMINY:
a) aktywują lipolizę przez receptory β1
b) utrudniają wiązanie insuliny z receptorem w błonie adipocytów
49. Lipazy wewnątrzkomórkowe - podział, mechanizm działania, regulacja aktywności
Lipazy to hydrolazy
1. Wewnątrzkomórkowe
2. Pozakomórkowe
wewnątrznaczyniowe
pozanaczyniowe
Lipazy przewodu pokarmowego
1. ślinianki podjęzykowej
2. żołądkowa
3. trzustkowa
Lipaza ślinianki podjęzykowej
5 - 7 wyższe powinowactwo do średniołańcuchowych
niż długołancuchowych kw. tł.
wyższe powinowactwo do glicerydów z nienasyconym kw. tł.
wyższe powinowactwo do wiązania estrowego w pozycji ၡ
nie hydrolizuje fosfolipidów i estrów cholesterolu
pH 2,2 - 6,0
nie jest inaktywowana w żołądku
inaktywowana pod wpływem trypsyny
Lipaza żołądkowa
aktywność lipazowa żołądka = lipaza ślinianki + żołądkowa
aktywacja wydzielania: pokarm, pentagastryna, histamina,
histalog, 2-dezoksyglukoza, pobudzenie nerwu błędnego
3 izoenzymy 50 - 300 kDa
wysokie powinowactwo do średnio- i długołańcuchowych
kw. tl.
nie hydrolizuje fosfolipidów i estrów cholesterolu
pH 2,2 - 7,4
duza aktywność w okresie noworodkowym
Lipaza trzustkowa
wydzielanie podstawowe i indukowane posiłkiem,
cholescystokininą i sekretyną
formy molekularne S (slow) i F (fast)
forma F = kolipaza + fosfolipidy + S
kolipaza - wydzielana w formie nieaktywnej
aktywowana przez trypsynę
glikoproteina 9 -13 kDa
umożliwia kontakt substratu i enzymu
optimum pH 6,0
szerokie spektrum aktywności lipolitycznej
wysokie powinowactwo do ၡ-glicerydów
nie hydrolizuje fosfolipidów i estrów cholesterolu
aktywacja: jony Ca i kwasy żółciowe
Lipazy mleka
lipaza lipoproteinowa (naczynia gruczołu sutkowego)
lipaza aktywowana kwasami żółciowymi
wydzielanie stymulowane prolaktyną
niska specyficzność substratowa TG, fosfolipidy,
estry cholesterolu)
funkcja: wstępne trawienie tłuszczy (niska aktywność lipazy
trzustkowej)
INHIBITOR LIPAZY - ORLISTAT
Lipoliza wewnątrzkomórkowa - schemat :
53. Biosynteza sfingozyny (ceramidu) i pochodne:
a) sfingozyna - nienasycony aminoalkohol o 18 atomach węgla
b) synteza (w siateczce śródplazmatycznej):
- palmitoilo-S-CoA reaguje z aktywowaną przez połączenie z fosforanem pirydoksalu seryną; reakcję katalizuje palmitoilotransferaza serynowa - odłącza się CoA-SH i CO2; powstaje 3-ketosfinganina
- 3-ketosfinganina jest redukowana z udziałem NADPH + H+ przez reduktazę 3-ketosfinganinową - powstaje dihydrosfingozyna
- n-acylotransferaza dihydrosfingozynowa przyłącza do grupy NH2 dihydrosfingozyny resztę acylową
- powstaje dihydroceramid
- desaturaza dihydroceramidowa odłącza dwa atomu węgla od dihydroceramidu - powstaje ceramid
c) ceramid - składa się ze sfingozyny połączonej wiązaniem amidowym przy udziale swojej grupy NH2 z resztą acylową
d) sfingomielina:
- ceramid w którym grupa OH przy węglu C1 sfingozyny ma przyłączoną cząsteczkę fosfocholiny, której dawcą jest fosfatydylocholina:
ceramid + fosfatydylocholina = sfingomielina + 1,2-diacyloglicerol
- sfingomieliny są ważnym lipidowym składnikiem tkanki nerwowej
- sfingomieliny w osłonkach mielinowych włókien zawierają długołańcuchowe kwasy tłuszczowe: lignocerynowy, nerwonowy; sfingomieliny istoty szarej - głównie kwas stearynowy
- synteza sfingomielin odbywa się w aparacie Golgiego
54. Glikosfingolipidy:
a) powstają z połączenia ceramidu z cząsteczkami cukrów
b) zawierają przede wszystkim kwasy tłuszczowe C24: lignocerynowy, nerwonowy, cerebronowy
CEREBROZYDY:
c) galaktozyloceramid:
- główny lipid mieliny
- powstaje w reakcji ceramidu z UDPGal
- może być sulfonowany przy udziale PAPS
d) glukozyloceramid:
- główny glikosfingolipid tkanek pozanerwowych
- prekursor bardziej złożonych glikosfingolipidów
GANGLIOZYDY:
e) syntezowane z ceramidu przez stopniowe, kolejne przyłączanie zaktywowanych cukrów i kwasu sialowego (najczęściej kwasu n-acetyloneuraminowego)
f) syntezowane w aparacie Golgiego
g) funkcje glikosfingolipidów:
- składniki zewnętrznej warstwy błony plazmatycznej
- uczestniczą w komunikacji między komórkami
- mają właściwości antygenowe (anytgen Forssmana, antygeny układu AB0)
- niektóre są receptorami toksyn bakteryjnych
56. Kwas arachidonowy - struktura, występowanie, źródła, tory metaboliczne, inhibitory enzymów przemian kwasu arachidonowego o istotnym znaczeniu klinicznym.
a) kwas 20 węglowy, 4 wiązania podwójne: cis-5,8,11,14-tetraenowy, szereg omega6
b) występuje w olejach roślinnych wraz z kwasem linolowym, duże ilości w oleju arachidowym, istotny składnik fosfolipidów zwierzęcych (5-15% kwasów tłuszczowych fosfolipidów)
c) jeden z niezbędnych wielonienasyconych kwasów tłuszczowych (składnik witaminy F)
d) funkcje:
- ważny składnik fosfolipidów błonowych
- substrat do syntezy eikozanoidów
- wtórny przekaźnik (LSM - lipid second messenger)
e) jako wtórny przekaźnik odgrywa rolę w OUN w utrwalaniu śladów pamięciowych:
- uwalniany przez fosfolipazę A2 po wyzwalającym napływ jonów wapnia do komórki pobudzeniu receptora NMDA
- kwas arachidonowy wiąże się z receptorami w szczelinie synaptycznej, do której jest uwalniany regulując wydzielanie kwasu glutaminowego przez zakończenie presynaptyczne na zasadzie sprzężenia zwrotnego
g) kwas arachidonowy w dużych ilościach uwięziony jest w pocycji 2 fosfolipidów błon biologicznych; mogą go stąd uwalniać:
- fosfolipaza A2 - enzym błonowy o konstytucyjnej i indukowanej aktywności
- w wyniku działania fosfolipazy C powstaje DAG, od którego lipaza odcina AA
- w wyniku działania fosfolipazy D powstaje kwas fosfatydowy, od którego lipaza również odcina AA
Substraty do syntezy prostanoidów:
- 8,11,14-ikozatrienoan - DHLA - dihomo-gamma-linolenian - powstaje z kwasu linolowego
- 5,8,11,14-ikozatetraenoan - arachidonian
- 5,8,11,14,17-ikozapentaenoan - powstaje z kwasu alfa-linolenowego albo spożywany z pokarmem
57. COX:
a) 3 izoenzymy:
- COX1
- COX2
- COX3
b) cyklooksygenaza - inaczej syntaza prostaglandynowa:
- przekształca kwas arachidonowy w prostaglandynę PGH2
c) aktywność:
- cyklooksygenazowa - przekształca kwas arachidonowy w PGG2
- peroksydazowa - wykorzystuje hem jako grupę prostetyczną, redukuje PGG2 do PGH2
d) COX1:
- ekspresja konstytutywna
- aktywny przede wszystkim w układzie pokarmowym, nerkach, płytkach krwi, ścianie naczyniowej
- bierze udział w wytwarzaniu prostanoidów o znaczeniu ochronnym (regulacyjnym)
- ekspresja nie jest hamowana przez glikokortykoidy
e) COX2:
- ekspresja indukowalna w odpowiedzi na cytokiny prozapalne
- pełni istotną rolę w procesach zapalnych
- indukcja hamowana przez glikokortykoidy
- aktywność enzymu hamowana selektywnie przez rofekoksyb i celekoksyb
f) COX3:
- jego transkrypt powstaje w wyniku alternatywnego splicingu mRNA COX1
- obecna w mózgu - rola w percepcji bólu
- odwracalnie hamowana przez acetaminofen (paracetamol)
58. Fosfolipaza A2:
a) hamowana przez:
- glikokortykoidy - indukują lipokortynę, białkowy inhibitor
b) aktywność pobudzają:
- angiotensyna II
- bradykinina
- adrenalina
- trombina
Prostacyklina:
a) PGI2
b) syntezowana w komórkach śródbłonka
c) działanie:
- wazorelaksacja
- antyadhezja
- antyagregacja
d) działa poprzez zwiększenie stężenia cAMP w komórce (mięśniowej), synergistycznie do NO
e) synteza katalizowana przez syntazę prostacyklinową:
- rozerwanie układu nadtlenkowego i utworzenie pierścienia furanowego
f) czas półtrwania - 3 minuty, potem przekształcana do 6-keto-PGF1, która jest niaktywna biologicznie
g) struktura:
- nasycony pierścień cyklopentanowy
- dodatkowy pierścień utworzony przez połączenie tlenu grupy karbonylowej przy C9 z węglem C6
- wiązanie podwójne C5-C6
h) działanie antagonistyczne do tromboksanu TXA2
Tromboksan:
a) syntezowany z prostaglandyny PGH2 w płytkach krwi
reakcja:
2PGH2 = TXA2 + dialdehyd malonowy + związek siedemnastowęglowy
b) enzym syntezujący: syntaza tromboksanowa
c) działanie przeciwstawne do prostacykliny i NO:
- wazokonstrykcja
- wzrost agregacji i adhezji płytek krwi
d) budowa nieprostaglandynowa
e) synteza hamowana przez imidazol i jego pochodne
f) okres półtrwania 32 sekundy, potem następuje przemiana w TXA2
g) stosowanie aspiryny w niewielkich dawkach (40-70 mg/dobę) hamuje cyklooksygenazę płytkową zmniejszając wytwarzanie tromboksanu, co wywiera pozytywny efekt na ścianę naczyniową i drożność naczyń
h) kwas 20:5, omega3 (ikozapentaenowy) jest wyjściowym związkiem do syntezy TX3 i PG3, które hamują uwalnianie kwasu arachidonowego i jego wykorzystanie do syntezy PG2 oraz TX2; PGI3 ma taką samą aktywność jak PGI2, TX3 jest natomiast słabszy pod względem działania od TX2; dlatego u osób spożywających duże ilości olejów rybnych (Eskimosi) przeważa przeciwagregacyjne działanie prostanoidów
i) struktura: pierścień sześcioczłonowy, w jego skład wchodzi atom tlenu; łańcuch omega ma podwójne wiązanie C13-C14 i grupę OH w pozycji C15
j) synteza: płuca i płytki krwi
Eikozanoidy:
a) powstają z kwasów C20 ikozanowych pochodzących od kwasu linolowego, alfa-linolenowego lub bezpośrednio od arachidonowego
b) kwas linolowy:
- traci dwa atomy wodoru przechodząc w gamma-linolenian
- zyskuje 2 atomy węgla przechodząc w 8,11,14-ikozatrienoan (dihomo-gamma-linolenian - DHLA) - wątroba
- prostanoidy które powstają z 8,11,14-ikozatrienoanu (dihomo-gamma-linolenianu): PGE1, PGF1, TXA1
- leukotrieny które powstją z 8,11,14-ikozatrienoanu: LTA3, LTC3, LTD3
- ikozatrienoan może zostać odwodorowany przechodząc w 5,8,11,14-ikozatetraenoan (arachidonian): przemiana DHLA w kwas arachidonowy
c) kwas alfa-linolenowy:
- odwodorowanie prowadzi do powstania oktadekatetraenoanu
- oktadekatetraenoan przyłącza 2C przechodząc w ikozatetraenoan, który tracą 2 atomy wodoru przechodzi w:
- 5,8,11,14,17-ikozapentaenoan (który znajduje się też w pokarmie)
- 5,8,11,14,17-ikozapenatenoan jest surowcem do syntezy:
prostanoidów: PGD3, PGE3, PGF3, PGI3, TXA3
leukotrienów: LTA5, LTB5, LTC5
Szlak syntezy prostaglandyn:
a) powstają przy udziale syntazy prostaglandyny H (PGHS) - dwa izoenzymy:
- PGHS-1, PGHS-2
- oba izoenzymy PGHS mają aktywność cyklooksygenazy jak i peroksydazy
b) aktywność cyklooksygenazowa PGHS przekształca arachidonian w PGG2, które przez aktywność peroksydazy przekształcany jest do prostaglandyny PGH2
c) prostaglandyna PGH2 (endoperoksyd) jest wyjściowym substratem do produkcji innych prostaglandyn oraz tromboksanów
d) izomeryzacja PGH2 prowadzi do powstania:
- PGD2
- PGE2
e) syntaza tromboksanowa (hamowana przez imidazol) przekształca PGH2 w TXA2
f) redukcja prostaglandyny PGH2 oraz PGE2 prowadzi do powstania:
- PGF2alfa
g) syntaza prostacyklinowa przekształca PGH2 w PGI2
h) PGHS hamowana przez:
- aspirynę - w mechanizmie acetylacji enzymu
- inne leki NSAID: indometacyna, ibuprofen hamują PGHS przez kompetycję z arachidonianami
i) transkrypcja PGHS-2 ale nie PGHS-1 jest całkowicie inhibowana przeciwzapalnym działaniem kortykosterydów!!
Prostaglandyny:
a) struktura:
- kwasy tłuszczowe zbudowane z 20 atomów węgla
- zawierają w swej strukturze pierścień cyklopentanowy
- masa: 332-356 Da
- pochodne kwasu prostanowego zawierającego cyklopentanowy pierścień i dwa łańcuchy boczne: α i ω
- łańcuch α zakończony grupą karboksylową od której rozpoczynamy liczenie atomów węgla
- łańcuch ω zakończony grupą metylową
- wszystkie naturalnie występujące prostaglandyny mają w łańcuchu ω podwójne wiązanie między C13 i C14 o konfiguracji trans i grupę hydroksylową w pozycji C15
b) różnice w budowie pierścienia cyklopentanowego między poszczególnymi klasami prostaglandyn:
- PGA - pierścień zawiera jedno wiązanie podwójne (C10-C11), grupa ketonowa przy C9
- PGE - pierścień nasycony, grupa ketonowa przy C9, hydroksylowa przy C11
- PGF - 2 grupy hydroksylowe: przy C9 i C11
c) syntezowane przez cyklooksygenazę z kwasu arachidonowego, który powstaje w głównej mierze z kwasu linolowego w wyniku jego elongacji i desaturacji (1mg z 10 g kwasu linolowego spożywanego dziennie, zamienia się w kwas arachidonowy)
d) synteza - szczegółowo:
- cyklooksygenaza: cyklizacja substratu między C8 a C12, wbudowanie czterech atomów tlenu do cząsteczki
- powstaje prostaglandyna PGG2 - endoperoksyd, zawiera 9,11-endonadtlenek i 15-hydronadtlenek
- redukcja grupy hydronadtlenkowej przy C15 przez 2 cząsteczki GSH w reakcji katalizowanej przez (hydro)peroksydazę prostaglandynową (syntazę PGH2) - powstaje PGH2
- cyklooksygenaza i hydroperoksydaza prostaglandynowa są składowymi kompleksu syntazy prostaglandyny H
f) synteza różnych klas prostaglandyn jest zróżnicowana narządowo:
- nerka i śledziona: PGE2 i PGF2α
- ściana naczyniowa: głównie PGI2
- serce: jednakowe ilości PGE2, PGF2α i PGI2
Prostaglandyny - działanie:
a) żołądek
- obecne w śluzie - działanie cytoprotekcyjne
- zwiększone wydzielanie śluzu
- zwiększony przepływ krwi w błonie śluzowej
b) trzustka - wpływ na wydzielanie
c) wpływ na napięcie ścian naczyń
d) wpływ na płytki krwi
e) wpływ na równowagę elektrolitową
f) regulacja cyklu menstruacyjnego
g) neurotransmisja
Wpływ na szlak wytwarzania aktywnych pochodnych kwasu AA mają:
a) glikokortykoidy - hamują fosfolipazę A2 i indukcję COX2
b) NSAID - niesterydowe leki przeciwzapalne - ibuprofen, aspiryna, indometacyna - hamowanie COX1 i COX2
c) pochodne imidazolu - hamowanie syntazy tromboksanowej
d) kwas hydroperoksyeikozatetraenowy i inne nadtlenkowe pochodne kwasów tłuszczowych hamują syntazę prostacyklinową
Prostaglandyny - działanie:
a) działają poprzez zwiększnie poziomu cAMP (zwykle), obniżają go w kanalikach nerkowych i tkance tłuszczowej
b) naturalne mediatory procesu zapalnego
- zaczerwienienie i wzrost temperatury:
- rozszerzenie małych naczyń krwionośnych
- wzrost przepuszczalności naczyń włosowatych
c) działanie kurczące na mięśnie gładkie macicy - wykorzystywane przy stymulacji akcji porodowej
d) hamowanie wydzielania soku żołądkowego - zmniejszenie produkcji HCl - przyspieszają gojenie się wrzodów trawiennych
e) PGE2 pobudza agregację płytek krwi
f) zatrzymanie Na+ i wody
g) hamowanie lipolizy
Prostaglandyny powodują:
a) podwyższenie stężenia cAMP w:
- płytkach krwi
- tarczycy
- ciałku żółtym
- kościach płodu
- części gruczołowej przysadki
- płucach
b) obniżenie stężenia cAMP w:
- komórkach kanalików nerkowych
- tkance tłuszczowej
59. Leukotrieny:
a) produkty przekształceń kwasu arachidonowego
b) w ich syntezie uczestniczą lipooksygenazy: dioksygenazy przekształcające kwas arachidonowy do różnych HPETE - kwasów hydroperoksyeikozatetraenowych; dzięki heterogenności grupy lipooksygenaz powstają różne HPETE (mają różne miejsca wiązania tlenu); specyficzną lipoksygenazą biorącą udział w syntezie leukotrienów jest 5-lipoksygenaza
c) HPETE redukowane są do hydroksypochodnych samoistnie lub pod wpływem peroksydaz - redukcja grup hydroksynadtlenkowych do grup hydroksylowych; kwasy HPETE przechodzą w kwasy HETE - hydroksyeikozatetraenowe
d) leukotrieny powstają z 5-HPETE w reakcji katalizowanej przez syntazę LTA4, która wprowadza wiązanie epoksydowe do C5 i C6
e) powstaje leukotrien LTA4, który może przekształcać się na dwa sposoby:
- rozpad epoksydu - przyłączenie cząsteczki wody, której wodór redukuje grupę epoksydową do grupy OH w pozycji 5, grupa OH przyłącza się w pozycji 12 - powstaje leukotrien LTB4
- przyłączenie glutationu zredukowanego poprzez C6, wodór z grupy -SH glutationu redukuje grupę nadtlenkową do grupy hydroksylowej, która pozostaje przy C5, siarka glutationu wiąże się z C6 - powstaje leukotrien LTC4
f) dalsze przemiany leukotrienu LTC4:
- odłączenie glutaminianu: przekształcenie LTC4 w LTD4
- odłączenie glicyny z pozostawieniem cysteiny - powstaje LTE4
g) działanie (dodatkowo z Bańka):
- pobudzanie degranulacji leukocytów i uwalnianie ich enzymów lizosomalnych
- HETE - wpływają na funkcję granulocytów poprzez wbudowywanie się do ich błon
h) synteza leukotrienów:
- powstają z kwasu arachidonowego pochodzącego z fosfolipidów błony okołojądrowej
- bodźce (np. alergenowe) powodują aktywację kinaz, która wyzwala kaskadę przemian katalizowaną przez fosfolipazę Cγ: powstaje DAG i IP3;
- te wtórne przekaźniki prowadzą do aktywacji fosfolipazy A2, która przemieszcza się w kierunku otoczki jądrowej, z której uwalnia kwas arachidonowy będący substratem dla 5-lipokygenaz
- 5-lipoksygenaza działa w kompleksie z białkiem FLAP (5-lipoxygenase activating protein) przy obecności jonów Ca i ATP; flap jest integralnym białkiem otoczki jądrowej
- 5-lipoksygenaza katalizuje przemianę kwasu arachidonowego do kwasu hydroperoksyeikozatetraenowego (HPETE), który przez peroksydazy lub samoistnie jest redukowany do HETE - kwasu hydroksyeikozatetraenowego
- LTC4 powstaje w mastocytach oraz granulocytach zasadochłonnych i kwasochłonnych z LTA4 przy udziale transferazy glutationowej
- poza komórką, GGTP przekształca LTC4 w LTD4
- dipeptydazy cysteinyloglicynowe przekształcają LTD4 w LTE4
- w granulocytach obojętnochłonnych i makrofagach płucnych hydrolaza epoksydowa przekształca LTA4 w LTB4
i) leukotrieny cysteinylowe: C, D, E
- receptory cys-LT4 - wysokie powinowactwo, cys-LT2 - niskie powinowactwo
j) leki przeciwleukotrienowe:
- zileuton, genleuton - przekształcanie HPETE w LTA4
- cinalukast, montelukast, pranlukast, verlukast, zafirlukast - blokada receptorów leukotrienowych (cys-LT)
Leukotrieny - funkcje:
a) skurcz mięśni gładkich oskrzeli - ich zwężenie:
- leukotrieny cysteinowe
b) skurcz naczyń krwionośnych
- LTD4, LTC4
c) zwiększone wydzielanie śluzu:
- leukotrieny cysteinowe
d) chemotaksja - naciek zapalny:
- LB4, LTD4, LTE4
e) wzrost przepuszczalności tkanek - obrzęk:
- leukotrieny cysteinowe
f) nadwrażliwość oskrzeli:
- LTD4
g) promowanie wzrostu komórek szpiku, fibroblastów, komórek nabłonka:
- LTC4, LTD4
h) stmulacja limfocytów do wytwarzania IFN-γ:
- LTD4
60. Lipoksyny:
a) sprzężone tetraeny powstające w leukocytach
b) synteza:
- powstają w wyniku działania więcej niż jednej lipoksygenazy - wprowadzają więcej atomów węgla do cząsteczki;
- 15-lipoksygenaza przekształca arachidonian w 15-HPETE, który w toku dalszych przemian przez 5-lipoksygenazę przekształcany jest do lipkoksyn
- leukotrieny pod wpływem 15-lipoksygenazy mogą być przekształcane do lipoksyn
szczegółowo
a) powstają przy udziale 12-lipoksygenazy i 15-lipoksygenazy
b) eikozanoidy tetraenowe, występują w postaci izomerów trans (all trans) oraz cis
c) syntezowane przez:
- megakariocyty
- płytki krwi
- granulocyty - wzrost wytwarzania lipoksyn towarzyszy spadkowi syntezy LTB4
- makrofagi
- komórki szpiku kostnego
- komórki mezangium nerkowego
- nabłonek przewodów oddechowych
c) synteza przebiega 2 torami:
- substratem kwas arachidonowy: synteza prowadzi przez 15-HPETE lub 5-HPETE (hydroperoksypochodne kwasu arachidonowego wytwarzane przez 15-LO i 5-LO), następnie 5,15-diHPETE, wreszcie kwas 5(6)-epoksytetraenowy przekształcany w LXA4 lub LXB4
- substratem substancja z grupy leukotrienów: LTA4, przekształcany do 15-OH-LTA4 lub kwasu 5(6)-epoksytetraenowego (np. płytki krwi syntezują LXA4 z LTA4 pochodzącego z PMN)
c) metabolizm:
- LXA4 i LXB4 hydroksylowane przy udziale monooksygenazy zależnej od cytochromu P-450 i NADPH
- izomery all-trans - redukcja jednego z podwójnych wiązań
d) mechanizm działania:
- związanie receptorem wpływa na metabolizm lipidów błonowych pobudzając bądź hamując uwalnianie wtórnych przekaźników lipidowych
- powoduje to zmiany wewnątrzkomórkowego stężenia jonów Ca2+
e) działanie LXA4 i LX4:
- rola regulacyjna w procesie zapalnym
- hamowanie chemotaktycznej odpowiedzi neutrofili indukowanej przez LTB4
- immunosupresja - hamowanie uwalniania LTB4 z neutrofili
- szpik kostny - działanie synergistyczne z GM-CSF - wzrost tworzenia kolonii pierwotnych komórek szpikowych; poza tym modulacja mielopoezy
- rozszerzanie naczyń krwionośnych
- wpływ na zależne od komórek śródbłonka rozluźnienie mięśniówki naczyń krwionośnych - wazodilatacja
- kurczące działanie na mięśniówkę przewodów oddechowych - bronchokonstrykcja
- modulacja wydzielania insuliny przez trzustkę: 12-HPETE, 12-HETE oraz LXB4 hamują wydzielanie insuliny
LXA4 - indywidualnie:
- LXA4 hamuje wiązanie LTD4 do komórek mezangium - wpływ na interakcje PMN i MS (mesangium cells) w zapaleniu kłębuszków nerkowych
- LXA4 hamuje proliferację komórek pnia pobudzaną przez LTC4
- LXA4 przeciwdziała spadkowi filtracji kłębuszkowej indukowanemu przez LTD4
- LXA4 jest możliwym przekaźnikiem w OUN
f) rola lipoksyn w patologii:
- spadek syntezy u pacjentów z przewlekłą białaczką szpikową
- rola w patologii układu oddechowego (wpływ bronchokonstrykcyjny)
- wpływ na zjawiska immunologiczne związane z chorobami płuc
61. Leki hipolipemizujące:
1.STATYNY:
a) odkryte po raz pierwszy w grzybach Penicillum citrinum
b) dziś używane: lowastatyna, simwastatyna, prawastatyna, ceriwastatyna, atorwastatyna
c) działają poprzez inhibicję reduktazy HMG-CoA hamując w ten sposób szlak mewalonianowy
d) zahamowanie szlaku mewalonianowego blokuje:
- syntezę cholesterolu
- syntezę dolicholu
- prenylację białek
- syntezę ubichinonu
e) efekty stosowania statyny w wątrobie/wpływ na gospodarkę lipidową:
- spadek stężenia cholesterolu w hepatocycie wskutek zmniejszenia jego syntezy, co powoduje:
- wzrost syntezy receptora apoB/E, którego zwiększone ilości w błonie hepatocytów powodują:
- zwiększone wyłapywanie LDL (import cholesterolu z osocza) i remnantów VLDL
- dodatkowo hamowana jest synteza apoB-100, co zmniejsza syntezę VLDL
- wraz z obniżeniem poziomu LDL obniża się poziom cholesterolu w osoczu, a VLDL - triglicerydów
f) działanie statyn w mięśniach:
- spadek syntezy dolicholu, ubichinonu, cholesterolu i prenylacji białek
- obniżenie stężenia dolicholu i zahamowanie glikozylacji białek zaburza szlaki przekazywania sygnału w komórce, przez co przestaje ona reagować na sygnały proliferacyjne
- obniżenie stężenia cholesterolu, który jest surowcem do produkcji błon komórkowych również hamuje podziały komórkowe
- spadek stężenia ubichinonu upośledza przemiany będące źródłem energii; niedobór energii również hamuje proliferację SMC
g) plejotropowe nielipidowe działanie statyn:
- hamowanie proliferacji miocytów gładkich
- hamowanie naciekania ściany naczyniowej przez LDL i VLDL
- stabilizacja blaszki miażdżycowej
- ochronny wpływ na śródbłonek
- modulacja wpływu czynników wazokonstrykcyjnych na miocyty gładkie
- hamowanie produkcji wolnych rodników
- hamowanie agregacji i adhezji płytek krwi
- zmniejszona ekspresja czynnika tkankowego (TF)
- spadek stężenia lipoproteiny (a)
2. ŻYWICE JONOWYMIENNE:
a) cholestyramina i kolestypol
b) wiążą kwasy żółciowe w jelicie prowadząc do ich zwiększonego wydalania
c) zmniejszenie ilości kwasów żółciowych powracających do wątroby w krążeniu jelitowo-wrotnym zmuszą ją do nasilenia ich syntezy; w efekcie wątroba wyłapuje więcej cholesterolu z osocza
d) w bliżej nieznanym mechanizmie żywice zwiększają syntezę VLDL podnosząc poziom triglicerydów w osoczu
3. FIBRATY:
a) gemfibrozil, fenofibrat, bezafibrat, ciprofibrat
b) hamują syntezę VLDL obniżając stężenie TG w osoczu
c) zwiększają aktywność LPL
d) obniżają stężenie remnantów VLDL
e) wywierają różny wpływ na LDL:
- mogą podwyższać lub obniżać ich stężenie w zależności od wyjściowej ich ilości w osoczu
f) podwyższają stężenie cholesterolu frakcji HDL
4. KWAS NIKOTYNOWY:
a) wpływa na gospodarkę lipidową w dawkach farmakologicznych (kilka gramów)
b) hamuje syntezę VLDL
c) powoduje wzrost poziomu cholesterolu HDL przez hamowanie CETP
5. PROBUKOL:
a) obniżenie stężenia LDL w mechanizmie niereceptorowym
b) działanie antyoksydacyjne
62. PPAR:
1. CHARAKTERYSTYKA OGÓLNA:
a) peroxisome proliferator activated receptors - receptory aktywowane proliferatorami peroksysomów
b) należą do rodziny receptorów jądrowych, do których zaliczamy:
- receptory kwasu retinowego
- receptory hormonów tarczycy
- receptory witaminy D
- receptory prostanoidów
- receptory glikokortykoidów
- PPAR
- orphan receptors - receptory sieroce (ich ligandy jeszcze nieznane)
c) wyróżniamy następujące typy PPAR:
- PPAR α
- PPAR δ
- PPAR γ
2. MECHANIZMY DZIAŁANIA PPAR:
a) działanie z ligandem:
- działanie genowe (genomowe) - transaktywacja: PPAR związany z ligandem dimeryzuje w jądrze z RXR i jako heterodimer łączy się z PPARre - PPAR response element w sekwencji DNA;
- działanie pozagenowe - transrepresja: PPAR połączony z ligandem hamuje inne czynniki transkrypcyjne (Nf-κB, STAT) - działanie bez łączenia się PPAR z sekwencją DNA
b) działanie PPAR bez liganda:
- PPAR połączony z RXR (ale nie z ligandem) przyłącza się do PPARre w DNA;
- do tych dwóch czynników przyłącza się następnie kompleks represora wyposażony w enzym - deacetylazę histonów; deacetylacja histonów prowadzi do przyjęcia przez DNA bardziej upakowanej struktury i w efekcie hamuje ekspresję genów
- jeśli przyłączy się ligand, wtedy dołącza też kompleks aktywujący związany z acetylazą histonów, która acetylując histony, stymuluje ekspresję genów
PPAR α (alfa):
a) występowanie:
- hepatocyty
- enterocyty
- kanaliki proksymalne nerki
b) PPAR α w połączeniu z ligandem powoduje:
- wzrost syntezy apoA-I, apoA-II - korzystnie na HDL
- wzrost beta-oksydacji w komórce wątroby
- przeciwzapalne hamowanie COX-2
- działanie pro- i przeciwapoptyczne w zależności od komórki
c) wpływ PPAR α na aterogenezę:
- spadek produkcji VLDL
- wzrost spalania FFA
- wzrost lipolizy
- zwiększenie ilości HDL
- wzrost transportu zwrotnego cholesterolu
- spadek stężenia małych gęstych LDL
- wpływ naczynioochronny - spadek ilości adhezyn śródbłonka
d) ligandami PPAR α są między innymi fibraty
PPAR δ (delta):
a) szeroko rozpowszechniony
b) działanie:
- indukcja syntezy HDL
- dojrzewanie oligodendrocytów
- tworzenie błon
- powstawanie raka jelita grubego
c) antagoniści:
- sulindak
d) agoniści:
- estry etylowe nasyconych i nienasyconych kwasów tłuszczowych
- fibraty (bezafibrat)
e) następstwa aktywacji PPAR δ:
- kardiomiocyt: wzrost transportu FFA oraz ich beta-oksydacji
- mięsień szkieletowy: wzrost transportu i oksydacji FFA
- tkanka tłuszczowa: wzrost termogenezy
- odtłuszczenie naczyń: wzrost syntezy HDL
- wątroba: spadek glukoneogenezy, nasilenie cyklu pentozowego
- spadek odpowiedzi zapalnej komórek nacieku zapalnego
PPAR γ:
a) 3 podtypy:
1. - szeroko rozpowszechniony
2. - biała tkanka tłuszczowa
3. - makrofagi
b) defekty PPAR γ - jedna z przyczyn insulinooporności
WPŁYW PPAR γ NA TRANSDUKCJĘ SYGNAŁU PO POBUDZENIU RECEPTORA INSULINOWEGO:
a) działając przez prowadzący do szkodliwych następstw szlak obejmujący czynnik MAPK, insulina powoduje:
- wzrost proliferacji miocytów gładkich
- zwiększoną proliferację monocytów
- wzrost syntezy PDGF
- wzrost syntezy angiotensyny II
- wzrost syntezy PAI-1
- wzrost syntezy endoteliny I
- wzrost ekspresji białek adhezyjnych
Ten szlak jest hamowany przez PPAR γ
b) PPAR γ aktywuje szlak, w którym uczestniczy 3-kinaza fosfatydyloinozytolu; jest to szlak korzystny, powoduje:
- wzrost ekspresji genu GLUT 4
- wzrost syntezy tlenku azotu
- spadek aktywności metaloproteaz (stabilizujący wpływ na płytkę miażdżycową)
- wzrost syntezy TIMP - tkankowych inhibitorów MMP
- hamowanie apoptozy np. endotelium
c) agoniści PPAR γ:
- prostaglandyny
- nienasycone kwasy tłuszczowe
- tiazolidenodiony: troglitazon, pioglitazon, parglitazon, ciglitazon, rosiglitazon - leki pozwalające na przełamanie insulinooporności
64. Regulacja aktywności reduktazy HMG-CoA:
a) regulacja na poziomie transkrypcji:
- gen posiada SRE - sterol response element, sekwencję, która w obecności dużych ilości steroli hamuje transkrypcję genu
b) regulacja na poziomie translacji:
- translacja mRNA reduktazy hamowana przez niesterydowe metabolity pochodzące od mewalonianu
c) kontrola rozpadu enzymu
- enzym złożony z dwóch domen: katalitycznej i błonowej (czujnika wrażliwego na stężenia pochodnych mewalonianu i cholesterolu); rozpada się przy dużym stężeniu wspomnianych substancji
d) regulacja aktywności przez fosforylację:
- fosforylacja reduktazy przez kinazę reduktazy powoduje inaktywację reduktazy
- kinaza reduktazy jest aktywna w formie ufosforylowanej
e) inhibicja przez produkt - mewalonian
f) regulacja hormonalna:
- hormony tarczycy i insulina zwiększają aktywność
- glikokortykoidy i glukagon obniżają aktywność enzymu
Regulacja aktywności 7alfa-hydroksylazy:
a) współdziała z NADPH + H+, cytochromem P-450, reduktazą cytochromu, O2
b) aktywna w formie ufosforylowanej - w formie defosfo jest nieaktywna
c) regulacja przez metabolity:
- hamowana przez kwasy żółciowe
d) synteza enzymu pobudzana przez cholesterol
e) aktywność zahamowana przy braku witaminy C
65. Przedziałowość komórkowa poszczególnych etapów biosyntezy cholesterolu.
- biosynteza cholesterolu zachodzi w cytozolu i siateczce śródplazmatycznej
- syntaza HMG-CoA ma 2 izoenzymy, z których jeden znajduje się w cytozolu i odpowiada za biosyntezę cholesterolu, a drugi znajduje się w mitochondrium i uczestniczy w ketogenezie
- redukcja HMG-CoA do mewalonianu przez reduktazę HMG-CoA zachodzi w siateczce środplazmatycznej
- enzymy, które przekształcają lanosterol do cholesterolu znajdują się w siateczce
POPRAWKA DO WERSJI WYŻEJ
hello donkey chcę cie wyprowadzić z błędu. Otóż w siateczce biosynteza zachodzi od etapu skwalen-2,3-epoksyd i ja tak będę pisać. Jak się wczytasz w Harpera to tak znajdziesz. A etap reduktazy HMG-CoA zachodzi defacto w cytosolu, ale enzym jest przyczepiony do siateczki ER, a miejsce aktywne ma skierowne w stronę cytosolu, tego właściwie jestem pewna.
69. Kwasy żółciowe:
a) syntezowane w wątrobie z cholesterolu - pierwotne kwasy żółciowe:
- kwas cholowy
- kwas chenodeoksycholowy
b) synteza z cholesterolu:
- 7α-hydroksylacja - katalizowana przez 7α-hydroksylazę - enzym mikrosomalny wymagający tlenu, NADPH i cytochromu P-450 - enzym jest monooksygenazą wymagającą witaminy C
- powstaje 3α,7α-dwuhydroksycholest-5-en, którego grupa hydroksylowa przy C3 zostaje utleniona do grupy ketonowej, natomiast wiązanie podwójne przemieszcza się z 5-6 na 4-5
- powstaje 7α-hydroksycholest-4-en-3-on
- wprowadzona zostaje grupa OH przy C12 (kwas cholowy, ten etap nie następuje w syntezie kwasu chenodeoksycholowego)
- pierścień B zostaje nasycony - redukcja wiązania C=C 4-5
- ketonowa grupa C3 zostaje przekształcona w grupę OH
- hydroksylacja przy C26 i następnie utlenienie tej grupy do grupy kwasowej
- przyłączenie CoA-SH
- hydroksylacja przy C24 i utlenienie
- odłączenie propionylo-S-CoA - skrócenie łańcucha bocznego o 3 węgle
c) powstałe kwasy żółciowe sprzęgane są z glicyną i tauryną przez acylotransferazę lizosomalną; zwiększa to ich rozpuszczalność; powstaje glikocholan i taurocholan
d) w ludzkiej żółci więcej kwasów związanych jest z glicyną (gliko:tauro - 3:1)
e) w jelicie kwasy pierwotne przekształcane są do kwasów wtórnych:
- 7α-dehydroksylacja i dekoniugacja
- kwas cholowy przechodzi w kwas deoksycholowy
- kwas chenodeoksycholowy przechodzi w kwas litocholowy
f) regulacja 7α-hydroksylazy:
- hamowana przez kwasy żółciowe
- cholesterol zwiększa jej syntezę
- aktywna w formie ufosforylowanej
- wymaga O2 i witaminy C
g) krążenie jelitowo-wątrobowe:
- całkowita pula kwasów żółciowych w organizmie: 3-5 g (średnio 4 gramy)
- ta pula przechodzi przez jelito 6-10 razy dziennie
- 97-99% kwasów żółciowych wydzielonych do żółci jest resorbowanych w jelicie krętym jako kwasy zarówno pierwotne jak i wtórne - odbywa się to przy udziale błonowego białka IBAT (ileal bile acid transporter)
- kwas litocholowy jest resorbowany w mniejszych ilościach jako substancja trudniej rozpuszczalna
- wchłonięte kwasy powracają do wątroby z krwią żyły wrotnej, tam są ponownie wydzielane do żółci
- 1-2-3% kwasów żółciowych wydzielonych do jelita nie ulega wchłonięciu i jest wydalane z kałem jako sterole kwaśne (0,3-0,4 g dziennie) - jest to główna droga eliminacji cholesterolu z organizmu
- z uwagi na tak niewielką dobową utratę kwasów żółciowych i synteza w wątrobie jest niewielka - sięga 0,3-0,4 g dziennie
h) funkcje:
- emulgacja lipidów - zwiększają powierzchnię kontaktu lipidu z wodą, co ułatwia lipazie strawienie całego tłuszczu
- tworząc micele ułatwiają transport produktów trawienia lipidów w wodnym środowisku oraz ich wchłanianie
- ostatnio udowodniono działanie kwasów żółciowych jako hormonów sterydowych: są one ligandami dla receptorów niedawno uznawanych za receptory sieroce: FXR (receptor farsenoid X), receptor LXRα oraz receptor CPA; w połączeniu z tymi receptorami, kwasy żółciowe mogą łączyć się z promotorami różnych genów blokując lub stymulując ich ekspresję;
przykłady:
1. kompleks kwas żółciowy-receptor FXR hamuje ekspresję genu 7α-hydroksylazy - sprzężenie zwrotne ujemne
2. kompeleks kwas żółciowy-receptor FXR wiąże się z promotorem genu IBAT w jądrze enterocytu stymulując jego ekspresję - prowadzi to do zwiększenia syntezy transportera IBAT i zwiększonego wchłaniania kwasów żółciowych w jelicie krętym
74. Oksydazy o funkcji mieszanej:
Wszystko z Harpera (rozdział 13 i 61):
a) są to inaczej monooksygenazy zależne od cytochromu P-450
b) nazywane oksydazami o funkcji mieszanej, ponieważ wbudowują tylko jeden atom tlenu z cząsteczki O2 do substratu jako grupę hydroksylową; drugi atom jest redukowany do cząsteczki wody;
reakcja:
RH+O2+NADPH+H = R-OH + H2O + NADP
c) u człowieka występuje co najmniej 14 rodzin monooksygenaz i 35-60 indywidualnych cytochromów P-450
d) oksydazy o funkcji mieszanej:
- są hemoproteinami
- katalizują ok. 60 rodzajów reakcji (=są wszechstronne)
- występują w gładkim ER oraz w mitochondriach
- wykorzystują NADPH: cytochrom P-450 jest redukowany przez reduktazę NADP-cytochrom P-450
d) występowanie oksydaz o funkcji mieszanej:
- najwięcej w wątrobie i jelicie cienkim
- nadnercza (steroidogeneza)
- jądra
- jajniki
- łożysko
- nerki
- płuca
e) oksydazy o funkcji mieszanej biorą udział w:
- syntezie sterydów
- syntezie kwasów żółciowych
- hydroksylacji witaminy D
- metabolizmie ksenobiotyków (odtruwaniu) - obecność grupy OH nadaje im bardziej hydrofilowy charakter, przez co stają się łatwiejsze do usunięcia z organizmu
f) znaczenie kliniczne:
- odpowiadają za interakcje między lekami - np. indukcja cytochromu P-450 przez fenobarbital powoduje szybsze metabolizowanie dikumarolu; również hamowanie tych enzymów odgrywa rolę w interakcjach lekowych
- niektóre ich produkty mają właściwości karcynogenne - rola w kancerogenezie
- aktywność zmienia się w niektórych stanach chorobowych
DOD. Wzory na energię
Kwasy parzystowęglowe: [ (n/2 - 1) * 5 ] + (n/2 * 12) - 2 = x [moli ATP]
Kwasy nieparzystowęglowe: [ (n/2 - 1) * 5 ] + [(n/2-1) * 12] - 2 = x [moli ATP]
gdzie n to liczba atomów węgla w łańcuchu
Zysk w kJ: y = X* 51,6
% energii spalania KT przetworzonej na wiązania wysokoenergetyczne w ATP:
%E - y / swobodna energia spalania kwasu tłuszczowego
Nie jestem pewien tych wzorów zwł. drugiego - sprawdźcie to w Harperze, str. 300
Prostanoidy niedienowe:
a) prostanoidy zawierające wiązania podwójne w liczbie innej niż dwa (w bocznych łańcuchach: alfa i omega)
b) prostanoidy monoenowe - jedno wiązanie podwójne:
- powstają z kwasu 8,11,14-ikozatrienowego (ikozatrienoan - powstaje w wyniku desaturacji i elongacji kwasu linolowego)
- należą tu: PGE1, PGF1, TXA1
c) prostanoidy trienowe - 3 wiązania podwójne:
- powstają z kwasu 5,8,11,14,17-ikozapentaenowego (ikozapentaenoan - obecny w pokarmie; powstaje też w wyniku podwójnej desaturacji i elongacji kwasu alfa-linolenowego)
- należą tu: PGD3, PGE3, PGF3, PGI3, TXA3
d) istotne znaczenie kliniczne mają PGI3 oraz TXA3:
- stwierdzono, że TXA3 ma znacznie słabsze działanie proagregacyjne niż TXA2; za to PGI3 ma tak samo silne pozytywne działanie antyagregacyjne jak PGI2
- zwiększone spożywanie kwasu 5,8,11,14,17-ikozapentaenowego sprzyja większej syntezie tych dwóch niedienowych prostanoidów, co wywiera pozytywny wpływ na drożność naczyń krwionośnych i ich ścianę - zjawisko stwierdzone u Eskimosów spożywających znaczne ilości kwasu ikozapentaenowego (kwas timnodonowy - omega3) w rybach (przede wszystkim rybich olejach)
Modyfikacje lipidowe białek:
a) mirystynylacja:
- dotyczy N-końca białka
- polega na przyłączeniu reszty kwasu mirystynowego, którego dawcą jest mirystynylo-S-CoA
- reakcja katalizowana przez N-mirystynylotransferazę
- w mirystynylowanym białku N-końcowy aminokwas to glicyna, pozycję 5 zajmuje seryna lub treonina, 6 i 7 aminokwasy zasadowe
- przykładem jest mirystynylacja białka wirusowego src, której zablokowanie pozbawia to białko właściwości onkogennych
b) palmitynylacja:
- dotyczy reszt cysteiny
- dawcą reszty palmitynylowej jest palmitynylo-S-CoA
- przykładem jest palmitynylacja rodopsyny
PRENYLACJA:
c) farnezylacja:
- dotyczy końca karboksylowego
- grupy farnezylowe (15 C) łączą się wiązaniami tioeterowymi z grupami -SH reszt cysteinylowych przy karboksylowym końcu białka
- farnezylowane są liczne białka biorące udział w przekazywaniu sygnałów, również białka kierowane
d) geranylogeranylacja:
- tak jak farnezylacja, dotyczy końca karboksylowego białka
- grupy geranylogeranylowe (20 C) poprzez wiązania tioeterowe łączą się z resztami cysteiny przy C-końcu białka;
DIETA KWAŚNIEWSKIEGO
Ponieważ to hasło zostało zrzucone na wykładzie jak i na seminarium u doktor Puch
śmiem podejrzewać że będzie na kolosie pytanko odnośnie diety Kwaś (Atkinsa)
Tyle znalazłam i trochę dopisałam, jeśli ktoś się ze mną nie zgadza, proszę o dopisanie swojego zdania, gdyż nie bardzo pamiętam jak to omówiła pani dr. Puchalska, bo zrobiła to w tempie błyskawicznym, tak błyskawicznym, że jak skończyłam pisać słowo "Kwaśniewskiego" ona skończyła omawiać tenże temat...
ehhhh.....
Dieta Kwaśniewskiego - jest propozycją nieracjonalnego żywienia
jest dietą ubogowęglowodanową i bogatotłuszczową
Analizowano dietę pod względem jakości spożywanych pokarmów i wysunięto takie wnioski:
1) dochodzi do ketogenezy -> zakwaszenie ustroju bo dochodzi do długotrwałej utraty zasad niezbędnych do zobojętnienia kwaśnych związków ketonowych wydalanych z moczem
2) wielokrotnie podwyższona dostawa cholesterolu pokarmowego np. wyniki... 2000mg%
przy normie ,300mg%...
3)przy małej dostawie błonnika
4) zaburzenie endogennej syntezy cholesterolu
5) dostawa zbyt dużej ilości wit A i Fe które odkładają się w hepar -> uszkodzenie hepatocytów
6) za duża dostawa KT -> hepar nie nadąża z wytwarzaniem VLDL -> to może doprowadzić do stłuszczenia wątroby
* pkt nr 6 to coś co zapamiętałam z seminarium ale pewna tego nie jestem na 100%. Jak ktoś wie coś więcej to proszę to skorygowa
41
Wyszukiwarka