1. Czynniki kształtujące skład chemiczny wód naturalnych.
Każdy składnik chemiczny jest zanieczyszczeniem dla wody, gdy jest szkodliwy to jest skażeniem - ma ujemny wpływ na wodę.
Podział:
czynniki naturalne - woda styka się z glebą, ze skałami, z minerałami, czyli z litosferą, w której skład wchodzą substancje rozpuszczalne w wodzie, składniki te krążą z wodą, później się w niej wytrącają. Wody powierzchniowe i podziemne zawierają pewne związki chemiczne, które występują w dużych ilościach w przyrodzie. Związki te występują w różnych wodach w różnych ilościach. Substancja jest rozpuszczana wskutek dodatkowej reakcji chemicznej. Naturalne procesy zachodzące w wodzie - reakcje chemiczne i biochemiczne - zachodzące z udziałem organizmów wodnych (obumieranie ich i dalszy rozkład materii organicznej, procesy wydalania związków).
czynniki antropogeniczne - woda dostaje się po zużyciu do kanalizacji i jest potem oczyszczana. Ścieki z oczyszczania są głównym powodem zanieczyszczeń wód powierzchniowych, dużo mniejszym jeśli chodzi o wody podziemne.
a) Ścieki miejskie (bytowe + przemysłowe) - punktowe źródło zanieczyszczeń, najpoważniejsze skażenie + źródło skażenia wody.
b) chemizacja rolnictwa - dostarczanie na pola nawozów i pestycydów - zanieczyszczenia obszarowe (wód powierz. i podz.)
c) transport (lądowy i wodny) - zanieczyszczenie liniowe - głównie materiały napędowe
2.Główne kationy występujące w wodach naturalnych
Na2+, Ca2+, Mg2+, Fe2+, Fe3+, Mn2+, NH4+, metale ciężkie
Na2+ - jon sodowy
główny składnik wód śródlądowych
źródło:
pokłady NaCl (woda morska)migracja jonu
wydobycie → użycie → do wód ze ściekamichemizacja rolnictwa (np. saletra sodowa)
stężenie: kilkadziesiąt mg/l → na ogół: 70÷100 mg/l
(kilkanaście lub >200 → rzadkie przypadki)
Ca2+ - kation wapniowy
w litosferze: CaCO3 - węglan wapnia (związek słabo rozpuszczalny)
obecność CO2 w wodach:
H2O + CaCO3 + CO2 ↔ Ca2+ + 2HCO3¯
reakcja aż do ustalenia stanu równowagi
stała równowagi (stężenie):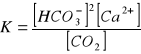
K = const, zatem jeśli rośnie w wodzie stężenie CO2, to musi również wzrastać ilość rozpuszczonego węglanu wapnia. Natomiast kiedy stężenie CO2 w wodzie maleje, węglan wapnia się wytrąca.
Ca2+ powstaje również w wyniku rozpuszczania (wypłukiwania) innych związków: takich, jak np. siarczan wapnia (= gips) (średnio rozpuszczalny)
inne źródła wapnia: ścieki (chlorek wapnia) i nawozy
stężenie: 60÷100 mg/l (trochę mniejsze niż sodu)
Mg2+ - kation magnezowy
bardzo często towarzyszy wapniowi
obecność w wodzie związana z podobnymi procesami, jak w przypadku Ca2+
obecny w wodzie poprzez rozpuszczanie węglanów → reakcja rozpuszczania MgCO3 (identyczna, jak w przypadku wapnia):
H2O + MgCO3 + CO2 ↔ Mg2+ + 2HCO3¯
pokłady: MgSO4, MgCl2 w glebie są źródłem Mg2+ w wodach
magnez może także dostawać się do wód ze ściekami ← MgSO4 stosowany w medycynie, w przemyśle
w wodach naturalnych są zachowane pewne proporcje między stężeniami Mg2+ i Ca2+ → w naszej strefie klimatycznej Mg2+ jest w wodzie średnio 4 razy mniej niż Ca2+
stężenie: zwykle kilka mg/l (rzadko >100mg) → ale w krajach Płn. Afryki magnez stanowi podstawowy składnik wód
Fe2+, Fe3+, Mn2+
występują w przyrodzie w rudach jako: węglany i tlenki
żelazo → dwa stopnie utlenienia: Fe2+, Fe3+ (żelazo często występuje jako mieszanina tych dwóch tlenków: MAGNETYT)
![]()
tlenki - podstawowa forma Fe
FeCO3, Fe2(CO3)3 - węglany te mogą rozpuszczać się w wodzie dając wodorowęglany
H2O + FeCO3 + CO2 ↔ Fe2+ + 2HCO3¯
3H2O + Fe2(CO3)3 + 3CO2 ↔ 2Fe3+ + 6HCO3¯
Fe2O3 + 3H2O + 6CO2 ↔ 2Fe(HCO3)3
w wodach podziemnych, gdy jest mała ilość O2, żelazo i mangan występują w postaci jonów Fe2+ i Mn2+ (żelazo Fe3+ przy małej ilości tlenu ulega redukcji)
gdy woda podziemna - zawierająca wodorowęglan (np. wodorowęglan żelaza) - wydostaje się na powierzchnię, zachodzi reakcja, w wyniku której wytrąca się wodorotlenek (np. wodorotlenek żelaza):
Fe(HCO3)2 + H2O → Fe(OH)2 + CO2
zielony
następnie żelazo w rozpuszczonym Fe(OH)2 pod wpływem tlenu utlenia się do Fe3+ → powstaje Fe(OH)3 (związek bardzo trudno rozpuszczalny w wodzie, wytrąca się w postaci brunatnego osadu)
4Fe(OH)2 + O2 + 2H2O → 4Fe(OH)3
brunatny
powyższe reakcje są powodem, dla którego w wodach powierzchniowych żelazo w postaci jonów praktycznie nie występuje → nie ma go zatem prawie w ogóle w wodzie, natomiast występuje w osadach dennych
w wodach żelazo może występować w postaci zawiesiny
żelazo może tworzyć połączenia ze związkami organicznymi, czyli tzw. związki kompleksowe
mangan występuje w wodach w postaci zawiesiny
stężenie żelaza w wodach: 0.1 mg/l ÷ kilka mg/l (nawet do 10mg/l)
manganu jest na ogół kilka razy mniej niż żelaza
NH4+ - jon amonowy
w wodach podziemnych praktycznie nie występuje
głównie w wodach powierzchniowych
nie ma pochodzenia naturalnego
pochodzenie antropogeniczne: główny składnik ścieków miejskich (bytowych) oraz składnik nawozów
coraz więcej oczyszczalni usuwa azot → coraz więcej oczyszczalni usuwa azot, coraz mniej azotu dostaje się do wód ze ściekami
azot amonowy, tiosiarczan amonu ← z nawozów azotowych
niekiedy NH4+ występuje w wodach podziemnych (z opadami dostaje się do gleby, ale gleba zatrzymuje dużą część jonów amonowych)
stężenie: bardzo małe ilości, ale bardzo duże znaczenie
Metale ciężkie
występują w bardzo małych ilościach: μg/l - z naturalnych źródeł (naturalny poziom metali w wodach)
zbyt wysokie stężenie jest szkodliwe
metale ciężkie mogą pochodzić ze ścieków przemysłowych, także z gleb i zanieczyszczenia powietrza
3.Główne aniony występujące w wodach naturalnych
|
Pochodzenie |
Występujące stężenia |
reakcje /znaczenie |
|
HCO3- węglowodany |
Utlenienie Węglanów wapnia, „ magnezu „ żelaza |
Śr>100 mg/l Nawet>200mg/l Istnieje w równowadze z CO2 w wodach; Największa ilość w porównaniu do innych anionów; 3x więcej niż jonów Ca2+ |
Wpływ na odczyn wody |
|
Cl- chlorki |
NaCl CaCl2 Występuje naturalnie w wodach (pokłady soli, ale również dostaje się do wód ze ścieków przemysłowych, wód kopalniane; także ze ścieków miejskich |
Od kilkunastu do kilkudziesięciu mg/l Śr>100mg/l Rzadko>200mg/l |
|
|
SO42- siarczany |
Naturalnie występowanie; Ścieki miejski; Przemysł |
Od kilkunastu do kilkudziesięciu mg/l Zdarza się, że stężenie jest większe od Cl-; >200mg/l |
|
|
NO3- azotany
|
Nie występują w przyrodzie (nie są wiązane w skałach) Zanieczyszczenia antropogeniczne: tj. rolnictwo (nawozy szuczne;), przemysł(zakłady azotowe)
Ścieki miejskie: nieznaczne ilości azotanów |
Duże stężenia na terenach użytkowanych rolniczo; nawet do kilkudziesięciu mg/l; (również w płytkich wodach podziemnych;) często: kilka mg/l (największe stężenie wiosną) |
NaNO3 saletra sodowa KNO3 saletra potasowa NH4NO3saletra amonowa |
Nawozy azotowe są bardzo mobilne (słabo absorbowane przez glebę -szybko wypłukiwane przez deszcz dostają się do wód |
Jony fosforanowe HPO42-wodorofosforany |
Główne źródło zanieczyszczenia -ścieki miejskie(polifosforany zawarte w środkach piorących); -rozkład białek (obecnie usuwany w oczyszczalniach) -nawozy fosforowe (wodorofosforany wapniowe); Nie występuje naturalnie; |
Mała ilość; średnio: kilka mg/l |
Składnik mało mobilny, wytrąca się fosforan wapnia |
|
(SiO3)2 krzemiany |
Składnik wielu minerałów Krzem w litosferze w ilości>20% |
Małe ilości; kilka mg/l |
Pierwiastek trudnowymywalny z gleb ; Słabo rozpuszczalny |
|
NO2- azotyny |
Redukcja azotanów, utlenianie jonów amonowych |
Występują bardzo rzadko w wodach w ilościach <1mg/l (μg/l)
|
Szkodliwy; Działanie toksyczne; Obecność w wodzie świadczy o dużym zanieczyszczeniu |
|
F- fluorki |
Pochodzenie naturalne ; Przemysł (trawienie szkła -HF) |
Szeroki granice od 0 do >1mg/l |
|
|
Mikrozanieczyszczenia tj.; Jodki, bromki, borany(czteroborany), seleniany
Małe ilości (μg/l) ; trudne do stwierdzenia
Pochodzenie naturalne
5.Mikroskładniki mineralne występujące w wodach naturalnych.
Żelazo - w wodach jako węglan oraz tlenki; występuje jako Fe2+ i Fe3+. Bardzo łatwo się utlenia. Występuje bardzo chętnie pod ziemią, gdyż jest tam dużo CO2. W wodach podziemnych wskutek redukcji mamy Fe2+. Dostaje się do wód przede wszystkim jako wodorowęglan. Woda podziemna po dostaniu się na powierzchnię wytrąca żelazo. Może tworzyć w wodach związki kompleksowe.
Mangan - Występuje bardzo chętnie pod ziemią, gdyż jest tam dużo CO2.W wodach podziemnych wskutek redukcji mamy Mn2+. Dostaje się do wód przede wszystkim jako wodorowęglan. Woda podziemna po dostaniu się na powierzchnię wytrąca mangan.
Jon amonowy - NH4 w wielu wodach podziemnych praktycznie nieobecny, występuje głównie w wodach powierzchniowych. Jest to ważny składnik wód.
Azotany - obecność w wodach z powodu stosowania nawozów sztucznych
Fosforany - Kiedyś dostawały się do wód ze ścieków miejskich. Dostają się tam też z rozkładu białka. Są głownie zanieczyszczeniem antropogenicznym, stosuje się je w nawozach. Jest to składnik mało mobilny.
Krzemiany - krzem w wodach naturalnych występuje w postaci koloidalnej jako kwas meta lub poli krzemowy oraz w postaci zjonizowanej jako rozpuszczony metakrzemian sodowy - produkt wietrzenia i erozji wodnej skał. Jest go w wodach bardzo mało.
Azotyny - pośredni produkt utleniania jonów amonowych. Jony b.szkodliwe, pochodzą od ścieków miejskich(gdy je wykryjemy można się spodziewać bakterii i zanieczyszczeń w wodzie).
Fluorki - dostają się do wody ze źródeł naturalnych (pokładów geologicznych) lub w wyniku odprowadzania ścieków. Najczęściej w postaci fluorku wapniowego.
Glin - niska zawartość w wodach naturalnych, nie jest to pierwiastek toksyczny, oznacza się go głównie w wodach technologicznych
Bromki - mikrozanieczyszczenia
Jodki - mikrozanieczyszczenia
Czteroborany - mikrozanieczyszczenia
Seleniany - mikrozanieczyszczenia
6.Substancje biogenne w wodach naturalnych.
Do biogennych zalicza się te substancje, które są niezbędne do życia i rozwoju żywych organizmów. Występują one w wodzie w postaci jonów oraz koloidów, których skład w większości przypadków nie jest wyjaśniony.
Związki azotu
Nieorganicznymi formami zw. azotu w wodzie nat. są jony amonowe(NH4+), azotanowe(III) (NO2-) i azotanowe(V) (NO3-). Cechuje je podobna geneza oraz to, że mogą ulegać wzajemnej transformacji, przechodząc z jednej postaci w drugą. Głównym źródłem jonów NH4+, NO2- i NO3- są substancje organiczne pochodzenia roślinnego i zwierzęcego zawierające w swym składzie białko. W wyniku tego skomplikowanego procesu biochem. zachodzącego przy udziale licznych bakterii i enzymów następuje wydzielanie się amoniaku
R-CHNH2-COOH + H2O->R-CHOH-COOH + NH3
Jon NH4+ może mieć również pochodzenie nieorganiczne. Zaobserwowano na przykład jego obecność w wodach bagiennych, bogatych w substancje humusowe, które zdolne są do reakcji azotanów do jonów amonowych. Azotany i azotyny mogą być redukowane do jonów NH4+ także przez inne substancje np. siarkowodór i Fe (II). Prócz tego jony amonowe często dostają się do zbiorników wodnych ze ściekami przemysłowymi np. z fabryk nawozów azotowych oraz ściekami bytowo-gospodarczymi i miejskimi.
W niewielkich ilościach jony amonowe wyst. w wodach podziemnych górnych poziomów wodonośnych szczególnie w pobliży gruntów uprawnych, osiedli ludzkich oraz w rzekach poniżej wielkich miast i obiektów przemysłowych. Z powodu stosunkowo łatwej sorbcji w glebie jego ilość rzadko osiąga maksymalną wartość do kilkudziesiątych mg/dm3.
W niezanieczyszczonych wodach podziemnych jony NH4+ występują prawie zawsze, jednakże w ilościach nieznacznych-setne, dziesiąte części mg/dm3.
Prócz stanu jonowego(w postaci NH4+) grupa amonowa występuje w wodzie naturalnej w złożonych zw., tworzących się podczas częściowego rozkładu substancji organ., mających charakter koloidalny(azot albuminowy).
W wodzie nat. NH4+ jest stosunkowo nietrwały i pod wpływem czynników fizyczno-chemicznych i biochemicznych przechodzi w inne postaci związków azotu. W warunkach tlenowych, wskutek działania tzw. bakterii nitryfikacyjnych(Nitrosomonas, Nitrobacter) jon NH4+ utlenia się do jonu azotanowego(III) a następnie azotanowego(V). Proces ten to nitryfikacja i może być przedstawiony za pomocą równań
2NH3 + 3O2->2HNO2 + 2H2O; 2HNO2 + O2->2HNO3.
Drugim ważniejszym źródłem bezpośrednio wód nat. w azotany są tlenki azotu, tworzące się podczas wyładowań atmosferycznych, które po pochłonięciu ich przez wodę atmosferyczną opadają na powierzchnię ziemi.
Jony azotanowe(III) są bardzo nietrwałe i w wyniku tego w wodach powierzchniowych, gdzie istnieją optymalne warunki dla procesów utleniania występują one w ilościach nieznacznych, podczas gdy w wodach podziemnych zawartość NO2- zwykle jest znacznie wyższa zwłaszcza w górnych poziomach wodonośnych.
Jony azotanowe(V) występują w wodach nat. w ilościach nieco większych niż jony NO2-. Proces nitryfikacji zachodzący w glebie decyduje o mniejszej lub większej zawartości NO3- w górnych poziomach wód gruntowych. W głębszych warstwach wodonośnych azotany(V) występują w ilościach bardzo nieznacznych.
Azotany (V) są substancjami pokarmowymi dla org. roślinnych, które przyswajają je z wodą, to też w ciągu okresu wegetacyjnego ilość azotanów w otwartych zbiornikach wodnych wyraźnie się zmniejsza aż do ich całkowitego zaniku. W okresie zimowym w związku z obumieraniem organizmów następuje mineralizacja subst. Organicznych a w jej wyniku regencja azotanów.
Związki fosforu
W stanie rozpuszczonym zw. fosforu występują w postaci jonów kwasu ortofosforowego oraz w postaci związków w zespole organicznym.
W stanie jonowym w wodach nat. fosfor mineralny występuje przede wszystkim w postaci HPO4-2 i w mniejszym stopniu jako H2PO- i PO4-3. Zawartość poszczególnych form jonowych fosforu mineralnego zależy od odczynu środowiska wodnego.
Rozpuszczony fosfor organiczny jest częścią składową złożonych zw. org. Pierwiastek ten może znajdować się w odzie nat. nie tylko w stanie rozpuszczonym lecz także w postaci cząstek zawieszonych zarówno pochodzenia mineralnego jak i organicznego.
Związki fosforu mimo iż znajdują się w odach nat. w ilościach znikomych to należąc do zw. biogennych mają duże znaczenie dla rozwoju świata roślinnego, a więc także dla produkcji biomasy w zb. Wodnych.
Związki żelaza
Często występują w wyniku przejścia ich w roztwór z różnych skał w składzie których żelazo jest bardzo rozpowszechnione. Przejście żelaza w roztwór może zachodzić pod wpływem zw. utleniających(tlen) lub kwasów(węglowego, organicznych). Także kwasy org. Występujące w wodach biogennych przeprowadzają żelazo w roztwór w postaci złożonych zespołów humusowych. Z tym związana jest stała obecność żelaza w wodach bogatych w substancje humusowe.
Postacie pod którymi żelazo znajduje się w war. nat.: w wodach podziemnych przeważa żelazo(II, Fe+2) które przechodzi w roztwór gł. w postaci wodorowęglanu żelaza. Przy zawartości CO2 poniżej stanu równowagi i przy pojawieniu się rozpuszczonego tlenu zachodzi hydroliza i żelazo przechodzi w trudno rozpuszczalny wodorotlenek żelaza.
Fe+2 + 2HCO3- + 2H2O<->Fe(OH)2 + 2H2CO3
Wodorotlenek żelaza(II) dalej łatwo utlenia się i przechodzi w wodorotlenek żelaza(III) nierozpuszczalny w wodzie.
4Fe(OH)2 + O2 + 2H2O<-> 4Fe(OH)3
Proces utleniania Fe(OH)2 w wielu przypadkach przebiega przy współudziale mikroorganizmów-bakterii żelazistych, które w procesie swej działalności życiowej wykorzystują energię wydzielaną przy utlenieniu zw. żelazawych do żelazowych.
Wody zawierające żelazo mają zwykle odczyn kwaśny.
Żelazo(III) występuje prawie wyłącznie w wodach powierzchniowych. Żelazo(II) gł. w podziemnych.
Związki krzemu
Krzem może występować w postaci zw. organicznych i mineralnych-kwas krzemowy (IV) i jego formy jonowe, jest on bardzo słaby i dysocjuje w nieznacznym stopniu:
H2SiO3<->H+ + HSiO3-
Dysocjacja na SiO3-2 może mieć znaczenie tylko przy odczynie silnie alkalicznym raczej nie spotykanym w wodach nat.
Cześć krzemu występuje w stanie koloidalnym-w cząsteczkach o składzie SiO2*H2O oraz w postaci kwasu polikrzemowego o składzie x*SiO2*y*H2O.
Powszechne rozprzestrzenianie krzemu w środowisku tworzącym większość skał skorupy ziemskiej zapewnia ciągły dopływ krzemu do wód nat. w postaci wyługowanego przez wodę kwasu krzemowego i jego soli alkalicznych. Dlatego też największa ilość krzemu występuje w wodach podziemnych, najmniejsza zaś w powierzchniowych warstwach otwartych zbiorników wodnych, gdzie krzem zużywany jest przez rośliny. W wodach słabo zmineralizowanych zwłaszcza w rejonach wysokogórskich krzem stanowi przeważający składnik wody.
7. Gazy występujące w wodach naturalnych.
Z gazów rozpuszczonych w wodzie największe znaczenie mają O2 iCO2.
Tlen.
Znajduje się w wodzie nat. w postaci rozpuszczonych cząsteczek. Jego obecność jest warunkiem istnienia większości organizmów żyjących w zbiornikach wodnych. Na zawartość O2 w wodzie wpływają dwa przeciwstawne procesy: jedne wzbogacają a drugie zmniejszają zawartość O2. Stężenie O2 powinno być równe wartości wynikającej ze stanu równowagi dynamicznej p=k*c. (k- rośnie wraz z temp., c- maleje ze wzrostem temp.). Przy dużym zanieczyszczeniu spada ono poniżej wartości stężenia nasyconego.
Do wzbogacających należą dyfuzja z atmosfery(reaeracja) oraz wydzielanie tlenu przez rośliny wodne w procesie fotosyntezy. Dyfuzję ograniczają temperatura i ciśnienie. Procesy te mogą zachodzić gł. w przypowierzchniowych warstwach zbiorników wodnych, w głąb tlen przekazywany jest wskutek przemieszczania wody przez falowanie, konwekcję i prądy poziome.
Zawartość O2 zmniejsza oddychanie organizmów, fermentacja- procesy te zachodzą bez przerwy, zwiększając swoją intensywność przy wyższej temperaturze.
Stosunek natężenia obu grup procesów decyduje o zawartości O2, przy czym dąży ona by osiągnąć ilość przy której woda w danej temp. i przy danym ciśnieniu byłaby nasycona tlenem. Zawartość rozpuszczonego w wodzie O2 waha się od 0-14 mg/dm3. Jego rozkład zależy od pionowej stratyfikacji- im bliżej dna tym go mniej, a w warstwach może występować jego deficyt. Tlen musi być dostarczany w sposób ciągły, bo jest ciągle zużywany(oddychanie, biochemiczny rozkład zanieczyszczeń).
Osady denne ulegając rozkładowi również zużywają O2, jeśli jest mało zanieczyszczeń, to O2 rozpuszcza się z powietrza i stężenie jest stale wyrównywane. W przypadku rzek stężenie wyrównuje się podczas mieszania wody. W wodach głębinowych mieszanie jest wolne więc liczy się głównie dyfuzja jest ona czynnikiem limitującym mieszanie.
Do oceny względnej zawartości tlenu stosowany jest stopień nasycenia wody O2 (%).
Dwutlenek węgla.
Znajduje się w wodzie przede wszystkim w postaci rozpuszczonych cząsteczek gazu(CO2). Część ich jednak wchodzi w reakcję z wodą, tworząc kwas węglowy: CO2 + H2O <-> H2CO3. Źródłem CO2 w wodach nat. powierzchniowych są procesy utleniania substancji organicznej i reaeracja, natomiast w wodach podziemnych procesy geochemiczne, zachodzące w głębokich warstwach skorupy ziemskiej, związane ze zmianami zachodzącymi w skałach osadowych.
Pochłanianie CO2 z atmosfery obserwuje się prawie wyłącznie w morzach. Do procesów zmniejszających zawartość CO2 w wodach nat. należą: 1. wydzielanie CO2 do atmosfery z powodu przesycenia wody tym gazem, 2. zużywanie CO2 w procesie rozpuszczania skał węglanowych CaCO3 + H2O + CO2 ->Ca2+ + 2HCO3-, 3. zużywanie CO2 przez roślinność wodną w procesie fotosyntezy.
Znane są różne formy występowania CO2 w wodzie. Ogólny CO2 dzieli się na wolny, związany, agresywny i równowagi. Wolny występuje w wodach nat. w postaci rozpuszczonego CO2 i kwasu węglowego. Stanowi on sumę CO2 równowagi i CO2 agresywnego, przy czym układ ten nie jest stabilny. CO2 równowagi stanowi część wolnego CO2 niezbędnego do utrzymania wodorowęglanów w roztworze, zgodnie z reakcją:
Ca(HCO3)2<-> CO2 + CaCO3 + H2O. Pozostały CO2(nadmierny) może wykazać działanie agresywne w stosunku do betonu, stali i niektórych tworzyw. Stąd określa się go jako agresywny. Dwutlenek węgla związany występuje w wodach nat. pod postacią(HCO3-).
Zawartość CO2 w wodach nat. waha się od kilku do kilkuset(wody podziemne) mg/dm3.
Siarkowodór.
Jego obecność w wodach powierzchniowych jest zjawiskiem stosunkowo rzadkim, gdyż łatwo ulega utlenieniu tlenem rozpuszczonym. Ze względu na zerowe ciśnienie cząstkowe H2S w atmosferze, dłuższe zatrzymanie go w wodach powierzchniowych jest niemożliwe. Występuje na ogół w warstwach przydennych, bo dobrze rozpuszcza się przy dużym ciśnieniu.
Metan.
Jest bardzo słabo rozpuszczalny, ulatnia się dlatego tylko część pozostaje w wodzie.
Azot.
Gaz obojętny, nietoksyczny, potrzebny organizmom wodnym, ale nie w postaci N2, a w związkach azotu.
Oprócz wyżej wymienionych w wodach nat. występuje wodór i gazy szlachetne.
8. Substancje humusowe i inne naturalne związki organiczne w wodach naturalnych.
Jest znacznie mniej niż związków mineralnych ze względu na dużo mniejsze rozpuszczanie.
Wśród zw. org. wyróżnia się: związki humusowe, białka, cukry (węglowodany), a także prostsze substancje powstałe w reakcjach biochemicznych np. proste kwasy organiczne (octowy, szczawiowy).
Pochodzenie:
- wypłukiwanie z wierzchnich, uprawnych części gleb,
- procesy życiowe w wodach: metabolity, produkty procesów rozkładu materii organicznej,
- antropogeniczne - ścieki, spływy powierzchniowe (ogólnie źródła punktowe, liniowe, powierzchniowe). Zazwyczaj są to skażenia.
Związki humusowe.
Są to pośrednie produkty procesów rozkładu materii organicznej - bakterie rozkładają ją częściowo, a powstałe (złożone jeszcze) związki są powtórnie łączone. Dzięki temu może powstać praktycznie niezliczona liczba różnych związków. Dlatego nie można ich jednoznacznie zidentyfikować, nie ma też jakiegoś dominującego.
Związki te tworzone są głównie w glebie, a następnie wypłukiwane przez wodę (w wodzie też może następować ich tworzenie). Mogą pochodzić również z oczyszczonych metodami biologicznymi ścieków (humus ściekowy).
Budowa: związki bardzo skomplikowane, zawierają wiele połączonych ze sobą pierścieni aromatycznych, między pierścieniami jest dużo łańcuchów alifatycznych, a na zewnątrz makrocząsteczki znajdują się różne grupy funkcyjne. Wśród grup funkcyjnych dominują grupy karboksylowe (COOH) oraz fenolowe. Ponadto są grupy alkoholowe (OH), ketonowe, aldehydowe oraz grupy estrowe (COOR).
Mają masę cząsteczkową: 600 - ponad 100 tys. g/mol. W zależności od masy dzieli się na:
- kwasy fulwowe (fulwokwasy), poniżej 3000 g/mol
- kwasy hymatomelanowe, średnie masy
- kwasy huminowe, najcięższe
90% zw. hum. w wodzie stanowią kw. fulwowe, pozostałe 10% to związki cięższe. Cięższe trudno się rozpuszczają i nie są dobrze wypłukiwane z gleby. Dlatego w wodzie dominują związki lżejsze.
Rozpuszczalność w wodzie: Generalnie im mniejsza jest cząsteczka, tym lepiej się rozpuszcza w wodzie. Rozpuszczalność ułatwia również obecność grup polarnych (ww. grupy funkcyjne). Im więcej tych grup, tym rozpuszcz. jest większa. Polarność jest większa w przypadku mniejszych cząsteczek, bo większy jest udział grup funkcyjnych w stosunku do pierścieni aromatycznych.
Związki humusowe mają właściwości słabych kwasów organicznych ze względu na obecność grup COOH. Związki z grupami COOH są dysocjowane, ale obniżenie pH zmniejsza stopień dysocjowania. Przy obniżeniu pH do ok. 2 następuje wytrącenie znacznej części zw. hum. Dysocjowane pozostaną tylko małe cząsteczki, które mają dużo grup karboksylowych (w praktyce są to kwasy fulwowe). Wszystkie większe nie rozpuszczają się w środowisku kwaśnym.
Barwa: żółta - ciemnobrązowa. W miarę wzrostu ciężaru cząsteczkowego barwa pogłębia się (staje się ciemniejsza). Barwa zależy też od pH. Gdy cząst. są w stanie dysocjowanym - mocniejsza, gdy w stanie niezdysocjowanym (niższe pH) - barwa jaśniejsza.
Zawartość: Najczęściej kilka mg/l. Na terenach bagnistych - ponad 10 mg/l. W jeziorkach po wydobyciu torfu - nawet powyżej 100 mg/l.
Białka.
Ogólnie są to długie łańcuchy aminokwasów powstałe podczas polikondensacji. Poszczególne aminokwasy połączone są wiązaniami peptydowymi. Oprócz C,O,H zawierają N,S,P.
Jeżeli białko występuje w wodzie, to ulega hydrolizie pod wpływem organizmów. Powstają aminokwasy, które ulegają dalszemu rozkładowi.
Zawartość: poniżej 1 mg/l, rzadko powyżej.
Cukry (węglowodany).
Pochodzenie - głównie jako składniki roślin (błonnik, celuloza, skrobia). Złożone węglowodany (polisacharydy) hydrolizują do węglowodanów prostych np. glukoza, fruktoza - dużo jest izomerów geometrycznych o tym samym wzorze sumarycznym.
Zawartość: poniżej 1 mg/l, ale może być więcej.
9. Surfaktanty w wodach naturalnych
Surfaktanty (detergenty, środki powierzchniowo-czynne), są stosowane w wielu dziedzinach chemii gospodarczej jako środki myjące, piorące, dyspergujące, emulgujące, pianotwórcze, itp. Związki te są związkami organicznymi zawierającymi co najmniej jedną grupę polarną (hydrofilową), wykazującą powinowactwo do wody, oraz grupę hydrofobową, nie wykazującą tego powinowactwa. Cząsteczki detergentów gromadzą się na granicy faz, zmniejszając napięcie powierzchniowe rozpuszczalnika. Pozwala to uzyskać większą zwilżalność oczyszczanego lub barwionego tworzywa. Ze względu na charakter chemiczny grupy polarnej rozróżnia się detergenty anionowe (sulfoniany alkilowe RSO3-Na+, estry alkilowe kwasu fosforowego ROPO32-), detergenty kationowe (aminy alifatyczne) oraz detergenty niejonowe (alkilo-, alkilofenylo-polietylenoglikole). Najczęściej stosowanymi surfaktantami są alkilobenzenosulfoniany, zawierające od 10 do 15 atomów węgla w cząsteczce.
Masowe stosowanie surfaktantów powoduje coraz większą ich obecność w ściekach komunalnych. Mimo że surfaktanty nie są silnie toksyczne w stosunku do ssaków, często okazują się wysoko toksyczne dla organizmów wodnych, w tym mikroorganizmów osadu czynnego w biologicznych oczyszczalniach ścieków. Odporność detergentów na biochemiczną degradację jest jedną z przyczyn zanieczyszczenia rzek i zbiorników wodnych tymi związkami
10. ChZT i indeks nadmanganianowy
Indeks nadmanganianowy i ChZT to dwa podstawowe wskaźniki utlenialności stosowane w badaniach mających na celu określenie ogólnej ilości związków organicznych zawartych w wodzie lub ściekach. W metodach tych, na podstawie ilości zużytego utleniacza możemy określić ilość związków organicznych zawartych w wodzie. Związki organiczne występują w dużej różnorodności, jedne z nich utleniają się łatwiej, inne trudniej, jedne potrzebują do utlenienia więcej utleniacza, a inne mniej. Dlatego w przypadku dużych wahań ich procentowej zawartości metody pomiaru utlenialności byłyby mało miarodajne. Na szczęście w wodach naturalnych procentowy skład związków organicznych jest bardzo podobny i dlatego metody te możemy stosować uzyskując dość dużą dokładność.
Metoda indeksu nadmanganianowego, zwana dawniej utlenialnością lub chemicznym zapotrzebowaniem tlenu metodą nadmanganianową polega na podgrzaniu roztworu wody i ściśle odmierzonej ilości utleniacza, którym jest nadmanganian potasu. W metodzie ChZT rolę utleniacza spełnia dwuchromian potasu. Po upływie określonej ilości czasu sprawdzamy, ile utleniacza uległo zużyciu w wyniku reakcji utleniania. Metodą miareczkową określamy ile utleniacza zostało w roztworze. Wiedząc, ile utleniacza dodaliśmy na początku, możemy określić ilość zużytego utleniacza. Następnie przeliczamy gramorównoważnikowo ilość zużytego utleniacza na ilość tlenu. W metodzie ChZT stosujemy o wiele ostrzejsze warunki, dlatego metody tej używa się dla ścieków i wód zanieczyszczonych trudno utleniającymi się związkami pochodzenia antropogenicznego, np. węglowodorami. Metodę indeksu nadmanganianowego stosujemy dla czystych i lekko zanieczyszczonych wód naturalnych.
11.BZTn - dotyczy głównie ścieków. Ścieki poddajemy biologicznemu oczyszczaniu .BZT jest miarą ładunku tych zanieczyszczeń, które mogą być usunięte w oczyszczalni i ile ten proces będzie potrzebował tlenu ( Ilość tlenu zużyta na procesy biochemicznego rozkładu. ). Szybkość procesów biochemicznych możemy zapisać za pomocą równań.
dL/dt = - K*L
Szybkość rozkładu całej materii organicznej jest wprost proporcjonalne do sumarycznego stężenia tych składników.
dL/dt = - K*L
L = L0*e^(-K1*t)
y = BZTn
y = L0-L
L-zapotrzebowanie tlenu w danym czasie
L0-całe zapotrzebowanie na tlen
dL/dt-przyrost zapotrzebowania na tlen
K-wartość tego zapotrzebowania
K1-przelicznik z e na 10
BZTn-BZT po dowolnym czasie n , ilość tlenu która została skonsumowana
y = L0-L0*e^(-K*t)
y = L0(1-e^(-K*t))
y = L0(1-10^(-K1
12.OWO - ogólny węgiel organiczny-jego zawartość w lepszym stopniu niż indeks określa zawartość pierwiastka węgla. W tym miejscu pojawia się problem różnej zawartości procentowej węgla. Oznaczenie wymaga drogiej aparatury co powoduje stosunkowo rzadkie oznaczanie.
13.AOX- adsorbujące się organiczne halogeny.
Mają złożona strukturę i są mało lotne i trudne do oznaczenia. Nie można ich oznaczyć met chromatografii gazowej. Zalicza się min chlorowane ligniny. Są to związki polarne. Oznacza się wszystkie związki zawierające halogeny. Stosowane są w przemyśle chemicznym i ze ściekami mogą dostawać się do wód. Powstają poprzez działanie halogenów na węglowodory (chlorowanie ścieków i wody, bielenie papieru i tkanin). Pomimo małego stężenia są bardzo ważne, ponieważ maja dużą toksyczność. Czasami zwane ubocznymi produktami dezynfekcji. Trihalometany (THM) CHX3 X-halogeny F, Cl, Br, I. [1-100 um/dm3]. Właściwości rakotwórcze
14.Ekstrakt eterowy- pewne związki pochodzenia antropologicznego jak tłuszcze i oleje mineralne, nie rozpuszczają się w wodzie, ale za to rozpuszczają się w węglowodorach (wyekstrahowuja się). Rozpuszczalność olejów maleje wraz że wzrostem masy cząsteczkowej, a rośnie wraz ze wzrostem zanieczyszczenia. Większe znaczenie mają oleje emulgowane. Oleje doprowadzane do wód tworzą cienka warstwę na powierzchni wód. Oleje wprowadzane do wód podlegają procesom stopniowej i powolnej biodegradacji. Przemiany te zależą od temperatury. Oblepiają przedmioty zanurzone w wodzie, a także zawiesiny, które tworząc większe aglomeracje, opadają na dno. Stężenie: 50-100 mg/dm3. Działanie: zmiany organoleptyczne wody, śmierć organizmów przydennych. Oznaczanie: polega na ekstrakcji olejów z wody za pomocą rozpuszczalnika organicznego (eter naftowy). Po odparowaniu rozpuszczalnika zawartość olejów określa się wagowo. Jest to metoda niespecyficzna. Oznacza się wszystkie substancje, które ulęgają ekstrakcji min. Oleje mineralne, roślinne, tłuszcze, inne związki organiczne. Gdy odparowujemy wodę w tym oznaczeniu, to brak suchej pozostałości oznacza, że woda nie jest zanieczyszczona tymi związkami, ale może być zanieczyszczona związkami innego pochodzenia.
15.Twardość - [mg CaCO3dm3] jest pojęciem umownym, określająca zawartość kationów wapnia i magnezu. Im twardość wody jest wyższa, tym mamy do czynienia z bardziej niekorzystnymi warunkami.
Rozróżniamy twardość:
-ogólna - 11 - - 11 - całkowita wartość jonów Mg2+ i Ca2+
-węglanowa- zawartość węglanów i wodorowęglanów wapnia i magnezu. Jest ona przemijającą, bo zanika pod czas gotowania wody, wytracając węglany wapnia, magnezu i żelaza, tworząc niepożądany kamień kotłowy.
-niewęglanowa- różnica miedzy twardością ogólną, a węglanową. Określa zawartość anionów dwuwartościowych.
Zasadowość - zdolność wody do zobojętniania silnego kwasu. Nadają ją zasady tj. HCO3-, CO32-, OH-. Oznaczenie: miareczkowanie próbki wody silnym kwasem wobec wskaźników lub posługując się pHmetrem.
CO32- + H+ HCO3-
HCO3 + H+ H2O +CO2
16. Równowaga węglanowa-
CaCO3 +H2O +CO2 Ca2+ +2 HCO3-
Reakcja ta nie przebiega do końca i stali się pewien stan równowagi. Reakcja zachodzi do momentu w którym nie przereaguje substrat (stary węglan wapnia, Co2), stężenia tak muszą się ułożyć, by nastąpiła równowaga. Ustalone wartości stężeń noszą nazwę Stałej równowagi chemicznej. Możemy założyć, ze stężenie H2O w wodach jest stałe. CaCo3 tworzy oddzielna fazę i jest to zw. czysty . jeżeli w H2O jest za dużo CO2, to stan równowagi nigdy nie zostanie osiągnięty z uwagi na brak węglanów. Nadmiar CO2 po wydobyciu wody może on być agresywny dla zanurzonych w wodzie elementów. CO2 agresywny (N) nadmiarowy- woda, która zawiera dużo CO2, tak aby podtrzymać równowagę. CO2 agresywny, to nadmiarowy. CO2 równowagi (R)- takie stężenie, które pozwala na zajście równowagi. Ilość CO2 jest ograniczona jego rozpuszczalnością. W wodach powierzchniowych CO2 może być ulatnianie do atmosfery. Po wystygnięciu wody może ulatniać się wolny węglan. Praktycznie reakcja może nie zachodzić, bo woda jest układem alkalicznym. Wartość K stałej równowagi zależy od temp. Dla różnych związków wartość stałej się zmienia dla rożnych wartości temp. W wyższych temp wartość K będzie mniejsza, szybkość reakcji się zwiększa. Cały wodorowęglan wapnia ulegnie rozkładowi i powstanie węglan wapnia. W wodzie może pozostać wapń z wolnymi zw. Reakcja wytrącania węglanu wapnia stwarza różne problemy- wytrącanie kamienia kotłowego.
17) Atmosfera. Skład powietrza troposfery.
Atmosfera - cienka powłoka gazowa otaczająca Ziemię i utrzymywana przez siły grawitacji. Gęstość atmosfery (największa przy powierzchni Ziemi) - maleje wraz ze wzrostem wysokości w końcu staje się nieodróżnialna od gęstości gazu między planetarnego.
W najniższej części atmosfery (troposfera do 10 - 17 km - w zależności od szerokości geograficznej ) powietrze jest dobrze wymieszane w wyniku konwekcji. Temperatura w tej strefie maleje z wysokością od 150 C średnia temp. na poziomie morza do około - 500 C. W warstwie tej znajduje się około 75% całkowitej masy atmosfery. W warstwie tej przebiegają wszystkie zjawiska związane z pogodą. Troposferę kończy tropopauza.
Stratosfera - temperatura wzrasta do około 00 C na wysokości 50 -55 km. Wzrost temp. z wysokością powoduje mniejszą podatność atmosfery na większych wysokościach do mieszania w kierunku pionowym. Stratosferę kończy stratopauza.
Kolejna warstwa to Mezosfera ( temp. spad do około -800C na wysokości około 80 - 85 km). Mezosferę kończy mezopauza. Mezopauza przechodzi w termosferę.
Atmosferę podzielić można na homosferę i heterosferę.
Homosfera - część atmosfery do około 120 km w której następuje stosunkowo dobre mieszanie się składników. W heterosfera (powyżej 120 km) mieszanie burzliwe jest bardzo słabe i cząstki gazu mogą się rozdzielać pod wpływem sił grawitacyjnych. Granica między homosferą i heterosfera nazywa się turbopauzą. Ze względu na separację grawitacyjną w górnych warstwach następuje względna koncentracja lekkich atomów (helu i wodoru), natomiast cięższych (azotu i tlenu) w niższych.
Skład chemiczny troposfery:
Ogólny skład nie zanieczyszczonego powietrza
Gaz |
Stężenie |
Azot N2 |
78,084% |
Tlen O2 |
20,946% |
Argon Ar |
0,934% |
Para wodna H2O |
0,5 - 4% |
Dwutlenek węgla CO2 |
360 ppm |
Neon Ne |
18,18 ppm |
Hel He |
5,24 ppm |
Metan CH4 |
1,7 ppm |
Krypton Kr |
1,14 ppm |
Wodór H2 |
0,5 ppm |
Ksenon Xe |
0,087 ppm |
ppm -część na milion
18.Związki siarki w powietrzu atmosferycznym
Dwutlenek siarki jest bezbarwnym gazem o ostrej duszącej woni. Jest cięższy od powietrza , charakt. się ograniczoną rozpuszcz. w wodzie. Stwierdzono toksyczność dla ludzi, zwierząt mikroorganiz., niektórych bakterii i pleśni oraz dla roślin.
SO2 jest emitowany ze źródeł naturalnych i antropogennych. Źródłami naturalnymi so wybuchy wulkanów , procesy rozkładu materii organicznej , pożary lasów i stepów oraz erozja gleb. SO2 pochodzący ze źródeł antropogennych powstaje główni w wyniku spalania paliw kopalnych w trakcie którego utleniana jest siarka w nich zawarta.
Dostaje się do atmosfery wraz ze spalinami , a następnie jest z niej usuwana w wyniku suchej i mokrej depozycji, a także depozycji przez mgłę i kropelki chmurowe .
Pod wpływem tlenu i obecnych w atmosferze katalizatorów , a także w reakcjach z rodnikami, ozonem, i nadtlenkiem wodoru , SO2 utlenia się do H2SO4 . Kwas siarkowy ulega dysocjacji elektr. w kropelkach wody , w wyniku czego powstają jony SO4-2
i H+ , które stanowią gł. związki zakwaszające. Aerozol siarczanowy (SO4-2) , działając w kierunku jego osłabienia, przyczynia się do zmniejszenia widzialności oraz ma bardzo szkodliwy wpływ na zdrowie ludzkie , wpływa również na zubożenie warstwy ozonowej w stratosferze.. Zimą w obszarach wysoko uprzemysłowionych SO2 przyczynia się do powstawania tzw. czarnego smogu , który ma bardzo niekorzystny wpływ na środow. a szczególnie na zdrowie ludzkie.
Siarkę zawartą w węglu , zwaną siarką całkowitą dzieli się na siarkę palną, biorącą udział w procesie spalania i na siarkę związaną wchodzącą w skład popiołu lotnego i żużla. Podział ten jest umowny , gdyż siarka palna utleniona do SO2 I SO3 łączy się częściowo w górnych partiach komory paleniskowej z cząstkami CaO i MgO zawartymi w popiele lotnym , stając się siarką związaną.
Związki siarki. Zanieczyszczenie atmosfery powodują gazowe związki siarki - SO2, SO3, H2S, kwas siarkowy H2SO4 i siarczany różnych metali.
Dwutlenek siarki (SO2 ) jest bezbarwnym, silnie toksycznym gazem o duszącym zapachu. Wolno rozprzestrzenia się w atmosferze ze względu na duży ciężar właściwy (2,93 kG/m3, gęstość względna 2,26). Powstaje m. in. w wyniku spalania zanieczyszczonych siarką paliw stałych i płynnych (np. węgla, ropy naftowej) w silnikach spalinowych, w elektrociepłowniach, elektrowniach cieplnych. Największy udział w emisji SO2 ma przemysł paliwowo-energetyczny. Opalana węglem elektrownia o mocy 1000 MW emituje do atmosfery w ciągu roku 140 000 ton siarki, głównie w postaci SO2. Dwutlenek siarki utrzymuje się w powietrzu przez 2-4 dni i w tym czasie może si przemieścić na bardzo duże odległości. W powietrzu SO2, utlenia się do SO3, a ten z kolei łatwo reaguje z wodą ( z parą wodną zawartą w powietrzu) tworząc kwas siarkowy - H2SO4, jeden ze składników kwaśnych deszczów.
SO2 + 1/2 O2 => SO3 +H2O+ H2SO4
PRZYCZYNY I SKUTKI KWAŚNYCH DESZCZY
PRZYCZYNY |
SKUTKI |
|
niszczenie lasów skażenie gleby wykluczając możliwość ponownego sadzenia drzew niszczenie roślinności szkodliwy wpływ na ludzi kamień, beton, farba i stal ulegają procesom przyspieszonego niszczenia w miastach o zasiarczonej atmosferze (niszczenie budynków) kwaśne deszcze powodują zakwaszenie jezior, zanim dotrą przechodzą przez warstwę gleby rozpuszczają wówczas glin, który normalnie byłby mocno związany z cząstkami gleby. Glin jest przenoszony do jeziora, gzie zatruwa niektóre organizmy wodne kwas zawarty w deszczu zmienia charakter gleby także w inny sposób, a mianowicie wpływa na mikroorganizmy, które w normalnych warunkach pomagają utrzymywać właściwości uprawne gleby, a także na korzenie włosowate, które służą roślinom do pobierania składników pokarmowych ekosystem poddawany codziennym dawkom dwutlenku siarki może uruchomić skomplikowany system odpornościowo- genetyczny i doprowadzić do rozwoju różnych zmutowanych formacji ryby w zakwaszonym jeziorze cierpią z powodu bezpośrednich uszkodzeń skrzeli i innych narządów ciała spowodowanych kwasem ilość i jakość ryb w jeziorach obniża się, wiele gatunków nie jest w stanie przeżyć |
Źródła emisji:
Uwalnia się przy spalaniu:
paliw kopalnych w elektrowniach
paliw kopalnych w paleniskach przydomowych
paliw w silnikach pojazdów.
Dwutlenek siarki jest przyczyną :
zwolnienia procesów fotosyntezy, oddychania i parowania,
małego przyrostu masy roślinnej,
uszkodzenia blaszek liściowych oraz zakłócenia ich rozwoju.
Dwutlenek siarki wchodząc w reakcję z parą W tak popularnych regionach turystycznych jak |
|
Karkonosze i Góry Izerskie obserwuje się
od pewnego czasu obumieranie lasów.
Badania powietrza w tym rejonie wykazały
bardzo dużą zawartość SO2.
Stopień zanieczyszczenia powietrza atmosferycznego dwutlenkiem siarki można określić na podstawie tzw. skali porostowej.
Pewne gatunki wykazują określoną wrażliwość na średnie roczne stężenie SO2 w powietrzu.
Na niektórych obszarach występują tzw. pustynie porostowe.
19. Związki azotu w powietrzu atmosferycznym.
Gleby Ziemi są bogate w różnorodne związki azotu, co powoduje wytwarzanie wielu śladowych substancji gazowych, zawierających ten pierwiastek. Można wymienić mocznik (diamid kwasu węglowego NH2CONH2), jako typowy związek organiczny wytwarzany biologicznie). W wyniku hydrolizy zachodzi przemiana NH2CONH2 w amoniak (NH3) i CO2.
NH2CONH2 + H2O → 2 NH3 + CO2
Jeśli gleba, w której przebiega hydroliza ma odczyn alkaliczny, może wydzielać się gazowy amoniak, podczas gdy w środowisku kwaśnym amoniak reaguje z wytworzeniem jonów amonowych(NH4+)
NH3 + H+ → NH4+
Rośliny mogą pochłaniać glebowy NH3 lub NH4+ bezpośrednio, a niektóre mikroorganizmy utleniają NH3, zużywając go jako źródło energii do oddychania. Jedną z możliwych reakcji jest
2 NH3 + 2 O2 → N2O + 3 H2O
Proces ten stanowi biologiczne źródło podtlenku azotu, trwałego gazu śladowego występującego w troposferze. W przyrodzie przebiega wiele innych reakcji związków azotowych w glebie, w których są wytwarzane gazy: NH3, N2, N2O i monotlenek azotu (NO).
Ważnym źródłem azotu są także procesy spalania paliw. Obecne w paliwach organiczne związki azotu podczas spalania łączą się z tlenem z powietrza tworząc tlenki azotu (NOX). Reakcja między ditlenkiem azotu (NO2) i rodnikiem OH prowadzi do powstania HNO3, który ma znaczący udział w kwaśnych deszczach.
20. Reakcje tlenków azotu w powietrzu atmosferycznym.
NO2 + hv (długość fali poniżej 310 nm) → O + NO
dysproporcja: 2 NO2 + H2O → HNO3 + HNO2
powstawanie kwasu azotowego: NO2 + OH˙ → HNO3
asocjacja: 2 NO2 → N2O4
21.Kwaśne deszcze.
W warunkach naturalnych pH opadu atmosferycznego wynosi 5,6. Wartość taką nadaje wodzie deszczowej proces rozpuszczania dwutlenku węgla.
CO2(g) + H2O(c)↔H2CO3(aq)
H2CO3(aq) ↔H+(aq)+HCO-3(aq)
HCO-3(aq) ↔ H+(aq)+HCO-3(aq)
Opad, którego wartość pH jest niższa od 5,6 to kwaśny opad.
Głównymi substancjami które powodują zakwaszanie atmosfery, poniżej naturalnego ph=5,6 a co za tymi idzie kwaśne deszcze są dwutlenek siarki i tlenki azotu.
SO2(g)+H2O(c) ↔H++HSO-3(aq)
2HSO-3(aq)+O2(aq)↔2H+(aq)+2SO2-4(aq)
H2O2+HSO-3→ SO42-+H++H2O
O3+HSO3_→SO42-+H++O2
NO2(g)+OH(g)↔HNO3(g)
N2O5+H20↔2HNO3
Naturalnym źródłem SO2 w atmosferze są wulkany. Istotną sprawą jest fakt, że wulkany stanowią bardzo nieciągłe (w czasie i przestrzeni) źródło. Z tego względu mogą one „jednorazowo” wprowadzać stosunkowo duże ilości SO2. Dodatkowym źródłem emisji są wulkaniczne pęknięcia i szczeliny łagodnie emitujące gazy przez długi czas.
Antropogenicznym źródłem SO2 są źródła stacjonarne: kotły, palniki (w tradycyjnych maszynach parowych), paleniska domowe, turbiny parowe oraz elektrownie. Spalane w nich paliwo zawiera siarkę (np: węgiel 0,2-7,0; koks 0,3 - 0,9 ; paliwa płynne 0,5 - 4,0% -masy) która utlenia się do SO2.Siarka częściowo występuje w postaci pirytu FeS2.
4FeS2+11O2→8SO2+2Fe2+O3. Dodatkowo zanieczyszczenia metaliczne zawarte w paliwach (żelazo lub mangan) katalizują przemianę rozpuszczonego SO2 do H2SO4.
W Europie przemysł „pompuje” rocznie do atmosfery 34 mln ton dwutlenku siarki i ok. 21 mln ton tlenków azotu. Obok spalania węgla, ropy naftowej, naturalnych gazów, innych paliw kopalnych, „producentami kwasów do atmosfery” są konwencjonalne elektrownie, rafinerie ropy naftowej, zakłady chemiczne oraz motoryzacja.
Kwaśne deszcze powodują zakwaszanie gleby, obumieranie dużej ilości drzew, niszczenie runa leśnego i innych roślin (hamowanie fotosyntezy), a także niszczenie budowli i innych obiektów stworzonych przez ciężką czasami niewolniczą pracę człowieka. Jak wiadomo H2SO4 działa korozyjnie w stosunku do elementów z żelaza i niszczy materiały budowlane.
Kwas siarkowy powoduje przemianę kamienia wapiennego w gips.
H2SO4+CaCO3+H2O→CO2+CaSO4*2H2O
Gips jest rozpuszczalny pod wpływem wody deszczowej, ponadto gips zajmuje większą objętość niż kamień wapienny i „eksploduje od wewnątrz”.
22.Ozon w troposferze i atmosferze
Ozon (O3) w naturalny sposób powstaje na wysokości 30-50 km. Na wysokości ok.25 km ozon już nie powstaje, jak również nie ulega rozpadowi. Powoli osiadając tworzy maksymalną koncentrację na wysokości ok. 23 km. Ta warstwa atmosfery to stratosfera, w której występuje prawie 90% ozonu. Do dolnych, gęstych warstw atmosfery ozon przenika w niewielkich ilościach, gdzie szybko ulega rozpadowi. Jest to troposfera, której pułap sięga (w zależności od szerokości geograficznej)wysokości 8-17 km, zawierająca około 10% ozonu. W stratosferze ozon stanowi swoistą "tarczę ochronną" chroniącą biosferę Ziemi. W troposferze jest niepożądany, gdyż należy do gazów cieplarnianych. Ozon troposferyczny jest zanieczyszczeniem wtórnym. Powstaje w wyniku reakcji fotochemicznych zachodzących w powietrzu zanieczyszczonym tlenkami azotu, węglowodorami i tlenkiem węgla (są to głównie reakcje transformacji tlenowych związków azotu) pochodzących ze źródeł antropogenicznych, głównie transportu drogowego. Formowaniu ozonu sprzyja wysoka temperatura, duże nasłonecznienie i duża wilgotność powietrza. Przy wyższych stężeniach ozon powoduje podrażnienia oczu, głównie zapalenie spojówek, zmiany w parametrach widzenia, zmiany czynności płuc (szczególnie u dzieci), zwiększoną częstotliwość ataków astmy oraz wzrost zachorowalności na raka skóry.
Substancje które przyczyniają się do tworzenia ozonu przyziemnego w powietrzu
tlenki azotu, tlenek węgla, etan, etylen, acetylen, propan, propylen, butan, i-butan, 1-buten, trans-2-buten, cis-2-buten, 1,3-butadien, pentan, i-pentan, 1-penten, 2-penten, izopren, heksan, i-heksan, heptan, oktan, i-oktan, benzen, toluen, etylobenzen, m+p-ksylen, o-ksylen, 1,2,4-trimetylobenzen, 1,2,3-trimetylobenzen, 1,3,5-trimetylobenzen, formaldehyd, węglowodory inne niż metan ogółem
Powstawanie ozonu jest procesem fotochemicznym, w którym zużywana jest energia, którą niesie światło. Dostateczną ilość energii , potrzebną do powstania ozonu, zawiera promieniowanie nadfioletowe o dl fali mniejszej niż 242nm.
O2(g) + hν O(g) + O(g)
hν -foton promieniowania nadfioletowego
pojedyncze atomy tlenu powstałe w tej reakcji mogą reagować z O2
O2(g) + O(g) O3(g)
Wytwarzanie ozonu może być zrównoważone reakcją rozkładu O3(g) :
O3(g) + hν O2(g) + O(g)
O3(g) + O(g) 2O2(g)
O(g) + O(g) + M(g) O2(g) + M(g)
gdzie M. - „trzecie ciało”(zazwyczaj cząsteczka O2 lub N2 , które unosi nadmiar energii wyzwalającej się podczas reakcji.
23. Utleniacze w powietrzu atmosferycznym
Utleniacze są zanieczyszczeniami wtórnymi, powstałymi na drodze reakcji chemicznych podstawowych zanieczyszczeń. Zalicza się do nich między innymi ozon, który tworzy się w atmosferze pod wpływem światła słonecznego na skutek reakcji pomiędzy tlenkami azotu a węglowodorami. Tlenek węgla (CO) powstaje wskutek niezupełnego spalania i w mniejszym lub większym stopniu powstaje we wszystkich procesach spalania. W atmosferze utlenia się do dwutlenku węgla (CO2). Głównym źródłem emisji tlenku węgla jest transport drogowy, a w szczególności pojazdy z silnikami benzynowymi. Innym dużym źródłem CO jest sektor komunalny i rolnictwo oraz niektóre procesy przemysłowe (produkcja stali). Jest on gazem silnie toksycznym jednak ze względu na małą gęstość (1,25 kg/m3) rozprzestrzenia się szybko w powietrzu atmosferycznym. Źródłem antropogenicznym dwutlenku węgla (CO2), będącego głównym gazem cieplarnianym, są wszystkie procesy spalania węgla, produktów naftowych, torfu i innych paliw.
Dwutlenek węgla nie jest gazem toksycznym i jego zawartość w powietrzu nie jest normowana. Wyniki zawartości CO2 w powietrzu dają jednak informację o ogólnym stanie jego zanieczyszczenia, ponieważ emisji dwutlenku węgla towarzyszy we wszystkich procesach przemysłowych emisja innych zanieczyszczeń powietrza.
Jest gazem, który w największych ilościach wśród zanieczyszczeń gazowych emitowany jest do atmosfery. Istnieje obawa, że dalszy wzrost koncentracji dwutlenku węgla (a także pary wodnej, która jest najskuteczniejszym gazem cieplarnianym) w powietrzu spowoduje podniesienie temperatury powierzchni ziemi w wyniku spotęgowania efektu cieplarnianego, co wywoływać będzie dalsze zwiększone parowanie, a to z kolei wymusza dalszy wzrost temperatury
Utleniacze fotochemiczne to grupa zanieczyszczeń powstających w wyniku reakcji chemicznych zachodzących pod wpływem promieniowania słonecznego w powietrzu zanieczyszczonym tlenkami azotu i węglowodorami. Głównym składnikiem tej grupy jest ozon. Podwyższone stężenia ozonu mogą u osób wrażliwych powodować podrażnienie górnych dróg oddechowych i oczu, kaszel i ból głowy. Zwiększa się też prawdopodobieństwo występowania ataków astmy. Możliwe są uszkodzenia części zielonych roślin. Stężenia ozonu w czystym powietrzu, w przyziemnej warstwie atmosfery, dochodzą chwilowo do 120 g/m3, natomiast w obecności tlenków azotu i węglowodorów (tzw. prekursorów ozonu) mogą być znacznie większe. Formowaniu ozonu w dolnej atmosferze sprzyja wysoka temperatura powietrza oraz duże natężenie promieniowania słonecznego. Stężenia ozonu zmieniają się cyklicznie w okresie doby, tygodnia i roku. Na ich poziom największy wpływ mają warunki meteorologiczne (zwłaszcza natężenie promieniowania słonecznego i temperatura powietrza) oraz zmiany natężenia emisji i stężeń tlenków azotu i węglowodorów.
24. Freony w powietrzu atmosferycznym - dziura ozonowa
Freony to gazy - chlorowe i fluorowe pochodne węglowodorów (CFC) - do niedawna powszechnie stosowane w dezodorantach i w lodówkach jako materiał chłodzący. CFC dzięki swej niemal całkowitej chemicznej bierności były uważane za idealny surowiec. Jednak uwalniane w coraz większej ilości zaczęły się przedostawać do wyższych warstw atmosfery ( stratosfery)- tam, gdzie jest ozon(ok. 90%). Pod wpływem pochodzącego ze Słońca promieniowania UV we freonach pękają wiązania chemiczne i rozpadają się one - podobnie jak tlen - na poszczególne atomy. Uwalniający się chlor powoduje, że cząsteczki ozonu rozpadają się. Przerwany zostaje łańcuch reakcji, dzięki którym promienie UV nie docierają do Ziemi. Reakcja fotodysocjacji dla przykładowego przebiega w następujący sposób:
CFCl3(g) + hν CFCl2(g) + Cl(g)
CF2Cl2(g) + hν CF2Cl2(g ) + Cl(g)
W reakcjach tych powstają atomy chloru, które reagują z O3
O3(g) + Cl(g) O2(g) + Cl(g) + ClO(g)
ClO(g) + O(g) O2(g) + Cl(g)
Co daje w sumie:
O3(g) + O(g) 2O2(g)
ClO które powstaje w wyniku reakcji atomów chloru z O3 nie musi reagowac z tlenem, a zamiast tego może wchodzić w interakcje ze związkami azotu
ClO(g) + NO2(g) + M ClONO2(g) + M. M. - „trzecie ciało”
W obecności cząstek stałych chlor może być uwolniony ponownie w reakcji:
ClONO2(g) + HCl Cl2(g) + HNO3(g)
Cl2(g) + hν 2Cl(g)
2Cl(g) + 2O3(g 2ClO(g) + 2O2(g)
2ClO(g) + M. Cl2O2(g) + M
Cl2O2(g) + hν ClO2(g) + Cl(g)
ClO2(g) + M. Cl(g) + O2(g) + M.
Po zsumowaniu wyżej wymienionych 6 reakcji otrzymujemy:
2O3(g) 3O2(g)
reakcje te przebiegają wyjątkowo szybko w niskiej temperaturze. Co w pewien sposób może tłumaczyć dramatyczny spadek ilości ozonu, obserwowanego nad kontynentem antarktycznym.
25. Budowa i skład skorupy ziemskiej.
W skład skorupy ziemskiej wchodzą:
- skały kwaśne krzemianowe
- skały zasadowe z piroksenu i oliwinu ??
Skład pierwiastkowy skorupy ziemskiej:
- minerały zawierające: O2 - 46.6%; Si - 27.7%; Al - 8.1%; Fe - 5.6%; Ca - 3.6%; Na - 2.8%; K - 2.6%; Mg - 2.1%; inne - 1.4%
- woda i powietrze glebowe
Woda jest higroskopijnie związana siłami Van der Waalsa z cząsteczkami glebowymi. Część wody jest związana trwale i nie może zostać pobrana przez rośliny, a często występuje jako woda wolna zawierająca rozpuszczone związki organiczne
Powietrze glebowe zawiera metan, siarkowodór, oraz zwiększoną ilość CO2
Skorupa ziemska od środka składa się z:
- jądra wewnętrznego
- jądra zewnętrznego
- płaszcza dolnego i górnego
- skorupy ziemskiej go 40km
Jądro wypełnione jest ciekłymi metalami: Ni, Si, Fe. W płaszczu dolnym są tlenki magnezu i żelaza, płycej występują krzemiany magnezu i żelaza.
26. Skład glebowy (ogólnie).
Gleba zbudowana jest nie tylko z ciał stałych - minerałów, ale także z wody i powietrza glebowego. Część wody jest higroskopijnie związana siłami Van der Waalsa z cząstkami glebowymi. Część wody jest związana trwale i nie może zostać pobrana przez rośliny, a część występuje jako woda wolna zawierająca rozpuszczone związki organiczne. Wodę glebową ze względu na stopień związania dzielimy na:
- wodę gruntową - wypełnia wolne przestrzenie w gruncie, podlega ruchowi
- wodę grawitacjną - wypełnia szerokie przestrzenie między cząsteczkami gleby, jest łatwo dostępna dla roślin, przyczynia się do przewietrzania gleb
- wodę kapilarną - wypełnia włosowate kanaliki glebowe i jest dostępna dla roślin
- wodę błonkowatą - tworzy się na powierzchni wody higroskopijnej, jest słabiej związana niż woda higroskopijna. Bierze udział w procesach glebowych.
Powietrze glebowe zawiera siarkowodór, metan i zwiększoną zawartość CO2.
Wierzchnia warstwa gleby podczas suszy zawiera tylko kilka % wilgoci, podczas gdy warstwa wodonośna do 50%.
Stałe składniki mineralne gleby:
- kwarc
- krzemiany
- glinokrzemiany
- skalenie
- miki
27. Woda i powietrze glebowe.
Powietrze glebowe występuje tylko w strefie areacji. Zawiera ono CH4, H2S, i wyższe w stosunku do atmosfery stężenie CO2.
Wodę higroskopijną można podzielić ze względu na stopień związania z glebą:
- wodę higroskopijną, związaną siłami Van der Waalsa z cząsteczkami glebowymi i nie może być pobrana przez organizmy żywe
- woda gruntowa - wypełnia wolne przestrzenie w gruncie i podlega ruchowi
- woda grawitacyjna - wypełnia średnie przestrzenie między cząsteczkami gleb. Jest łatwo dostępna dla roślin i przyczynia się do przewietrzania gleb.
- woda kapilarna - wypełnia włosowate kanaliki glebowe i jest dostępna dla roślin
- woda błonkowa - tworzy się na powierzchni wody higroskopijnej, jest słabiej związana niż woda higroskopijna. Bierze udział w procesach glebowych.
Powietrze glebowe zawiera siarkowodór, metan i zwiększoną zawartość CO2.
Wierzchnia warstwa gleby podczas suszy zawiera tylko kilka % wilgoci, podczas gdy warstwa wodonośna do 50%.
25. Skład gleby gleba - kilkudziesięciocentymetrowa warstwa gruntu; struktura gleby: porowata (dużo wolnych przestrzeni pomiędzy cząsteczkami (ziarnami) ciała stałego)oprócz części stałych w glebie zawarte również powietrze i woda - ich zawartość różni się w zależności od rodzaju gleby, woda penetruje glebę, wypełnia wolne przestrzenie i zatrzymuje się na pewnych głębokościach (np. na glinie) tworząc warstwę wodonośną (lustro wody) powyżej warstwy wodonośnej również znajduje się woda, a także powietrze glebowe - o zmienionym składzie (zawiera m.in. produkty rozkładu substancji organicznej np. CO2);
CZĘŚCI STAŁE GLEBY: substancje mineralne - na pewnej głębokości w ogóle nie ma części organicznych, tylko same mineralne np. piasek kwarc: SiO2 (krzemionka) krzemiany: augit, glaukonit glinokrzemiany:
skalenie; ortoklaz: K2O · Al2O3 · 6SiO2;albit: Na2O · Al2O3 · 6SiO2;anortyt: CaO · Al2O3 · 6SiO2; miki (budowa blaszkowa);muskowit (illit, welmikulit): K2O · Al2O3 · 6SiO2 · 2H2O;kalcyt, dolomit: CaCO3 · MgCO3;magnezyt: MgCO3;syderyt: FeCO3; gips: CaSO4; hematyt: FeO3; magnetyt: FeO4 (FeO · Fe2O3); diaspor: Al2O3
substancje organiczne - obecność związana z procesami biologicznymi, bo w glebie żyją organizmy i rośliny, które ulegają rozkładowi - powstają produkty przemiany m.in. związki humusowe (te są trwałe); może być nawet do 50% związków organicznych (np. torf, czarnoziemy); piaski - b. mało substancji organicznych
minerały: związki o budowie krystalicznej z ewentualnymi zanieczyszczeniami (domieszkami); rozpad takich skał - nazywany procesem wietrzenia - powoduje powstawanie gleby
gleby: zawierają minerały powstałe z rozpadu minerałów pierwotnych oraz z wietrzenia skał
kwarc: ostateczny produkt wietrzenia; trwały związek chemiczny; podstawowy składnik piasku
RODZAJE WIETRZENIA: utlenianie: ulegają mu siarczki i żelazo; karbonatyzacja: w wyniku tego procesu powstają węglany CaCO3; kaolinizacja: w wyniku tego procesu powstaje kaolinit Al2O3 · 2SiO2 · 2H2O; desilikacja: w wyniku tego procesu powstaje SiO2 ; hydratacja: w wyniku procesu cząsteczki wody przyłączają się do cząsteczek związku
27. Woda i powietrze w glebie Zawartość wody i powietrza w glebie jest różna w czasie. Gleba ma strukturę porowatą, ma dużo wolnych przestrzeni pomiędzy granulkami ciała stałego. Są one zajmowane przez wodę albo powietrze. Woda na pewnej głębokości ma swoje lustro (warstwa wodonośna). Powyżej tez jest woda, ale z powietrzem glebowym, które częściowo zostało przekształcone pod wpływem procesów( głownie rozkład materii organicznej). Pod czas utleniania tlenu powstaje CO2( pod wpływem temperatury nie zmienia objętości). Woda obecna w warstwie wodonośnej jest niej lub bardziej związana z cząstkami stałymi.
W zależności od stopnia związania wodę dzielimy na:
-woda gruntowa- swobodna, nie związana z cząstkami gleby, płynąca, wypełnia przestrzenie
-woda grawitacyjna- wypełnia szerokie przestrzenie pomiędzy cząstkami gleby, jest łatwo dostępna dla roślin i przyczynia się do przewietrzania gleb. Znajduje się nad lustrem wód gruntowych.
-woda kapilarna- wypełnia włoskowate kawałki glebowe, jest dostępna dla roślin (siły kapilarne). Woda przylega do ścianek. Siły włoskowate powodują, ze ta woda nie może spłynąć. Zostaje ona zatrzymana.
-woda błonkowata- tworzy się na powierzchni błonek wody higroskopijnej. Jest słabiej związana niż woda kapilarna. Bierze udział w procesach glebowych.
-woda higroskopijna- jest związana siłami Van der Waalsa z cząstkami glebowymi i nie może być pobrana przez mikroorganizmy i rośliny wyższe.
W wierzchniej warstwie gleby jeśli gleba jest sucha, to mamy tylko higroskopijną ( parę wodna %), która zawsze może wyparować. A przy słabym parowaniu wartość jej może sięgać do 20%. Im głębiej tym zawartość wody w glebie rośnie. W warstwie wodonośnej może jej być 50%, w zależności od porowatości gruntu.
28. Substancje mineralne gleb. Skorupa Ziemska składa się z wielu związków chemicznych o różnych strukturach. Minerały obecne w skorupie : (zaw. 46,8% tlenu); krzem - 27,7% (krzemionki, krzemiany, glinokrzemiany); glin - 8,1%; żelazo - 5%; wapń - 3,6%; sód - 2,8%; potas - 2,5%; magnez - 2,1%;pozostałe ( fosfor, mangan, bór, miedź, molibden, kobalt)- 1,4%. W zależności od stopnia zwietrzenia części mineralne gleby dzieli się według wielkości na: części szkieletowe (kamienie, żwir) - powyżej 1mm średnicy, części ziemiste - poniżej 1mm średnicy (piasek, pył), części spławiane - poniżej 0,02 lub 0,01mm, a wśród nich ważne części ilaste (to znaczy ił koloidowy) - poniżej 0,0001 lub 0,0002mm. Cząstki ziemiste, najczęściej połączone w agregaty, tworzą strukturę gleby, określającą stosunki wodno-powietrzne w glebie. O strukturze gleby decyduje układ cząsteczek mineralnych, zlepionych substancjami koloidowymi, tworzących różne agregaty.
Minerały wchodzące w skład gleby ( pod względem chemicznym):
1)Krzemiany ( połączenie krzemionki z tlenkiem innego metalu) :kwarc ( SiO2) - nie rozkłada się już do żadnego związku; augit; glaukonit 2)Glin ( tlenek glinu - może tworzyć połączenie z tlenkiem zasadowym)W krzemianach w wyniku różnych procesów krzem może być zastępowany glinem. Połączenia tlenków metali, glinu, krzemu - to glinokrzemiany, proporcje tych połączeń są różne: a) skalenie np.: ortoklaz K2O·Al2O3·6SiO2; albit Na2O·Al2O3·6SiO2; anostyt CaO·Al2O3·6SiO2 b) miki, o budowie warstwowej np.: muskowit K2O·3Al2O3·6SiO2·2H2O. Wśród glinu bardzo istotną rolę odgrywają substancje ilaste, są to glinokrzemiany pozbawione trzeciego tlenku, charakteryzują się drobnymi rozmiarami < 0,2÷0,4 μm >. Ze skupisk iłów glinokrzemianowych powstają masy łupków ilastych. Gleba powstaje ze skały macierzystej w procesie wietrzenia, które obejmuje między innymi hydrolizę kwaśną , zachodzącą w obecności CO2. Końcowym etapem takiej reakcji mogą być różne tlenki, przy czym tlenki glinu i krzemu są nierozpuszczalne w wodzie. Z minerałów ilastych powstałych w wyniku wietrzenia glinokrzemianów najważniejszymi są : kaolinit, haloizyt, montmorylonit, nontronit, beidelit, pirofilit i alofauny. Minerały te stanowią główne składniki gleby. Kaolinit, wchodzący w skład gliny, przedstawia glinokrzemian o stosunku krzemu do glinu 1:1. W strefie podzwrotnikowej hydroliza glinokrzemianów doprowadza do powstania koloidalnych osadów wodorotlenku glinowego i uwodnionej krzemionki, wskutek czego tworzy się boksyt bądź lateryt (boksyt zawierający znaczny procent tlenków żelaza) i opal (uwodniona krzemionka). 3)Węglany wapnia i magnezu : kalcyt ( węglan wapnia); dolomit CaCO3·MgCO3 (jest to mieszanina chemiczna, nie fizyczna); magnezyt MgCO3 ; gips CaSO4;diaspor ( wolny tlenek glinu ) Al2O3 4) Żelazo - 5% zawarte w litosferze
W wyniku hydrolizy soli żelazowych powstają skupienia koloidalnego osadu Fe(OH)3. Osad ten przeobraża się w limonit (towarzyszy mu często kwas fosforowy) i hematyt.- syderyt (żelazo) FeCO3→ z wodą daje rozpuszczalny wodorowęglan i to jest źródło żelaza w wodach podziemnych;- hematyt Fe2O3; - magnetyt Fe2O4. W trakcie procesu wietrzenia może zachodzić utlenianie żelaza i manganu np.: Fe2O3 · FeO + 2O2 → 6Fe2O3 (magnetyt przechodzi w hematyt)
Redukcja żelaza zachodzi w obecności reduktorów np.: siarkowodoru. Duża część żelaza nie występuje w postaci krystalicznej tylko w koloidalnej. Skład mineralny gleby ma wpływ na jej barwę np.: gleba zasobna w związki żelaza - barwa czerwona.
29. glinokrzemiany. Materiały ilaste W przyrodzie występuje około 800 znanych minerałów krzemianowych co czyni z nich najliczniejszą grupę minerałów. Ilościowy udział krzemianów i glinokrzemianów w budowie skał wynosi około 75% wagowych. Krzemiany, oprócz dominującego znaczenia skałotwórczego, mogą być także źródłem wielu cennych metali (np. krzemiany Ni, Zn, Zr, Li), tworzą również złoża wielu ważnych surowców mineralnych (kaolin, azbest, skalenie). Krzemiany, w których część anionów [SiO4]4 - zastąpiona jest anionami [AlO4]5 - , określane są jako glinokrzemiany. Glinokrzemiany, krzemiany glinu, podwójne tlenki o ogólnym wzorze mAl2O3⋅nSiO2. Najprostszemu wzorowi (Al2O3⋅SiO2) odpowiadają: andaluzyt, sylimanit, dysten. Ważniejszymi glinokrzemianami są także: kaolinit Al2O3⋅2SiO2 i topaz Al2SiO4(OH,F)2.Andaluzyt, Al2[SiO]5, minerał, polimorficzna odmiana krzemianu glinu. Krystalizuje w układzie rombowym, kryształy słupkowe, prążkowane, w przekroju poprzecznym kwadratowe. Barwa różowawa, czerwonawobrązowa, różowoczerwona, biała, białoszara, żółtawa, zielonkawa . Używany do wyrobu ceramiki szlachetnej oraz materiałów ogniotrwałych i kwasoodpornych. Znajduje również zastosowanie w jubilerstwie. Sylimanit, sillimanit, Al2SiO5, krzemian(IV) glinu, minerał, modyfikacja polimorficzna krzemianu glinu. Krystalizuje w układzie rombowym, w formie włóknistych lub igiełkowych skupień. Barwa najczęściej biała, szarawa, brunatnawa lub zielonawa. Częsty składnik skał metamorficznych (głównie gnejsów i łupków łyszczykowych).Dysten, cyjanit, minerał, krzemian glinu (glinokrzemiany), o barwie niebieskiej, zazwyczaj o szklistym połysku, składnik wielu skał metamorficznych. Dysten stosowany jest do wyrobu materiałów kwasoodpornych i ogniotrwałych. Kaolinit (Al4(OH)8Si4O10), pospolity minerał, składnik iłów i glin. Powstaje podczas wietrzenia (kaolinizacji) skał zawierających znaczne ilości skaleni. Topaz, Al2SiO4(OH, F)2, przezroczysty minerał o różnych odcieniach (głównie żółty lub złocisty, czasem zielonawy, niebieski, czerwony). Wzorzec mineralogiczny w skali twardości Mosha. Mullit, 3Al2O3⋅2SiO2, składnik materiałów ogniotrwałych i elektroizolacyjnych (tzw. mullitowych). Otrzymywany w procesie wypalania minerałów zawierających tlenek glinu i dwutlenek krzemu w odpowiednich ilościach, np. sylimanitu, cyjanitu, andaluzytu, a także mieszanin kaolinu i tlenku glinu.
MINERAŁY ILASTE Minerały ilaste są uwodnionymi glinokrzemianami Al, Mg i Fe, należącymi do krzemianów warstwowych. W zależności od wzajemnego układu warstw oktaedrów i tetraedrów mogą one należeć do krzemianów: * dwuwarstwowych o typie budowy 1:1, gdzie warstwa oktaedrów jest trwale i jednostronnie połączona z warstwą tetraedrów ; * trójwarstwowych o typie budowy 2:1, gdzie warstwa oktaedrów zamknięta jest między dwoma warstwami tetraedrów zwróconych do siebie wierzchołkami. Minerały ilaste na podstawie budowy dzieli się na: dwuwarstwowe: grupa kaolinitu (kandyty) - kaolinit, hydrohaloizyt i dickit. trójwarstwowe: grupa hydromik: illit i hydromuskowit, grupa montmorillonitu (smektyty): montmorillonit i beidelit, grupa wermikulitu: wermikulit. allofany - formy bezpostaciowe, Al2 . SiO2 . nH2O. Minerały ilaste powstają głównie w wyniku wietrzenia chemicznego innych glinokrzemianów, mogą także krystalizować z roztworu. Rodzaj powstających minerałów ilastych zależy od składu chemicznego wietrzejącego minerału pierwotnego oraz od warunków środowiska (odczynu, obecności różnych jonów itp.). Odczyn kwaśny sprzyja powstawaniu kaolinitu, obojętny lub alkaliczny - montmorillonitu. Grupa minerałów ilastych wyróżnia się zespołem charakterystycznych cech, do których należą: 1. Powierzchnia właściwa - cząstki ilaste ze względu na ich drobne wymiary posiadają dużą powierzchnię zewnętrzną , 2.Elektroujemne ładunki - cząstki ilaste posiadają zazwyczaj ładunek ujemny. Powoduje on przyciąganie przez powierzchnię minerału ilastego ogromnej ilości kationów. W ten sposób powstaje tzw. podwójna warstwa jonowa, 3.Właściwości fizyczne - w zależności od uwilgotnienia minerały ilaste wykazują odmienne właściwości. W stanie wilgotnym są one plastyczne i maziste, zaś w trakcie suszenia kurczą się, przechodząc w zwięzły i silnie scementowany materiał. Właściwości te decydują o szeregu właściwości fizycznych gleb, takich jak pęcznienie i kurczliwość, czy plastyczność i lepkość KAOLINIT Al4(OH)8(Si4O10) -najczęściej jest produktem wietrzenia glinokrzemianów (głównie skaleni) w środowisku wilgotnym i w obecności CO2 (odczyn kwaśny). Proces kaolinizacji rozwija się zwłaszcza w skałach granitowych i pokrewnych. Kaolinit jest głównym składnikiem glin i iłów, występuje powszechnie w zwietrzelinach i glebach, w których wchodzi w skład frakcji ilastej. ILLIT-najczęściej jest produktem wietrzenia glinokrzemianów (głównie skaleni). Może również powstawać w procesach przemian innych minerałów ilastych i muskowitu. Illit jest pospolitym składnikiem iłów, występuje powszechnie w zwietrzelinach i glebach, w których wchodzi w skład frakcji ilastej. MONTMORILLONIT - powstaje w strefie wietrzenia ciemnych skał magmowych: diabazów, bazaltów i gabra w warunkach alkalicznych. Jest on głównym składnikiem iłów bentonitowych. W glebach występuje jedynie tam, gdzie zaistniały warunki alkaliczne, niezbędne przy jego tworzeniu. WERMIKULIT jest produktem wietrzenia lub hydrotermalnego rozkładu biotytu. Występuje powszechnie w zwietrzelinach i glebach, w których wchodzi w skład frakcji ilastej. Część substancji ilastej występuje w glebach w postaci amorficznej. Odnosi się to do niektórych uwodnionych tlenków żelaza i glinu oraz części krzemionki, zwłaszcza w glebach powstałych z popiołów wulkanicznych. Grupę tych związków określa się wspólnym mianem allofanów, będących słabo dotychczas poznaną kombinacją krzemionki i półtoratlenku glinu. Pojemność sorpcyjna allofanów jest bardzo duża. Na szczególną uwagę zasługuje fakt, iż pojemność ta odnosi się nie tylko do kationów (jak w przypadku pozostałych minerałów ilastych), ale również do anionów. Mechanizm powstawania niezrównoważonych ładunków w tego typu minerałach nie został jeszcze poznany, wiadomo jednak, iż mają one charakter zależny od pH
30. Substancje organiczne gleb Zawartość w glebie 10%/suchą masę gleby; różna dla różnych gleb. Powstają z procesów zachodzących na powierzchni Ziemi: biochemicznych, życiowych, rozkładu resztek organizmów gł. roślin. Materia organiczna w glebie: 1)substancje próchnicze (humusowe) 2)subst bitumiczne 3)węglowodory 4)białka 5)aminokwasy 6)nie do końca rozłożone cząstki org, bakterie, fauna w ogóle. Rozkładające się rośliny rosnące na glebie powodują powstanie subst humusowych. Są one produktami syntezy przejściowych produktów utleniania zw org. Zanim do tego dojdzie, gleba zawiera wiele cząstek org, nieorg, resztek organizmów, głównym składnikiem gleby jest próchnica. Próchnicę stanowią: a)związki humusowe (głównie) i b)bituminy. Związki humusowe w glebie zawierają: 1)kwasy fulwowe (łatwo wymywane > mało) 2)kwasy huminowe (znacznie większy udział niż w wodzie) 3) huminy (zw o podobnej budowie zawierające więcej części mineralnych czyli połączeń z metalami oraz mniej związków hydroksylowych decydujących o rozpuszczalności). W kwasach huminowych jest większy udział pierścieni aromatycznych, większa jest masa cząsteczkowa i rozmiary, a mniejsza jest rozpuszczalność w wodzie (od kilku tys do ponad stu tys), zawierają dużo połączeń z metalami. Jest to frakcja najmniej przekształcona związków humusowych. Związki humusowe mogą ze sobą reagować tworząc związki mieszane z iłem. W ile jest glin, a w zw humusowym wodór. Cząstka humusowa może łączyć się z ilastą. Bituminy są to naturalne składniki organiczne gleb, stanowią mieszaninę kilku różnych typów związków. Skład: węglowodory alifatyczne (wysoka masa cząsteczkowa), w. skondensowane aromatyczne, wielkocząsteczkowe estry. Węglowodory o m. cz. 40 i więcej: mogą posiadać grupy COO- i rodnik; nierozpuszczalne w wodzie. Są to utlenione węglowodory. Od typowych subst humusowych różnią się większą zawartością C, a mniejszą tlenu.
31. Właściwości sorpcyjne gleb. Kompleks sorpcyjny. Właściwości sorpcyjne gleby polegają na zatrzymywaniu w niej cząstek stałych, niektórych gazów i par, a przede wszystkim różnych związków chemicznych i jonów. Rola zdolności sorpcyjnych: odżywianie się roślin (pobieranie składników pokarmowych gleby); nawożenie gleb (jego efektywność, wielkość strat składników i przenikanie jonów do wód gruntowych i powierzchniowych; zatrzymywanie różnych związków, np. metali ciężkich, pestycydów itp. 5 rodzajów sorpcji wg Giedroycia: 1.Sorpcja biologiczna - polega na przetwarzaniu składników pokarmowych na własną materię organiczną przez organizmy żywe. 2.Sorpcja mechaniczna - jest to zatrzymywanie cząstek stałych, mineralnych mineralno-organicznych i organicznych w porach glebowych. 3.Sorpcja fizyczna - polega na pochłanianiu gazów, par i związków niedysocjujących przez ich zagęszczanie na powierzchni fazy stałej. 4.Sorpcja chemiczna - jest to zatrzymywanie nierozpuszczalnych wodzie soli powstających wyniku reakcji chemicznych zachodzących w glebie. 5.Sorpcja wymienna - polega na adsorbowaniu związków dysocjujących, tzn rozpadających się w wodzie na jony, przez aktywną część fazy stałej gleby, zwaną kompleksem sorpcyjnym, z zachowaniem zdolności do wymiany zasorbowanych jonów z roztworem glebowym. Kompleks sorpcyjny - koloidalna część fazy stałej gleby wraz z zaadsorbowanymi wymiennie jonami. Skład kompleksu sorpcyjnego: 1.koloidy mineralne-glinokrzemiany;- krystaliczne wodorotlenki i tlenki żelaza oraz glinu-minerały amorficzne- czyli niekrystaliczne 2.koloidy organiczne: -głównie próchnica glebowa; 3.koloidy organiczno-mineralne - tj kompleksowe połączenia próchnicy z minerałami ilastymi
32. Metale w środowisku Metale ciężkie - przyjmuje się, że to te których gęstość przekracza 6kg/dm3. Inne też mogą mieć znaczenie ekologiczne. O istotności zanieczyszczenia decyduje jego szkodliwość i ilość. Dużo jest: Zn (zużycie 1mln t/rok), Ni(300tys), Cd(22tys), Hg(8tys), Cu, Pb, Cr(800tys). Mobilność pierwiastków: - ołów jest najmocniej wiązany w glebie, jego mobilność jest b. mała; - Cd, Cu, Zn są najbardziej mobilne. Źródła: 1)naturalne (geochemiczne): pokłady rud, metali, z których są wymywane i rozprzestrzeniane oraz wulkany. 2)antropogeniczne: przemysł wydobywczy, kopalnictwo, przetwórstwo, metalurgia, użytkowanie, odpady (składowiska odpadów przemysłowych i wysypiska śmieci), transport samochodowy (>metale w powietrzu). Hg (naturalne stężenia: gleba 1mg/kg sm, wody: rzeki 0,01(g/l, morza 0,005 (g/l, powietrze 0,06-0,6(g/m3), Cd (g. ponad 2, w. 0,1-5, p. 0,003-0,6), As (w. do 22), Pb (g. do 40, w. do 20). Stężenia naturalne rosną wskutek migracji antropogenicznych zanieczyszczeń. Im większe zużycie tym poziom zanieczyszczenia będzie większy np. Zn (w Polsce kilkaset mg/kg sm) występuje w ilościach, proporcjach większych niż wynikałoby to z zużycia (Cu - kilkadziesiąt; Cr zbliżony do Pb). Formy występowania: 1)jonowa. Tak jak każdy metal w roztworze wodnym lub glebowym. 2)zw nierozpuszczalne (cząstki stałe) -wodorotlenki, węglany, fosforany (mniej), siarczki (w osadach dennych wód, w warunkach beztlenowych), siarczan ołowiu. 3)organiczne połączenia metali (Hg, Pb, As, cyna). Wskutek procesów biochemicznych w osadach dennych powstaje. aminy org. reagujące z jonami metali, gr. CH3 odrywa się z tej struktury i powstają połączenia org. np. (CH3)2Hg dimetylortęć, CH3Hg(+) kation dimetylortęciowy, CH3HgOH, (CH3Hg)2S siarczek metylortęci, CH3HgCl chlorek m. Zdolność do tworzenia połączeń metylowo-metanowych zależy od warunków utleniająco-redukujących i od pH (niższe - mniej przyłączeń). Przemiany metali: przenikanie Hg do różnych elementów środowiska: łatwo przenika do rtęci metalicznej. Schemat: (atmosfera<)Hg<->Hg(2+) (woda) <-> 1) HgS, 2)CH3Hg(+) (woda-osady) <-> (CH3)2Hg (>atm). Zdolność rtęci dostawania się do atmosfery jest większa niż innych metali. Ołów:1)SO3(2-)+8H(+)+8e(-)>S(2-)+3H2O. 2)Pb(2+)+S(2-) > PbS. 1 i 2 to redukcja siarczanów do siarczków (zamiast Pb może być dowolny metal występujący w wodzie). 3) 3(CH3)3PbX >2(CH3)4Pb+CH3X+PbX2 -dowolna sól trimetyloołowiu> tetrametyloołów + sól ołowiu. 4) 2(CH3)3Pb(+)+S(2-) >[(CH3)3Pb]2S. 5) 3[(CH3)3Pb]2S >3(CH3)4Pb+3(CH3)2PbS. Ołów występuje na drugim i 4-tym stopniu utleniania. Ołów może dawać sole i inne produkty. Metale mogą reagować ze zw hum. Chrom. Może występować w anionie i w kationie. W praktyce dostaje się do środowiska jako anion dwuchromianotlenkowy (6-wartościowy > bardziej toksyczny niż 3- w.) Chromiany są silnymi utleniaczami, ulegają redukcji (szczególnie w środowisku. wodnym) w Cr(3+) i wytrącają się do wodorotlenków. Arsen. Tworzy aniony i kationy. Może być 3-w. i 5-w. As(3+), AsO4(3-), As)3(3-). As(3+) jest bardziej szkodliwy od 5-w. Chętnie tworzy kation metylowy -trimetyloarsen. Ulega redukcji dając arsenowodór AsH3 - tak może dostawać się do atmosfery.
33. Ropopochodne węglowodory w środowisku Produkty naftowe stanowią mieszaninę związków, głównie węglowodorów, o zróżnicowanych właściwościach fizycznych, chemicznych i biologicznych. Obecność w środowisku węglowodorów, nawet nietoksycznych, jest przyczyną niszczenia życia biologicznego wskutek odcięcia dostępu powietrza i wody. Poszczególne ekosystemy wodne bądź lądowe stanowią jednostkę ekologiczną, w skład której wchodzą zespoły organizmów tworzące pewną całość. Wprowadzenie do ekosystemów produktów naftowych powoduje zaburzenia w przebiegu naturalnych cykli obiegu materii i energii.
Skład węglowodorowy ropy naftowej. W ropie naftowej w największej ilości występują węglowodory naftenowe (cykloparafinowe) - zawiera ona ok. 50% naftenów, a ich ilość rośnie wraz z temperaturą wrzenia. Następne w kolejności to węglowodory parafinowe (CnH2n+2]), które dominują we frakcjach benzynowych i są podstawową grupą węglowodorów najstarszych rodzajów ropy. Parafiny o prostym łańcuchu, zwane n-parafinami są chemiczne inertne, nie stwarzają zatem żadnego zagrożenia dla środowiska. Kolejna grupa węglowodorów występujących w ropie naftowej to węglowodory aromatyczne (areny). Zawierają one przynajmniej jeden pierścień benzynowy. Jest to płaski, 6-węglowy pierścień zawierający naprzemiennie podwójne i pojedyncze wiązania z sąsiednimi atomami węgla. Ropa naftowa rzadko zawiera więcej niż 15% węglowodorów aromatycznych. Kancerogenność benzenu oraz duża toksyczność jego alkilowych pochodnych, które są dobrze rozpuszczalne w wodzie powodują, że jest to grupa węglowodorów bardzo niebezpieczna dla środowiska.
działaniu mutagennym i kancerogennym. Biodegradacja produktów naftowych Zanieczyszczenie gruntów i wód produktami naftowymi wpływa niekorzystnie na produkcję roślinną. Stwarza zagrożenie dla zdrowia ludzi i zwierząt (większość produktów naftowych działa toksycznie na organizmy żywe). Dlatego konieczne jest przeprowadzenie szeregu zabiegów o charakterze fizycznym, chemicznym lub biologicznym mających na celu oczyszczenie skażonych terenów.
Podstawowe metody postępowania z zanieczyszczeniami ropopochodnymi w środowisku gruntowo-wodnym to:
unieruchomienie substancji ropopochodnych przez zastosowanie metod izolacji (bariery fizyczne, bariery hydrauliczne, zagęszczenie materiału gruntowego)
likwidacja, inaczej dekontaminacja (m.in. utylizacja wysokotemperaturowa czyli termiczny rozkład substancji ropopochodnych, płukanie i ogrzewanie gruntu).
W ostatnich latach coraz szersze zastosowanie znajdują technologie remediacji oparte na metodach biologicznych. Podstawową rolę w procesie biologicznego oczyszczania spełniają mikroorganizmy zdolne do wykorzystywania węglowodorów (szczególnie niebezpiecznych składników produktów naftowych) w charakterze źródła węgla i energii.
Węglowodory obecne w środowisku są degradowane biologicznie głównie przez bakterie i grzyby. Mikroorganizmy, które rozkładają tę grupę związków występują w środowisku naturalnym w większych ilościach, przede wszystkim w miejscach zanieczyszczonych produktami naftowymi. Stwierdzono, że około 100 gatunków mikroorganizmów ma zdolność rozkładu węglowodorów. W środowisku wodnym dominują bakterie jako drobnoustroje biorące udział w rozkładzie węglowodorów. Grzyby odgrywają nieco mniejszą rolę. W glebie - zarówno bakterie, jak i grzyby rozkładające węglowodory występują licznie i biorą aktywny udział w procesie biodegradacji.
34. Lotne węglowodory aromatyczne w środowisku. Węglowodory aromatyczne mają wiązania podwójne, benzen, toluen występują do 20%w składzie ropy naftowej. Benzen C6H6. zjawisko rezonansu umocnienie struktury benzenu= ciągły obieg pary elektronów. Rezonans- krążenie wiązań podwójnych dookoła. rodzaje ksylenów: 1-3 ksylen, 1-2 ksylen, para ksylen. Zastosowanie: rozpuszczalniki, styren- substrat do produkcji wielu tworzyw sztucznych . BTX- lotne węglowodory aromatyczne ( benzen, toluen, ksylen).Węglowodory aromatyczne są bardziej toksyczne od węglowodorów alifatycznych. W szczególności benzen, który ma właściwości rakotwórcze. Obecnie najczęściej stosowany ksylen. Węglowodory aromatyczne dobrze rozpuszczają się w wodzie bo mają budowę polarną np. Benzen 700mg/l. Stanowią głównie zagrożenie dla środowiska wodnego i powietrza. W środowisku glebowym pozostają krótko, gdyż zostają szybko wypłukane. Pokrywa na powierzchni wody z np. ropy naftowej ogranicza dostępność tlenu.
35. WWA w środowisku. (Uwaga! Rysunki związków znajdują się na papierowym załączniku!!!) Wielopierścieniowe węglowodory aromatyczne to związki składające się z co najmniej kilku pierścieni aromatycznych, tzn. pierścieni, w których każdy atom węgla wytwarza podwójne wiązanie z innym atomem węgla w pierścieniu. WWA występują w sadzy powstającej przy spalaniu z niedoborem tlenu. Oznacza to, że WWA emitowane są wszędzie, gdzie następuje spalanie paliw: zakłady energetyczne (ponad połowa emisji), przemysł, stare silniki (kopcące), a także DYM Z PAPIEROSÓW.WWA zawarte są też w ropie naftowej. Podczas destylacji pozostają w najcięższej frakcji. Frakcja ta to smoła zawierająca oprócz WWA asfalteny, węglowodory alifatyczne i in. Służy ona do wyrobu asfaltu drogowego ( ścieranie powierzchni asfaltowych powoduje ich emisję do powietrza. W powietrzu utrzymują się w pyle, a wraz z nim mogą trafiać do gleby oraz do wód. Ze względu na minimalną rozpuszczalność trafiają do osadów dennych, gdzie się kumulują (w Polsce stężenie rzędu μgramów/ kg osadu). Ich rozkład nie jest szybki, więc można je wykryć. Ilość w środowisku jest dość stała, gdyż na miejsce rozłożonych związków dochodzą nowe z pyłu. WWA występują również w ściekach z przemysłu petrochemicznego, rafinerii, baz transportowych. Podstawowe WWA nie są szkodliwe: piren (zał.), fluoranten (zał.). Są też nieaktywne chemicznie. Jednak ich kolejne pochodne są bardzo szkodliwe. Niektóre WWA mają silne właściwości rakotwórcze, dlatego ich obecność w środowisku jest oznaczana. Najbardziej niebezpieczny i najsilniej rakotwórczy jest benzo(a)piren (zał.). Następny to benzo(b)fluoranten (zał.). Średnio aktywne/szkodliwe: benzo(a)antracen (zał.), perylen, benzo(e)piren, chryzen. Działanie rakotwórcze polega na tym, że tlen może rozrywać podwójne wiązania między węglami, a prze to cząsteczka WWA może się połączyć z łańcuchem DNA.
36.Lotne węglowodory chlorowane w środowisku Jeżeli wprowadzimy atomy pewnego pierwiastka do związku chemicznego to rośnie toksyczność tego związku. Jeżeli zamiast H wprowadzimy Fluor albo chlor to ta chlorowcopochodna lub fluorowcopochodna jest bardziej groźna dla środowiska. Chlorowcopochodne są bardzo toksyczne jednak potrzebne m.in. chlorowane pochodne węglowodorów alifatycznych(gazy: metan, etan, propan, butan) Jeżeli w metanie zastąpimy jeden wodór chlorem to będzie to ciecz. Pochodne chlorowane są często stosowane jako rozpuszczalniki. Każdy rozpuszczalnik paruje, stanowiąc zagrożenie dla powietrza atmosferycznego. One dostają się do ścieków ( pralnia chemiczna, zakłady metalowe - do odtłuszczania metali czy do wielu innych). Są składnikami klejów, do tych celów te rozpuszczalniki tj. :
CH3Cl - chlorek metylu ( bardzo lotny, małe zastosowanie ); CH2Cl2 - chlorek metylu ( często w laboratorium chem. Do ekstrakcji ); CHCl3 - chloroform w przemyśle częściej ( kiedyś do narkozy, stosowano go też jako rozcieńczalnik, do farb, też w laboratorium); CCl4 - był stosowany w pralniach chem., w gaśnicach tetrowych. W skutek powodowania dziury ozonowej został wyeliminowany. Powyższe związki powstają na skutek chlorowania metanu. Związki chlorowane pochodzące od etylenu : CHCl = CCl2 - trichloroetylen dobrze rozpuszcza tłuszcze, jest mniej szkodliwy; CCl2 = CCl2 - nadchloroetylen; CCl3 = CH3 - trichloroetan mało toksyczny, stosowany do odtłuszczania i produkcji kleju. Wszystkie związki są bardzo trudno rozkładalne, głównie tetrachlorek węgla ( najbardziej odporny na rozkład biologiczny ) Mobilność w środowisku - im więcej atomów chloru tym rozpuszczalność mniejsza, np. CH2Cl2 dobrze rozpuszczalny. CCl4 - słabo rozpuszczalny. Bo ma budowę najmniej polarną. Najmniej lotny, ma duże ciepło parowania. W skutek lotności związki te nie utrzymują się długo w środowisku. Dostają się do atmosfery, są cięższe od wody dlatego idą pod wodę. Trwałe w środowisku.
38. Herbicydy to syntetyczne substancje chemiczne stosowane do zwalczania chwastów upraw rolnych i leśnych oraz do oczyszczania z niepożądanych roślin torów kolejowych i poboczy dróg. Najczęściej stosowane herbicydy można podzielić na dwie grupy: 1. herbicydy triazynowe - Są to związki chemiczne będące pochodnymi triazyny. Najczęściej stosowane to simazyna i atrazyna
2. pochodne kwasu fenoksyoctowego, np. kwas 2,4 dichlorofenoksyoctowy Czasem kwas octowy zastępuje się kwasem propionowym Związki te powodują nadmierny przyrost części naziemnej rośliny przy jednoczesnym zahamowaniu wzrostu systemu korzeniowego, który nie nadąża z dostarczaniem składników pokarmowych i roślina usycha. Herbicydy są przeważnie dobrze rozpuszczalne w wodzie, co wpływa na ich dużą mobilność. Charakteryzują się średnim poziomem toksyczności. Współcześnie stosowane preparaty są ponadto podatne na biodegradację co zapobiega ich akumulowaniu się w środowisku.
39.Polichlorowane bifenyle w środowisku.(PCB) Wykorzystuje się je jako składniki farb ,zwłaszcza drukarskich, jako plastyfikatory tworzyw sztucznych oraz materiały izolujące w transformatorach, materiałów przylepnych, kalki maszynowej, smarów. Na rynku są dostępne PCB o nazwach handlowych :aroclor, phenoclor, clophen. Ogółem można wyróżnić 209 różnych |PCB, poczynając od chlorobifenylu do całkowicie podstawionego dekachlorobifenylu.W1966r. stwierdzono, że mieszaniny PCB są substancjami szkodliwymi, przy czym ich chromatogramy gazowe wykazują duże podobieństwo do chromatogramów DT. Szacuje się, że na świecie wyprodukowano dotychczas ok.1mln. ton PCB, z czego ok.40% odprowadzono do środowiska. Polichlorowane bifenyle są związkami niejonowymi bardzo małej rozpuszczalności w wodzie. Mają one właściwości lipofilne i są rozpuszczalne w wielu rozpuszczalnikach organicznych. Polichlorowane bifenyle są związkami wolno rozkładającymi się i długo utrzymują się środowisku. Właściwości hydrofobowe powodują, że PCB mogą gromadzić się na granicy faz woda-powietrze co powoduje ich szybsze utlenienie się. Przy normalnym poziomie narażenia PCB nie są bardzo toksyczne dla ludzi, przy większych stężeniach mogą doprowadzić np. do chorób skóry.
40.Dioksyny w środowisku. DIOKSYNA - toksyczny związek chemiczny obcy żywym organizmom dostający się do środowiska naturalnego wskutek działalności produkcyjnej człowieka lub z odpadami. Toksyczne działanie dioksyn polega na powolnym, ale skutecznym uszkadzaniu narządów wewnętrznych: wątroby, płuc, nerek, rdzenia kręgowego lub kory mózgowej. Niebezpieczne dioksyny powstają jako uboczny produkt podczas produkcji: pestycydów, herbicydów. Dioksyny powstają również podczas spalania tworzyw sztucznych i olejów w naszych piecach. Spalanie śmieci w spalarniach także powoduje powstawanie dioksyn i ich emisję do atmosfery. Dioksyny są doskonale rozpuszczalne w tłuszczach, dlatego akumulują się w tkankach tłuszczowych, ryb, ptaków, węży, a także ssaków. Jedyną metodą wykrywania zawartości dioksyn jest monitoring środowiska. Na świecie spala się rocznie setki milionów ton śmieci, jest to kolejne źródło ogromnego skażenia środowiska dioksynami na całym globie. Badania kliniczne wykazały , że dioksyny działają silnie mutagennie, naruszając właściwą strukturę kodu genetycznego, rozmnażających się komórek żywych organizmów.
Z dotychczasowych badań wynika, że polichlorowane dioksyny ulegają rozkładowi w świetle nadfioletowym (obecnym w świetle słonecznym) do dużo mniej toksycznych związków. Główne źródła wnoszące niebezpieczne dioksyny do środowiska to: 1. Odpady atmosferyczne (emisja szkodliwych gazów) 2. Ścieki przemysłu tekstylnego i skórzanego 3. Przemysł metalowy. 4. Spalanie odpadów szpitalnych. 5. Pralnie chemiczne. 6. Ruch uliczny (ścieranie opon i gazy spalinowe)7. Przemysł papierniczy 8. Środki impregnacji drewna. 9. Środki ochrony roślin. 10. Produkcja chloru przy użyciu elektrod grafitowych. 11. Produkcja miedzi.
.
bezpośrednie
pośrednie
HCO3¯ - wodorowęglan wapnia (dobrze rozpuszczalny)
reakcje zachodzą trudniej, niż w przypadku wapnia
Wyszukiwarka
Podobne podstrony:
Egzamin chemia środowiska, PW, SEM III, Chemia Środowiska, Egzaminy
Pytania i zagadnienia egzaminacyjne z chemii ogólnej (4), PW, SEM III, Chemia Środowiska, Egzaminy
Gleba połówkowa, PW, SEM III, Gleboznawstwo, Egzamin
Gleba połówkowa pierwsza, PW, SEM III, Gleboznawstwo, Egzamin
gleba odp 57-80 (bez 59, PW, SEM III, Gleboznawstwo, Egzamin
gleba 1-14, PW, SEM III, Gleboznawstwo, Egzamin
gleba pytania od 121-150, PW, SEM III, Gleboznawstwo, Egzamin
gleba 2, PW, SEM III, Gleboznawstwo, Egzamin
egzaqm3-wyn-t, nauka, PW, sem 6, PKM 2, PKM2, PKM 2, Egzaminy
egzam - 4 zadania, nauka, PW, sem 6, PKM 2, PKM2, PKM 2, Egzaminy
egzaqm1-wyn-t, nauka, PW, sem 6, PKM 2, PKM2, PKM 2, Egzaminy
Pytania-egzamin, Inżynieria Środowiska [PW], sem 1, chemia
laboratorium5pop, Inżynieria Środowiska [PW], sem 2, Chemia, 2, sprawka
laboratorium3pop, Inżynieria Środowiska [PW], sem 2, Chemia, 2, sprawka
chemia - poprawka - wszystkie zestawy - odpowiedzi, Inżynieria Środowiska [PW], sem 1, chemia, chemi
Chemia sem I, Inżynieria Środowiska [PW], sem 1, chemia, chemia
więcej podobnych podstron