Tadeusz Trzmiel, Radosław Olejnik
Instytut Biochemii Technicznej
Biodegradacja związków heterocyklicznych
1. WSTĘP
W dobie gwałtownego rozwoju przemysłu, stale wzrasta liczba substancji chemicznych trafiających do środowiska. Można tu wyróżnić ksenobiotyki (substancje egzogenne), zarówno celowo wprowadzone (pestycydy) jak i te, które znalazły się w nim przypadkowo. Najlepszym tego przykładem jest przemysł chemiczny, farmaceutyczny, tekstylny i inne. Na szczególną uwagę zasługują związki heterocykliczne, których w środowisku naturalnym wyodrębniono tak wiele, że w podręcznikach do chemii z braku miejsca nie można wymienić nawet najważniejszych, stanowią bowiem one więcej niż połowę wszystkich znanych związków organicznych [3].
Związkami heterocyklicznymi są połączenia o budowie pierścieniowej, w których jeden lub kilka atomów w pierścieniu stanowią atomy pierwiastków innych niż węgiel. Atomy te noszą nazwę heteroatomów, a najczęściej spotykanymi są atomy azotu, tlenu i siarki. Oprócz tych pierwiastków w pierścieniach związków heterocyklicznych mogą występować atomy fosforu, krzemu, boru, arsenu, antymonu i innych pierwiastków [1]. Należy podkreślić bardzo duże znaczenie tego typu związków, zarówno wśród produktów naturalnych, jak i szeregu cennych preparatów otrzymywanych na drodze syntetycznej [2].
Z uwagi na wysoką toksyczność wielu związków, należałoby ograniczyć ich zużycie w przemyśle. Ponieważ jest to nie zawsze możliwe, należy szukać procesów powodujących ich usuwanie ze środowiska. Metody niszczenie związków heterocyklicznych przy pomocy wysokiego ciśnienia i temperatury są kosztowne. Okazuje się jednak, że praktycznie dla każdego typu związku chemicznego można dobrać mikroorganizm zdolny go biodegradować. Liczebność drobnoustrojów w środowisku naturalnym jest teoretycznie nieograniczona, stąd jedynym praktycznym problemem jest znalezienie mikroorganizmu o uzdolnieniach w kierunku degradacji konkretnego ksenobiotyku. Można także, na drodze inżynierii genetycznej, zmodyfikować dany szczep w taki sposób, aby przejawiał interesujące nas cechy. Zaletą mikroorganizmów są niskie koszty utrzymania drobnoustrojów i stosunkowo proste, ewentualne, procesy technologiczne. Podatność ksenobiotyków na biodegradację zależy od dostępności tych związków dla drobnoustrojów oraz od liczebności i aktywności populacji mikroorganizmów. Obecność grup reaktywnych np. -COOH, -OH, czy -NH2 może ułatwiać atak drobnoustrojów, ale niektóre ugrupowania, takie jak halogenowe czy alkilowe czynią cząsteczkę bardziej odporną na degradację [15].
Także ogromne znaczenie mają biologiczne predyspozycje gleby do biodegradacji, które zależą od żyjących w niej drobnoustrojów, ich rodzaju czy też liczebności populacji. Najliczniejszą grupę stanowią bakterie. W glebie występują bakterie z rodzajów: Acinetobacter, Agrobacterium, Alcaligenes, Arthrobacter, Bacillus, Brevibacterium, Caulobacterium, Cellulomonas, Clostridium, Corynebacterium, Flavobacterium, Pseudomonas, Staphylococcus, Streptococcus i Xanthomonas [15].
Największą grupę drobnoustrojów wyizolowanych z gleby, zanieczyszczonych wód czy osadów, stanowią należące do rodzaju Pseudomonas. W glebie występują również grzyby, z których najbardziej liczne to: Aspergillus, Geotrichum, Penicillium i Trichoderma. Także w glebie znajdują się enzymy w stanie wolnym oraz zewnątrzkomórkowe enzymy uwalniane przez drobnoustroje lub dostające się tam po lizie komórek. Zwykle już w 1g gleby ogrodowej można znaleźć bakterie, które zdolne są do rozkładu każdego naturalnego substratu [2].
2. Podstawowe dane o związkach heterocyklicznych
W przeciwieństwie do związków izocyklicznych (karbocyklicznych) takich jak benzen, naftalen, w których wszystkie atomy pierścienia są atomami węgla, pierścienie związków heterocyklicznych zbudowane są z atomów różnych pierwiastków. Pierścienie takie zawierają zwykle obok atomów węgla również jeden lub więcej atomów tlenu, azotu lub siarki. Ponieważ kąty między wiązaniami tych atomów nie różnią się zbytnio od kątów wartościowości węgla, pierścienie heterocykliczne zbudowane z pięciu lub sześciu atomów są praktycznie beznapięciowe i w związku z tym bardzo trwałe. Za najprostszy związek heterocykliczny można uważać tlenek etylenu, ponieważ jego pierścień zawiera dwa atomy węgla i jeden atom tlenu. Związek ten jest jednak bardzo nietrwały i pozbawiony charakteru aromatycznego, który cechuje większość związków heterocyklicznych.
Związki heterocykliczne występują w znacznych ilościach w przyrodzie. Często wykazują specjalne działanie fizjologiczne (m.in. witaminy, alkaloidy). Niektóre z tych produktów naturalnych, jak również wiele innych połączeń otrzymuje się syntetycznie. Znanych jest około 3600 typów pierścieni heterocyklicznych. Jednak tylko część z nich produkowana jest masowo i rozpatrywana jest w kategorii ksenobiotyków.
Związki heterocykliczne możemy podzielić na dwie podstawowe grupy:
Heterocykliczne związki jednopierścieniowe z atomami tlenu, siarki i azotu.
Heterocykliczne związki wielopierścieniowe.
Do pierwszej z tych grup zaliczamy:
związki heterocykliczne pięcioczłonowe z jednym heteroatomem (najważniejszymi podstawowymi układami w tej klasie są: tiofen, furan i pirol),
związki heterocykliczne sześcioczłonowe z jednym heteroatomem (najważniejszym przedstawicielem tej grupy połączeń jest pirydyna, czyli azotowy analog benzenu),
związki heterocykliczne jednopierścieniowe zawierające kilka heteroatomów (z pięcioczłonowym pierścieniem np. histydyna; z sześcioczłonowym pierścieniem np. pirazyna).
W drugiej grupie klasyfikuje się heterocykliczne układy jednopierścieniowe, które mogą być skondensowane z pierścieniami aromatycznymi, hydroaromatycznymi lub z innymi pierścieniami heterocyklicznymi. W tej grupie przykładem może być; indol, tryptofan, chinolina czy akrydyna [1].
Poniżej przedstawiono wzory chemiczne najważniejszych związków heterocyklicznych:
4. Mechanizmy biodegradacji związków heterocyklicznych
4.1. Biodegradacja pięcioczłonowych związków pierścieniowych z jednym heteroatomem.
W grupie pięcioczłonowych związków pierścieniowych z jednym heteroatomem przedstawiono mechanizmy mikrobiologicznej degradacji pirolidyny, proliny, hydroksyproliny, kwasu furano-2-karboksylowego i tiofeno-2-karboksylowego.
Pirolidyna
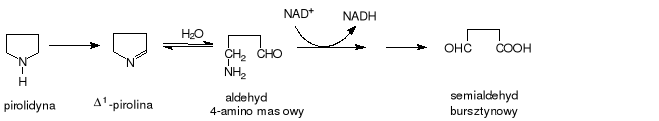
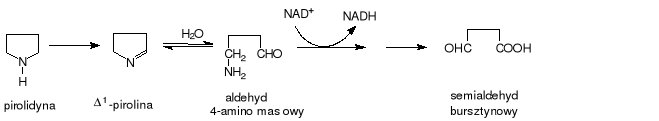
Pirolidyna (tetrahydropirol) zaliczana jest do laktamów, czyli wewnątrzcząsteczkowych imidów kwasowych. Powstaje w wyniku redukcji pirolu. Wykorzystywana bywa w produkcji włókien syntetycznych. Degradacja pirolidyny u Pseudomonas fluorescens [5] zaczyna się od przekształcenia do Δ1-piroliny (rys.1.). Enzym uczestniczący w tej przemianie nie został jeszcze poznany. Δ1-pirolina jest związkiem bardzo charakterystyczny z uwagi na to, że pod wpływem wody przemienia się spontanicznie w aldehyd 4-aminomasłowy. Oba te związki w wodzie występują w równowadze. Kolejnym etapem w degradacji pirolidyny u Pseudomonas fluorescens jest przemiana powstałego aldehydu 4-aminomasłowego dzięki dehydrogenazie aminomasłowej (EC 1.2.1.19) do 4-aminomaślanu, a następnie do semialdehydu bursztynowego. W tej reakcji biokatalizatorem jest transaminaza 4-aminomasłowa (EC 2.6.1.19). Ostatnią reakcją jest przemiana semialdehydu bursztynowego do bursztynianu, a ten może być „włączony” do cyklu Krebsa [5]. Tej przemiany szczep Pseudomonas fluorescens jednak nie przeprowadza, prawdopodobnie dlatego, że brak już w tym związku azotu potrzebnego mikroorganizmowi do wzrostu.
Rys.1. Degradacja pirolidyny przez Pseudomonas fluorescens.
Prolina i hydroksyprolina
W nieco inny sposób katabolizowane są prolina jak i hydroksyprolina. Podane związki to aminokwasy występujące w dużych ilościach w białkach tkanki łącznej: kolagenie i elastynie [6]. Zarówno prolina jak i hydroksyprolina zostają przekształcane do kwasu 2-oksoglutarowego przez drożdże Hansenula subpelliculosa. W przypadku 4-hydroksy-L-proliny pierwszą przemianą jest przekształcenie jej do 4-hydroksy-D-proliny (rys.2). Odbywa się to zapewne dzięki epimerazie 4-hydroksyprolinowej (EC 5.1.1.8). Powstały związek jest następnie przeprowadzany najpierw w Δ1-pirolino-4-hydroksy-2-karboksyloan przez nie sklasyfikowaną reduktazę, a potem w semialdehyd 2-oksoglutarowy (przy udziale deaminazy EC 3.5.4.22.) [5].
Ostatnią reakcją jest przekształcenie semialdehydu w kwas 2-oksyglutarowy pod działaniem dehydrogenazy (EC 1.2.1.26). Ten ostatni związek uczestniczy w przemianach cyklu Krebsa.
Rys.2. Katabolizm hydroksy-L-proliny
Sytuacja z biodegradacją L-proliny jest bardziej skomplikowana. Bardzo trudno znaleźć mikroorganizm degradujący L-prolinę ponieważ wiele z nich syntetyzuje ją, gdyż jest potrzebna do biosyntezy białka. Tym niemniej wykazano [5], iż drożdże Hansenula subpelliculosa mogą prowadzić degradację L-proliny dwoma drogami (rys.3a. i 3b.). W obydwu przypadkach końcowym produktem jest kwas 2-oksoglutarowy.
Pierwszy etap transformacji L-proliny jest podobny w obu drogach. Pod działaniem odpowiednich reduktaz następuje odwodornienie i pojawia się kwas pirolino-2- lub pirolino-3-karboksylowy. W kolejnym kroku pęka wiązanie N-C6 (droga a) lub N-C1 (droga b) w wyniku czego powstaje semialdehyd glutaminowy lub kwas α-oksoaminowalerianowy. Semialdehyd glutaminowy powstaje po działaniem dehydrogenazy Δ1-pirolino-5-karboksylowej (EC 1.5.1.12.), natomiast dehydrogenaza występująca w drodze b) nie została jeszcze sklasyfikowana w nomenklaturze enzymów z 1992 rok (aktualnie obowiązującej).
Rys.3. Dwie drogi katabolizmu L-proliny u Hansenula subpelliculosa
Kwas furano-2-karboksylowy
Związki posiadające w swojej budowie pierścień furanowy w tym kwas furano-2-karboksylowy (znany również jako kwas pirośluzowy) są szeroko rozpowszechnione w przyrodzie, między innymi wchodzą w skład żywic furanowych używanych w przemyśle chemicznym [6].
Kwas furano-2-karboksylowy, według dostępnej obecnie wiedzy, jest wykorzystywany przez Aerobacter aerogenes, Aspergillus niger i różne szczepy Pseudomonas jako substancja wzrostowa. Degradacja tego związku (rys.4.) przebiega według schematu rzadko spotykanego w przypadku innych związków organicznych - wszystkie związki przejściowe tego szlaku są estrami koenzymu A [5].
Pierwszą reakcja jest powstanie estru CoA. Enzym tej reakcji nie został w literaturze omówiony. W kolejnym kroku powstały 2-furanoylo-CoA jest transformowany przez hydrolazę, która powoduje przyłączenie hydroksylowej grupy pochodzącej z wody dając 5-hydroksyfurano-2-karboksylo-CoA. Ta hydroksylacja jest bardzo podobna do tej spotykanej przy biodegradacji kwasu nikotynowego. Następnymi etapami tego szlaku są: powstanie 5-okso-Δ2-dihydro-2-furanoyloCoA (którego budowa chemiczna jest typu laktonu) przekształcanego przez nieznaną hydrolazę poprzez formę enolową do α-oksoglutarylo-CoA, z którego później CoA jest odrywany [5]. Enzymy powyższych reakcji nie zostały dotychczas bliżej poznane i sklasyfikowane.
Rys.4. Degradacja kwasu furano-2-karboksylowego przez Pseudomonas sp.
Kwas tiofeno-2-karboksylowy
Związki zawierające w swojej budowie tiofen znajdują się w smole węglowej czy też w nie zużytych olejach. Jednym z ich przedstawicieli jest kwas tiofeno-2-karboksylowy. Podczas biodegradacji tego kwasu i innych związków zawierających atom siarki jest on uwalniany jako siarkowodór. Mikroorganizmy potrzebujące tego kwasu jako źródła węgla są nadal poszukiwane. Jednym ze znanych jest Flavobacterium sp. [5]. Droga degradacji kwasu tiofeno-2-karboksylowego nie jest dokładnie poznana. Sugeruje się, że metabolizm prowadzony przez Flavobacterium sp. (rys.5.) jest niemal analogiczny do degradacji kwasu furano-2-karboksylowego omawianej powyżej. Także i w tym przypadku enzymy uczestniczące w procesie nie zostały poznane.
Rys.5. Degradacja kwasu tiofeno-2-karboksylowego
4.2. Degradacja histydyny jako przedstawiciela pięcioczłonowych związków pierścieniowych zawierających więcej niż jeden heteroatom
Histydyna jest aminokwasem, składnikiem budulcowym białek. W przypadku ustrojowego rozkładu tego typu związków chemicznych mówi się o ich metabolizmie lub katabolizmie, raczej unikając pojęcia „biodegradacja”, zarezerwowanego dla mikrobiologicznego rozkładu ksenobiotyków. Tym niemniej w niniejszej pracy zdecydowano się przedstawić szlak katabolitycznego rozkładu histydyny.
Chociaż histydyna może podlegać transaminacji, reakcja ta nie należy do głównego szlaku katabolizmu tego aminokwasu, ponieważ powstający 2-oksokwas nie może być dalej rozkładany i musi ulec ponownej aminacji do L-histydyny [22]. Pierwsza reakcja degradacji L-histydyny (rys.18.), katalizowana przez histydazę (amoniako-liazę histydynową, EC 4.3.1.3.), jest w warunkach fizjologicznych nieodwracalna. Podczas tej reakcji uwalniany jest amoniak i tworzy się podwójne wiązanie między atomami węgla w łańcuchu bocznym histydyny (powstaje kwas urokanianowy) [22,23]. Przyłączenie cząsteczki wody i przegrupowanie podwójnych wiązań daje imidazolonopropionian (4-imidazolono-5-propionian). Enzymem przeprowadzającym tą reakcję jest hydrataza urokanianowa (EC 4.2.1.49.) [23].
Kolejnym etapem jest hydroliza wiązania amidowego w pierścieniu imidazolonowym. Reakcję tę katalizuje propionaza imidazolonowa .(EC 3.5.2.7.) Powstający N-formiminoglutaminian jest przekształcany przez enzym (EC 2.1.2.5) przenoszący grupę formiminową na tetrahydrofolian (THFA); w wyniku tej reakcji powstają glutaminian i N5-formimino-THFA. Następnie od N5-formimino-THFA odłącza się amoniak i związek ten przekształca się w N5,N10-metyleno-THFA. Udział THFA w przemianach formiminoglutaminianu pozwala na wykrywanie niedoborów folianu w organizmie. W warunkach normy nadmiar histydyny dodany do pożywienia jest przekształcany w glutaminian i amoniak, a uwalniany fragment jednowęglowy przyłączany do THFA; wszystkie te związki są łatwo wykorzystywane w organizmie [22]. Jeżeli poziom folianu jest niewystarczający, formiminoglutaminian nie może być przekształcany w glutaminian tak szybko, jak zachodzą pierwsze reakcje metabolizmu histydyny. W rezultacie nagromadza się we krwi i jest wydalany z moczem. Powyższe reakcje tworzą główny pod względem ilościowym szlak degradacji histydyny. Ponieważ jego produktem jest glutaminian, który może być wykorzystywany do syntezy glukozy, histydynę zalicza się do aminokwasów glukogennych [22]. Dalej glutaminian przekształcany jest dzięki dehydrogenazie glutaminianowej (EC 1.4.1.2) w α-oksoglutaran.
Rys.18.Reakcje rozkładu histydyny [wg 22-23].
4.3. Biodegradacja związków heterocyklicznych sześcioczłonowych
4.3.1. Pierścienie o charakterze aromatycznym z jednym heteroatomem
W rozdziale tym omówiono mikrobiologiczne przekształcenia pirydyny i jej niektórych pochodnych, kwasu nikotynowego, nikotyny, nornikotyny oraz anabizyny.
Pirydyna
Pirydyna używana jest jako rozpuszczalnik w niektórych reakcjach chemicznych oraz przy ekstrakcji węgla kamiennego. Jej pochodne odgrywają ważną rolę biologiczną (NAD, witamina B6). Pochodne pirydyny mają działanie bakteriobójcze i grzybobójcze. Stosowane są jako leki rozkurczowe, przeciwgruźlicze, antydepresyjne [15]. Zanieczyszczenie środowiska pirydyną jest wynikiem przetwarzania oleju silnikowego i używania pestycydów. Związek ten jest niezwykle toksyczny [7]. Pirydyna jest rozpuszczalna w wodzie i stanowi główne zanieczyszczenie beztlenowych, podpowierzchniowych osadów. Mimo przeprowadzonych badań, mechanizm przemian nie jest do końca poznany [15]. Degradacja pirydyny przebiega tak samo w warunkach tlenowych jak i beztlenowych. Transformacje pirydyny przebiegają według alternatywnych, redukcyjnych szlaków. Jeden z tych szlaków przebiega z udziałem szczepu Bacillus 4. Podobnie, pirydyna jest przemieniana przez Corynebacterium i Brevibacterium [7,15]. Przeprowadzone badania (z oznakowaną 214 -C) pirydyną) dowodzoną, że otwarcie pierścienia pirydyny następuje między drugim a trzecim atomem węgla. Jednakże nie bardzo są znane etapy pośrednie w tej reakcji (rys.6.-ścieżka a) [15].
Nieco inną drogą przebiega transformacja pirydyny przy udziale Nocardia Z1. Zidentyfikowano pięciowęglowe produkty pośrednie, co świadczyć może o pęknięciu pierścienia pirydyny między atomem azotu a węglem w pozycji 2. Również w tym szlaku pierwszym etapem jest uwodornienie. Dalsze reakcje przebiegają jednak według innego mechanizmu otwarcia pierścienia pirydyny i do powstania innych produktów (rys.15.- ścieżka b). W obu szlakach degradacyjnych następuje deaminacja, po czym powstają produkty mogące wchodzić w różne szlaki metaboliczne drobnoustrojów [15]. Jeszcze inny szczep Azoarcus evansi, ma zdolność degradować pirydynę zarówno w warunkach beztlenowych jak i tlenowych. Degradacja pirydyny z udziałem tego szczepu, w obu typach przemian (tlenowe i beztlenowe), przebiega w podobny sposób (schemat 1b dla Nocardia Z1). Zmianie ulega jedynie końcowy akceptor elektronów. Z uwagi na szerokie działania tego szczepu na różne pochodne pirydyny może być on wykorzystany na szeroką skalę do usuwania związków tych ze środowiska [15].
Rys.6. Degradacja pirydyny przez szczepy Bacillus 4 oraz Nocardia Z1
2-hydroksypirydyna, 3-hydroksypirydyna i 2-karboksypirydyna
Przekształcenia 2-hydroksypirydyny i 3-hydroksypirydyny następują na tej samej drodze (rys.7.). W pierwszym kroku obydwa te związki są przemieniane w 2,5-dihydroksypirydynę, która z kolei jest następnie przemieniana poprzez szereg reakcji do pirogronianu.
Pierwszy etap szlaku degradacji 2-hydroksypirydyny katalizuje prawdopodobnie 5-monooksygenaza 2-hydroksypirydynowa, enzym wydzielony z Arthrobacter sp., który aktualnie jest bliżej badany [15]. Brak natomiast wiadomości o enzymie działającym na 3-hydroksypirydynę.

Rys.7. Schemat zbiorczy szlaków degradacji 2-hydroksypirydyny, 3-hydroksypirydyny oraz 2-karboksypirydyny
Dalsze przemiany 2,5-dihydroksypirydyny do pirogronianu nie są poznane. Nie udało się znaleźć pośrednich związków powstających w tym szlaku, ani enzymów, które mogły by w nim uczestniczyć. Być może przemianę 2,5-dihydroksypirydyny do 2,3,6-trihydroksypirydyny katalizuje jakaś oksygenaza. Hipotetyczny szlak kolejnych reakcji tego szlaku przedstawiono na rys. 7.
4-Hydroksypirydyna u Arthrobacter sp. nie ulega podobnym przemianom [15], co świadczy zarówno o innej jej drodze przemiany, jak i o potrzebie znalezienia innych mikroorganizmów ją degradujących.
Natomiast podobnym transformacją u Arthrobacter sp. [15] ulega 2-karboksypirydyna (kwas 2-pikolinowy). Ustalono, że pierwszym intermediatem jest kwas 6-hydroksy-2-pikolinowy, który jest dalej hydroksylowany w pozycji trzeciej (rys.7), a następnie dekarboksylowany z wytworzeniem 2,5-dihydroksypirydyny. Brak natomiast wiadomości o enzymach uczestniczących w tych reakcjach [15]. Przekształcenia kwasu pikolinowego w powyższy sposób dokonuje również szczep Alcaligenes faecalis [8].
3-metylopirydyna i 3-etylopirydyna
Do pochodnych pirydyny należą alkilopirydyny. Alkilopirydyny są toksycznymi zanieczyszczeniami środowiska występującymi w wielu wodach, zarówno powierzchniowych jak i gruntowych. Szczególnie w wodach znajdujących się niedaleko przemysłu paliwowsego. Metabolizm alkilopirydyn może podążać jedną z trzech inicjujących reakcji: redukcja aromatycznego pierścienia, oksydacja aromatycznego pierścienia lub oksydację grupy alkilowej.
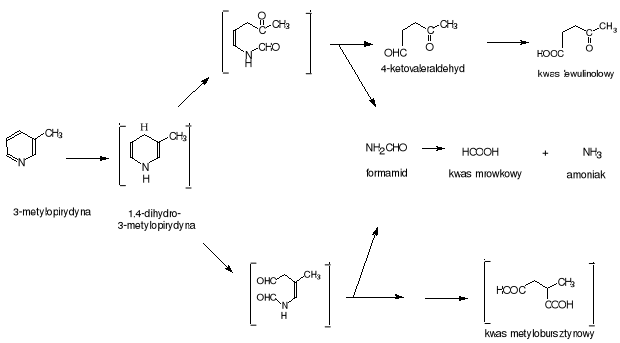
W maju 2001 roku doniesiono [9] o wyizolowaniu bakterii degradujących 3-metylopirydynę (3-mp) i 3-etylopirydynę (3-ep). Bakterie te zidentyfikowano jako Gordonia nitida LE31 (nie podano jednak danych o środowisku bytowania tych mikroorganizmów). Gordonia nitida LE31 w procesie tlenowym używa 3-mp i 3-ep jako wyłączne źródło węgla, azotu i energii. Podczas hodowli w ciekłych podłożach z dodatkiem 3-mp i 3-ep nie udało się zidentyfikować żadnych aromatycznych produktów przejściowych. Natomiast na podstawie pojawiania się jonu amonowego w podłożu hodowlanym omawianej kultury bakteryjnej podejrzewa się, że pierścień aromatyczny zostaje rozerwany z jednoczesną eliminacją azotu. Uwolniony azot stanowi 64% masy 3-mp lub 3-ep, pozostała część związku prawdopodobnie zostaje wbudowana w komórkę bakteryjną [9].
Podczas degradacji 3-mp i 3-ep przez szczep LE 31 w podłożu hodowlanym pojawia się formamid, przekształcany pod działaniem formamidazy (EC 3.5.1.49) w kwas mrówkowy, który nagromadza się jako produkt końcowy. Może to wskazywać na słabą aktywację dehydrogenazy mrówczanowej (EC 1.2.1.2), enzymu degradującego ten kwas. Wykryty w podłożu formamid raczej wyklucza inicjację degradacji 3-mp i 3-ep poprzez oksydację pierścienia pirydynowego a raczej wskazuje na rozerwanie pierścienia C2-C3 lub C5-C6, choć nie jest to pewne. Teoretyczne mogą istnieć cztery drogi degradacji 3-metylopirydyny, z czego dwie zostały pokazane na rys.8. Jeśli przejściowo powstaje kwas metylobursztynowy, to w pierwszej reakcji pęka prawdopodobnie wiązanie C5-C6, jeśli natomiast powstaje kwas lewulinolowy (4-oksopentanowy), to pęka wiązanie C2-C3 [9]. Enzymy, które mogą katalizować te przemiany to dehydrogenaza aldehydu lewulinolowego (gdy pęka wiązanie C2-C3) lub dehydrogenaza aldehydu metylobursztynowego (przy rozerwaniu wiązania C5-C6), jednakże ich aktywności nie udało się wykazać.
Dwie pozostałe domniemane drogi degradacji 3-mp i 3-ep są mniej prawdopodobne z uwagi na nie znalezienie w podłożu hodowlanym Gordonia nitida LE31 odpowiednich produktów przejściowych, choć z kolei wykryta aktywność dehydrogenazy aldehydu 2-metyloglutarynowego, może sugerować rozerwanie wiązania N-C1 lub wiązania N-C6 [9].
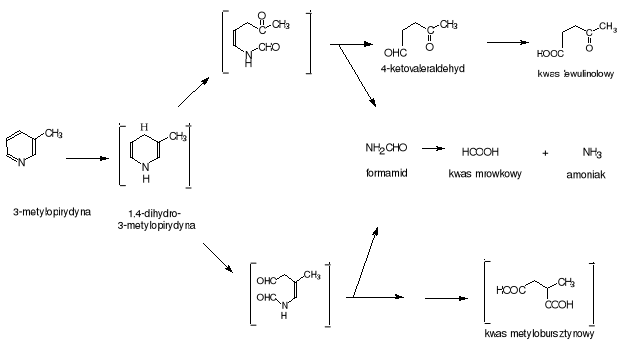
Rys.8. Proponowana droga degradacji 3-mp przez Gordonia nitida LE 31 [wg 9]
Kwas nikotynowy
Kwas nikotynowy (3-karboksypirydyna), zwany też niacyną, produkowany jest w skali światowej w znacznych ilościach. Jego pochodna kwas 6-hydroksynikotynowy jest wszechstronnie wykorzystywana jako podstawowa jednostka budulcowa wielu syntetycznych, nowoczesnych insektycydów [10].
Dowiedziono, że wiele różnych bakterii może wykorzystywać kwas nikotynowy jako źródło węgla i energii, a szczególną aktywność w tym kierunku wykazuje Pseudomonas. Mikroorganizmy mogą degradować kwas nikotynowy wykorzystując różne szlaki metaboliczne. Pierwszy etap szlaku degradacji tego związku obserwowany u Pseudomonas fluorescens rozpoczyna hydroksylacja do kwasu 6-hydroksynikotynwego katalizowana przez kompleks enzymatyczny sprzężony z cytochromem [10]. W kolejnym etapie zachodzi oksydacyjna dekarboksylacja kwasu 6-hydroksynikotynwego, której produktem jest 2,5-dihydroksypirydyna. Rozszczepienie jej pierścienia prowadzi do utworzenia kwasu mrówkowego i kwasu maleaminowego, który dalej, w wyniku hydrolitycznej deaminacji, przechodzi w kwas maleinowy. Następnie ma miejsce jego izomeryzacja w kwas fumarowy. Końcowym produktem tego szlaku jest pirogronian (rys.9) [5,10,15].
Rys.9. Paradiolowa droga degradacji kwasu nikotynowego u Pseudomonas fluorescens
Rys. 10. Degradacja kwasu nikotynowego przez Bacillus sp.
Niektóre gatunki bakterii z rodzaju Bacillus degradują kwas nikotynowy, traktując go jako źródło węgla, azotu i energii. Na rysunku 10 przedstawiono szlak degradacji tego związku znaleziony u Bacillus sp.. Jego przebieg jest bardzo podobny do omówionego poprzednio. Enzymy biorące w nim udział, podobnie jak to ma miejsce w przypadku degradacji pirydyny, nie są dotychczas jednoznacznie ustalone. Z kwasu nikotynowego, prawdopodobnie pod działaniem dehydrogenazy nikotynowej (hydroksylującej EC 1.5.1.13.), powstaje w pierwszym etapie kwas 6-hydroksynikotynowy, który przekształcany jest w kwas 2,6-dihydroksynikotynowy i w kolejnych etapach szlaku do 2,3,6-trihydroksypirydyny. W wyniku działania dioksygenazy następuje rozszczepienia pierścienia między atomami C1 i C2, w wyniku czego powstaje kwas mrówkowy oraz kwas maleaminowy; który z kolei pod działaniem amidazy przekształcany jest w kwas fumarowy, a ten włączany bezpośrednio w katabolizm komórkowy [5,10,15].
Kwas nikotynowy może ulegać degradacji mikrobiologicznej także w warunkach beztlenowych (rys.11.). W wyniku jego enzymatycznej hydroksylacji powstaje jak poprzednio kwas 6-hydroksynikotynowy, który przy udziale ferrodoksyny jest przekształcany do kwasu 6-oksonikotynowego. Ten spontanicznie ulega przemianie w kwas N-formyloglutarowy i w wyniku deaminacji powstaje kwas 2-metylenoglutarowy, który z kolei przekształcany jest enzymatycznie w kwas dimetylomaleinowy. Końcowym produktem tej drogi degradacji są kwas pirogronowy, kwas octowy, CO2 i amoniak. Taką drogę odnaleziono u Clostridium sp.[5,10,15]. Wyizolowano także z mułów morskich szczep Desulfococcus niacini posiadający zdolność wykorzystywania nikotynianu jako źródła węgla i akceptora elektronów. Degraduje on ten związek do CO2 i amoniaku. W procesie tym nie powstaje propionian ani octan [15].
Rys.11. Wstępne etapy beztlenowej drogi degradacji kwasu nikotynowego przez Clostridium sp. [wg 5].
Nikotyna, nornikotyna i anabizyna
Nikotyna jest przekształcana na wiele sposobów. Przemianom może ulegać zarówno pierścień pirydynowy jak i pirolidynowy. Wyizolowano z gram dodatnich bakterii glebowych Arthrobacter oxydans oksydazę nikotyny, która wbudowuje atom tlenu pochodzący z wody w strukturę nikotyny dając 6-hydroksynikotynę. Z 6-hydroksynikotyny powstaje 6-hydroksypseudonikotyna dzięki oksydazie hydroksynikotynowej. Enzym ten określany i klasyfikowany jest jako enzym podwójny, tzn. jeżeli degradacja nikotyny znajduje się w logarytmicznej fazie wzrostowej Arthrobacter oxydans to wydzielana jest oksydaza 6-hydroksy-L-nikotynowa (EC 1.5.3.5). Natomiast jeżeli degradacja nikotyny znajduje się w fazie stacjonarnej (faza druga) to mikroorganizm wydziela oksydazę 6-hydroksy-D-nikotynową (EC 1.5.3.6) [25]. Dalej formuje się 2,6-dihydroksypseudonikotyna, a potem 2,6-dihydroksypirydyna. 3-Monooksygenaza katalizuje transformację tego intermediatu do 2,3,6-trihydroksypiridyny. W warunkach tlenowych i przy braku enzymu, ta trihydroksypiridyna ulega spontanicznemu utlenieniu i formuje się niebieski pigment. Barwnik ten nie powstaje w obecności ekstraktu komórkowego A.oxydas i trójhydroksypochodna jest przekształcana enzymatycznie do kwasu maleaminowego (rys.12.). Ta oksydaza jest enzymem indukcyjnym i wymaga obecności zredukowanego nukleotydu pirydynowego, co oznacza, że jest monooksygenazą. Sugeruje się, że przekształcenie 2,6-dihydroksypirydyny do kwasu maleaminowego wymaga obecności dwóch enzymów-oksydazy i enzymu rozrywającego pierścień aromatyczny [15]. Dalej kwas ten może ulegać przekształceniom jak przy przekształceniach kwasu pikolinowego. Podczas degradacji rozpoczynającej się od przemian w pierścieniu pirolidynowym wstępnie powstaje N-metylomyozmina, która jest przekształcana do pseudonikotyny. Dalej powstaje 3-bursztynylopirydyna i taki układ ulega dopiero hydroksylacji do 6-hydroksy-3-bursztynylopirydyny [15]. Ostatni związek został wykryty tylko przy degradacji nikotyny przez Achromobacter nicotinophagum [5]. Później (rys. 12) ten związek ulega przemianie prawdopodobnie nie bezpośredniej do 2,5-dihydroksypirydyny [15].
Inne związki nikotynowe, nornikotyna (w miejscu grupy metylowej nikotyny znajduje się wodór), oraz anabizyna (pierścień pirolidynowy w nikotynie zastąpiony jest drugim pierścieniem pirydynowym bez wiązań podwójnych) ulegają niemal identycznym przemianom jak sama nikotyna. Degradację nornikotyny prowadzą mikroorganizmy z cytochromem P-34 a anabizyny Arthrobacter. Zmianie w przypadku nornikotyny ulega jedynie trzeci intermediat, w którym brak wiązania podwójnego [5].
Rys.12. Metabolizm nikotyny przez Arthrobacter oxydans oraz szczepy P-34 i JTS-0006 wg [5 i 15].
Szybkość degradacji pochodnych pirydyny zależy od typów podstawników. Najłatwiej ulegają degradacji karboksylowe pochodne, dalej w kolejności: monohydroksy-, metylo-, aminopirydyny. Natomiast najtrudniej degradowane są chloropirydyny [15].
4.3.2. Pierścienie sześcioczłonowe o charakterze niearomatycznym
W rozdziale tym omówiono wspólnie, z uwagi na podobny mechanizm biochemiczny, biodegradację morfoliny, tiomorfoliny i piperydyny.
Biodegradacja przemysłowych zanieczyszczeń organicznych, zwłaszcza związków heterocyklicznych takich jak morfolina jest niezwykle potrzebna środowisku. Morfolina ma bowiem wielkie znaczenie przemysłowe i szeroką rangę zastosowań. Między innymi jest używana jako antykorozyjny czynnik w systemach podgrzewania wody i jako chemiczny pośrednik (katalizator, rozpuszczalnik, przeciwutleniacz) w wytwarzaniu gumowych dodatków oraz w przemyśle tekstylnym [11]. Pomimo wielu poszukiwań, poznano tylko szczepy należące do Mycobacterium oraz dwa szczepy Arthrobacter, które degradując morfolinę wykorzystują ją jako wyłączne źródło węgla, azotu i energii. Szczepy te to: Mycobacterium aurum MO1, Mycobacterium sp. RP1 i Mycobacterium sp. MORG [11,12,13,14]. Szczepy MO1 i RP1 względnie szybko degradują morfolinę [14].
Nie są znane wszystkie produkty przejściowe podczas degradacji morfoliny, tiomorfoliny i piperydyny (rys.13.). Lecz wiadomo na pewno, że pierwszym krokiem w czasie degradacji tych związków jest rozerwanie pierścienia (pęka wiązanie N-C1), co prowadzi prawdopodobnie do utworzenia kwasu aminowego lub oksydacja atomu siarki w przypadku tiomorfoliny, zanim nastąpi otwarcie pierścienia [11]. Powyższe reakcje są inicjowane przez występujący w Mycobacterium cytochrom P450 [11,13,14]. W kolejnym kroku, podczas powstawania kwasów dikarboksylowych wydzielany jest amoniak. W przypadku piperydyny prawdopodobnie powstaje kwas 5-aminowalerianowy, który jest szybko deaminowany i oksydowany, w wyniku czego powstaje kwas glutarowy. Następnie uzyskany kwas glutarowy jest włączany do centralnego metabolizmu komórki bakterii. Degradacja tiomorfoliny przebiega podobnie. Także tutaj powstaje kwas dikarboksylowy - kwas tiodiglikolowy, który jednak jest akumulowany w komórkach bakterii [11].
Jeśli zaś chodzi o degradację morfoliny, to właśnie te badania okazały się najważniejsze z uwagi na to, że tylko tutaj został wykryty produkt przejściowy przed powstaniem kwasu diglikolowego. Był nim kwas 2-(2-aminoetoksy )-octowy, który jest deaminowany i oksydowany do kwasu diglikolowego z wydzieleniem się amoniaku. Dalej kwas ten jest rozszczepiany do kwasu glikolowego, który jest mineralizowany przez komórki Mycobacterium aurum MO1 i Mycobacterium sp. RP1 [11-14].
Reakcja otwarcia pierścienia nie zachodzi w przypadku, gdy pozycja jest zajęta na przykład przez grupę metylową. Testowane związki to dioksan i 2,6-dimetylomorfolina, które nie były degradowane przez powyższe szczepy [11].
Rys.13. Degradacja azotowych związków heterocyklicznych przez Mycobacterium aurum MO1 [wg 11].
Rys.14. Domniemana część drogi degradacyjnej morfoliny przez Mycobacterium sp. szczep RP1.
Prawdopodobne etapy przejściowe przekształcenia morfoliny do kwasu diglikolowego mogą przebiegać w sposób przedstawiony na rys.14., przy czym enzymy katalizujące te przemiany nie są znane [14].
4.3.3. Związki s-triazynowe
Atrazyna i związki pokrewne
Pierścienie s-triazynowe są obecne w wielu związkach użytych przy wytwarzaniu barwników, żywic i herbicydów. Atrazyna (2-chloro-4-etyloamino-6-izopropyloamino-1,3,5-triazyna) jest powszechnie używa w rolnictwie całego świata na szeroką skalę. Za jej pomocą kontroluje się „chwasty”. Niestety, często jest odnajdywana w wodach gruntowych oraz w glebie powierzchniowej, co stanowi ich poważne skażenie. Z dwóch farm wyizolowano grzyby i bakterie, które mineralizują atrazynę i inne S-triazynowe związki. Należą one do Pseudomonas, Rhizobium, Acinetobacter, Alcaligenes, Ralstonia, Agrobacterium, Clavibacterium i inne, które jeszcze nie zakwalifikowano do żadnego gatunku, a znane są tylko z symboli [16-18].
Degradacja atrazyny może być zarówno tlenowa jak i beztlenowa. Spośród licznych izolowanych drobnoustrojów dziewięć gram ujemnych szczepów jest zdolnych używać atrazyny jako wyłącznego źródła azotu [17]. 8 z izolowanych szczepów degraduje atrazynę do CO2, chlorku oraz NH3. Z tych 8 szczepów zidentyfikowano jedynie szczep C147, opisany jako Pseudoaminobacter [16]. 6 spośród izolowanych szczepów transformuje atrazynę do hydroksyatrazyny, która nagromadza się w podłożu. Dwa z nich: C157 i C190 zidentyfikowano jako Nocardioides simplex i Nocardioides plantarum [17].
Degradacja atrazyny następuje poprzez serię hydrolitycznych reakcji (rys.15.) inicjowanych przez dechlorację, a następnie dealkilację. Bardzo istotnym faktem, a wręcz decydującym jest występowanie genów atzA, atzB i atzC. Mikroorganizmy, które nie posiadają choćby jednego z nich nie degradują atrazyny całkowicie tzn. do CO2, lecz tylko do 6-hydroksyatrazyny [16]. Natomiast te, które nie mają genu atzA w ogóle nie są zdolne do degradacji.
Wspomniany Pseudoaminobacter traktują atrazynę jako wyłączne źródło węgla. Chlorohydrolaza atrazynowa kodowana przez gen atzA inicjuje pierwszą reakcję szlaku. Dwa geny atzB i atzC odpowiedzialne są razem za kolejne dwie reakcje dealkalizacji, które prowadzą do powstania kwasu cyjanurowego. Dalej następuje rozerwania pierścienia (prawdopodobnie w wyniku działania enzymu alkiloaminohydrolazy [16]), powstaje karbamylomocznik i na końcu mocznik (karbamid) [16,17]. W wyniku szeregu reakcji wytwarzana jest także etyloamia i izopropyloamina podtrzymująca wzrost komórek mikroorganizmu.
Według innego źródła [18], degradacja atrazyny następuje nie poprzez dwie, lecz trzy następujące kolejno reakcje hydrolityczne, w których w kolejności odrywane są podstawniki: chlorek, N-etylamina, N-izopropyloamina. Enzymy katalizujące te reakcje to kolejno: chlorohydrolaza atrazynowa oznaczona jako AtzA, etyloaminohydrolaza hydroksyatrazynowa (AtzB) i izopropyloaminohydrolaza izopropyloammelidowa (AtzC). Wszystkie te enzymy zostały rozpoznane jako amidohydrolazy proteinowe przez komputer na podstawie ich analizy sekwencji genowej [18], co nie zostało bliżej wyjaśnione.
Rys.15. Prawdopodobna droga degradacji atrazyny [wg 17-19 i autora].
Opis w tekście.
Inne związki S-triazynowe (nazwy podano dalej) także są degradowane przez opisane powyżej bakterie, lecz degradacja zachodzi trudniej. Ponieważ procesy biodegradacji atrazyny i innych pochodnych związków S-triazynowych są słabo poznane, to tylko można przypuszczać, jakie produkty pośrednie mogą się pojawiać w tych przemianach i jakie enzymy mogą uczestniczyć w tym procesie. Wiadomo z całą pewnością, że między innymi u Rhodococcus sp. TE1 i N 186/21, które inicjują proces przez odłączenie chloru lub innego podstawnika z pozycji 2 pochodnych S-triazyny następuje dalej dealkilacja i deaminacja z udziałem cytochromu P-450 [16-18].
Na rysunku 16 przedstawiono reakcję inicjacji przemian pochodnych S-triazyny u drobnoustrojów wykazujących zdolność dechloracji atrazyny pod działaniem chlorohydrolazy atrazynowej.
X= F,Cl X= F, Cl, CN, N3 ,OCH3, SCH3
Pseudomonas ADP Clavibacter michiganensis
Rhizobium PATR
Alcaligenes SG1
Agrobacterium J14a
Ralstonia picketti D
Rys.16. Inicjacja transformacji związków S-triazynowych przez różne szczepy dechlorujące atrazynę
Atrazine 2-Chloro-4-(N -ethylamino)-6-(N -isopropylamino)-1,3,5-s-triazine
Deethylatrazine 2-Chloro-4-amino-6-(N-isopropylamino)-1,3,5-s-triazine
Deisopropylatrazine 2-Chloro-4-amino-5-(N- ethylamino)-1,3,5-s-triazine
Simazine 2-Chloro-4,6-di(N-ethylamino)-1,3,5-s-triazine
Propazine 2-Chloro-4,6-di(N-isopropylamino)-1,3,5-s-triazine
Terbuthylazine 2-Chloro-4-(N- ethylamino)-6-(N-t-butylamino)-1,3,5-s-triazine
Seffernick i wsp. [18] podają, że:
chlorohydrolaza atrazynowa (AtzA) katalizuje dechlorację atrazyny i propazyny (2-chloro-4,6-di (N-izopropyloamino)-1,3,5-triazyna);
hydrolaza triazynowa (TrzA) znaleziona u Rhodococcus corallinus NRRL B-154444R katalizuje deaminację niealkilowych aminotriazyn, jak np. CAAT (2-chloro-4,6-diamino-1,3,5-triazyna);
TrzA z Rhodococcus corallinus NRRL B-154444R katalizuje powolną dechlorację CEAT (2-chloro-4-amino-6-(N-etyloamino-)-1,3,5-triazyna), ale nie może katalizować dwóch reakcji deaminacji i dechloracji atrazyny;
TrzA katalizuje deaminację diaminotriazyn i pirymidyn oraz dechlorację triazyn posiadających pojedynczą grupę alkilową na jednym z dwóch azotowych podstawników;
niezdolność katalizowania przez AtzA reakcji deaminacji w aminoatrazynach np. CAAT, umiejscawia tenże enzym w nowej dehalogenującej podgrupie „superrodziny” amidohydrolaz.
We wszystkich S-triazynowych związkach najpierw następuje hydrolityczna dechloracja i zawsze powstaje odpowiedni 6-hydro związek [16]. Obrazowo widoczne jest to na rys.16. Dalsze reakcje nie są obecnie poznane. Szczep C190, o którym wcześniej już wspominałem, degraduje wszystkie chloro- i metylo- S-triazynowe związki beztlenowo [17].
Heksahydro-1,3,5-trinitro-1,3,5-triazyna
Kolejnym związkiem o budowie triazynowej jest RDX, czyli heksahydro-1,3,5-trinitro-1,3,5-triazyna. Ponieważ o tym związku i jego biodegradacji w literaturze naukowej niewiele jest informacji, dlatego poświęcono mu osobny podrozdział.
RDX jest wysoko energetycznym związkiem chemicznym, który ma szerokie zastosowanie w wojsku i wśród cywilnej ludności. RDX znaleziono w wodzie, którą wykorzystywano w fabrykach do produkcji żywności oraz w szlamie z okolic tych fabryk. Związek ten wchodzi w połączenia z glebą, a także przenika do wód podziemnych, co stanowi jej zanieczyszczenie. Ponieważ nitroaminy cykliczne są niezwykle toksyczne [20], należy je eliminować ze środowiska naturalnego w sposób naturalny, który jest na pewno tańszy i bardziej bezpieczniejszy.
RDX i HMX (octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazyna) są degradowane w anaerobowy (beztlenowy) sposób w szlamie, poprzez redukcję azotanu, jednak o szlaku degradacyjnym HMX brak jest jakichkolwiek danych. W szlamie niezbędne są warunki sulfidogenne i/lub metanogenne. Sama droga degradacyjna RDX może przebiegać na dwa sposoby [20]. Pierwszy (droga b, rys.17.) bazuje na kolejnych redukcjach RDX do MNX (heksahydro-1-nitrozo-3,5-dinitro-1,3,5-triazyna), DNX (heksahydro-1,3-dinitrozo-5-nitro-1,3,5-triazyna), TNX (heksahydro-1,3,5-trinitrozo-1,3,5-triazyna); przy czym ostatni związek nie został wykryty i jest tylko sugestią. Także żadnych enzymów ani produktów przejściowych rozszczepienia pierścienia na tej drodze nie udało się wykryć. TNX przemienia się z kolei w pochodną RDX, prawdopodobnie w aminohydro-RDX i dopiero w tym miejscu następuje reakcja, bądź reakcje rozerwania pierścienia; końcowymi produktami tych reakcji, tym razem już wykrytymi są formaldehyd, metanol, NH2NH2 oraz (CH3)2NNH2. Proces ten jest całkowicie beztlenowy, a do eksperymentów użyto RDX ze szlamu z trzech różnych fabryk [20]. Brak natomiast wiadomości o jakichkolwiek mikroorganizmach uczestniczących w tym procesie. Degradację RDX mogą prowadzić prawdopodobnie Methanosaeta concilii i Methanosarcina barkeri występujące w badanych ściekach [21].
Rys.17.Propozycja beztlenowej biodegradacji RDX w szlamie pochodzącym z trzech fabryk produkujących żywność [wg 20].
Druga droga biodegradacji RDX przebiega nieco w inny sposób (droga a, rys.17.). Także jest to proces beztlenowy, w którym jako pierwszy krok, następuje rozerwanie pierścienia N-C-N, co jest prawdopodobnie spowodowane działaniem hydrolazy. Jednak i tutaj obecność enzymów nie została stwierdzona. W tym szlaku pojawiają się dwa nowe metabolity powstałe w wyniku bezpośredniego rozerwania pierścienia metylenodinitroamina i bis(hydroksymetylo)nitroamina. W kolejnym kroku z tych dwóch związków powstają dwutlenek węgla, N2O i formaldehyd, który jest utleniany do kwasu mrówkowego HCOOH i dalej transformowany do CO2 i CH4 [20]. Mikroorganizmy także i na tej drodze nie są jednoznacznie określone.
Sugeruje się [21], iż transformację RDX może prowadzić Pseudomonas pseudoalcaligenes JS45 (który wytwarza enzym nitroreduktazę redukującą nitrobenzen) lub Stenotrophomonas maltophilia PB1, choć w tym przypadku proces jest tlenowy. Ze wstępnych danych wynika, że także i w tym przypadku powstają dwa produkty rozszczepienia pierścienia: z ang. methylene-N-nitroamino-N'-acetoxyammonium i methylene-N-(hydroxymethyl)-hydroxylamine-N'-(hydroxymethyl)nitroamine. O dalszych losach tych związków jednak nic nie wiadomo [21]. Innym mikroorganizmem, który być może tlenowo degraduje RDX i HMX jest Phanerochaete chrysosporium [20].
4.4. Biodegradacja dwupierścieniowych związków heterocyklicznych.
Tryptofan
Tryptofan jest dla człowieka substancją egzogenną i dlatego stanowi niezbędny składnik pokarmowy. Podobnie, jak w przypadku histydyny, poprawniej jest rozpatrywać wewnątrzkomórkowy rozkład tryptofanu w kategoriach metabolizmu czy katabolizmu komórkowego.
Aminokwas ten może podlegać transaminacji, ale reakcja ta, podobnie jak w przypadku histydyny, stanowi ślepą uliczkę w jego metabolizmie. Powstały w wyniku transaminacji oksokwas może ulec procesom katabolicznym dopiero po ponownej aminacji do tryptofanu [22]. Główny szlak degradacji tryptofanu (rys.19.) rozpoczyna się reakcją rozerwania pierścienia pięcioczłonowego katalizowaną przez oksygenazę tryptofanową (2,3-dioksygenazę tryptofanową,). Następnie, powstała N-formylokinurenina ulega hydrolizie do kinureniny i mrówczanu. W wyniku kolejnych przemian dochodzi do rozerwania łańcucha bocznego cząsteczki i uwolnienia alaniny. Kolejnymi ważnymi metabolitami pośrednimi degradacji tryptofanu są 3-hydroksyantranilan powstający dzięki kinureninazie, a następnie semialdehyd amino-karboksymukonowy [23] W tym miejscu następuje rozgałęzienie szlaku; główna droga prowadzi do acetylokoenzymu A, a droga mniej wydajna ilościowo jest szlakiem syntezy niacyny (kwasu nikotynowego). Okazuje się, że nie więcej niż jedna sześćdziesiąta całej ilości tryptofanu może być przetworzona w niacynę [22].
Wiadomo także, że parę szczepów Pseudomonas używa do degradacji tryptofanu drogi poprzez kwas kinureinowy, lecz dotychczas nie udało się ustalić pośrednich produktów tego szlaku [24].
5-Hydroksytryptofan
Inną drogę metabolizmu tryptofanu, nie tak istotną ilościowo jak szlak omówiony poprzednio, lecz mającą wielkie znaczenie jakościowe, rozpoczyna jego hydroksylacja w pozycji 5. Reakcja ta może zachodzić w wielu tkankach, lecz jest szczególnie ważna w mózgu i szyszynce. W wyniku hydroksylacji tryptofanu powstaje 5-hydroksytryptofan, który może ulegać dekarboksylacji katalizowanej przez karboksylazę tryptofanową, wymagającą udziału fosforanu pirydoksalu (PALP). Produktem tej dekarboksylacji jest 5-hydroksytryptamina, znana także jako serotonina. Serotonina jest jednym z przekaźników w ośrodkowym układzie nerwowym [22].
Dlatego jakiekolwiek zaburzenia tej drogi metabolizmu tryptofanu mogą prowadzić do nieprawidłowości w pracy mózgu. Serotonina może być degradowana enzymatycznie przez oksydazę monoaminową (zawierającą flawinę), która katalizuje powstawanie amoniaku i aldehydu 5-hydroksyindolilooctowego. Reakcja ta jest nieodwracalna. Wytwarzany aldehyd jest szybko utleniany enzymatycznie przy udziale NAD+ do 5-hydroksyindolilooctanu (5-hydroksyindolilooctowego), który jest zazwyczaj wydalany [22].
Rys.19. Katabolizm tryptofanu
Enzymy uczestniczące w katabolizmie tryptofanu:
1 .2,3-dioksygenaza tryptofanowa EC 1.3.11.11
2. amidohydrolaza arylo-formyloaminowa EC 3.5.1.9
3. 3-monooksygenaza kinureninowa EC 1.14.13.9
4. kinureninaza EC 3.7.1.3
5. 3,4-dioksygenaza 3-hydroksyantranilowa EC 1.13.11.6
6. dekarboksylaza aminokarboksymukono-semialdehydowa EC 4.1.1.45
7. dehydrogenaza mainomukono-semialdehydowa EC 1.2.1.32
8. hydrataza
9. dehydrogenaza
10. dehydrogenaza 2-oksokwasu
11. dehydrogenaza glutarylo-CoA EC 1.3.99.7
12. dekarboksylaza glutakonylo CoA EC 4.1.1.70
13. hydrataza enoylo-CoA EC 4.2.1.17
14. dehydrogenaza 3-hydroksyacylo-CoA EC 1.1.1.211
15. tiolaza 3-oksoacylo-CoA
Indol
Indol jest bezbarwnym związkiem krystalicznym o zapachu podobnym do naftalenu. Jego pochodna - skatol (3-metyloindol) ma nieprzyjemny zapach kału. Indol występuje w środowisku, do którego jest uwalniany podczas palenia tytoniu oraz wraz z niektórymi farmaceutykami, pestycydami czy barwnikami [25,26]. Można go również wykryć w smole węglowej i ściekach. Indol jest toksyczny i wraz z innymi związkami N-heterocyklicznymi, stanowi odpady w dziedzinach przemysłu związanych z obróbką termiczną materiałów organicznych (rafinacja ropy, produkcja koksu). Wiele alkaloidów zawiera w swej strukturze część indolową [15].
Eksperymentalnie dowiedziono [15,27], że indol i jego 3-metylowa pochodna ulegają degradacji w warunkach metanogennych do metanu i CO2. Transformacje w obu przypadkach zaczynają się hydroksylacją i powstają pochodne 2-hydrokslowe [15,25,27,28]. W przypadku indolu powstaje oksyindol, a następnie izatyna. Umożliwia to rozerwanie pierścienia pirolowego między drugim a trzecim atomem węgla [15] (rys.20.). Natomiast degradacja 3-metyloindolu przebiega według odmiennego szlaku. Z uwagi na zajętą przez podstawnik metylowy pozycję C-3 niemożliwe jest formowanie izatyny. Degradacja tej pochodnej tym samym szlakiem wymagałaby wcześniejszej demetylacji [15,27]. Takie przemiany, jak się wydaje, nie mają jednak miejsca. Przemawia za tym fakt, że drobnoustroje degradujące 3-metylową pochodną nie są w stanie utylizować indolu. Przypuszcza się, że mogą istnieć dwa różne szlaki degradacyjne indolu [15].
Inny szlak biodegradacji indolu został zaproponowany przez Andreoni`ego i wsp. [19] dla Aspergillus niger. Indol jest metabolizowany przez 2,3-dihydroksyindol. Dihydroksyindol jest dalej przeprowadzany w kwas antranilowy prawdopodobnie przez 2,3-dioksygenazę 2,3-dihydroksyindolową EC 1.13.11.23, która występuje tylko wtedy, gdy mikroorganizm rośnie na indolu [19,28]. Kwas antranilowy następnie jest przekształcany do pirogronianu i kwasu maleinowego przez 1,2-dioksygenazę gentyzynianową (EC 1.13.11.4) obecną w komórkach rosnących na indolu.
Rys.20. Proponowany szlak metanogennej fermentacji indolu przez mikroorganizmy występujące w przefermentowanych osadach [wg 19,28].
Innym mikroorganizmem degradującym indol są gramujemne bakterie siarkowe Desulfobacterium indolicum In04, wyizolowane z morskich osadów [27,28]. U tych drobnoustrojów w pierwszym kroku przekształceń indolu powstaje indoksyl (3-hydroksyindol - rys.21), który następnie jest transformowany do kwasu N-formyloantranilowego przez dioksygenazę (prawdopodobnie chodzi tu o 1,2-dioksygenazę antranilową, deaminująco-dekarboksylującą - EC 1.14.12.1). Dalej związek ten, przekształcany jest do katecholu. U Desulfobacterium indolicum In04 podczas degradacji indolu znaleziono kilka enzymów [19]: deformylazę N-formyloantranilową; bliżej jeszcze nie określoną hydroksylazę antranilanową (być może EC 1.14.12.1. lub EC 1.14.13.35); nie sklasyfikowaną dekarboksylazę dihydroksybenzoesową oraz oksygenazę katecholową (EC 1.13.11.1 lub EC 1.13.11.2).
Oksyindol - pierwszy związek przejściowy w czasie degradacji indolu (w obecnie proponowanych szlakach), nie był dotychczas wykrywany w podłożach w trakcie wzrostu drobnoustrojów[15,19]. Dopiero niedawno pojawiły się doniesienia o wykryciu tego związku m.in. w podłożach Alcaligenes oraz podczas metabolizmu tlenowego prowadzonego przez Pseudomonas putida, u których wykryto również kwas antranilowy [27].
Rys.21. Szlak degradacji indolu przez denitryfikujące mikroorganizmy zawarte w ściekach [wg 19,24,29].
Chinolina i jej pochodne
Chinolina i jej pochodne są głównymi składnikami wielu środowisk wodnych zanieczyszczonych przez odpady pochodzące z konserwacji drewna (kreozot), kopalni i przetwórni paliw kopalnych. Chinolina wchodzi też w skład oleju silnikowego. Są to związki toksyczne i szkodliwe już w bardzo małych stężeniach. Stwierdzono, że sama chinolina jest przyczyną powstawania raka wątroby u myszy i szczurów. Powoduje też zmiany w płucach u niektórych zwierząt doświadczalnych i raka skóry u myszy [15]. Przypuszcza się, że powodem takiej toksyczności jest formowanie N-tlenku chinoliny. Teorię tę popiera się doświadczeniami nad toksycznością 2-chlorochinoliny. Nie wykazuje ona wprawdzie działania kancerogennego, ale powoduje marskość wątroby u szczurów. Brak aktywności kancerogennej 2-chlorochinoliny tłumaczy się przeszkodami przestrzennymi w tworzeniu 2-chlorochinoliny [15].
Z uwagi na wysoką toksyczność przeprowadzono już wiele działań dotyczących degradacji chinoliny i jej pochodnych i to zarówno w warunkach tlenowych jak i beztlenowych. Tak kompleksowe badania są konieczne ponieważ warunki degradacji bardzo łatwo mogą ulec zmianie z beztlenowych na tlenowe i ważna jest, z przyczyn oczywistych, dobra znajomość szlaków metabolicznych tych związków w każdych warunkach. Do takich sytuacji dochodzi, na przykład, podczas zabiegów hydrologicznych. Beztlenowe osady w płytkich wodach stają się nagle tlenowymi jako wynik spadku poziomu wody i aktywność zyskują inne drobnoustroje niż w strefach beztlenowych [15].
Większość z mikroorganizmów zdolnych degradować chinolinę to Pseudomonas sp., ale także Rhodococcus, Nocardia, Moraxella, Desulfobacterium [30-33]. Miedzy innymi są to szczepy: Pseudomonas aeruginosa QP, Pseudomonas putida QP oraz Pseudomonas sp. MPQ [32]. Bakterie degradujące chinolinę w warunkach tlenowych izolowano z gleby ogrodowej i ścieków [30]. Kultury tlenowe izolowano także z próbek ziem zawierających resztki kreozotu [32].
Na podstawie przeprowadzonych badań stwierdzono, że pierwszym etapem w degradacji chinoliny i to zarówno w warunkach tlenowych jak i beztlenowych, jest hydroksylacja węgla C1, w wyniku której formuje się 2-hydroksychinolina (rys.22.) [30-32]. Atom tlenu potrzebny do tej reakcji pochodzi od cząsteczki wody [31]. W warunkach aerobowych, tlen jest potrzebny jako ostateczny akceptor elektronów w łańcuchu oddechowym, a także do późniejszych hydroksylacji prowadzących do rozcięcia pierścienia. Reakcje te prowadzone są przez oksygenazy [15], lecz w nomenklaturze enzymów nie zostały jeszcze sklasyfikowane.
W celu wykrycia produktów pośrednich lub enzymów katalizujących te przemiany w doświadczeniach użyto tzw. ciężkiej wody (H2O18 ). Po wykonaniu analiz okazało się, że pochodna chinoliny (2-hydroksy-chinolina) zawiera w swej cząsteczce „ciężki” atom tlenu O18 [15].
Dalsza degradacja 2-hydroksychinoliny może przebiegać według alternatywnych szlaków metabolicznych. Stwierdzono, że bakterie wykorzystujące chinolinę jako źródło węgla i azotu, mogą po wstępnej hydroksylacji (rys.22.), dalej metabolizować ten związek albo przez 2,6-dihydroksychinolinę i dalej przez 2,5,6-trihydroksychinolinę, lub przez tworzenie 2,8-dihydroksychinoliny i 8-hydroksykumaryny. Druga z tych ścieżek metabolicznych przebiega z udziałem bakterii Pseudomonas sp. Produktem przejściowy jest tu kwas 2,3-dihydroksyfenylopropionowy [15]. Transformacje chinoliny prowadzone przez populację Pseudomonas sp. oraz szczep Rhodococcus B1 przedstawiono na rysunku 22. Inny, gramujemny Pseudomonas ayucida szczep IGTN9m wykorzystuje chinolinę jako wyłączne źródło azotu, lecz co jest charakterystyczne nie wyłączne źródło węgla. Inicjującym enzymem jest 2-oksoreduktaza chinolinowa [33] (w nomenklaturze enzymów z 1992 roku, brak o niej i innych enzymach biorących udział w tych reakcjach wiadomości).
Rys.22. Degradacja chinoliny przez Pseudomonas sp. lub Rhodococcus B1 [wg 15].
Z przedstawionych szlaków metabolicznych wynika, że otwarcie pierścienia może nastąpić zarówno w homocyklicznej jak i w heterocyklicznej części cząsteczki. Szczep Pseudomonas putida transformuje oba pierścienie: homocykliczny i heterocykliczny [15]. Kiedy atakowi ulega pierścień benzenowy powstają cis-dihydrodiole. Sugeruje się, że za powstawianie tych związków są odpowiedzialne dioksygenazy, a wynikiem działania monooksygenaz jest hydroksylowy pierścień pirydyny. Tworzenie hydroksylowych pochodnych pierścienia heterocyklicznego tłumaczy się niestabilnością cis-dihydrodioli tego pierścienia i ich spontaniczną dehydratacją. Chinolina ulega degradacji także w procesie metanogenezy [33]. Po okresie adaptacji (6 miesięcy), drobnoustroje są w stanie całkowicie degradować chinolinę do metanu, produkty pośrednie, jak dotąd nie są znane.
Z głęboko położonych osadów podpowierzchniowych wyizolowano dwa typy gramujemnych bakterii, które są w stanie mineralizować chinolinę w warunkach tlenowych. W warunkach beztlenowych, natomiast, tworzą się rozpuszczalne produkty pośrednie. Różnicę tę interpretuje się w następujący sposób. W obecności tlenu, potrzebnego do hydroksylacji pierścienia benzenowego, zachodzi pęknięcie tego pierścienia i zupełna degradacja chinoliny [15]. Natomiast, gdy brakuje tlenu nie może dojść do rozcięcia pierścienia i wówczas nagromadzają się rozpuszczalne produkty pośrednie [30].
Rys.23. Inicjacja degradacji mikrobiologicznej 4-metylochinoliny [wg 15]
1) u Methylobacterium extorquenes, 2) u Pseudomonas putida
Typ transformacji, jakim ulegają pochodne chinoliny, w dużej mierze zależy od usytuowania podstawnika w cząsteczce tego związku. Degradację 2-karboksy-4-hydroksychinoliny badano wykorzystując ekstrakt komórkowy bakterii Pseudomonas putida. Drobnoustroje te degradują tą pochodną chinoliny w obecności NAD, a jako produkty reakcji powstają: L-glutaminian, L-alanina, kwas octowy i dwutlenek węgla. Stwierdzono, że produktami pośrednimi są: 7,8-dihydro-(2-karboksy-4-hydroksychinolina)-7,8-diol oraz 7,8-dihydroksy-(2-karboksy-4-hydroksychinolina). Podobną transformację 2-karboksy-4-hydroksychinoliny przeprowadzają bakterie Aerococcus sp. [15].
Drobnoustroje wykazują różną specyficzność substratową [15], gdyż większość z populacji zdolnych degradować pochodne metylowe, nie jest w stanie przekształcać 2-metylochinoliny[23]. Wynika to z faktu zajęcia pozycji C-2 przez podstawnik metylowy. Okazuje się jednak, że istnieją drobnoustroje zdolne degradować tą pochodną chinoliny. Opisano szlak metaboliczny 2-metylochinoliny u Arthrobacter sp. oraz u Pseudomonas sp. szczep MPQ [32]. Wyodrębniono następujące produkty pośrednie: 1H-4-oxochinolinę, kwas N-acetyloizatowy, kwas N-acetyloantranilowy, kwas antranilowy, kwas 3-hydroksy-N-acetyloantranilowy, katechol [15]. Szczep MPQ traktuje chinolinę jako wyłączne źródło węgla [32].
Także podczas degradacji pochodnych chinoliny (podstawniki w pierścieniu pirydynowym) rozerwaniu ulega pierścień benzenowy. Nie obserwuje się alternatywnej ścieżki, w której w wyniku przemian pęka pierścień heterocykliczny [15]. Zasada ta stosuje się również do transformacji 4-metylochinoliny. Związek ten został odnaleziony w wodach gruntowych do których przeniknął [32,34]. Jeden z mikroorganizmów degradujących ten związek, określany jako Pseudomonas lub Xantomonas, posiada zdolność degradacji 4-metylochinoliny zarówno jako czysta populacja jak i w mieszaninie z innymi drobnoustrojami [15,34]. Inny szczep bakterii zdolny wykorzystywać 4-metylochinolinę to Methylobacterium extorquenes [34]. Szlak metaboliczny biodegradacji 4-metylochinoliny u tych bakterii przedstawiono na rys.23 ścieżka 1. Pierwszym krokiem jest hydroksylacja pozycji C-2 metylochinoliny. W wyniku dalszych przemian powstaje hydroksy-4-metylokumaryna. Inni autorzy [15] opisują degradację tej samej pochodnej chinoliny przez szczep Pseudomonas putida, z pęknięciem pierścienia między C-7 a C-8 (rys.23. ścieżka 2). Wspomniany już Pseudomonas aeruginosa QP jest zdolny do hydroksylacji 6-metylochinoliny, 7-metylochinoliny i 8-metylochinoliny [33].
4.5. Biodegradacja trójpierścieniowych związków heterocyklicznych.
Karbazol
Karbazol jest używany w wielu dziedzinach przemysłu (produkcja barwników, insektycydów, przemysł medyczny) dlatego jego rozpowszechnienie w środowisku jest dosyć powszechne. Niewiele natomiast wiadomo o jego losach w środowisku. Związek ten charakteryzuje się wysoką toksycznością i przejawia aktywność mutagenną.
W głównej mierze za wysoką toksyczność tego związku odpowiadają jego hydroksynitrowe pochodne, natomiast za aktywność mutagenną odpowiedzialne są pochodne N-metylowe i N-hydroksymetylowe. Obecność tych pochodnych może prowadzić do zatrucia katalizatorów używanych w krakingu nafty do użytku komercyjnego [15]. Generalnie, karbazol dosyć trudno ulega degradacji, jednakże wyizolowano pewną ilość drobnoustrojów posiadających zdolność transformowania tego związku. Przemiany karbazolu rozpoczyna, podobnie jak i w innych przypadkach, hydroksylacja. Pochodna powstała, w wyniku pęknięcia pierścienia heterocyklicznego ulega przekształceniu do 2'-aminobifenylo-2,3-diolu. Związek ten, w wyniku dalszych przemian (otwarcie pierścienia), jest przekształcany do kwasu antranilowego. Przemiany mogą zostać wstrzymane na tym etapie, i powstała pochodna może zostać wykorzystana do produkcji tryptofanu lub w wyniku dalszych przekształceń, karbazol może ulec całkowitemu rozkładowi do CO2 i wbudowaniu w biomasę [8,35]. Przemiany karbazolu zilustrowano na rysunku 24.
Wiele pochodnych karbazolu ulega przekształceniom do produktów fenolowych. Mikroorganizmem zdolnym do prowadzenia takich przemian jest Cunninghamella echinulata Tymi pochodnymi są N-metlokarbozol i hydroksykarbazol [8].
Akrydyna
Akrydyna wraz z 7,8-benzochinoliną i karbazolem, wchodzi w skład zanieczyszczeń, pochodzących z odpadów chemicznych przemysłu drzewnego (impregnacja drewna). Akrydyna swą budową i właściwościami przypomina zarówno pirydynę jak i chinolinę. Związek ten ulega degradacji w warunkach beztlenowych przez populacje metanogenne, denitryfikacyjne i redukujące siarczany. Jednym z produktów pośrednich jest kwas benzoesowy. Według zaproponowanego, teoretycznego szlaku w warunkach beztlenowych utlenieniu ulega zarówno benzenowa jak i pirydynowa część cząsteczki. Dalej ulega przekształceniom typowym dla utlenionych związków aromatycznych: utlenieniu, redukcji, dekarboksylacji, rozerwania pierścienia i rozpadu na produkty alifatyczne [15].
Rys.24. Proponowany szlak degradacyjny karbazolu przez Cunninghamella echinulata [wg 8,35].
Dibenzofuran
Dibenzofuran jest niezwykle toksycznym związkiem dostającym się do środowiska w herbicydach [37]. Drogi degradacji tego związku nie zostały jeszcze poznane, gdyż nie udało się wyizolować produktów pośrednich. Przypuszczalne intermediaty (rys.25) zostały określone na podstawie wykrytych końcowych produktów: kwasu salicylowego i pochodnej kwasu 2-hydroksyfenyloheksadienowego. Mikroorganizmy zdolne do degradacji dibenzofuranu zidentyfikowano jako Ralstonia sp. SBUG 290 [36], Staphylococcus auriculans DBF 63, Brevibacterium DPO 1361 i Pseudomonas HH69 [37]. Zarówno dla pierwszego gram-ujemnego jak i dla pozostałych gram-dodatnich dibenzofuran stanowi wyłączne źródło węgla i energii [36-38].
Podczas degradacji prowadzonej przez szczep Ralstonia musi być dodany do pożywki 4-chlorobifenyl jako substancja wzrostowa, inaczej dibenzofuran nie zostanie zużyty. Nie jest wiadome dlaczego tak się dzieje. Sam proces degradacji następuje poprzez oksygenolityczny atak na narożną pozycję 4 i 4a przylegającą do eterowego mostka (rys.25.). W rezultacie formuje się 2,2`3-trihydroksyfenyl. Ten intermediat jest przemieniany przez meta rozszczepienie do pochodnej kwasu 2-hydroksyfenyloheksadienowego i kwasu salicylowego [36].
Rys.25. Prawdopodobna droga degradacji dibenzofuranu przez Ralstonia sp. SBGU 290 [wg 36].
Pochodne dibenzofuranu takie jak 2-,3-,4-hydroksy; 2-,3-,4-metoksy; 2-,3-,4-acetooksy; 2-,3-nitro są degradowane przez szczep Sphingomonas sp HH69, a ich produktami są odpowiednie kwasy hydroksy-; metoksy-; acetooksy- i nitro-salicylowe (każdy z osobna) [38].
Dibenzo-p-dioksyn
Podobnie jak dibenzofuran, dibenzo-p-dioksyn także jest wykorzystywany w rolnictwie. Poniższy schemat obrazuje degradację tego związku przez bakterie, ale co jest charakterystyczne Pseudomonas sp. szczep HH69 może także degradować dibenzofuran w identyczny sposób (uwzględniając oczywiście budowę) [37]. Enzymy także i w tym przypadku nie zostały wykryte i sklasyfikowane (prawdopodobne na rysunku 26) [39].
Sądzi się, że mechanizm podany na rysunku jest wspólny dla wszystkich podwójnych eterów biarylowych, ponadto halogenowe związki aromatyczne są bardziej oporne na biodegradację niż inne [39].
Rys.26. Degradacja dibenzo-p-dioksynu przez Pseudomonas sp. szczep HH69 [wg 38,39].
5. Podsumowanie
W przyrodzie występuje ogromna różnorodność drobnoustrojów przystosowana do bytowania w najróżniejszych warunkach środowiska. Z drugiej strony, większość z ksenobiotyków będących pochodnymi aminowymi związków organicznych wykazuje pewne podobieństwo do naturalnie występujących w przyrodzie substancji. Nic też dziwnego, że teoretycznie jedynie kwestią czasu jest znalezienie mikroorganizmu zdolnego do biodegradacji lub detoksyfikacji (np. na drodze transformacji) praktycznie każdego z takich ksenobiotyków. Sprawą zasadniczą jest jedynie wykształtowany ewolucyjnie zasób enzymów o odpowiednio szerokiej specyficzności substratowej i odporności na mogące powstawać w wyniku jego działania związki o właściwościach inhibitorów. W badaniach laboratoryjnych wykazano, że drobnoustroje zdolne do biodegradacji i biotransformacji związków heterocyklicznych oraz ich pochodnych są szeroko rozpowszechnione w przyrodzie. Zdziwienie może jedynie budzić różnorodność enzymatycznych mechanizmów jakie wykorzystują do tych celów. Niektóre z nich są zdolne bezpośrednio rozszczepiać pierścień związków heterocyklicznych. Inne, inicjują proces biodegradacji na drodze hydroksylacji. Można przypuszczać, iż w środowiskach naturalnych również zachodzą identyczne przemiany do tych, jakie obserwuje się w laboratoriach.
Niektóre drobnoustroje nie są jednak zdolne do mineralizacji związków heterocyklicznych i w ich podłożach hodowlanych kumulują się produkty pośrednie biotransformacji, jaką prowadzą (np. podczas degradacji chinoliny przez Rhodococcus B1 w podłożach nagromadza się pochodna pirydonu).
Należy przyznać, że w kilku przypadkach biochemiczny mechanizm biodegradacji związków heterocyklicznych nie został jeszcze do końca rozszyfrowany. Wiele z enzymów uczestniczących w tych przemianach nie zostało dotychczas sklasyfikowanych, a niekiedy nawet bliżej określonych.
Z występujących w przyrodzie mikroorganizmów, w reakcjach biodegradacji związków heterocyklicznych najczęściej uczestniczą bakterie z rodzaju Pseudomonas (np. w degradacji kwasu nikotynowego, chinoliny, pirolidyny i wielu innych).
Innym mikroorganizmem niezwykle uniwersalnym pod kątem degradacji związków heterocyklicznych jest niedawno odkryty rodzaj Gordonia, z którym wiąże się bardzo wielkie nadzieje. Szczepy należące do tego rodzaju bakterii (np. Gordonia nitida) potrafią mineralizować bardzo wiele związków heterocyklicznych (m.in. 3 metylo- i 3-etylopirydynę, dibenzofuran) i niektóre związki cykliczne pochodzące z ropy naftowej (np. naftalen).
Wiele związków heterocyklicznych (m.in. S-triazynowe, dibenzofuran) degradowanych jest zarówno przez bakterie gram-ujemne jak i gram-dodatnie.
Niektóre związki heterocykliczne mogą ulegać przemianom zarówno w warunkach tlenowych jak i beztlenowych. Okazuje się, że biochemiczny mechanizm biodegradacji pirydyny u Nocardia sp. Z1 jest identyczny w obu przypadkach.
Podczas degradacji związków heterocyklicznych prowadzonych przez różne mikroorganizmy, pierwszym etapem jest najczęściej hydroksylacja. Taką transformację obserwuje się m.in. podczas degradacji chinoliny u Pseudomonas sp., nikotyny u Arthrobacter oxydans, itp. W przypadku degradacji pirydyny przez Bacillus sp. 4, przemiany rozpoczyna reakcja uwodornienia, a już powstałe pochodne tego związku ulegają w dalszych przemianach hydroksylacji.
6. Literatura
1. Joule J.A. 1985. Chemia związków heterocyklicznych.
2. Schlegel H.G. 1996. Mikrobiologia ogólna.
3. Bojarski J. Chemia organiczna
4. Russel S. 1990. Biotechnologia.str.355-361.
5. Progress in the industrial microbiology vol.14..
6. Encyklopedia PWN 1995
7. Rhee S.K., Lee G.M., Yoon J.H., Park Y.H., Bae H.S., Lee S.T. 1997. Anaerobic and areobic Degradation of Pyridine by a Newly Isolated Denitrifying Bacterium. Appl. Environ. Microbiol. 63:2578-2585.
8. Reed G., Rehm H.J. 1993.Bioprocessing. Biotechnology.vol 3:499-502.
9. Lee J.J., Rhee S.K., Lee S.P. 2001. Degradation of 3-Methylopyrioline and 3-Ethylpyridine by Gordonia nitida. Appl. Environ. Microbiol. 67:4342-4345.
10. Trzmiel T. PŁ Wydział CHSPiB, Instytut Biochemii Technicznej.
11. Combourieu B., Besse P., Sancelme M., Godin J.P., Monteil A., Veschambre H., Delort A.M. 2000. Common Dagradative Pathways of Morpholine, Thiomorpholine, and Piperidine by Mycobacterium aurum MO1:Evidence from 1H-Nuclear Magnetic Resonance and Ionspray Mass Spectrometry Performed Directly on the Incubation Medium. Appl. Environ. Microbiol. 66:3187-3193.
12. Cech J.S., Hartman P., Slosarek M., Chudoba J. 1988. Isolation and Identification of a Morpholine-Degrading bacterium. Appl. Environ. Microbiol. 54:619-621.
13. Combourieu B., Besse P., Sancelme M., Veschambre H., Delort A.M., Poupin P., Truffaut N. 1998. Morpholine degradation pathway of Mycobacterium aurum MO1:direct evidence of intermediates by in situ 1H-Nuclear Magnetic Resonance. Appl. Environ. Microbiol. 64:153-158.
14. Poupin P., Truffaut N. Combourieu B., Besse P., Sancelme M., Veschambre H., Delort A.M. 1997. Degradation of Morpholine by an Environmental Mycobacterium strain Involves a Cytochrome P-450. Appl. Environ. Microbiol. 64:159-165.
15. Brzezińska-Rodak M., Peczyńska-Czoch W. 2000. Biodegradacja związków N-heterocyklicznych. Biotechnologia 48:102-116.
16. Topp E., Zhu H., Nour S.M., Houot S., Lewis M., Cuppels D. 2000. Characterization of an Atrazine-Degrading Pseudoaminobacter sp. Isolated from Canadian and French Agricultural Soils. Appl. Environ. Microbiol. 66:2773-2782.
17. Topp E., Mulbry W.M., Zhu H., Nour S.M., Cuppels D. 2000. Characterization of S-Triazine Herbicide Metabolism by a Nocardioides sp. Isolated from Agricultural Soils. Appl. Environ. Microbiol. 66:3134-3141.
18. Seffernick J.L., Johnson G., Sadowsky M.J., Wackett L.P. 2000. Substrate Specificity of Atrazine Chlorohydrolase and Atrazine-Catabolizing Bacteria. Appl. Environ. Microbiol. 66:4247-4252.
19. Andreoni V., Baggi G., Bernasconi S. Progress in Industrial Microbiology. Vol 32:14-20.
20. Hawari J., Halasz A.M., Sheremata T., Beaudet S., Groom C., Paquet L., Rhofir CH., Ampleman G., Thiboutot S. 2000. Characterization of Metabolites during Biodegradation of RDX with Municipal Anaerobic Sludge. 66:2652-2657.
21. Binks P.R., Nicklin S., Bruce N.C. 1995. Degradation of RDX by Stenotrophomonas maltophilia PB1. Appl. Environ. Microbiol. 61:1318-1322.
22. Matthews H.R., Freeland R.A., Miesfield. Biochemia i biologia molekularna p:283-305.
23. Voet D., Voet J.G. 1995. Biochemistry.p:740-744.
24. Hockenhull D.J.D. 1963. Progress in industrial microbiology, vol. IV:18-20.
25. Mauch L., Krauss B., Brandsch R. 1989. Growth stage-dependent expression of 6-hydrokxy-D-nicotine oxidase of the nicotine regulon of Arthobacter oxidans. Arch. Microbiol. 152:95-99.
26. Wang Y.T., Suidan T.M., Pfeffer J.T. 1984. Anaerobic Biodegradation of Indol to Methane. Appl. Environ. Microbiol.48:1058-1060.
27. Bak F., Widdel F. 1986. Anaerobic degradation of indolic compounds by sulfate-reducing enrichment cultures, and description of Desulfobacterium indolicum gen. nov., sp. nov. 146:170-176.
28. Berry D.F., Madsen E.L., Bollag J.M. 1987. Conversion of Indol to Oxindole under Methanogenic Conditions. Appl. Environ. Microbiol. 53:180-182.
29. Madsen E.L., Bollag J.M. 1989. Pathway of Indole metabolism by a denitrifying microbial community. Arch. Microbiol. 151:71-76.
30. Brockman F.J., Denovan B.A., Hicks R.J., Fredrickson J.K. 1989. Isolation and Characterization of Quinoline-Degrading Bacteria from Subsurface Sediments. Appl. Environ. Microbiol. 55:1029-1032.
31. Pereira W.E., Rostad C.E., Leiker T.J., Updegraff D.M., Bennett J.L.1987. Microbial Hydroxylation of Quinoline in Contaminated Groundwater:Evidence for Incorporation of the Oxygen Atom of Water. Appl. Environ. Microbiol. 54:827-829.
32. Aislabie J., Bej A.M., Hurst H., Rothenburger S., Atlas R.M. 1989. Microbial Degradation of Quinoline and Methylquinolines. Appl. Environ. Microbiol. 56:345-351.
33. Kilbane J.J., Ranganathan R., Cleveland L., Kayser K.J., Ribiero C., Linhares M.M. 1999. Selective Removal of Nitrogen from Quinoline and Petroleum by Pseudomonas ayucida IGTN9m. Appl. Environ. Microbiol. 66:688-693.
34. Sutton S.D., Pfaller S.L., Shann J.R., Warshawsky D., Kinkle B.K., Vestal J.R. 1996. Aerobic Biodegradation of 4- Methylquinoline by a Soil Bacterium. Appl. Environ. Microbiol. 62:2910-2914.
35. Benedik M.J., Gibbs P.R., Riddle R.R., Willson R.C. 1998 Microbial denitrogenation of fossil fuels. Microbiological rewievs. 16:390-395.
36. Becher D., Specht M., Hammer E., Francke W., Schauer F. 2000. Cometabolic Degradation of Dibenzofyran by Biphenyl-Cultivated Ralstonia sp. Strain SBUG 290. Appl. Environ. Microbiol. 66:4528-4531.
37. Monna L., Omori T., Kodama T. 1992. Microbial Degradation of Dibenzofuran, Fluorene, and Dibenzo-p-Dioxin by Staphylococcus auriculans DBF63. Appl. Environ. Microbiol 59:285-289.
38. Harms H., Wilkes H., Wittich R.M., Fortnagel P. 1995. Metabolism of Hydroxydibenzofurans, Methoxydibenzofurans, Acetoxydibenzofurans, and Nitrodibenzofurans by Sphingomonas sp. Strain HH69. . Appl. Environ. Microbiol. 61:2499-2505.
39. Progress in Industrial Microbiology. Vol 27:108-110.
40. Dong Gu J., Berry D.F. 1992. Metabolizsm of 3-Methyloindole by Methanogenic Consortium. Appl. Environ. Microbiol. 58:2667-2669.
And all enzymes used in this research are comming from Enzyme Nomenclature.1992. Academic Press New York.
Dziękuję
Panu profesorowi Tadeuszowi Trzmielowi
za pomoc przy tworzeniu tej pracy.
0
12
Wyszukiwarka