Typowa komórka ssaka zawiera ok. 10-5μg RNA, z czego 80-85% to rRNA. Większość z pozostałych 15-20% składa się z różnorodnych niskocząsteczkowych odmian (np. tRNA). Te RNA występujące w dużych ilościach o zdefiniowanej masie i sekwencji mogą być wyizolowane w teoretycznie czystej postaci za pomocą elektroforezy w żelu (a jakże! xP), wirowaniu w gradiencie gęstości, chromatografii jonowymiennej lub HPLC. Dla kontrastu mRNA, które zawiera się w 1-5% całości RNA, jest heterogenne co do długości i sekwencji. Jednakże, większość eukariotycznego mRNA zawiera na końcu 3' łańcuch poliadenylowy, który jest dostatecznie długi, by pozwolić na oczyszczenie mRNA za pomocą chromatografii powinowactwa na celulozie oligo(dT). Otrzymana heterogenna populacja molekuł wspólnie koduje całość polipeptydów syntetyzowanych przez komórkę.
Ponieważ reszty rybozy zawierają grupy hydroksylowe w miejscach 2' i 3', RNA jest chemicznie znacznie bardziej reaktywne niż DNA i jest łatwą ofiarą dla zanieczyszczających preparat RNaz, które je potną bez litości. RNazy są wielospecyficzne i mają zdolność do hydrolizy wiązań dwuestrowych łączących rybonukleotydy. Ponieważ RNazy są uwalniane podczas lizy komórek i są obecne na skórze, wymagana jest ciągła czujność, by zapobiec zanieczyszczeniu szkła, blatów i generowania aerozolu RNaz. Problem jest złożony, ponieważ nie ma prostej metody inaktywacji RNaz. Dzięki zawartości wewnętrznych mostków disulfidowych, wiele RNaz jest opornych na przedłużone gotowanie i łagodne środki denaturujące. Są także zdolne szybko renaturować. W przeciwieństwie do wielu DNaz, RNazy nie wymagają dwuwartościowych kationów do działania, więc nie mogą być łatwo hamowane poprzez dodawanie środków chelatujących w buforach. Najlepszym sposobem na uniknięcie problemów z RNazami jest niedopuszczenie do kontaminacji.
PROTOCOL 1
Oczyszczanie RNA z komórek i tkanek za pomocą ekstrakcji kwasem fenolowym - tiocyjanianem guanidyny - chloroformem.
Kluczem do pomyślnego oczyszczenia nie uszkodzonego RNA z komórek i tkanek jest szybkość (naprawdę!). Komórkowa RNaza powinna być inaktywowana najszybciej, jak to możliwe, najlepiej w pierwszym kroku ekstrakcji. Gdy endogenna RNaza zostaje zniszczona, zagrożenie integralności RNA jest znacznie zredukowane a oczyszczanie może przebiegać w spokojniejszym tempie.
Z powodu konieczności izolacji nie uszkodzonego RNA z komórek używa się silnych substancji denaturujących jak hydrochlorek guanidyny lub tiocyjanian guanidyny do jednoczesnego rozerwania komórek, rozpuszczania ich składników i denaturacji wewnętrznej RNazy. Wzmianki o użyciu izotiocyjanianu guanidyny w wytrącaniu RNA pojawiają się po raz pierwszy u Ullricha (1977), a zostało ono udokumentowane i porządnie opisane przez Hana (1987) i Chirgwina (1979). Metoda Hana jest metodą pracochłonna, obejmuje rozpuszczanie granulek RNA w malejącym poziomie 5 M tiocyjanianu guanidyny. W metodzie Chirgwina kultura komórek lub tkanka jest homogenizowana w 4M izotiocyjanianie guanidyny, a lizat zostaje rozdzielony na warstwy w gradiencie gęstości CsCl. Ponieważ RNA pływa w CsCl o o wiele większej gęstości (1.8g/ml) niż inne komponenty komórki, rRNA i mRNA wędrują w dół probówki podczas ultrawirowania. Jak długo gradient nie zostanie przeładowany białka pozostają w lizacie guanidynowym, a z kolei DNA unosi się w gradiencie CsCl. Otrzymane dzięki metodzie Chirgwina RNA jest bardzo wysokiej jakości i czyste a także metoda ta nie jest bardzo żmudna - dlatego stała się standardową techniką izolacji wysokocząsteczkowego RNA we wczesnych latach 1980. Jednakże ta metoda ma jedną wadę: nie jest właściwa dla jednoczesnego badania wielu próbek. Z tego powodu została prawie kompletnie zastąpiona metodą pojedynczego kroku Chomczanskiego i Sacchi (1987), w którym homogenizacja tiocyjanianem guanidyny jest zastąpiona ekstrakcją fenol:chloroform w zredukowanym pH. Eliminacja kroku z ultrawirowaniem pozwoliła na jednoczesne badanie wielu próbek i szybkie obniżenie kosztów bez strat zawartości lub jakości RNA. Dla wielu badaczy, technika pojedynczego kroku opisana w protokole 1 jest metodą z wyboru w izolacji RNA z komórek i tkanek zwierząt.
Procedura pojedynczego kroku nie jest zalecana w dwóch przypadkach. Po pierwsze, nie pozwala na wytrącenie RNA z tkanki tłuszczowej bogatej w trójglicerydy. RNA z tego źródła jest zdobywane zmodyfikowaną metodą Chirgwina (1979), opisaną przez Tavangara (1990). Po drugie, RNA bywa zanieczyszczone niewielkimi ilościami polisacharydów i proteoglikanów. Można temu zapobiec poprzez rozpuszczenie RNA po precypitacji alkoholem. Te zanieczyszczenia mogą hamować odwrotną transkrypcję łańcuchowej reakcji polimerazy (RT- PCR) i wiązanie z membraną podczas blottingu RNA. Jeśli zanieczyszczenia proteoglikanami i polisacharydami stają się problemem, to włączając krok organicznego wtrącania zmienia się warunki precypitacji RNA jak w protokole 2.
Stężenie całkowitego RNA zależy od źródła tkanek czy komórek, ale generalnie jest to 4-7µg/mg z tkanek lub 5-10µg/106 komórek. Stosunek A260/A280 wytrąconego RNA wynosi 1.8-2.0
Przechowywanie RNA
Po wytrąceniu etanolem, przechowywać RNA można:
- Rozpuszczając precypitat w dejonizowanym formamidzie i przechowując w -20C.
Formamid powoduje stabilizację chemiczną środowiska, która chroni RNA przed degradacją RNazą. Czyste, wolne od soli RNA szybko rozpuszcza się w formamidzie w stężeniu 4mg/ml. Takie stężenie pozwala na analizowanie RNA bezpośrednio przez elektroforezę, RT-PCR lub ochronę przed RNazą, co pozwala zaoszczędzić czas i uniknąć potencjalnej degradacji. Jeśli to konieczne, RNA można go oczyścić z formamidu przez wytrącenie 4 objętościami etanolu lub przez rozcieńczenie formamidu z 0.2 M NaCl i wtedy należy dodać 2 objętość etanolu.
- Rozpuszczając precypitat w wodnym buforze i przechowując w -80C.
Bufor zwyczajowo używany do tego celu zawiera SDS (0.1-0.5%) w TE (pH 7.6) lub w wodzie, potraktowanej DEPC zawierającym 0.1mM EDTA (pH 7.5). SDS powinien być usunięty przez wytrącenie chloroformem i etanolem przed traktowaniem RNA enzymami.
- Przechowując wytrącony RNA w zawiesinie etanolu w -20C.
Próbki RNA mogą być przenoszone w razie potrzeby automatyczną pipetą. Jednakże wytrącony RNA jest guzowaty i lepki a częściowo przez straty na powierzchni tipsów jednorazowego użytku, odzysk RNA bywa różny. (is inconsistent?..)
PROTOKÓŁ 2
Jednostopniowa metoda na jednoczesne oczyszczanie DNA, RNA i białek z komórek i tkanek
Niniejszy protokół, odmiana jednostopniowej metody opisanej w Protokole 1, pozwala na jednoczesne odzyskanie RNA, DNA i białek z alikwoty tkanek lub komórek. Tak jak metoda poprzednika, ta metoda zawiera w sobie lizę komórek za pomocą jednofazowego roztworu izotiocyjanianu guanidyny i fenolu. Dodatek chloroformu tworzy drugą (organiczną) fazę, w której umiejscawiają się DNA i białka, pozostawiając RNA w uwodnionym supernatancie. DNA i białka mogą być wyizolowane z fazy organicznej poprzez sekwencyjną precypitację odpowiednio etanolem i izopropanolem. DNA odzyskany z fazy organicznej ma ok. 20 kpz i nadaje się jako matryca do PCR. Białka jednakże zostają zdenaturowane, co jest wynikiem ich ekspozycji na guanidynę i są wykorzystywane przede wszystkim w immunoblottingu. RNA wytrącony z fazy wodnej za pomocą izopropanolu może być oczyszczony poprzez chromatografię na kolumnach oligo(dT)-celulozowych i/lub użyty do hybrydyzacji typu Northern, odwrotnej transkrypcji lub RT-PCR.
Wydajność odzyskiwania RNA zależy od źródła tkanki czy komórek, ale zwykle wynosi 4-7 μg/mg tkanki lub 5-10 μg/106 komórek. Stosunek A260/A280 ekstraktu RNA wynosi 1,8-2,0.
MATERIAŁY
Ważne!!! Wszystkie reagenty należy przygotować z użyciem wody traktowanej DEPC (szczegóły w części „jak wygrać walkę z RNazą”).
Bufory i roztwory:
- chloroform, etanol, izopropanol, ciekły azot
- jednofazowe reagenty do lizy - nie opublikowano składu owych reagentów do jednoczesnej izolacji DNA, RNA i białek. Jednakże dostępna jest duża liczba komercyjnych reagentów o różnorodnych nazwach (Trizol Reagent, TRI Reagent, Isogen, RNA-Stat-60). Kiedy używamy komercyjnych reagentów należy postępować zgodnie z instrukcją dostarczoną przez producenta, ponieważ w większości przypadków to co tam napisali różni się od tego, co jest napisane w tym protokole. Jednakże należy zwrócić uwagę na to, że modyfikacje technik opisanych tutaj zmniejszają kontaminację RNA DNA, polisacharydami i proteoglikanami. W czasie powstania tego tekstu nie wszystkie instrukcje producenta zawierały te modyfikacje
- lodowaty PBS - wymagany, by komórki rosły w zawiesinie i tylko w jednej warstwie
- roztwór do precypitacji RNA - 1,2 M NaCl, 0,8 M cytrynian disodowy * 15 H2O, pH jakie się nam podoba
- octan sodu (3 M, pH 5,2)
Oprócz tego jakieś źródło komórek i tkanek, wiróweczki oraz specjalne wyposażenie, w skład którego wchodzą kuwety do pomiaru absorbancji, homogenizator, schłodzony moździerz i tłuczek przemyte wodą z dodatkiem DEPC, polipropylenowe probówki snap-cap (o_O)(ja - Martin - postuluję, że to te małe probówki do PCRa z zamykanym wieczkiem.), łaźnia wodna nastawiona na 65oC.
METODA
1. Przygotowanie komórek lub próbek tkanek do izolacji DNA:
- tkanki - najlepiej pociąć tkankę na mniejsze kawałki (100 mg), a następnie umieścić je błyskawicznie w ciekłym azocie, żeby enzymy itp. nie szalały. Fragmenty takich tkanek mogą być przechowywane w temperaturze -70oC przez kilka miesięcy albo użyte natychmiast do ekstrakcji RNA. Tkanki, które nie mają dużo RNazy, mogą zostać pocięte i umieszczone bezpośrednio w polipropylenowych probówkach. Tkanki mogą być nadal zamrożone podczas rozbijania ich w homogenizatorze; homogenizacja przebiega w temperaturze pokojowej
- komórki ssaków rosnące w zawiesinie - zwirować komórki, usunąć pożywkę i ponownie zawiesić komórki w lodowatym PBSie, znowu zwirować, usunąć PBD i dodać jednofazowego reagentu do lizy komórek, znowu homogenizować
- komórki ssaków rosnące w postaci monolayer - usunąć medium, dodać PBS, usunąć PBS, zrobić lizę komórek, przenieść „zlizowane” (zlizane?...) komórki do probówek, homogenizować. Inkubować homogenat w temperaturze pokojowej przez 5 minut (by pozwolić na zakończenie dysocjacji kompleksów nukleoproteinowych). Dodać chloroform, vortexować. Zwirować (uzyskamy w ten sposób dwie fazy), fazę wodną przenieść do czystej probówki.
A dalej nie wiem bo nie ma stron ;)
PROTOCOL 3
Selekcja poli(A+)RNA chromatografią na oligo(dT)celulozie.
W odróżnieniu od rRNA, 5S RNA, 5,8S RNA i tRNA, większość eukariotycznego mRNA zawiera trakty poliA na końcu 3'. mRNA może być zatem separowane spośród komórkowego RNA dzięki chromatografii powinowactwa na celulozie z oligo(dT). Metoda wykorzystuje zdolność ogonów poliA na mRNA do formowania hybryd RNA-DNA z krótkimi łańcuchami oligo(dT) (generalnie 18-30 nt długości) przyczepionymi do podpory matrix celulozowego. Ponieważ formowanych jest tylko kilka par dT-A, do wstępnego buforu chromatograficznego muszą być dodawane wysokie stężenia soli dla stabilizacji duplexów kwasów nukleinowych. Po wymyciu z macierzy niepoliadenylowanego RNA, jest stosowane niskie stężenie buforu do destabilizacji dwuniciowej struktury i do elucji poli(A)RNA z zawiesiny. Poli(A) RNA mogą być rozdzielone przez chromatografie na oligo(dT) kolumnach przez elucję zestawem składników chemicznych. Kolumna chromatograficzna jest metodą preferowaną dla oczyszczenia dużych ilości (>25mikrogram) nie radioaktywnego poli(A)+RNA wyekstrahowanego z komórek ssaków. Dla jednoczesnego przetwarzania wielu próbek RNA ssaków, znakowanego radioaktywnie bądź nie, elucja chemiczna jest lepszym wyborem, ponieważ zbieranych jest mniej frakcji, co przyspiesza proces; oraz ponieważ można użyć lepszej klasy oligo(dT)-celulozy (typ III), co zwiększa wydajność wiązania oraz elucji RNA. Generalnie, od 1% do 10% RNA nakładanego na oligo(dT) kolumnę jest odzyskiwane w postaci poli(A)+RNA. Jednakże jest trudno usunąć w całości niepoliadenylowany rodzaj RNA, nawet po 5ciu do 6ciu cykli chromatografii powinowactwa.
Oligo(dT) - celulozowa chromatografia jest zasadniczym krokiem w preparowaniu mRNA do użycia jako matrycy w konstrukcji bibliotek cDNA. Dodatkowo poli(A)+RNA często dostarczają lepszych rezultatów niż całościowy RNA, gdy jest analizowany przez hybrydyzację typu blot, PCR, nukleazę S1 i analizę ochrony przed RNazą. To ulepszenie jest związane z 10-30 złożonym oczyszczaniem mRNA uzyskanego z chromatografii na oligo(dT) - celulozie.
Tu dużo procedur, których nie tłumaczymy :P
4. Rozpuścić RNA w podwójnie destylowanej, wysterylizowanej wodzie i podgrzać roztwór do 65o C przez 5 min. Szybko schłodzić roztwór do temperatury pokojowej i dodać 1 objętość 2x stężonego buforu.
Ogrzewanie RNA zakłóca strukturę drugorzędową , co może dotyczyć także ogona poli(A). Dla RNA, które zawierają złożoną strukturę drugorzędową, włączenie 10% (v/v) dimetylosiarczanu (DMSO) do buforu może poprawić zatrzymywanie (chyba tego RNA w kolumnie, co?) i późniejsze odzyskiwanie.
Sól sodowa N-metyloglicyny laurylu (??) jest stosunkowo nierozpuszczalna i przez to może utrudniać przepływ przez kolumnę w temperaturze poniżej 18'C. Można uniknąć tego problemu przez dodawanie do buforu LiCl zamiast NaCl.
Odzyskiwanie poli-(A) + RNA z kolumny
5. Dodaj roztwór RNA na kolumnę i niezwłocznie zacznij zbierać materiał przepływający przez kolumnę do sterylnych próbówek. Kiedy cały roztwór RNA wejdzie do kolumny, przemyj ją 1 objętością 1x stężonego buforu, kontynuując zbieranie materiału.
6. Kiedy cały płyn wypłynie z kolumny, podgrzej go do 65oC przez 5 min i ponownie podaj na szczyt kolumny. Ponownie zbierz materiał przepływający przez kolumnę (i tak do końca laborki...).
Inaczej, niż wiele złóż chromatograficznych (chromatography resins?...), oligo(dT)celuloza nie pęcznieje po nawilżeniu, ani nie pęka, gdy się ją wysuszy. Nie trzeba zatem koniecznie utrzymywać przepływu przez kolumnę, by matryca była wilgotna. (`damp' - to ma tysiące znaczeń. Z „przygnębiony” W odpowiedzi włącznie :P)
7. Przemyj kolumnę 5-10 objętościami 1x stężonego buforu, zbierz 1 ml frakcji do plastikowych, sterylnych próbówek.
*W niektórych protokołach po kroku 7 kolumna jest przemywana 5 objętościami 1x stężonego buforu zawierającym 0,1 M NaCl. Bardzo mały i poli-(A)- RNA jest wymywany z kolumny podczas przemywania, które nie może być pominięte.
8. Użyj kuwety do zmierzenia absorbancji dla każdej frakcji przy 260 nm dla rozcieńczenia 1:20, użyj 1x stężonego buforu jako próby kontrolnej.
*Roztwór zawierający 38 ug/ml RNA daje absorbancję 1 dla 260 nm i 0,4-0,5 przy 280 nm. Początkowo OD260 dla niepoliadenylowanego RNA będzie wysokie dla przepływającego przez kolumnę RNA. Późniejsze frakcje powinny mieć bardzo małą lub nie mieć wcale absorbancji przy 260 nm.
9. Osady frakcji zawierają większość OD260 materiału po dodaniu 2,5 objętości etanolu. (wiem, dziwne. Ale prawdziwe.)
*To tzw. poli-(A)-RNA jest użyteczną kontrolą w dalszych eksperymencie.
Elucja poli-(A)+ RNA z kolumny
10. Wymyć poli-(A)+ RNA z oligo(dT)-celulozy za pomocą 2-3 objętości sterylnego, pozbawionego RNaz buforu. Zbierać ekwiwalenty frakcji w ilości 1/3 do ½ objętości kolumny.
11. Użyć kuwety do zmierzenia absorbancji przy 260 nm dla każdej frakcji zebranej z kolumny. Zgromadzone frakcje zawierają wymyty RNA.
Dalsze oczyszczanie RNA (do wyboru, dowolne)
Materiał uzyskany po 1 chromatografii na oligo(dT-celulozie zawiera w przybliżeniu jednakowe ilości poliadenylowanych i nie poliadenylowanych rodzajów RNA. Poliadenylowany RNA może by dalej oczyszczany następującymi krokami:
12. Oczyszczony poli-(A)+ RNA ogrzać do 65oC przez 5 minut i potem schłodzić szybko do temperatury pokojowej. Ustawić stężenie NaCl w wymywanym RNA na 0,5 M używając 5 M NaCl i przeprowadzi© 2 rundę chromatografii na tej samej kolumnie oligo(dT)-celulozowej.
13. Do wymytego w 2 rundzie chromatografii RNA dodać 3M octanu sodu (pH=5,2) tak, aby uzyskać końcowe stężenie 0,3M. Dobrze wymieszać. Dodać 2,5 objętości zimnego etanolu, wymieszać i przechowywać roztwór w lodzie przez conajmniej 30 min.
14. Odzyskać poli-(A)+ RNA przez wirowanie 10000 g (9000 rpm w Sorvall SS-34 rotor) przez 15 minut w 4oC. Delikatnie wylać supernatant, przemyć osad (często niewidoczny) 70% etanolem. Zwirować, usunąć supernatant przez zasysanie (???)(Martin obstawia zasysanie pipetką) i trzymać otwartą probówkę w odwróconej pozycji przez kilka minut aby pozwolić wyparować pozostałościom etanolu. Nie pozwolić osadowi wyschnąć.
15. Rozpuścić wilgotny osad RNA w małej objętości sterylnej, traktowanej DEPC wody. Użyć kuwety do zmierzenia absorbancji przy 260 nm dla każdej frakcji zebranej z kolumny. Zebrać frakcje zawierające RNA.
*Przyjmuje się że całość odzyskanego poli-(A)+ RNA równa się 5-10% początkowego RNA i mierzy się absorbancję w rozcieńczonym roztworze RNA. Roztwór dla którego OD260=1 zawiera 38 ug/ml RNA.
16. Jak przechowywać poli-(A)+ RNA jest napisane w protokole 1.
PROTOKÓŁ 4
Selekcja Poli(A)+ RNA przez Batch Chromatography
(od korektora: batch adsorption = adsorpcja w zawiesinie, batch culture = hodowla w podłożu płynnym itd. Ale z kolei batch distillation = destylacja periodyczna... Po krótkim researchu przyjmuję, że Nat ma rację.)
Kiedy musimy przetworzyć wiele próbek RNA, albo kiedy pracujemy z małymi ilościami (<50 μg) całego ssaczego RNA, techniką z wyboru jest chromatografia okresowa na oligo(dT)-celulozie. Metoda ta przeprowadzona jest na oligo(dT)-celulozie o wysokim stopniu czystości, w temperaturze optymalnej dla wiązania i elucji. Wiele metod oczyszczania poli(A)+ RNA przez chromatografię okresową zostało opisanych w ciągu ostatnich lat. Technika opisana poniżej jest modyfikacją metody Celano. W celu zapoznania się z dodatkowymi technikami zajrzyj do rozdziału DODATKOWE METODY SELEKCJI POLI(A)+ RNA.
MATERIAŁY
Ważne! Wszystkie reagenty należy przygotować używając wody traktowanej DEPC.
Bufory i roztwory - bufor myjący (TES z 0,5 M NaCl), octan amonu (10 M), etanol, lodowata woda, NaCl (5 M), TES
Specjalne wyposażenie - oligo(dT)18-30-celuloza (należy użyć celulozy typu III o zdolności wiązania 100 OD260/g) wykalibrowanej w buforze absorbującym/myjącym i zawieszonej w tym samym buforze i stężeniu 100 mg/ml).
Łaźnia wodna - 55 i 65oC
METODA
- uzupełnia się probówkę z RNA (max 1mg) do 600 μl za pomocą TES, podgrzewa, chłodzi, dodaje NaCl, dodaje oligo(dT)-celulozę i inkubuje, potem się wiruje i przenosi supernatant do świeżej probówki
- do granulek oligo(dT) pozostawionych w pierwszej probówce dodaje się lodowatego roztworu absorbującego/myjącego, vortexuje, inkubuje, wiruje, po czym powtarza się do wszystko 2x (blablabla)
- ponownie zawiesza się granulki oligo(dT) w lodowatej, podwójnie destylowanej, autoklawowanej wodzie (w której po tym wszystkim to już chyba nawet wody nie będzie, taka będzie czysta o.O) przez delikatne vortexowanie, wiruje i usuwa ostrożnie supernatant
- odzyskanie związanego poli(A)+ RNA przez ponowne zawieszenie granulek oligo(dT)-celulozy w naszej uberpr0czystej wodzie, inkubować, wirować, przenieść supernatant do świeżej probówki, powtórzyć wszystko raz jeszcze, blablablabla
- dodać octanu amonowego i etanolu do supernatantu, schłodzić
- odzyskać wytrącone poli(A)+ RNA przez wirowanie, zlać supernatant, przemyć granulki (które są często niewidzialne) etanolem. Znowu sobie chwilkę wirować (ale tylko chwilkę), zlać supernatant i przechowywać otwarte probówki w pozycji odwróconej, żeby powiedzieć etanolowi bye-bye, a potem rozpuszczamy RNA w naszej uperpr0czystej wodzie :p
PROTOCOL 9
Hybrydyzacja Dot Slot oczyszczonego RNA
Dot slot blotting to technika polegająca na immobilizacji przygotowanych próbek RNA na stałym nośniku, zazwyczaj na nylonowej membranie. Koncentracja sekwencji docelowej może być ustalona poprzez hybrydyzację immobilizowanych próbek z właściwą sondą. Ilość sekwencji może być ustalona poprzez porównanie intensywności sygnałów emitowanych przez prążki zawierające testowane próbki z standardem zawierającym znaną ilość sekwencji docelowej.
Przez kilka lat, dot blotting i slot blotting były traktowane nieprzychylnie przez badaczy przez to, że otrzymywano losowe sygnały hybrydyzacji z identycznych próbek naniesionych na tą samą membranę, w szczególności gdy analizowano złożone populacje RNA i DNA. Mimo, że problem nie został do końca rozwiązany, nadejście dodatnio naładowanych, zmodyfikowanych membran nylonowych doprowadziło do znaczącego zmniejszenia ilości błędów hybrydyzacji. W przypadku DNA, oczyszczone preparaty albo alkaliczne lizaty komórek i tkanek mogą być naniesione na membranę w środowisku alkalicznym.
Analiza dot blot RNA jest trochę bardziej skomplikowana. W pewnym momencie badacze eksperymentowali z dot i slot blotem przy pomocy nieoczyszczonej cytoplazmy przygotowanej z świeżo zebranych lub mrożonych kultur komórkowych, a także z tkanek zwierzęcych. Jednakże efekty hybrydyzacji nieoczyszczonych preparatów RNA nie zawsze pokrywały się z tymi uzyskanymi po hybrydyzacji oczyszczonego RNA. Z tego powodu Dot i Slot blotting jest przeprowadzany z użyciem oczyszczonych preparatów RNA, które zostało zdenaturowane aldehydem szczawiowym, lub formaldehydem tuż przed naniesieniem na membranę.
Nakładanie na membranę.
Jakkolwiek próbki RNA mogą być nałożone na membranę manualnie za pomocą automatycznej pipety, to odległości i wielkość plamek są często są różne. Otrzymane obrazy mogą być zniekształcone, rozmazane lub nawet niemożliwe do odczytania. Preferowaną metodą nakładania próbek na membranę jest ta z użyciem kolektora próżniowego. Wiele komercyjnie dostępnych kolektorów ma możliwość wyboru sposobu w jaki zostaną nałożone próbki na membranę. (doty, sloty, co tam chcecie) To zapewnia, że im mobilizowane próbki mają ten sam kształt, powierzchnię i odstępy, co ułatwia porównanie intensywności sygnału hybrydyzacji.
Standardy
Aby uzyskać ilościowe wyniki, ważnym jest by uwzględnić pozytywne i negatywne kontrole, które będą miały fizyczne właściwości zbliżone do kwasów nukleinowych poddawanych badaniu. Dla przykładu, kiedy analizuje się RNA ssaków, negatywna kontrola powinna zawierać RNA z komórki lub tkanki w której wiadomo, że nie występuje badana sekwencja. Pozytywna kontrola powinna zawierać preparat RNA zmieszany ze znaną ilością standardu RNA, komplementarnego do badanej próbki. Te standardy i znakowane radioaktywnie próbki są najlepiej syntetyzowane z szablonów DNA, które zostało wklonowane w wektory plazmidowe. Miejsce wklonowania jest flankowane przez dwa różne promotory bakteriofagowe w odmiennych orientacjach. Nic kodująca RNA może być użyta jako standard do hybrydyzacji, może być syntetyzowana przy pomocy tylko jednego promotora; wyznakowana radioaktywnie (nić matrycowa) próbka może być syntetyzowana przy pomocy drugiego promotora.
Kiedy tworzy się standardy, syntetyczna nić kodująca RNA jest mieszana z niespecyficznym RNA tak, że w rezultacie otrzymana masa jest identyczna z badanymi próbkami. Niespecyficzne RNA powinno być przygotowane w ten sam sposób jak testowe RNA. Te środki ostrożności są niezbędne do kontroli obecności nieczystości w cytoplazmatycznym RNA które zmniejsza intensywność sygnału hybrydyzacji oraz aby kontrolować spadki efektywności hybrydyzacji w próbkach zawierających duże ilości oczyszczonego RNA.
Normalizacja
Aby uniknąć przeładowania membrany, nie więcej niż 5ug całkowitego RNA powinno zostać użyte w slocie standardowej wielkości. Taka sama ilość RNA musi zostać umieszczona we wszystkich slotach. Jednakże, jest zawsze pewna niepewność co do ilości RNA które zostało zatrzymane na membranie. Ten problem może zostać rozwiązany poprzez zabarwienie membrany błękitem metylenowym już po tym jak RNA zostało krzyżowo związane z dodatnio naładowaną membraną poprzez ekspozycję na UV. Alternatywnie, zawartość Poli(A) RNA zatrzymanego na membranie może zostać zmierzone poprzez hybrydyzację z oligo(dT). Ta metoda jest wyjątkowo użyteczna kiedy nakłada się małe ilości oczyszczonego Poli(A)+ RNA do każdego slotu.
Pomiar intensywności sygnału
W wielu przypadkach wizualne szacowanie intensywności sygnału hybrydyzacji jest wystarczające. Jednakże, bardziej dokładne szacunki ilości sekwencji docelowej w każdej próbce mogą być uzyskane poprzez skanowanie densytometryczne, bezpośrednie obrazowanie fosforem oraz luminometrię. Cieczowa spektroskopia scyntylacyjna także zapewnia bezpośrednie oszacowanie koncentracji docelowej sekwencji DNA. Jednakże ta metoda wymaga pocięcia próbki na kawałki i umieszczenia we fluorze scyntylacyjnym(?)(taki promieniotwórczy może?), tym samym eliminując możliwość ponownego badania próbek.
PROTOKÓŁ 10
Edycja, ufam Ci. Nie mam siły, umieram, nie jestem w stanie nic przełknąć od doby. Nic nie umiem! Zatem nie czytam Twojej części tłumaczenia.
Mapowanie RNA nukleazą S1
Do namnażania cząsteczek RNA, mapowania pozycji intronów i identyfikacji regionów 5' i 3' w mRNA na klonowanych matrycach DNA stosuje się trzy różne nukleazy - nukleazę S1, rybonukleazę i egzonukleazę VII. Nukleazę S1 używa się w testach ochrony, kiedy testowe RNA jest hybrydyzowane z matrycą DNA. Rybonukleaza jest używana w hybrydyzacji testowego RNA z RNA skopiowanym z matrycy DNA (Protokół 11.). Egzonukleaza VII służy do bardziej wyspecjalizowanych celów - mapowania krótkich intronów oraz rozwiązywania problemów powstających w testach ochrony przez nukleazą S1.
Metody ze wszystkimi trzema enzymami są rozszerzeniem klasycznej techniki [testu] ochrony przed nukleazą S1 opisanej przez Berka i Skarpa (1977). Preparaty RNA zawierające pożądane mRNA inkubuje się z komplementarną sondą DNA lub RNA w warunkach faworyzujących powstawanie hybrydów. Pod koniec reakcji podaje się enzym, który degraduje nieshybrydyzowane jednoniciowe RNA lub DNA. Pozostałe hybrydy DNA-RNA lub RNA-RNA rozdziela się w elektroforezie i uwidacznia autoradiograficznie lub za pomocą hybrydyzacji Southerna (Favaloro i wsp. 1980; Calzone i wsp. 1987). Gdy w reakcji hybrydyzacji sonda jest w nadmiarze molowym, natężenie sygnału jest proporcjonalne do stężenia badanego mRNA. Dokładne stężenie można oszacować na podstawie krzywej wzorcowej, gdzie nadmiar sondy jest hybrydyzowany ze znanymi ilościami sekwencji docelowej.
Głównym problemem testu ochrony przez nukleazą S1 w jego oryginalnej formie (Berk i Sharp 1977) było użycie dwuniciowych DNA jako sond. Aby zapobiec reasocjacji sondy DNA w trakcie hybrydyzacji, niezbędne (ale nie zawsze możliwe) było ustalenie takich warunków hybrydyzacji, które faworyzowałyby tworzenie hybrydów RNA-DNA, a nie DNA-DNA. Ponieważ hybrydy DNA-RNA są trochę stabilniejsze niż DNA-DNA, etap annealingu był zazwyczaj przeprowadzany w 80% formamidzie, w temperaturze wyższej niż obliczona temperatura topnienia dwuniciowego DNA (Casey i Davidson 1977; Dean 1987). Jednakże w tych warunkach tempo hybrydyzacji jest zmniejszone ok. 10 razy, a stabilność hybrydów DNA-RNA nie jest możliwa do przewidzenia. Problemy te można było ominąć dzięki sondom jednoniciowym. Etap annealingu można było przeprowadzić w standardowych warunkach hybrydyzacji, ponieważ w mieszaninie reakcyjnej nie było nici komplementarnej mogącej konkurować z docelowym RNA o sondę. W czasach Berka i Sharpa (kiedy to było!) nie było pewnej metody przygotowania ssDNA wolnego od nici komplementarnej. Jedyną dostępną techniką była elektroforeza żelowa rozdzielająca nici (strand-separating gel electrophoresis; Hayward 1972), ale nie do końca można było na niej polegać. Rozdział był możliwy tylko z ~70% fragmentów DNA i, nawet w wypadku powodzenia, dawał w rezultacie preparaty zanieczyszczone komplementarną nicią DNA. Ze względu na te trudności, wzory prążków otrzymanych po elektroforezie były różne w kolejnych eksperymentach. Problemy te rozwiązano w późnych latach 80. dzięki rozwojowi metod wytwarzania jednoniciowych sond DNA lub RNA.
RYS. 7-4 Mapowanie RNA nukleazą S1
Gdy jako sond używa się klonowanych segmentów genomowego DNA, umiejscowienie intronów można wywnioskować na podstawie rozmiaru prążków po trawieniu nukleazą S1. Hybrydy utworzone pomiędzy transkrybowaną nicią genomowego DNA i mRNA zawierają pętle jednoniciowego DNA (introny). Trawienie hybrydów nukleazą S1 w 20°C generuje cząsteczki, w których nici RNA są nietknięte, ale DNA zawierają luki w miejscu intronów. Cząsteczki te migrują jako pojedynczy prążek w trakcie elektroforezy w warunkach niedenaturujących (żel A). Jednakże w żelach zasadowych (żel B) RNA ulega hydrolizie, a poszczególne fragmenty DNA rozdzielają się w zależności od swoich rozmiarów. Gdy trawienie nukleazą S1 przeprowadza się w 45°C, obie nici DNA i RNA rodzicielskiego hybrydu są cięte na serie małych hybrydków DNA-RNA, które można rozdzielić w elektroforezie w warunkach niedenaturujących (żel C). Cząsteczki DNA w tych hybrydach (żel D) są tego samego rozmiaru, co te z żelu B.
RYS. 7-5 Mapowanie końców 5' i 3' mRNA
Hybrydy utworzone pomiędzy sondami mRNA i DNA znakowanymi radioznacznikami na końcach 5' lub 3' trawi się nukleazą S1. Umiejscowienie radioznaczników jest pokazane za pomocą gwiazdek. Lokalizację końców 5' i 3' docelowego RNA można wywnioskować poprzez zmierzenie długości fragmentów DNA odpornych na nukleazę i oszacowanie odległości pomiędzy radioznacznikami na końcach 5' i 3' mRNA. (Ścieżka a) sonda znakowana na końcu 5' przed trawieniem nukleazą S1; (ścieżka b) sonda znakowana na końcu 5' po trawieniu nukleazą S1; (ścieżka c) sonda znakowana na końcu 3' przed trawieniem nukleazą S1; (ścieżka d) sonda znakowana na końcu 3' po trawieniu nukleazą S1. W zewnętrznych ścieżkach znajdują się standardy masy cząsteczkowej. W układach eukariotycznych, koniec 5' mRNA określany poprzez mapowanie nukleazą S1 zwykle odpowiada miejscu startu transkrypcji, podczas gdy koniec 3' reprezentuje miejsce poliadenylacji. Podobną strategię można zastosować do mapowania lokalizacji 3' i 5' miejsc splicingu.
SONDY STOSOWANE W TESTACH OCHRONY PRZED NUKLEAZĄ S1.
Sondy o znanej polarności i wysokiej specyficznej aktywności wytwarza się poprzez rozdzielanie nici fragmentu dsDNA lub, częściej, przez syntezę de novo albo RNA komplementarnego do jednej nici dwuniciowej matrycy DNA, albo DNA komplementarnego do jednoniciowej matrycy.
Sondy powstałe przez rozdzielenie nici (strand-separated probes) przygotowuje się za pomocą enzymów restrykcyjnych typu II, pojedynczo lub w połączeniu, w celu utworzenia fragmentu DNA o odpowiedniej długości (zwykle 100-500 nukleotydów) z przedłużeniem na końcu 5' i 3'. Tym samym, jedna z nici będzie o ok. 8 nukleotydów dłuższa niż druga. Ta różnica w długości wystarczy do rozdziału obu nici w elektroforezie w żelu poliakrylamidowym, w warunkach denaturujących. Przed lub po elektroforezie, koniec 5' pożądanej nici jest defosforylowany i znakowany radioznacznikiem in vitro przez transfer znakowanej reszty fosforanowej z [γ-32P]ATP, reakcję katalizowaną przez kinazę polinukleotydową faga T4.
W syntezie de novo wytwarza się znakowane na końcu lub jednolicie znakowane sondy DNA in vitro. Sondy znakowane na końcu (end-labeled probes) przygotowuje się przez fosforylację końca 5' primera oligonukleotydowego; w jednolicie znakowanych sondach (uniformy labeled probes) w rosnącą nić DNA wbudowuje się znakowane nukleotydy. W obydwu przypadkach nowo zsyntetyzowane nici DNA mogą być oddzielone od matrycy przez trawienie enzymem restrykcyjnym, który rozpoznaje unikalne miejsce w nowo utworzonym dsDNA. Znakowana sonda może być wówczas oddzielona od zlinearyzowanego ssDNA w elektroforezie w żelu poliakrylamidowym w warunkach denaturujących.
Sondy znakowane radioznacznikami faworyzujące jedną nić DNA są wytwarzane w reakcjach PCR, w których stężenie jednego primera przewyższa stężenie drugiego 20-200 razy. W czasie początkowych cykli PCR, przyrost dwuniciowego DNA zachodzi w trybie wykładniczym. Jednakże, w momencie gdy stężenie jednego z primerów staje się limitujące, reakcja generuje ssDNA akumulowane w tempie arytmetycznym. Pod koniec reakcji, stężenie jednej z nici DNA jest 3-5 razy wyższe od stężenia drugiej nici.
Sondy znakowane radioznacznikami, składające się całkowicie z jednej nici DNA, syntetyzuje się w termicznych reakcjach cyklicznych (thermal cycling reactions), które zawierają dwuniciową matrycę DNA, ale tylko jeden primer. Dwuniciowa matryca DNA (20 μg) generuje ok. 200 μg jednoniciowej sondy w trakcie 40 cykli. Długość sondy można ustalić poprzez przecięcie matrycy DNA w miejscu restrykcyjnym poniżej miejsca wiązania startera.
Jednolicie znakowane sondy RNA (rybosondy - riboprobes) generuje się przez transkrypcję dwuniciowej matrycy DNA dołączonej do promotora bakteriofagowego. Matryca DNA jest generowana albo przez trawienie rekombinowanego plazmidu enzymem restrykcyjnym, który rozcina DNA wewnątrz lub poniżej klonowanej sekwencji, albo przez namnożenie za pomocą PCR. Zlinearyzowana matryca jest transkrybowana przez odpowiednią bakteriofagową polimerazę RNA zależną od DNA, w obecności [α-32P]NTPs. Powstaje znakowany RNA, wydłużany od miejsca inicjacji w promotorze do końca fragmentu DNA. Promotor i sekwencja DNA są zorientowane w taki sposób, że powstająca rybosonda jest antysensowna (komplementarna) do badanego mRNA. Dobrze jest oczyścić sondy RNA w elektroforezie denaturującej. Oczyszczanie może być przeprowadzone łatwo i szybko w miniżelach z 5% poliakrylamidem/8M mocznikiem, w aparacie miniprotein gel (np. Bio-Rad Mini-Protean).
Nawet, gdy używa się jednoniciowych sond, analiza struktury eukariotycznych RNA przeprowadzana z nukleazą S1 nie jest wolna od artefaktów. Na przykład, małe niesparowania w heterodupleksach DNA:RNA są stosunkowo odporne na działanie nukleazy. I odwrotnie, regiony pełnego heterodupleksu bogate w sekwencje rU:Da są wrażliwe na cięcie. Pojedyncza cząsteczka DNA może być często chroniona przed działaniem nukleazy dzięki jednoczesnej hybrydyzacji z dwoma różnymi RNA. W końcu, nukleaza S1 nieskutecznie rozcina segment DNA, znajdujący się naprzeciwko pętlowego regionu RNA. Wiele z tych problemów można rozwiązać poprzez użycie gamy stężeń nukleazy S1, przeprowadzenie trawienie w innych temperaturach, zastosowanie innej nukleazy (np. nukleazy z fasoli mung), albo użycie kilku różnych nukleaz (np. RNaza H z nukleazą S1). Ważne jest, aby zrozumieć, że rozcięcie nukleazą S1 niekoniecznie odzwierciedla zróżnicowanie sekwencji pomiędzy nićmi dwóch kwasów nukleinowych, a odporność na trawienie nie musi oznaczać zbieżności. Mapowanie nukleazą S1 powinno być zatem uznane za użyteczne, ale nie niezawodne, w poznawaniu struktury RNA. I tak, kiedy używamy nukleazy S1 do mapowania, na przykład, końców 5' i 3' mRNA, ważne jest, aby potwierdzić wynik inną, niezależną techniką, taką jak wydłużanie startera.
Nukleaza z fasoli mung może zastąpić nukleazę S1 w wielu eksperymentach mapujących. Dla niektórych sond DNA potrzeba o wiele mniej nukleazy z fasoli mung niż S1, aby osiągnąć całkowite trawienie jednoniciowych regionów. Na przykład, nukleaza S1 jest stosowana w stężeniu 1000 U/ml, aby całkowicie strawić nadmiarową jednoniciową sondę DNA odpowiadającą ludzkiemu receptorowi lipoproteiny o niskiej gęstości, podczas gdy identyczny wynik można osiągnąć stosując 10 U/ml nukleazę z fasoli mung. Wadą tej nukleazy jest to, że jest 20 razy droższa niż S1. Nukleaza z fasoli mung może zastąpić S1 na etapie 22 w tym protokole. Trawienie jest przeprowadzane w buforze zawierającym 10 mM octan sodu (pH 4.6)/50 mM NaCl/1 mM Znal/1 mM β-merkaptoetanol/0.001% (v/v) Tryton X-100.
Protokół ten przedstawia metodę mapowania RNA nukleazą S1 z zastosowaniem jednolicie znakowanej, jednoniciowej sondy DNA. Procedury mapowania RNA z użyciem rybonukleazy i sond RNA oraz mapowanie RNA przez wydłużenie startera są opisane odpowiednio w Protokołach 11 i 12.
JAK WYGRAĆ BITWĘ Z RNazą
Wiele eksperymentów zostało niepotrzebnie zrujnowanych przez zanieczyszczenie RNazą. Problemów z egzogeniczną RNazą można uniknąć, wystarczy zdrowy rozsądek (a łyżka na to: „Niemożliwe!”). Z naszego doświadczenia wiemy, że zanieczyszczenie egzogeniczną RNazą bierze się z:
Zanieczyszczonych buforów: bufory mogą zostać zanieczyszczone mikroorganizmami przez niedbałe techniki aseptyki Wzrost drobnoustrojów zwykle nie jest widoczny gołym okiem. RNazy nie da się pozbyć w autoklawie, także zanieczyszczone roztwory trzeba po prostu wywalić.
Urządzenia do automatycznego pipetowania: Nie ma sensu używać jednorazowych końcówek wolnych od RNazy, jeśli pipeta automatyczna została wcześniej użyta do roztworów zawierających RNazę, np. w trakcie obróbki plazmidów albo, co gorsza, w testach ochrony przez rybonukleazą. Jeśli wypychacz pipety wejdzie w kontakt ze ścianami końcówki (???), staje się bardzo efektywnym zakażaczem RNazą.
W wielu laboratoriach uparcie wierzy się w odpędzającą problemy z RNazą moc gumowych rękawiczek. Prawda jest taka, że zmiana jednej czy dwóch par rękawiczek daje tyle, co noszenie przy sobie łapki królika. Bardziej prawdopodobne jest, że zanieczyszczenia będą przeniesione na włosach albo brodach badaczy, a co więcej, rękawiczki chronią co najwyżej do momentu, w którym nie dotkną powierzchni mającej wcześniej kontakt ze skórą. Żeby z rękawiczek był jakikolwiek pożytek, trzeba je wymieniać po każdym dotknięciu aparatu, otwarciu lodówki, wypełnieniu wiaderka z lodem, zapisania czegoś w notatniku… Nie jest to ani mądre, ani praktyczne Możesz założyć rękawiczki, ale nie myśl, że ochronią cię przed RNazą! Bardziej rozsądnie jest:
Trzymać specjalny zestawi pipet automatycznych służących do nakładania RNA.
Odłożyć na bok sprzęt szklany i plastikowy, oraz bufory, mające służyć tylko do posługiwania się RNA.
Przechowywać roztwory/bufory w małych porcjach i każdą wyrzucać po użyciu. Unikać materiałów i roztworów używanych wcześniej w innych celach.
Odłożyć na bok aparaty do elektroforezy mające służyć do rozdzielania RNA. Czyścić aparaty detergentem, płukać wodą,, suszyć etanolem i wypełnić 3% H2O2. Po 10 minutach, w temperaturze pokojowej, dokładnie przepłukać aparat wodą potraktowaną DEPC (diethylopyrocarbonate - dietylopirowęglan).
Wszystkie roztwory i bufory przygotowywać w szkle wolnym od RNazy, wodą potraktowaną DEPC i związkami chemicznymi zarezerwowanymi do pracy z RNA, obsługiwanymi jednorazowymi łopatkami, albo nakładanymi prosto z butelki. Jeśli to możliwe, potraktować roztwory 0,1% DEPC przez co najmniej 1 h w 37°C a potem wstawić do autoklawu na 15 minut przy 15 psi (1,05 kg/cm2) w cyklu płynnym.
Wrzucenie sprzętu do autoklawu może nie wystarczyć do inaktywacji RNazy. Sprzęt szklany należy wypalać przez 4h w 300°C. Sprzęt plastikowy należy przemyć DEPC albo kupnymi środkami inaktywującymi RNazę (np. RNaseZap z Ambion Inc.).
Używać jednorazowych końcówek i probówek od dobrego producenta. Aby zmniejszyć prawdopodobieństwo kontaminacji, dobrze jest używać sterylnych pincet w trakcie przenoszenia ich z oryginalnych opakowań do stojaków.
Używać inhibitorów RNazy w trakcie izolacji RNA.
INHIBITORY RNaz
RNazy są wydajnymi i silnymi enzymami - mogą zagrozić integralności RNA na każdym etapie izolacji i charakteryzacji. Do zatrzymania aktywności RNazy powszechnie stosuje się trzy typy inhibitorów:
Dietylopirowęglan (DEPC), wysoko reaktywny związek alkilujący, stosowany do inaktywowania RNaz w buforach i na sprzęcie szklanym. DEPC modyfikuje zarówno białka, jak i RNA, nie można go więc stosować w trakcie izolowania i oczyszczania RNA, poza tym jest niemieszalny z niektórymi buforami (np. z Tris).
Rybonukleozydowe kompleksy wanadylu (vanadyl ribonucleoside complexes) są analogami stanu pośredniego, wiążą się do miejsc aktywnych wielu RNaz i inhibują ich aktywność katalityczną prawie całkowicie. Ponieważ rybonukleazy wanadylowe nie modyfikują RNaz kowalencyjnie, trzeba ich używać na wszystkich etapach ekstrakcji i oczyszczania RNA. Jednakże, ponieważ kompleksy te inhibują polimerazy RNA i translację in vitro, trzeba usunąć je z ostatecznego preparatu RNA przez wielokrotną ekstrakcję z fenolem zawierającym 0,1% hydroksychinolinę.
Inhibitory białkowe. Wiele RNaz wiąże się bardzo silnie, aczkolwiek niekowalencyjnie, z ~50 kD białkami znajdującymi się w cytoplazmie praktycznie wszystkich tkanek ssaków, można je wyizolować w dużych ilościach z łożyska. In vivo, białka te pełnią funkcje inhibitorów białek należących do nadrodziny RNaz trzustkowych, szczeólnie angiogenina - indukująca powstawanie naczyń krwionośnych pochodząca z eozynofilów neurotoksyna. Powinowactwa tych inhibitorów do ich cząsteczek docelowych są jednymi z najwyższych opisanych (1-70 fM).
Archetypowym inhibitorem RNazy jest cząsteczka w kształcie podkowy, zawierająca w sobie siedem przemiennych powtórzeń bogatych w lucyny, o długości 28 i 29 reszt. Inhibitor posiada także wiele reszt cysternowych, wszystkie w formie zredukowanej. Powierzchnia przylegania pomiędzy rybonukleazą a inhibitorem jest niezwykle duża i zawiera reszty zlokalizowane w wielu domenach obu białek. Miejsca kontaktu ważne pod względem energetycznym dotyczą tylko regionu karboksy-końcowego inhibitora i centrum katalitycznego rybonukleazy, w tym kluczowej reszty lizynowej.
Inhibitory białkowe wywiedzione z kilku źródeł są sprzedawane przez wielu producentów pod różnymi nazwami handlowymi (np. RNAsin, Promega; Prime Inhibitor, 5 Prime 3 Prime). Chociaż różnią się wymaganiami dla reagentów tiolowych, wszystkie wykazują szerokie spektrum aktywności inhibitorowych przeciwko RNazom, ale nie inhibują innych nukleaz, polimeraz czy układów translacyjnych in vitro.
Ponieważ inhibitory nie tworzą kowalencyjnych kompleksów z RNazą, nie można ich stosować w obecności związków denaturujących, takich jak SDS i guanidyna, które są powszechnie używane do lizy komórek ssaków w początkowych etapach ekstrakcji RNA. Jednakże inhibitory można podać na każdym kolejnym etapie oczyszczania. Inhibitory trzeba kilka razy uzupełniać w czasie oczyszczania, jako że są usuwane w ekstrakcji fenolem.
DIETYLOPIROWĘGLAN
DEPC stosuje się w klonowaniu do inaktywacji śladowych ilości RNaz, które mogą zanieczyszczać roztwory, sprzęt szklany i plastikowy, który potem ma być użyty do preparatyki RNA jądrowego lub mRNA. DEPC jest wysoko reaktywnym związkiem alkilującym - niszczy aktywność enzymatyczną RNazy przez etoksyformylację reszt histydynowych.
Sprzęt szklany i plastikowy powinno się napełnić 0,1% wodnym roztworem DEPC i zostawić tak na 1 h w 37°C lub na noc w temperaturze pokojowej. Przedmioty przepłukać kilka razy DEPC-owaną wodą, potem wrzucić na 15 minut do autoklawu.
W roztworze wodnym DEPC natychmiast hydrolizuje do CO2 i etanolu, z czasem półtrwania w buforze fosforanowym ok. 20 minut, pH 6 i 10 minut w pH 7. Hydroliza jest mocno przyspieszana przez Tris i inne aminy, które same zużywają się w tym procesie. DEPC nie może być zatem stosowany w roztworach które zawierają te bufory. Próbki DEPC, wolne od nukleofilów (np. wody i etanolu) są bardzo stabilne, ale nawet małe ilości tych rozpuszczalników mogą spowodować całkowitą DEPC do dietylowęglanu. Z tego powodu DEPC powinno chronić się przed wilgocią. DEPC powinno się przechowywać w małych porcjach w suchych warunkach i przed otwarciem butelki poczekać, aż osiągnie temperaturę otoczenia.
Mimo że woda oczyszczana przez dobrze utrzymane, nowoczesne systemy odwróconej osmozy jest wolna od RNazy, źle utrzymywane systemy oczyszczania mogą zostać zanieczyszczone przez mikroby. Taka sytuacja często ma miejsce w dużych scentralizowanych układach, gdzie woda może ulegać stagnacji. W takich wypadkach do wody należy dodać 0,1% DEPC na 1h w 37°C…
Inne zastosowania dla DEPC
Oprócz reagowania z resztami His w białkach, DEPC może tworzyć niestabilne alkaliczne związki addycyjne z N7 pierścienia imidazolowego. W efekcie zostaje przecięte wiązanie glikozydowe. Ze względu na wysoką reaktywność i specyficzność, DEPC był używany jako sonda chemiczna dla drugorzędowej struktury DNA i RNA. Niesparowane reszty adeninowe są silnie reaktywne, podobnie jak reszty guaninowe w formie Z-DNA. Spadek reaktywności puryn z DEPC może być użyty do zmierzenia wiązania między Z-DNA i specyficznymi białkami.
Problemy w stosowaniu DEPC
Usuwanie DEPC przez degradację termiczną generuje małe ilości etanolu i CO2, które mogą zwiększać siłę jonową i obniżać pH niebuforowanych roztworów. DEPC może karboksymetylować niesparowane reszty adeninowe w RNA. mRNA wystawione na działanie DEPC ulegają translacji z obniżoną wydajnością w układach syntetyzujących białka in vitro. Jednakże zdolność DEPC-owanego RNA do tworzenia hybrydów DNA-RNA i RNA-RNA nie jest poważnie zmniejszona, chyba że zmodyfikowane zostaną duże ilości reszt purynowych.
RYS. 7-7 Budowa dietylopirowęglanu
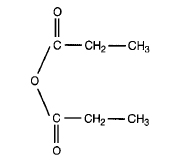
SOLE GUANIDYNOWE
Sole guanidynowe są chaotropami - niszczą trójwymiarową strukturę białek. Najsilniejszym z tych związków denaturujących jest izotiocyjanian guanidyny i chlorek guanidyny - konwertują one białka do losowo zwiniętych stanów. Pierwszą solą użytą jako związek deproteinizujący w izolacji RNA był chlorek (rys. 7-8).
Mimo że chlorek guanidyny jest silnym inhibitorem rybonukleazy, nie denaturuje wystarczająco mocno, aby można było ekstrahować nietknięte RNA z tkanek bogatych w RNazę (jak np. trzustka). Izotiocyjanian guanidyny zawiera reaktywne grupy kationowe i anionowe, tworzące silne wiązania wodorowe (rys. 7-9). Jest on stosowany w obecności czynnika redukującego, aby zerwać wiązania disulfidowe białek, oraz w obecności detergentu takiego jak Sarkosyl (zakłócanie oddziaływać hydrofobowych).
Izotiocyjanian guanidyny, lub jego bliski krewny tiocyjanian amonu, są składnikami komercyjnych zestawów które używają reagentów monofazowych (zawierających zakwaszony fenol, guanidynian, lub tiocyjanian amonu ze środkiem zwiększającym rozpuszczalność fenolu, np. glicerolem) aby zoptymalizować szybkość i zakres inaktywacji RNazy.
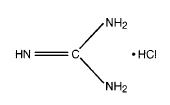
RYS. 7-8 Budowa hydrochlorku guanidyny
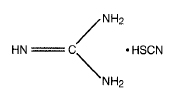
RYS. 7-9 Budowa izotiocyjanianu guanidyny
PROTOCOL 11
Obecne metody syntezy cDNA.
Podstawowa strategia enzymatycznego przekształcania mRNA na cDNA niewiele zmieniła się od połowy lat 70., ale wprowadzono wiele usprawnień, które podniosły efektywność całego procesu. Co więcej, obecnie można wybierać spomiędzy kilku typów enzymów na wielu etapach.
Synteza pierwszej nici cDNA. (Imho, ta pierwsza nić, która się będzie co jakiś czas pojawiała, to jest dokładnie pierwsza nić, powstała podczas pierwszego cyklu PCRa)
Pierwsza nić cDNA jest syntetyzowana przez RNA-zależną polimerazę DNA (odwrotna transkryptaza) z użyciem poli(A)RNA (albo mRNA) jako matrycy i oligo(dT) lub losowych oligonukleotydów jako primerów.
Odwrotna transkryptaza
W sprzedaży jest dostępnych kilka różnych form odwrotnej transkryptazy:
- Ptasia odwrotna transkryptaza (A może z kryla??), która jest oczyszczana z cząsteczek wirusa ptasiej białaczki (ALV).
- Mysia odwrotna transkryptaza, izolowana ze szczepu E. coli z wklonowanym genem odwrotnej transkryptazy wirusa mysiej białaczki Moloneya (wirus jest Moloneya, nie białaczka. Białaczka jest mysia.) Skrót na takiego wirusa to: Mo-MLV.
Zarówno ALV jak i MLV przygotowywane w celach komercyjnych jest obecnie wolne od zanieczyszczenia RNazą, która wcześniej była główną przeszkodą w syntezie dużych cDNA. Jednak niektóre preparaty zawierające ptasi enzym są zanieczyszczone endonukleazą, która przecina DNA, choć nie jest to już obecnie taki problem jak w latach 80. Ów ptasi enzym składa się z dwóch niejednakowych podjednostek polipeptydowych, które zawierają kilka aktywności enzymatycznych: RNA-zależna synteza DNA (odwrotna transkryptaza), endonukleolityczne trawienie DNA (integraza) i endonukleolityczny atak na RNA w hybrydach DNA-RNA, z następującym po nim egzonukleolitycznym usunięciem rNTPs (RNaza H). Enzym mysi składa się z pojedynczej podjednostki polipeptydowej, która prowadzi zarówno RNA- jak i DNA-zależną syntezę DNA, ale ma zmniejszoną zdolność degradowania RNA w hybrydach RNA-DNA. Mysia odwrotna transkryptaza nie posiada też „zanieczyszczającej” aktywności endonukleazy.
Ptasi enzym dostępny od wielu producentów jest odpowiedni do rutynowego tworzenia bibliotek cDNA. Jednakowoż, wysoki poziom aktywności RNazy H owego enzymu powoduje ograniczenie ilości i długości cDNA. Na początku reakcji hybrydy utworzone między matrycowym mRNA i primerem stanowią substrat dla RNazy H. Zatem, na początku syntezy cDNA degradacja mRNA współzawodniczy z inicjacją syntezy DNA. Co więcej, aktywność RNazy H może przecinać matrycę RNA niedaleko końca 3'-OH rosnącej nici DNA, jeżeli polimeraza zatrzyma się podczas syntezy. Tym sposobem, mysi enzym ze swoimi osłabionymi endogennymi aktywnościami degradującymi (mam nadzieję, że też to zrozumiecie) jest bezpieczniejszym wyjściem, jeżeli zależy nam na otrzymaniu pełnej długości cDNA, dłuższego niż 2-3kb. Mysie i ptasie enzymy mają różne optima temperaturowe i stężenia soli, dlatego ważne jest, by modyfikować odpowiednio warunki syntezy.
Odwrotne transkryptazy pozbawione aktywności RNazy H są zmodyfikowanymi (genetycznie?..) wersjami mysiego enzymu, posiadającego mutację drastycznie obniżającą ową aktywność, bez naruszania aktywności polimerazy DNA. Całkowity uzysk pierwszej nici cDNA jest znacznie wyższy, gdy używa się takich enzymów, niż wtedy, gdy korzystamy z enzymów niezmodyfikowanych, a i długość fragmentów cDNA jest znacznie większa. Na dokładkę, jako że polimerazowa aktywność enzymu nie jest hamowana wysokimi temperaturami, synteza pierwszej nici cDNA może być prowadzona nawet przy 50'C. Jest to niewątpliwą zaletą, gdy RNA matrycowe jest upakowane w struktury drugorzędowe.
Primery dla syntezy pierwszej nici cDNA
Lista poniżej przedstawia cztery typy primerów używanych w tej syntezie.
- Oligo(dT) długości 12-18nt, które przyłączają się do ogona poli(A) na 3' końcu eukariotycznych mRNA. Primer dodawany jest do mieszaniny reakcyjnej w dużym nadmiarze, więc każda cząsteczka mRNA wiąże kilka cząsteczek oligo(dT). Sekwencja sklonowanego cDNA pokazuje, że priming syntezy pierwszej nici zaczyna się prawdopodobnie od najbardziej proksymalnych z tych związanych primerów i jest bardzo wydajny.
- Adaptory dla primerów (primer adaptors), zawierające ciąg oligo(dT) na końcu 3' i miejsce restrykcyjne na końcu 5' używane są w przypadku sekwencji, które mogą być zakończone ciągiem dC. Ta procedura pozwala na ligację DNA z wektorem przed syntezą drugiej nici, lub też pozwala użyć drugiego primer-adaptor jako primera dla syntezy drugiej nici cDNA.
- Primery połączone z plazmidem. Priming syntezy pierwszej nici jest przeprowadzany za pomocą ciągu oligo(dT), który jest kowalencyjnie przyłączony do plazmidu. Często nie jest możliwe włączenie do reakcji plazmidu z odpowiednio długim „ogonem”, aby osiągnąć wysoki stosunek zawartości primera do matrycy, dlatego wiązanie primera jest zazwyczaj etapem ograniczającym reakcji. Owo ograniczenie skutkuje mniej efektywnym wykorzystaniem matryc mRNA podczas syntezy pierwszej nici cDNA. Jednakowoż, zmniejszenie ilości uzyskiwanej pierwszej nici cDNA jest przynajmniej częściowo równoważone przez wzrastającą wydajność klonowania, wynikającą z bezpośredniego kowalencyjnego przyłączenia cDNA do wektora. Biblioteki sporządzone zgodnie z oryginalnym protokołem Okayamy-Berga są generalnie średnio na jeża kompletne, ale za to mają dużo cDNA o pełnej długości. Główną wadą tego protokołu jest to, że jest dłuuugi, wymagający i angażujący (a to nie to samo?) Nowsze procedury, także używające asymetrycznie zakończonego plazmidu jako primera dla syntezy pierwszej nici cDNA są znacznie prostsze i dostarczają bardziej kompleksowych bibliotek.
- Losowe primery. Nawet najlepsze biblioteki cDNA zawierają jedynie małą ilość pełnowymiarowych klonów. Jeżeli synteza pierwszej nici cDNA była niekompletna, klonom będzie brakowało sekwencji, która była na 5' końcu mRNA. Jeżeli synteza drugiej nici była przerwana lub zahamowana, to z kolei kopii końca 3' mRNA będzie za mało w tworzonej bibliotece. Powyższym problemom można zaradzić używając losowych oligonukleotydów (zwykle heksamerów) jako primerów. Jednak, chociaż ta strategia pozwala rozwiązać jeden problem, stwarza ona kolejne. Klony cDNA uzyskane z użyciem losowych primerów bywają mniejsze, niż te otrzymane z oligo(dT). Co więcej, efektywność primingu różni się w zależności od różnych RNA. Zależy to od rozmiaru mRNA i stopnia skomplikowania oraz składu losowych oligonukleotydów. Gdy stosowanie losowych primerów stwarza problemy, istnieją alternatywne sposoby na generowanie klonów cDNA zawierających regiony 3' i 5' końcowe mRNA - na przykład, 5' RACE i 3' RACE.
Obrazek ze strony 17:
Priming cDNA z użyciem sekwencji oligo(dT).
Reakcja syntezy pierwszej nici cDNA jest zapoczątkowywana przez startery oligo(dT) przyłączone do adaptora dla primera, zawierającego miejsce restrykcyjne dla endonukleazy. Sekwencja mRNA jest reprezentowana przez kolorową nić (chyba na rysunku była w oryginale kolorowa...), natomiast adaptor oligo(dT) jest czarny. Potraktowanie hybrydy (witaj, gramatyko!) RNA-DNA RNazą H i polimerazą DNA I powoduje rozcięcie jej rybonukleotydowej połowy z utworzeniem końców 3'-OH. Są one wykorzystywane przez polimerazę DNA T4 do ukończenia syntezy drugiej nici. Włączenie adaptorów EcoRI do dwuniciowego produktu cDNA i jego cięcie NotI pozwala na włączenie w określonej orientacji do odpowiedniego wektora.
Drugi obrazek, z następnej strony:
Randomowy priming cDNA.
Synteza pierwszej nici cDNA jest zapoczątkowana przez losowe oligonukleotydy, zwykle heksamery (pokazane jako czarne strzałki), które są wydłużane przez odwrotną transkryptazę (produkty tego wydłużania pokazane są jako kolorowe strzałki). Działanie RNazą H i polimerazą DNA I przecina RNA i uzupełnia przerwy w cDNA. Synteza drugiej nici jest prowadzona z użyciem polimerazy DNA T4.
Tabela ze strony 18:
Primery oligo(dT)
Zalety: Prosty i wydajny.
Pozwala na użycie procedury Gublera i Hoffmanna (1983) do syntezy drugiej nici cDNA.
Na każdym końcu cDNA mogą zostać przyłączone inne linkery i adaptory, co umożliwia ukierunkowane klonowanie i duży wybór wektorów.
Wady: Linkery, „ogony” i adaptory muszą być dodane zanim wszczepimy DNA w wektor.
Wiele procedur wymaga, by dsDNA było strawione enzymami restrykcyjnymi przed klonowaniem. Ten krok może być ryzykowny, ponieważ może zostać strawione cDNA, które nas interesuje. Potencjalne miejsca restrykcyjne w „naszym” fragmencie cDNA można ochronić przez metylację albo inkorporację metylocytozyn podczas syntezy pierwszej nici cDNA. W każdym przypadku, jako gospodarza warto użyć nici E. coli niezawierającej układu restrykcyjnego mcr. Nici mcr(+) powodują cięcie DNA nawet, gdy zawiera ono metylocytozyny.
Wyższy sygnał dawany przez „puste” klony, niż ten otrzymany, niż przy inicjacji za pomocą plazmidu.
Większość bibliotek DNA skonstruowanych za pomocą primerów oligo(dT) zawiera nadmiar sekwencji leżących na 3' końcu mRNA. (??)
Adaptory primerów
Zalety: Synteza drugiej nici może być przeprowadzona po włączeniu do wektora cDNA z homopolimerycznym ogonem.
Alternatywnie, synteza drugiej nici może być przeprowadzona z użyciem metody Gublera-Hoffmana. W tym przypadku, bezpośrednie klonowanie cDNA może być osiągnięte przy użyciu linkera-primera i adaptora dla enzymu restrykcyjnego SfiI.
„Tło” z powstałych pustych klonów jest generalnie niższe przy użyciu primer-adaptorów, niż przy primerach oligo(dT). Mogą zostać użyte zarówno wektory plazmidowe, jak i bakteriofagi λ.
Wady: Wiele protokołów wymaga eliminacji małych oligonukleotydów i niezużytych primerów z pierwszej nic cDNA przez elektroforezę w alkalicznym żelu agarozowym. Odzyskiwanie DNA na tym etapie zwykle wychodzi lipnie.
Primery plazmidowe
Zalety: Selekcja klonów o pełnej długości, lub prawie pełnej długości, być może dlatego, że długie cDNA są preferowanymi substratami dla terminalnej transferazy.
Podczas, gdy oryginalny protokół dawał biblioteki o umiarkowanej kompleksowości, bardziej współczesne wersje są, jak głosi plotka, tak superwydajne, że generują prawie 107 do 109 klonów/μg primera.
Wady: Oryginalna procedura była dłuuuga i trudna. Nowsze wersje są nieco łatwiejsze, choć nadal wymagające.
Wektory mogą być jedynie plazmidowe.
Priming za pomocą losowych oligonukleotydów
Zalety: Taki priming może niekiedy poprawić efektywność klonowania sekwencji na końcach 5' mRNA.
Wady: cDNA utworzone przez losowy priming lubi być małe i musi być ostrożnie rozdzielane na podstawie wielkości, by wyeliminować niepotrzebną część populacji malutkich cząsteczek. Losowe heksamery są zwykle usuwane pomiędzy syntezą pierwszej nici cDNA a syntezą drugiej.
RT-PCR
Zalety: „Uniwersalne primery” mogą być używane do amplifikacji wszystkich mRNA, niezależnie od ich sekwencji. Są one używane do tworzenia bibliotek cDNA, gdy dostępna jest jedynie mała ilość poli(A) RNA (<200ng). Inne protokoły PCR polegają na primerach specyficznych dla genów, które są unikalne lub wybiórcze dla konkretnego docelowego cDNA. Na przykład metoda 5' RACE pozwala zamplifikować koniec 5' mRNA, gdy tylko znana jest jakaś część sekwencji blisko tego końca.
Wady: Klony generowane przez standardowy PCR zwykle zawierają większą liczbę mutacji, niż produkowane innymi drogami. Metody wymagające unikalnego primera mogą być użyte tylko wtedy, gdy dysponujemy sekwencją przynajmniej części cDNA albo kodowanego przez nie białka.
Synteza drugiej nici cDNA.
Przez wiele lat nie było innego skutecznego sposobu na syntezę drugiej nici cDNA, poza używaniem jako primera pierwszej nici cDNA, powstałej w pierwszym cyklu PCR. (self-priming); i niemal wszystkie klony cDNA powstałe przed 1982r. zostały otrzymane przy użyciu tej właśnie reakcji enzymatycznej. Taka „samoinicjująca się” synteza drugiej nici cDNA była w najlepszym wypadku słabo kontrolowana, a następujące po niej trawienie struktury spinki do włosów nukleazą S1 prawie zawsze prowadziło do utraty albo rearanżacji sekwencji sąsiadującej z końcem 5' mRNA. Ten schemat syntezy dwuniciowych cząsteczek cDNA został obecnie w dużej mierze zastąpiony bardziej wydajnymi metodami, które niosą mniejsze ryzyko zniszczenia DNA. Dostarczają one 10-200 razy więcej kopii cDNA niż opisany powyżej priming.
Obecnie, synteza drugiej nici jest zwykle katalizowana przez polimerazę DNA I i RNazę H, obie z E. coli. Jednak, inne enzymy, w tym termostabilne polimerazy DNA, jak Tth i Taq również są w użytku, aczkolwiek (albeit!! Nie however!) rzadko. Warunki stosowane do osiągnięcia syntezy drugiej nici cDNA o pełnej długości zależą od użycia konkretnej polimerazy DNA. Reakcje z polimerazą DNA I E. coli zwykle prowadzi się w pH 6.9, by zminimalizować aktywność 5'=>3' egzonukleazową enzymu i w 15'C dla zmniejszenia możliwości powstania DNA z popękanym końcem 5' (`snapback'). Powyższych problemów można całkowicie uniknąć przez użycie odwrotnej transkryptazy albo fragmentu Klenowa, którym to enzymom brakuje aktywności 5'=>3' egzonukleazy i są dostępne w czystej postaci ze źródeł komercjalnych. Ligaza DNA E. coli jest niekiedy włączana do reakcji, gdyż są dowody na to, że poprawia ona wydajność klonowania dłuższych cDNA. Nierzadko, polimeraza DNA T4 albo termostabilna polimeraza, jak Pfu jest dodawana pod koniec syntezy drugiej nici, by wyrównać (??polish??) końce gotowego dsDNA. Jednak, tworzenie tępych końców w ten sposób, może zaowocować utratą sekwencji z końca 5' - wtedy klony cDNA będą 5-30 nukleotydów krótsze, niż oryginalne mRNA.
Synteza zastępcza drugiej nici
W tej metodzie, wprowadzonej przez Okayamę i Berga, a zmodyfikowanej przez Gubblera i Hoffmana produkt pierwszej syntezy - hybryd cDNA-mRNA - jest używany, jako matryca do reakcji nick-translacji. RNaza H tworzy nacięcia i przerwy w nici mRNA, dostarczając zestawu primerów RNA, które są używane przez polimerazę DNA I E. coli podczas syntezy drugiej nici cDNA. Reakcja ma trzy główne plusy:
+ Jest bardzo wydajna.
+ Można ją przeprowadzać bezpośrednio, z użyciem produktów reakcji syntezy pierwszej nici, które nie wymagają żadnego dalszego oczyszczania i przetwarzania.
+ Eliminuje konieczność używania nukleazy S1 do cięcia jednoniciowej struktury spinki do włosów w dwuniciowym DNA, co jest trudne w kontrolowaniu i często skutkuje ogromnymi stratami cDNA.
Rysuneczek: Schemat podstawowej strategii Gublera-Hoffmana.
Seria trzech reakcji enzymatycznych daje dwuniciowy DNA. Synteza pierwszej nici jest inicjowana z użyciem starterów poli(dT), matryca mRNA jest usuwana przez trawienie RNazą H, a druga nic jest syntetyzowana z użyciem polimeraz DNA: I i T4. Powstały produkt dwuniciowego cDNA posiada tępe końce, odpowiednie do następującego potem przyłączenia linkerów i adaptorów, mających ułatwić klonowanie.
Synteza drugiej nici ze starterów oligonukleotydowych
Dla standardowej konstrukcji bibliotek cDNA, zwykła reakcja zastąpienia opisana przez Gublera i Hoffmana nadaje się doskonale. Jednakże, gdy mamy na celu utworzenie biblioteki ekspresji (?...) (wymagających cDNA o pełnej długości), lub sklonowanie sekwencji z końca 5' eukariotycznego mRNA z wysoką wydajnością, metody opisane poniżej oferują pewne zalety.
- Po ukończeniu syntezy pierwszej nici, można użyć terminalnej transferazy, by dodać homopolimeryczne ogony dC do wolnych grup 3'-OH. Takie ogony są następnie hybrydyzowane z oligo(dG), które to służą jako primery dla syntezy drugiej nici cDNA. Efektywność tej metody wydaje się zależeć od konkretnego mRNA, jaki badamy. W najlepszych przypadkach (np. lizozymu kurczaków), można z dużą wydajnością otrzymać klony cDNA pełnej długości. Inne mRNA dostarczyły jednak bardziej zróżnicowanych wyników, być może odzwierciedlając względną wydajność, z jaką terminalna transferaza wykorzystuje 3' końce różnych nici cDNA z pierwszego cyklu, jako substraty do przyłączania ogonów. Obecność owych ogonów na każdym końcu pierwszej nici cDNA daje możliwość amplifikacji cDNA in vitro PCRem. Jest to użyteczne, gdy ilość dostępnego mRNA jest za mała, by wytworzyć cDNA na drodze standardowych procedur. Jednak, z powodu nieefektywności amplifikacji długich cDNA, następuje ich ostra selekcja.
- Okayama i Berg (1982) wykorzystali fragment plazmidowego DNA, zawierający krótki ogon poli(dG) jako primer dla syntezy drugiej nici. Nukleotydy dG zostały związane z homopolimerycznym traktem dC, dodanym na 3' końcu pierwszej nici cDNA przez terminalną transferazę. Choć generalnie protokół ów sprawdza się, jest pracochłonny i wymaga trawienia dupleksu mRNA-cDNA endonukleazą restrykcyjną, a następnie przyłączenia fragmentu z ogonem cG, dla zapoczątkowania reakcji naprawy drugiej nici. Opisano kilka modyfikacji mających usprawnić klonowanie, poprzez pominięcie wymaganego osobnego linkera upstream, używanego do zainicjowania syntezy drugiej nici i do włączenia dsDNA do plazmidu. Obejmują one:
Użycie syntetycznych adaptorów dla primerów, z oboma ogonami homopolimerycznymi do primingu pierwszej i drugiej nici cDNA i miejsc restrykcyjnych, do wklonowania do plazmidów i bakteriofagów λ. Użycie adaptorów ma trzy zalety. Po pierwsze, zmniejszona jest ilość etapów syntezy i klonowania cDNA. Na przykład, w protokole opisanym przez Hana (Solo?....:P) w 1987, hybryda cDNA-mRNA jest przyłączona do wektora bakteriofagowego syntetycznymi adaptorami przed syntezą drugiej nici cDNA. To eliminuje kilka kroków, które są wymagane, gdy klonujemy dsDNA za pomocą syntetycznych linkerów. Po drugie, wzrastająca efektywność primingu syntezy drugiej nici, dostarcza bibliotek zawierającą porównywalne ilości cząstek cDNA o pełnej długości. Po trzecie wreszcie, ta metoda może być zaadaptowana tak, by pozwolić na amplifikację pierwszej nici cDNA przez PCR. Istnieją jednak także i wady: wszystkie klony utworzone przez homopolimeryczny priming syntezy drugiej nici zawierają trakt dG:dC, bezpośrednio powyżej sekwencji komplementarnej do matrycy mRNA. Obecność owych dodatkowych sekwencji może hamować transkrypcję DNA zarówno in vivo, jak i in vitro. Co więcej, mogą one stanowić barierę dla fragment Klenowa polimerazy DNA I E. coli podczas sekwencjonowania DNA metodą dideoksy, co wymagałoby użycia innej polimerazy DNA (np. odwrotnej transkryptazy) i odpowiednich dla niej warunków
Obrazek 11-5: Homopolimeryczny priming drugiej nici cDNA. Synteza pierwszej nici jest inicjowana przez primer-linker oligo(dT), pokazany na czarno. Po usunięciu matrycy mRNA, ogon homopolimeryczny jest dodawany do jednoniciowego produktu. Druga nić jest zapoczątkowywana przez oligomery komplementarne do wspomnianego ogona, a powstały produkt jest trawiony endonukleazami restrykcyjnymi, dla których miejsca restrykcyjne zostały włączone na końcach dwuniciowych cząstek cDNA.
Malunek 11-6: Schemat strategii Okayamy i Berga. Przygotowanie primera plazmidowego i linkera DNA. Niezaciemniona część każdego kolistego wektora to pBR322 DNA, zaś zaciemnione segmenty reprezentują miejsca trawienia endonukleazą.
Użycie zlinearyzowanego plazmidu, niosącego symetryczny ogon (dT) na jednym końcu 3' i syntetycznego ogona (dC) na drugim. Taka konstrukcja jest tworzona przez ligację adaptorów niosących odpowiednie ogony homopolimeryczne i plazmidu, przeciętego dwoma enzymami restrykcyjnymi. Ogon dC zawiera grupę 3'-fosforanową, która blokuje dalszą addycję na końcu 3'. Po zastosowaniu ogona dT do zapoczątkowania syntezy pierwszej nici cDNA, terminalna transferaza dodaje nukleotydy dG na wolnym końcu 3' cDNA. Blokujący 3'-fosforan jest następnie usuwany przez alkaliczną fosfatazę (:)) a druga nić DNA jest syntetyzowana przez wspólne działanie RNazy H, polimerazy DNA I i ligazy DNA.
Powinno się uważnie rozważać metodę, używaną do syntezy drugiej nici cDNA, gdyż może to określać wybór wektora i sposób włączenia do niego cDNA.
KLONOWANIE MOLEKULARNE dsDNA
Wklonowanie dwuniciowego cDNA jest ułatwione przez dodawanie różnych ogonków, linkerów albo adaptorów na końcach cDNA.
Syntetyczne linkery i adaptory DNA.
Sztuczne linkery zawierające jedno lub więcej miejsc restrykcyjnych zapewniają efektywne łączenie dsDNA zarówno z wektorami plazmidowymi, jak i z bakteriofagiem λ i wyparły inne metody klonowania DNA. Dwuniciowe DNA traktuje się polimerazą DNA faga T4 albo polimerazą DNA I E. coli - enzymami usuwającymi wystające jednoniciowe końce 3' dzięki aktywności 3'=>5' egzonukleolitycznej i wypełniają końce 3'-OH dzięki aktywności polimerazowej. Otrzymujemy cDNA o tępych końcach, które są inkubowane z dużym nadmiarem cząstek linkera w obecności ligazy DNA bakteriofaga T4, enzymu, który może katalizować ligację DNA o tępych końcach. Produktami reakcji są cząsteczki cDNA zawierające polimeryczne sekwencje linkerowe na końcach. Cząsteczki są trawione w miejscu restrykcyjnym linkera, oczyszczane i włączane do wektora, uprzednio przeciętego enzymem restrykcyjnym, generującym lepkie końce kompatybilne z końcami linkera.
Dwuniciowe cząsteczki cDNA zawierające syntetyczne lepkie końce oczywiście będą się łączyć ze sobą nawzajem, tak samo, jak z DNA wektora. Ważne jest, by tak dobrać warunki reakcji, by zminimalizować tworzenie się cząsteczek chimerycznych, ponieważ strasznie pracochłonne jest sprawdzanie, czy klony cDNA zawierające konkretne miejsca restrykcyjne zostały utworzone przez ligację końców dwóch niezwiązanych cDNA. Mieszanina ligacyjna powinna wobec tego zawsze zawierać duży nadmiar molowy DNA wektora w stosunku do cDNA. Jednak, takie warunki faworyzują odtwarzanie wektora przez samoligację, co prowadzi do niemożliwego do zaakceptowania, wysokiego tła niezrekombinowaych klonów. Ten problem można zmniejszyć przez inkubację strawionego wektora z fosfatazą, przed ligacją z cDNA.
Jeżeli dwuniciowe cDNA zawiera conajmniej jedno miejsce rozpoznawane przez enzym restrykcyjny, będzie ono strawione i sklonowane jako dwa czy więcej fragmentów DNA, co zapobiegnie izolacji i analizie cDNA o pełnej długości. Starania, by złagodzić ten problem, obejmują:
- Użycie enzymów restrykcyjnych, które rzadko tną DNA ssaków. Nie ma gwarancji, że nawet małe fragmenty cDNA nie zawierają miejsca rozpoznawalnego dla poszczególnego enzymu restrykcyjnego. Jednak, gdy rozmiar badanego DNA jest znacznie mniejszy, niż rozmiar przeciętnego fragmentu genomowego DNA, jaki możemy otrzymać przez trawienie wybranym enzymem, jest spora szansa na to, że nie będzie on zawierał miejsca restrykcyjnego. Na przykład, szanse znalezienia sekwencji oligonukleotydowej bogatej w G/C, rozpoznawanej przez NotI w klonie długości 5kb są mniejsze niż 1/200. Dla SalI, którego miejsca restrykcyjne występują średnio raz na 100kb w ssaczym DNA, szanse owe rosną już do 1/20 - wciąż jest to ryzyko do zaakceptowania.
brzmi mądrze, aczkolwiek to tylko porcja roztworu
wydaje mi się że chodzi o chromatografię okresową, ale nie jestem pewna o.O
Wyszukiwarka