Victor W. Rodweli, PhD
WPROWADZENIE
Żywe komórki wytwarzają makrocząsteczki (białka, kwasy nukleinowe, polisacharydy), które służą jako składniki strukturalne, katalizatory, hormony, receptory lub magazyny informacji genetycznej. Te makrocząsteczki są biopolime-rami, utworzonymi z jednostek monomerycz-nych lub cegiełek budulcowych. Jednostkami monomerycznymi w kwasach nukleinowych są nukleotydy, w złożonych polisacharydach — pochodne cukrowe, a w białkach — L-a-amino-kwasy.
Chociaż białka mogą także zawierać poza aminokwasami, dodatkowe substancje (np. hem, cukrowce, tłuszcze), ich trójwymiarową strukturę i właściwości biologiczne warunkuje w głównej mierze rodzaj aminokwasów, kolejność, w jakiej łączą się ze sobą w łańcuchu polipeptydowym oraz ich przestrzenne ułożenie,, względem siebie.
Aminokwasy pełnią dodatkowe funkcje w komórkach. W tabeli 4-4 i 4-5 przedstawiono niektóre biologicznie ważne substancje, które wywodzą się z aminokwasów.
genne) muszą być dostarczane w pożywieniu, ponieważ nasz organizm nie może ich syntetyzować w ilościach niezbędnych do podtrzymania wzrostu (dzieci) i utrzymania zdrowia (dorośli). Metabolizm aminokwasów przyczynia się do powstania wielu biomedycznie ważnych związków. Na przykład dekarboksyla-cja pewnych aminokwasów prowadzi do powstania amin, wśród których cześć pełni ważne funkcje biologiczne (np. histamina i kwas 7-a-minomasłowy [GABA]). Liczne choroby są spowodowane nieprawidłowościami transportu aminokwasów do komórek. W pewnych warunkach może dojść do pojawienia się w moczu znacznej ilości jednego lub wielu aminokwasów — takie stany określa się jako aminoacydurie.
WSZYSTKIE AMINOKWASY ZAWIERAJĄ PRZYNAJMNIEJ 2 GRUPY FUNKCYJNE
Aminokwasy zawierają 2 grupy funkcyjne, tj. aminową i karboksylową. W a-aminokwasach obie te grupy połączone są z tym samym (a)
Ryc. 4-1.
kwasu.
ZNACZENIE BIOMEDYCZNE
Niektóre aminokwasy wydają się być zaangażowane w przenoszeniu impulsów w układzie nerwowym, czego przykładem są glicyna i kwas glutaminowy. Aminokwasy podstawowe (egzo-
H
\a
OH
R-C-NHj I COOH
Dwie formy przedstawienia j.-amino-
36 / ROZDZIAŁ 4
atomem węgla (ryc. 4-1). Chociaż w przyrodzie występuje ok. 300 aminokwasów, tylko 20 z nich występuje w białkach (tab. 4-3). Całkowita hydroliza* biatek dostarcza 20 L-a-ami-nokwasów. Należy podkreślić, że białka wszystkich form życia — roślin, zwierząt, drobnoustrojów — zawierają te same 20 aminokwasów. Przyczyną tego jest uniwersalność kodu genetycznego, co stanie się oczywiste dla Czytelnika w toku omawiania tego zagadnienia później (p. rozdz. 30). W skład niektórych białek wchodzą pochodne aminokwasów, utworzone po ich włączeniu w cząsteczkę białka (p. lab. 6-4).
Z wyjątkiem glicyny, dla której R (R — łańcuch boczny aminokwasu) stanowi atom wodoru (ryc. 4-1), wszystkie 4 podstawniki związane z atomem węgla et-aminokwasów są różne. Tetrąędryczne. ułożenie 4 różnych podstawników wokół atomu węgla a (tzw. węgiel asymetryczny) nadaje aminokwasom właściwość op-.tyczna. (zdolnośćTjio_ _skręcania . płaszczyzny światła spolaryzowanego). Chociaż pewne aminokwasy występujące w białkach są prawo-skrętne, a niektóre lewoskrętne, w pH 7,0 wszystkie mają bezwzględną konfigurację porównywalną z konfiguracją aldehydu L-glicery-nowego, stąd opisuje się je jako L-a-amino-kwasy.
Ogólne reakcje chemiczne aminokwasów można przewidzieć znając właściwości grup funkcyjnych
Grupy funkcyjne aminokwasów — karbok-sylowa i aminowa — wykazują typowe dla nich reakcje, np. tworzenie soli, estryfikację, acyla-
RÓWNOWAGI PROTONOWE AMINOKWASÓW
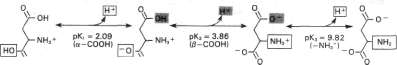
W zależności od pH otaczającego środowiska aminokwas może mieć ładunek dodatni, ujemny lub nie mieć żadnego ładunku
Aminokwasy zawierają co najmniej 2 zjoni-zowanc, słabo kwaśne grupy: —COOH i —NHt- W roztworze obie formy tych grup.
jedna naładowana, a druga obojętna, występują w równowadze protonowej:
R—COOH « R—COO" + H+ H+
W równowadze tej R—COOH i R—NH^ reprezentują związki protonowe lub kwaśne, natomiast R—COO~ i R—NH2 — zasady sprzężone (protonobiorców) z odpowiednimi kwasami. Chociaż zarówno R—COOH jak i R NH; są słabymi kwasami, R—COOH jest znacznie silniejszym kwasem niż R—NHr W pH osocza krwi czy przestrzeni śródkomór-kowej (pH odpowiednio 7,4 i 7,1) grupy kar-boksylowe istnieją prawie całkowicie jako jony karboksyjowe — R—COO~. Przy tych wartościach pH większość grup aminowych występuje w formie zasocjowanej (protonowej) — R—NH3. Ze względu na przeważającą postać zjonizowaną aminokwasów we krwi i większości tkanek, należy przedstawić ich budowę tak, jak na ryc. 4-2A. Należy pamiętać, że struktura B (ryc. 4-2) nie może istnieć w żadnym pH. Przy dostatecznie niskim pH, w którym cofa się jonizacja grupy karboksylowej, znacznie słabsza kwaśna grupa aminowa będzie występować także w formie protonowej. Przybliżone wartości pKa dla grup a-karboksylowej i oc-aminowej wynoszą odpowiednio 2 i 10 (tab. 4-1). Przy pH wynoszącym 2 jednostki poniżej wartości pKa, kwas w ok. 99% ma formę protonową. Jeżeli pH stopniowo wzrasta, to proton z grupy karboksylowej odłącza się znacznie wcześniej niż z grupy R—NH 3 • W każdym wystarczająco wysokim pH do utworzenia przewagi grupy aminowej R—NH2 musi być również obecny jon karboksylowy (R—COO~). Jednakże w wielu reakcjach, poza równaniami równowagi protonowej, stosuje się wzory aminokwasów w formie B.
NHj
![]()
NH,+
II O
8
0-
* Hydroliza = rozerwanie wiązania kowalencyjnego przy udziale cząsteczki wody.
Ryc. 4-2. Poprawna struktura ^jonizowanego aminokwasu w pH fizjologicznym lub blisko pH fizjologicznego (A). Postać B (niezjonizowana) nie występuje w żadnym pH, ale jest często' używana, dla wygody, przy omawianiu chemii aminokwasów.
AMINOKWASY / 37
Tabela 4-1. Słabo kwaśne grupy aminokwasów występujących w białkach
|
Sprzężony kwas |
Sprzężona zasada |
Przybliżona wartość pK |
a-Karboksylowa |
R—COOH |
R—COO" |
2.1 ±0,5 |
Nie a-kar boksy Iowa (aspara-ginian, glutaminian) |
R—COOH |
R—COO" f 1 R |
4,0 + 0,3 |
Imidazolowa (histydyna) |
|
r v |
6,0 |
^-Aminowa |
R-NH + |
R—NH2 |
9,8 ±1,0 |
s-Aminowa (lizyna) |
R-NH^ |
R—NHa |
10,5 |
Fenolowa OH (tyrozyna) |
|
|
10,1 |
Guanidynowa (arginina) |
H NH2 1 11 + R—N—C—NH2 |
H NH j II R—N—C—NH2 |
12,5 ■ |
Sulf hydry Iowa (cystein3) |
R—SH |
R-S- |
8,3 |
Względną moc słabych kwasów wyrażają wartości pK,
Względną moc słabych kwasów wyraża się przez ich stale dysocjacji Ka lub przez ich pK — ujemny logarytm ze stałej dysocjacji:
Punkt izoelektryczny aminokwasu (pl) jest to takie pH, przy którym nie jest on obdarzony żadnym ładunkiem i nie porusza się w polu elektrycznym
Strukturę aminokwasu alifatycznego, takiego jak alanina, w pH odpowiadającym punktowi izoelektrycznemu przedstawia ryc. 4-3.
NrV
W tabeli 4-1 przedstawiono wartości pK dla grup funkcyjnych 20 aminokwasów spotykanych w białkach.
Całkowity ładunek (algebraiczna suma wszystkich dodatnio i ujemnie naładowanych grup) aminokwasu zależy od pH lub stężenia protonów otaczającego środowiska. Możliwość zmiany ładunku aminokwasów i ich pochodnych, przez umiejętne sterowanie pH, ułatwia fizyczny rozdział aminokwasów, peptydów i białek.
Ryc. 4-3. Struktura postaci zjonizowanej lub jonu obojnaczego alaniny. Chociaż jon obojnaczy alaniny ma grupy obdarzone ładunkiem elektrycznym, nie wędruje w polu elektrycznym.
♦ Dla wygody zapis Ka lub pKtt będzie stosowany, w przypadku aminokwasu z tylko dwiema
lecz opuszczony później w przypisach dotyczących grupami zjonizowanymi (np. alanina) nie ma
symboli. niejasności w wyliczeniu pl. Ponieważ wartość
38 / ROZDZIAŁ 4
W mocnym kwasie
(pH ponlżeji); całkowity ladunek= + l
O
B
pH ok. 3; całkowity ładunek = o
pH ok. 6 - ft całkowity ładunek = -1
D
W mocno) zasadzie
(pH powyżej 11);
całkowity ładunek = -2
Ryc. 4-4. Równowagi protonowe kwasu asparaginowego.
pK, (R—COOH) = 2,35 natomiast pK2 (R—NH*) = 9,69, to punkt izoelektryczny (pl) alaniny wynosi:
2,35+9,69
= 6,02
Pl
Wyliczenie wartości pl dla aminokwasu z dodatkowymi grupami zjonizowanymi, poza tymi związanymi z węglem ot, jest bardziej skomplikowane. Na rycinie 4-4 przedstawiono różne formy jonowe kwasu asparaginowego. Jakie będzie zatem pH odpowiadające punktowi izo-elektrycznemu dla tego aminokwasu? W poszukiwaniu pl dla kwasu asparaginowego trzeba napisać wszystkie możliwe formy jonowe tego związku w kolejności, w której pojawiają się w roztworach — od silnie kwaśnych, poprzez obojętne do zasadowych (porównaj przykład kwasu asparaginowego na ryc. 4-4). Następnie należy rozpoznać formę izojonową, jon oboj-naezy lub obojętny (jak na ryc. 4-4B). Punkt izoelektryczny (pl) jest to pH w środku pomiędzy wartościami pK po obydwu stronach form izojonowych. W omawianym przykładzie:
2,09+ 3,86
2,98
Pl =
Takie rozwiązanie jest również stosowane do obliczania wartości pl aminokwasów z innymi, dodatkowymi grupami dysocjującymi, np. lizy-ny lub histydyny. Po napisaniu wszystkich możliwych form elektrycznie naładowanych aminokwasów zasadowych — lizyny i argininy — należy zapisać, że:
pK2+ pK3
P'= «
Wartość pl dla lizyny wynosi 9,7; dla argininy 10,8. Czytelnik powinien umieć określić pl dla histydyny.
Określenie wartości pK po każdej stronie jonu obojnaczego przez sprawdzenie form naładowanych, nie ogranicza się do aminokwasów. Można ją stosować do obliczania ładunku cząsteczki z każdą liczbą grup dysocjujących. Możliwość wykonywania takich obliczeń okazuje się bardzo przydatna w laboratorium klinicznym, gdyż pozwala przewidzieć ruchliwość elektro-foretyczną związków w polu elektrycznym i dobrać właściwe bufory do ich rozdziału. Na przykład, w buforze o pH 7,0 można rozdzielić 2 typy cząsteczek: z pl 6,0 i 8,0, ponieważ cząsteczka z pI = 6,0 będzie mieć większy całkowity ujemny ładunek przy pH 7,0 niż cząsteczka z pI = 8,0. Podobne rozważania stosują się do rozdziałów na złożach jonowych zarówno dodatnio, jak i ujemnie naładowanych polimerów (np. DEAE-celuloza, żywica Dowex 1),
Rozpuszczalność i temperatura topnienia aminokwasów odzwierciedlają ich charakter jonowy
Obecność elektrycznie naładowanych grup większości aminokwasów przyczynia się do ich rozpuszczalności. W formie jonowej rozpuszczają się one łatwo w rozpuszczalnikach polarnych, takich jak woda i etanol, ale nie rozpuszczają się w rozpuszczalnikach niepolarnych, jak: benzen, heksan i eter. Wysoka temperatura topnienia (punkty topnienia powyżej 200DC) odzwierciedla ilość energii potrzebnej do pokonania sił jonowych stabilizujących strukturę sieci kryształów.
AMINOKWASY / 33
Tabela 4-2. Podział L-at-aminokwasów występujących w białkach na podstawie względnej polar-ności ich grup R. W grupie niepolarnej jest mała bądź nie ma wcale różnicy w ładunku elektrycznym między regionami, podczas gdy w grupie polarnej różnica ta jest względnie duża
Aminokwasy |
Aminokwasy |
niepolarne |
polarne |
Alanina |
Arginina |
Izoleucyna |
Asparagina |
Leucyna |
Kwas asparaginowy |
Metionina |
Cysteina |
Fenyloa Janina |
Kwas glutaminowy |
Prolina |
Glutamina |
Tryptolan |
Glicyna |
Walina |
Histydyna |
|
Lizy na |
|
Sery na |
|
Treonina |
|
Tyrozyna |
PODZIAŁ AMINOKWASÓW WYSTĘPUJĄCYCH W BIAŁKACH NA PODSTAWIE WZGLĘDNEJ POLARNOŚCI ICH GRUP R
Aminokwasy występujące w białkach można podzielić na 2 duże grupy na podstawie poiar-ności grup R. przyłączonych do atomu węgla a. W celu ułatwienia przedstawiania niezwykle długich sekwencji amin o kwasowych niektórych białek, praktykuje się stosowanie jednoliterowych symboli aminokwasów (tab. 4-3).
Aminokwasy w stanie wolnym lub związanym (nie będące składnikami białek) odgrywają ważną rolę w procesach przemiany materii (tab. 4-4 i 4-5). Na przykład ornityna, cytrulina i kwas argininobursztynowy uczestniczą w biosyntezie mocznika, katanie naturalnym występuje ponad 20 D-aminokwasów. Należą do nich D-alanina i kwas D-glutami nowy ścian komórkowych niektórych bakterii oraz różne D-ami-nokwasy wchodzące w skład antybiotyków.
Tabela 4-3. L-s-aminokwasy występujące w białkach*
Nazwa
Symbol
Wzór strukturalny
Aminokwasy z. alifatycznymi łańcuchami bocznymi
Glicyna
Gly [G]
H—CH™COCT
Alanina
Ala [A]
CH3
Walina
Vaf [V]
,CH—CH—COO"
t
Leucyna
Leu [L]
H,C
X
Izoleucyna
Ile [1]
7>
CH—CB—CGO"
40 / ROZDZSAŁ 4
cd. tab. 4-3
Nazwa
Symbol
Wzór strukturalny
Aminokwasy z łańcuchem bocznym zawierającym grupy hydroksylowe (OH)
Sery na |
Ser [S] |
CH^CH^rC&O- .---.Li ^łJLJf- |
Treonina |
Thr[T] |
CH,-CH—OH—COCT |
|
|
OH *NHj |
Tyrozyna |
Tyr [Y] |
patrz niżej |
Aminokwasy z łańcuchem bocznym zawierającym atomy siarki
SH fl^ CH;—CH3-CH—COO"
Cysterna*
Metionina
Cys [C]
Met [M]
CH SH
;CH3HCOO
O^~Cn3 rwij -^
Aminokwasy z łańcuchem |
bocznym zawierającym |
grupy kwaśne lub ich amidy • |
|
Kwas asparaginowy |
Asp[D] |
|
"OOC—C Hs —CH—COO" t 1L 1 M flfTJ .$. |
Asparagina |
Asn [N] |
|
HjN-C-CH,-CH—COO" 0 ^H, |
Kwas glutaminowy |
Glu ! El |
|
"OOC—C H 2—C H3 —CH—COO" |
Glutamina |
Gin [Q] |
|
H,N—C—CH 2—C H2 ~CH—COO- |
|
|
|
|
Arg [R] Lys [K]
His |h|
Arginina
Aminokwasy z łańcuchem bocznym zawierającym grupy zasadowe
H—N—CH2—CH,—CH?—CH—COO"
I
+NH5
r
Lizy na
NH,
Histydyna
Hs-CH,—CH,—C H S-CM—COO"
H3 +NH,
j I C H,—GH—COO*
AMINOKWASY / 41
cd. tab. 4-3
Nazwa
Symbol
Wzór strukturalny
Aminokwasy zawierające pierścień aromatyczny
-Histydyna |
His [H] |
patrz |
wyżej |
Fenyioaianina |
Phe {F] |
\—/ |
>—CHj—CH^COO" |
Tyrozyna |
Tyr[Yl |
|
|
|
|
|
|
Tryptofan |
Trp[W] |
|
pCH;—ch—cocr |
|
|
H |
|
Immokwasy |
|
i 1 |
|
|
|
Ptoiina |
Pro [P] |
|
*. Z wyjątkiem hydroksyl i zyny (Hyl) thydroksyproliny (Hyp), które są włączane w wiązania peptydowe jako lizyna i proiina, a następnie hydroksylowane, swoiste tRNA istnieją dla wszystkich aminokwasów przedstawionych w tej t3be/i. Ich włączanie w białka pozostaje pod bezpośrednią kontrolą genetyczną. ** Cystyna składa się z 2 reszt cystetn połączonych wiązaniem disiarczkowym:
I
"OOC—CH—CHZ—S—S—CH2—CH—COO"
NH +
Tabela 4-4. Przykłady oc-aminokwasów nie występujących w białkach, ale spełniających istotną rolę w metabolizmie ssaków
Nazwa po tuczna i systematyczna
Wzór strukturalny w pH obojętnym
Znaczenie
Homocysteina (kwas 2-amino--4-merkaptoma słowy)
Kwas cysteinosulfinowy {kwas 2-amtno-3-sulfi-nopropionowy)
Homoseryna
(kwas 2-amino-4-hydro-
ksymasłowy)
CH2— CH?—CH—COCf
SH łNH3
H2—CH—COO"
CH2—C
so t^
CHS—CHS OH
Związek pośredni w biosyntezie metiortiny
Związek pośredni w kata-botizmie cysteiny
Związek pośredni w metabolizmie treoniny, aspara-ginianu i metioniny
42 / ROZDZIAŁ 4
cd. tab. 4-4
Nazwa potoczna i systematyczna
Wzór strukturalny w pH obojętnym
Znaczenie
Ornityna
(kwas 2,5-diaminoamy-
lowy)
C H2—CH2—CH2—bi—COO"
Cytrulina
(kwas2-amtno-5-ureidoa-
mylowy)
I
NH,
Związek pośredni w biosyntezie mocznika
Kwas argininobursztyno-wy
■NH CH2~™CHj11 "CHj HN—C—NH-
JW.
"COO—CH2—C—COO*
3—T V—CH,
CH,—
HO
![]()
Dopa {3,4-dihydroksyfe-nyloalanina)
3-Monojodotyrozyna
HO
I
Prekursor meianiny
Prekursor hormonów tarczycy
3,5 - D i j od oty r ozy n a
HO
-CH,
3,5,3'-Trijodotyronina (T3) .
jw.
Tyroksyna (3,5,3',5'-tetra-jodotyronina; T4}
HO
CH,
jw.
AMINOKWASY / 43
Tabela 4-5. Przykłady nie a-aminokwasów ważnych dla metabolizmu ssaków |
||
Nazwa potoczna i systematyczna |
Wzór strukturalny |
Znaczenie |
B-alanina |
CH2—CH2—COO" |
Składnik koenzymu A i witaminy |
(kwas 3-aminoproprionowy) |
[ |
pantoteiny |
|
+ NH3 |
|
Tauryna |
CH2—CH2—SO3" |
Występuje w żółci w połączeniu |
{kwas 2-amtnoerytosu)fonowy) |
1 1 |
z kwasami żółciowymi |
Kwas y-aminomasłowy (GA8A) |
On2—v*r12—^rl2—UUU |
Neuroprzekażnik powstający z |
(kwas 4-aminomasłowy) |
|
glutaminianu w tkance mózgo- |
|
|
wej |
Kwas fi-aminuizumasłowy |
H3N+—CH2—CH—COO" |
Końcowy produkt katabolizmu |
(kwas 2-metylo-3-aminopropio- |
|
pirymidyny, występujący w mo- |
nowy) |
ĆH3 |
czu niektórych osób |
WŁAŚCIWOŚCI POSZCZEGÓLNYCH AMINOKWASÓW ZALEZĄ ÓD ICH GRUP R PRZY ATOMIE WĘGLA a
Glicyna, najmniejszy aminokwas, może „dopasować się" łatwo w obszary trójwymiarowej struktury białek, niedostępne dla innych aminokwasów i występuje w obszarach, w których łańcuchy peptydowe ulegają silnemu zwinięciu.
Alifatyczne grupy R jłaniny, waliny, leucyny jjzoleucyny oraz aromatyczne grupy R fenylo-ajaniny, tyrozyny i tryptofanu są hydrofobowe i ta właściwość ma ważne znaczenie w uporządkowaniu cząsteczek wody w białkach, w bezpośrednim sąsiedztwie tych grup. Te aminokwasy są charakterystyczne przede wszystkim dla wnętrza łańcuchów białek cytozolowych.
Naładowane grupy R aminokwasów zasadowych odgrywają kluczową rolę w utrzymywaniu swoistej konformacji białka przez tworzenie wiązań typu soli. Przerwanie i odtworzenie wiązań typu soli towarzyszy utlenowaniu i od-tlenowaniu hemoglobiny {p. rozdz. 7). Ponadto aminokwasy z dodatnio lub ujemnie naładowanymi grupami R uczestniczą w układach „przekazywania ładunku" (ang, „charge relay"). które przesyłają ładunki na znaczne odległości podczas katalizy enzymatycznej. Histydyna ponadto pełni wyjątkową i ważną funkcję w katalizie enzymatycznej, gdyż wartość pK jej formy protonowej układu imidazolowego dopuszcza w pH 7,0 działanie jej jako katalizatora zasadowego lub kwasowego.
Pierwszorzędowa grupa alkoholowa seryny i pierwszo rzędowa grupa tioalkoholowa (—SH) cysteiny są doskonałymi czynnikami nukleofi-lowymi, których funkcja jest ważna dla katalizy. Z kolei drugorzędowa grupa alkoholowa treoniny jest dobrym czynnikiem nukleofiio-wym, aczkolwiek nie jest znany jej udział w katalizie. Dodatkowo, poza katalityczną rolą grupy —OH w przypadku seryny i tyrozyny, odgrywa ona rolę w regulacji aktywności niektórych enzymów, których aktywność katalityczna zależy od stanu ufosforylowania reszt serylowych lub tyrozylowych.
6-1
5"
Tryptofan
L
u
I
<3
I |3H
1-
<H
Fenyioalanlna
"r- 1 ^ 1 1 r
240 260 280
DłusjoSć fali [nm|
Ryc. 4-5. Widma absorpcyjne tryptofanu, tyrozyny
i fenyloalaniny.
44 / ROZDZIAŁ 4
Aminokwasy nie absorbują światła widzialnego (tj. są one bezbarwne) i, z wyjątkiem aminokwasów aromatycznych: tryptofanu, tyrozyny, fenyloalaniny i histydyny, nie absorbują światła nadfioletowego o długości powyżej 240 nm. Jak przedstawiono na ryc. 4-5 większość światła nadfioletowego o długości fali powyżej 240 nm, absorbowanego przez białko, pochłania zawarty w nim tryptofan.
Ryc. 4-7. Fluorescamma.
REAKCJA Z NINHYDRYNĄ LUB FLUORESCAMINĄ SŁUŻY DO WYKRYWANIA AMINOKWASÓW
Ninhydryna (ryc. 4-6). Powoduje oksydacyjną dekarboksylację aminokwasów, w wyniku czego powstają COZ, NH3 i atdehyd uboższy o jeden atom węgla w porównaniu z macierzystym aminokwasem. Zredukowana ninhydryna reaguje wtedy z uwolnionym amoniakiem, tworząc niebieski kompleks, który absorbuje światło o długości 570 nm. Natężenie barwy niebieskiej powstałego kompleksu stanowi podstawę ilościowej metody oznaczania aminokwasów, za pomocą której można wykryć mikrogramowe ilości aminokwasów. Aminy, inne niż a-amino-kwasy, także reagują z ninhydryna, tworząc barwny niebieski kompleks, ale bez tworzenia CO2.
Powstawanie CO? wskazuje zatem na obecność a-aminokwasów. Peptydy i NH$ reagują także z ninhydryna, lecz znacznie wolniej niż a-aminokwasy.
Pro lina i 4-hydroksyprolina tworzą z ninhydryna kompleks o barwie żółtej.
RÓŻNORODNOŚĆ TECHNIK ROZDZIAŁU AMINOKWASÓW
Chromatografia
We wszystkich rodzajach chromatografii cząsteczki rozdzielają się między fazę stacjonarną a ruchomą (tab. 4-6). Rozdział zależy od względnej tendencji cząsteczek mieszaniny do ich silniejszego wiązania się z jedną z tych faz. Chociaż przedstawione techniki rozdziału dotyczą głównie aminokwasów, ich zastosowanie nie ogranicza się tylko do tych cząsteczek.
Tabela 4-6. Relacje zachodzące między fazami w chromatografii
Typ chromatografii |
Faza stacjonarna |
Faza ruchoma |
Chromatografia podziałowa na stałych sorbentach; filtracja żelowa Chromatografia jonowymienna Chromatografia podziałowa między cienką warstwą ciekłego złoża i przepływającym gazem |
Ciekła Stała Ciekła |
Ciekła Ciekła Gazowa |
Ryc. 4-6. Ninhydryna.
Fluorescamina (ryc. 4-7). Jest czulszym związkiem, za pomocą którego można wykryć nano-gramowe ilości aminokwasów. Podobnie jak ninhydryna, fluorescamina tworzy kompleks z aminami innymi niż a-aminokwasy.
Chromatografia bibułowa
Chromatografia bibułowa, pomimo wypierania jej przez bardziej wyrafinowane metody, wciąż znajduje zastosowanie w rozdziałach aminokwasów. Próbki aminokwasów nanosi się na pasek bibuły, w określonym punkcie, w odległości 5 cm od jego końca. Następnie pasek zawiesza się w szczelnym naczyniu (komorze) zawierającym rozpuszczalnik (ryc, 4-8).
AMINOKWASY / 45
TnT
Kierunek
Pasek iblbutyj
wędrówki
rozpuszczalnika
1 Naczynia 2 rozpuszczalnikiem
Ryc. 4-8. Chromatografia bibułowa zstępująca (przekrój komory).
_ Start Cys
His Arg Ser
Glu
Ala'
Tvr
Met
Rozpuszczalniki stosowane do rozdzielania aminokwasów są mieszaninami wody, alkoholi, kwasów lub zasad. Bardziej polarne składniki rozpuszczalnika wiążą się z celulozą i stanowią fazę stacjonarną, zaś mniej polarne składniki — fazę ruchomą. Tak jest w klasycznej chromatografii podziałowej. W chromatografii podziałowej w odwróconej fazie polarności fazy ruchomej i stacjonarnej są odwrócone (np. przez
Kierunek |
* |
|
wędrówki |
|
■ |
rozpuszczalnika |
|
|
Lys Asp Glv |
• |
1 |
Thr |
|
|
|
|
1 |
Pro |
|
|
Val |
i |
i |
Trp |
|
|
Phe Leu |
|
# |
Ryc. 4-9. identyfikacja aminokwasów występują
cych w białkach. Po rozdziale mieszaniny amino
kwasów techniką chromatografii bibułowej zstępu
jącej w układzie n- butanol -kwas octowy-woda
plamy aminokwasów wywołano za pomocą nin-
hydryny.
pierwotne zanurzenie bibuły w roztworze silikonu). Chromatografię podziałową w odwróconej fazie stosuje się do rozdziału niepolarnych peptydów lub lipidów, niepolarnych związków, takich jak niektóre aminokwasy. Wyróżnia się 2 typy technik stosowanych do rozwijania chro-mato gram u; chromatografię wstępującą i zstępującą, tj. odpowiednio gdy rozpuszczalnik wędruje przez bibułę z naniesioną próbą w górę lub w dół. Kiedy rozpuszczalnik zbliży się prawie do końca bibuły, wtedy wyjmuje się ją, suszy i przeprowadza reakcję identyfikacji badanych związków (w przypadku aminokwasów przeprowadza się reakcję z 0,5% roztworem ninhydryny w acetonie, po czym ogrzewa przez kilka minut w temp. 90—110°C. Aminokwasy z dużymi niepolarnymi łańcuchami bocznymi (Leu, Ile, Phe, Trp, Val, Met, Tyr) wędrują dalej niż te z krótszymi niepolarnymi łańcuchami bocznymi (Pro, Ala, Gly) lub z polarnymi łańcuchami bocznymi (Thr, Glu, Ser, Arg, Asp, His, Lys, Cys) (p. ryc. 4-9). Odzwierciedla to większą względną rozpuszczalność cząsteczek polarnych w hydrofilowej fazie stacjonarnej, zaś cząsteczek niepolarnych w rozpuszczalnikach organicznych. W grupie cząsteczek niepolarnych (Gly. Ala, Val, Leu) zwiększeniu długości niepolarnego łańcucha bocznego towarzyszy zwiększenie ruchliwości tych cząsteczek.
Stosunek odległości przebytej przez aminokwas do odległości przebytej przez czoło rozpuszczalnika, mierzonych od miejsca naniesienia mieszaniny aminokwasów, opisuje się jako wartość Rf tego aminokwasu (względną ruchliwość). Wartości Rf dla danego aminokwasu zmieniają się w zależności od warunków doświadczalnych, np. od stosowanego rozpuszczalnika. Chociaż możliwa jest próbna identyfikacja aminokwasu za pomocą samej wartości Rf, bardziej wskazane jest rozdzielenie chromatograficzne nieznanej mieszaniny aminokwasów równocześnie ze znanymi aminokwasami wzorcowymi. Wobec tego ruchliwość może być porównana do ruchliwości wzorców (np. jako RAia raczej niż jako Rf). Ruchliwości, przedstawione jako względne wobec standardowych, różnią się mniej niż wartości Rf z kolejnych doświadczeń.
Zawartość aminokwasów można oznaczyć ilościowo, wycinając każdą plamę i eluując odpowiednim rozpuszczalnikiem, a następnie przeprowadzając analizę kolorymetryczną (z ninhydryną). W innym rozwiązaniu bibułę
46 / ROZDZIAŁ 4
spryskuje się ninhydryną, a natężenie barwy wyeluowanych plam mierzy się w fotometrze z zapisem natężenia światła przepuszczanego lub odbitego.
W innej technice — chromatografii bibułowej dwuwymiarowej, próbkę nanosi się w jednym rogu kwadratowego arkusza bibuły i rozdziela w odpowiednio dobranej mieszaninie rozpuszczalników. Po wysuszeniu i usunięciu rozpuszczalnika arkusz obraca się o 90°C i rozdziela w innej mieszaninie rozpuszczalników (ryc. 4-10).
Butanol-kwas octowy-woda (4:1:5).
Ryc. 4-10. Dwuwymiarowa chromatografia aminokwasów (przerysowano, nieznacznie zmodyfikowano z: Levy A. J. i Chung D.: Two-dimensional chi-omatographyof aminoacidsonbuffered papers. Aria!. Chem. 1953; Copyright ©1953 by American Chemical Society; zamieszczono za zgodą).
— ATLC), nie przypomina chromatografii bibułowej i opiera się na innych zasadach. W tej technice rozpuszczalnik (który nie musi być dwuskładnikowy lub bardziej złożony) eluuje składniki próby z zaadsorbowanych miejsc na aktywowanym sorbencie, takim jak prażony żel krzemionkowy. Metoda ATLC znajduje zastosowanie do rozdziału związków niepolarnych, takich jak lipidy, i stąd nie jest przydatna dla części aminokwasów i większości peptydów.
Chromatografia jonowymienna
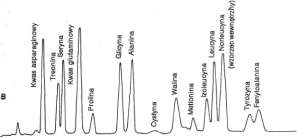
Całkowita analiza reszt aminokwasowych po hydrolizie łańcucha peptydowego wymaga na ogół automatycznej chromatografii jonowymiennej. Pełen rozdział, identyfikacja i oznaczenie ilościowe hydrolizatu białkowego nie trwa dłużej niż 3 h. W metodzie Moore'a i Steina stosuje się kolumny (krótką i długą) zawierające formę Na+ sulfonowanej żywicy polistyrenowej. Kiedy kwaśny hydrolizat w pH 2,0 zostanie naniesiony na kolumny, aminokwasy wiążą się z jonem Na+ wymieniacza kationowego. Następnie kolumny eluuje się cytrynianem sodu w zaprogramowanych warunkach pH i temperatury. Krótką kolumnę eluuje się jednym buforem, zaś długą — dwoma, W eluatach z kolumn wywołuje się reakcję z odczynnikiem ninhydrynowym, a odczyty natężenia barwy przeprowadza się w kolorymetrze przepływowym. Dane są rejestrowane na katodowej lampie obrazowej ze sprzężonym komputerowym odczytem integracji powierzchni pól rozdzielonych aminokwasów (ryc. 4-11).
570 nm
Chromatografia cienkowarstwowa
Wyróżnia się 2 odrębne klasy chromatografii cienkowarstwowej (ang. thin-layer chromato-graphy; skrót — TLC). Pierwsza — chromatografia cienkowarstwowa podziałowa (ang. par-tition thin-layer chromatography: skrót — PTLC), w pełni przypomina chromatografię bibułową podziałową. W metodzie PTLC wykorzystuje się te same układy rozpuszczalników i sposoby identyfikacji badanych związków, co w chromatografii bibułowej. Natomiast bibułę zastępuje płytka pokryta cienką warstwą sproszkowanej celulozy lub innego względnie obojętnego złoża. Możliwy jest również wariant PTLC w odwróconej fazie.
Z kolei druga klasa — adsorpcyjna TLC (adsorption thin-layer chromatography; skrót
Ryc. 4-11A
AMINOKWASY / 47
570 nm
-pH 3,25-
-pH 4,25-
55 °C
c. 4-11. Automatyczna analiza kwaśnego hydrolizatu białkowego na kolumnach Dowex 50 Moore'a iSteina (w temp. 55°C). A; Do rozdziału aminokwasów zasadowych użyto krótkiej kolumny (5,0x0,9 cm), stosując elucję roztworem o pH 5,28; potrzebny czas = 60 min. B: Do rozdziału aminokwasów obojętnych i kwaśnych użyto dłuższej kolumny (55 x 0,9 cm), stosując elucje najpierw buforem o pH 3,25, a potem buforem o pH 4,25. Norleucynę użyto jako substancję wzorcową {tzw. wzorzec wewnętrzny). Aminokwasy zasadowe pozostają związane na kolumnie; potrzebny czas =180 min. Eluowane próbki reagują z odczynnikiem ninhydrynowym, a pomiar ich gęstości optycznej jest dokonywany przy długości fal światła 570 nm i 440 nm. Przy 440 nm wykrywa się jedynie prolinę i hydroksyprolinę. Oś rzędnych — gęstość optyczna w skali logarytmicznej: oś odciętych — czas w min (reprodukowano za zgodą: Prof. ET. Mert, Pardue University).
Elektroforeza wysokonapięciowa
Elektroforeza wysokonapięciowa (ang. high--voltage electrophoresis; skrót — HVE) rozdzielająca aminokwasy, polipeptydy i inne am-folity (cząsteczki, których ładunek całkowity zależy od pH otaczającego środowiska) w polu prądu stałego ma wiele zastosowań w biochemii. W analizie aminokwasów najczęściej używanymi nośnikami są arkusze bibuły albo cienkowarstwowe płytki powleczone sproszkowaną celulozą. W rozdziałach wielkocząsteczkowych polipeptydów i białek używa się usieciowanego żelu poliakryloamidowego. W analizie oligo-merów nukleotydowych wykorzystuje się głównie 2 złoża — agarozę i żel poliakryloamidowy.
Rozdział amfolitów, umieszczonych w polu elektrycznym o napięciu 2000—5000 V w ciągu 0,5—2 h, zależy od ich ładunku elektrycznego i masy cząsteczkowej, W przypadku cząsteczek obdarzonych jednakowym ładunkiem elektrycznym, szybciej będą wędrować cząsteczki o mniejszej masie. Ładunek elektryczny jest jednak ważniejszym czynnikiem w określeniu stopnia osiągniętego rozdziału. Elektroforeza HVE znajduje zastosowanie w analizie aminokwasów, małocząsteczkowych polipeptydów,
niektórych białek, nukleotydów i fosfocukrów. Próbki nanosi się na nośnik zwilżony buforem o odpowiednim pH i łączy się ten nośnik ze zbiornikami buforu za pomocą bibułowych łączników, a cały układ z bibułą można przykryć płytą szklaną lub zanurzyć w chłodziwie węglowodorowym. Po podłączeniu prądu cząsteczki obdarzone ładunkiem dodatnim wędrują w roztworze o wybranym pH w kierunku katody, a cząsteczki z ładunkiem ujemnym — w kierunku anody. W celu zidentyfikowania rozdzielonej próby elektroforegram wybarwia się odczynnikiem ninhydrynowym (aminokwasy, peptydy), bromkiem etydyny i ogląda w świetle lampy UV (oligomery nukleotydowe) itd. Wybór pH „narzucają" wartości pK grup zjonizo-wanych cząsteczek w mieszaninie.
NAJWAŻNIEJSZĄ REAKCJĄ AMINOKWASÓW JEST TWORZENIE WIĄZANIA PEPTYDOWEGO
W zasadzie tworzeniu wiązania peptydowego towarzyszy odłączenie 1 mola wody, powstającego z grupy a-aminowej jednego aminokwasu
48 / ROZDZIAŁ 4
Alanina
— NH
OH + H
O
Walina
HSO
hydrolizy wiązania peptydowego. Aby przeprowadzić biosyntezę wiązania peptydowego między dwoma aminokwasami, najpierw musi ulec aktywacji grupa karboksylowa. Chemicznie grupę karboksylowa można przekształcić w chlorek kwasowy. W przyrodzie, aktywację zapoczątkowuje kondensacja z ATP (p. rozdz. 30).
CHpeptyd; alanyiowaltna (Ala-Val)
Ryc. 4-12. Aminokwasy połączone wiązaniem peptydowym (obszar zaciemniony).
i grupy oc-karboksylowej drugiego aminokwasu (ryc. 4-12). Reakcja ta nie przebiega jednak tak, jak przedstawiono na tej rycinie, gdyż stała równowagi reakcji jest przesunięta w kierunku
PIŚMIENNICTWO
Barrett GC: Chemistry and Biochemistry ofthe Amino
Acids. Chapman & Hal], 1985. Davies JS: Amino Acids and Peptides. Chapman
& Hali. 1985. Gehrke CW, Kuo KCT, Zumwalt RW, Kenneth CT:
Amino Acid Analysis by Gas Chromatography.
3 vols. CRC Press, 1987. Hancock WS: Handbook ofHPLCfor the Separation
of Amino Acids, Peptides. and Proteins. 2vols. CRC
Press, 1984. Hugli TE: Techniaues in Protein Chemistry. Academic
Press, 1989. Rattenbury JM: Amino Acid Analysis. Halstead Press,
1981.
Peptydy
5
Victor W. Rodweli, PhD
WPROWADZENIE
Po połączeniu grup aminowych i karbo-ksylowych aminokwasów z utworzeniem wiązań peptydowych, składowe ami no kwasowe określa się resztami aminokwasowymi. Peptyd składa się z dwóch lufo więcej reszt aminokwaso-wych połączonych wiązaniami peptydowymi. Peptydy zawierające więcej niż 10 reszt amino-kwasowych nazywa się polipeptydarni,
ZNACZENIE BIOMEDYCZNE
Peptydy są związkami budzącymi szerokie zainteresowanie, szczególnie w endokrynologii. Wiele hormonów jest peptydami i mogą być podawane chorym w celu korygowania ich niedoborów (np. wstrzyknięcia insuliny chorym na cukrzycę). Pewne antybiotyki są peptydami (np. walinomycyna i gramicydyna A), a nieliczne z nich substancjami przeciwnowotworowymi (np. bleomycyna). Szybka synteza chemiczna i technologia rekombinacji DNA ułatwiły produkcję znacznych ilości hormonów peptydowych. Wiele z nich występuje w organizmie w stosunkowo małych stężeniach, co utrudnia możliwość ich wydzielenia w ilościach wystarczających do leczenia. Ta sama technologia pozwala syntetyzować inne peptydy, dostępne z naturalnych źródeł w niezwykle małych ilościach (np. niektóre peptydy i białka wirusowe), wykorzystywane do produkcji szczepionek.
PEPTYDY TWORZĄ SIĘ Z L-k-AMINOKWASÓW POŁĄCZONYCH WIĄZANIAMI PEPTYDOWYMI
Na rycinie 5-1 przedstawiono tripeptyd utworzony z reszt aminokwasowych alaniny, cys-teiny i waliny. Należy odnotować, że tripeptyd składa się z 3 reszt aminokwasów ych. ale nie zawiera 3 wiązań peptydowych. Umownie strukturę peptydu zapisuje się rozpoczynając od lewej strony N-końcową resztą aminokwasów ą (z wolną grupą a-aminową), a kończąc po prawej stronie C-końcową resztą aminokwasową (z wolną grupą a-karboksylową). Przedstawiony peptyd ma jedną wolną grupę a-aminową oraz 1 wolną grupę a-karboksylową. Jednakże w niektórych pepiydach końcowe grupy aminowe lub karboksylowe mogą być podstawione (np. N-formyloamina lub amid grupy karboksylo-wej) i w ten sposób nie są wolne.
Alanyb- cysteinylo- walina
Ryc. 5-1. Wzór strukturalny tripeptydu {wiązania peptydowe zaciemniono).
4 — Biochemia
Prosta metoda przedstawiania wzorów strukturalnych peptydów
Aby w prosty sposóbzapisać wzór strukturalny peptydu, w pierwszej kolejności rysuje się jego „szkielet" z połączonych grup oc-NHU
50 / ROZDZIAŁ 5
i a-COOH oraz atomów węgla a. Następnie wpisuje się w odpowiednie miejsca łańcucha atomy węgla a (patrz niżej). Należy więc:
1. Narysować szkielet (zyg-zag) i dodać N-końcową grupę aminokwasową:
2. Dopisać atomy węgla a, grupy a-karbok-sylowe i a-aminowe:
G!u-A!a-Lys-Gly-Tyr-Ala E A K G Y A
Ryc. 5-2. Przykład zapisu jednoliterowymi i trój-I(terowymi symbolami reszt aminokwasowych przedstawiający pierwszorzędową strukturę heksa-peptydu, zawierającego, jako N-końcowy aminokwas, kwas glutaminowy (Glu, E) oraz jako C-końcowy — alaninę {Ala, A).
COO-
+H3N
3. Dopisać właściwe grupy R i atomy wodoru a do atomów węgla a:
SH
0 H CH,
H
00
II V -
H \ H
O H
CH,
CH,
Kolejność aminokwasów określa strukturę pierwszorzędową peptydu
Pierwszorzędową strukturę peptydu określa liczba, budowa chemiczna i kolejność (sekwen-
Symboli trój- i jednoliterowych używa się w zapisie aminokwasów w peptydach
Ponieważ polipeptydy (białka) mogą zawierać 100 i więcej reszt aminokwasowych, w celu przedstawienia ich struktury pierwszorzędowej (ryc. 5-2) stosuje się symbole albo trój- albo jednoliterowe (tab. 4-3, kolumna 2).
Glu-Lys-(A!a,Gly,Tyr)-His-Ala
Ryc. 5-3. Przykład heptapeptydu, zawierającego fragment o niepewnej strukturze pierwszorzędowej (w nawiasie).
Zapis struktury pierwszorzędowej za pomocą trójliterowych symboli, połączonych kreskami reprezentującymi wiązania peptydowe, jest zrozumiały i jednoznaczny. Kreski zwykle pomija się w zapisie sekwencji peptydów przy użyciu symboli jednoliterowych. W przypadku niepewności w ustaleniu kolejności fragmentu polipep-tydowego, wątpliwą sekwencję reszt aminokwasowych umieszcza się w nawiasie i oddziela przecinkami (ryc. 5-3).
Zmiana w pierwszorzędowej strukturze peptydu może zmieniać jego aktywność biologiczną
Podstawienie pojedynczego aminokwasu przez inny, w liniowej sekwencji polipeptydu o 100 lub więcej resztach aminokwasowych, może zmniejszyć lub znieść jego aktywność biologiczną oraz powodować potencjalnie poważne następstwa (np. niedokrwistość sierpo-wata). Wiele dziedzicznych wad metabolicznych wynika ze zmiany pojedynczego aminokwasu w określonym białku. Wprowadzenie nowych metod badania struktury białek i DNA przyczyniło się do poszerzenia wiedzy o biochemicznych podstawach wielu dziedzicznych chorób metabolicznych.
Peptydy są potielektrolitami, których ładunek zależy od pH otaczającego środowiska
Wiązanie peptydowe (amidowe) nie ma ładunku w żadnym ważnym fizjologicznie pH. Powstawaniu peptydów z aminokwasów w pH
PEPTYDY / 51
7,4 towarzyszy utrata jednego ładunku dodatniego i ujemnego przy utworzeniu jednego wiązania peptydowego. Jednakże peptydy są cząsteczkami obdarzonymi ładunkiem elektrycznym w pH fizjologicznym dzięki ładunkom ich grup C- i N-końcowych oraz grup funkcyjnych występujących w polarnych resztach ami-nokwasowych przyłączonych do atomów węgla a (p. tab. 4-1).
Liczba możliwych konformacji peptydu jest wymuszona siłami niekowalencyjnymi
Polipeptydy mogą wykazywać różną konformację (ułożenie przestrzenne), jednak w roztworze przeważa tendencja do niewielkich zmian konformacyjnych. Konformację pepty-dów utrwalają takie czynniki, jak zawada przestrzenna, oddziaływania kulombowskie, wiązania wodorowe i hydrofobowe (p. rozdz. 6). Tak jak w przypadku białek, do fizjologicznej aktywności polipeptydów jest nieodzowna specyficzna konformacja.
WIELE MAŁO CZĄSTECZKOWYCH PEPTYDÓW WYKAZUJE AKTYWNOŚĆ FIZJOLOGICZNĄ
Komórki zwierząt, roślin i bakterii zawierają różnorodne małocząs tęcz k owe polipeptydy (3—100 reszt aminokwasowych) o istotnej aktywności fizjologicznej. Niektóre z nich, łącznie z większością polipeptydowych hormonów ssaków, zawierają tylko wiązania peptydowe, utworzone między grupami a-aminowymi i 7-karboksylowymi, 20 L-a-arninokwasów występujących w białkach. Jednakże w polipep-tydach mogą również występować dodatkowe, ,,niebiałkowe" aminokwasy, lub też pochodne aminokwasów budujących białka.
Krótki polipeptyd — bradykinina, czy.anik zj32ui£^szaJ3«y.jłapica.e.inięśni^ła<ikicfe — uwalnia się ze swoistych białek osocza przez proteoliżę.
Arg-Pro-Pro-Giy-Phe-Ser Pro-Phe Arg Bradykinirta
Glutation (ryc, 5-4). Jest nietypowym tripep-tydem, w którym N-końcowy kwas glutaminowy wiąże się z cysteiną wiązaniem nie-ot-pep-tydylowym. Jest związkiem niezbędnym do działania niektórych enzymów. Glutation i en-
0
SH CH2
N I H
CHj I
coo-
CHj H-C-NHS
coo-
Ryc. 5-4. Giutation (y-glutamylocysteinyloglicy-na).
zym — reduktaza glutationowa — uczestniczą w powstawaniu prawidłowych wiązań disiarcz-kowych w wielu białkach i hormonach polipeptydowych (p. rozdz. 6).
Antybiotyki jolipeptydowe. Są wytwarzane przez grzyby i zawierają zarówno D- jak ...LLcaminokwasy, a także aminokwasy nie występujące w białkach. Zalicza się do nich np. tyrocydynę i gramicydynę S, cykliczne polipeptydy zawierające D-fenyłoalaninę oraz niebiał-kowy aminokwas — ornitynę. Należy podkreślić, że wymienione polipeptydy nie są syntetyzowane na ry boso mach.
Hormon tyreotropowy (TRH). W tym wariancie peptydowym N-końcowy kwas glutaminowy- ulega, cyklizacji do kwasu piro glutamino we-.gOjjiatomiast N-końcowa grupa karboksylowa jjroliny występuje jako amid (ryc. 5-5).
Ryc. 5-5. Piroglutamylohistydyloprolinoamid (TRH).
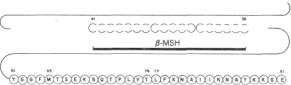
W niektórych przypadkach polipeptydy ssaków w swojej cząsteczce mogą zawierać więcej niż 1 fizjologicznie czynny peptyd, np, (3-lipo-tropina. Ten hormon przysadki pobudza uwalnianie kwasów tłuszczowych z tkanki tłuszczowej. W strukturze pierwszorzędowej pMipo-tropiny występują fragmenty łańcucha wspólne dla innych hormonów peptydowych o odrębnych właściwościach fizjologicznych (ryc. 5-6). Długi polipeptyd jest prekursorem krótszych polipeptydów.
52 / ROZDZIAŁ 5
«DLV»AEK K OrETSXPlTrftYMl €Y HTfYRYW G / S YPYP YKYDl K R
GL TSORLRNGDSPNAG ANOGEGPNALEH S L !_■»
0-Endorflna
...-
y-Endorfina
a-Endorflna
Enkefallna metloninowa
Byc. 5-6. Pierwszorzędowa struktura fMipotropiny. Reszty 41-58 reprezentują hormon pobudzający melanocyty (p-MSH). Reszty 61—91 zawierają sekwencje odpowiadające wskazanym endorfinom.
ZŁOŻONĄ MIESZANINĘ PEPTYDÓW MOŻNA ROZDZIELIĆ PODCZAS ELEKTROFOREZY LUB CHROMATOGRAFII
Chromatografia i elektroforeza wysokonapięciowa (HVE)
Techniki, które jako podstawę rozdziału wykorzystują ładunek elektryczny, znajdują zastosowanie zarówno w badaniach polipepty-dów, jak i aminokwasów (p. rozdz. 4). Wartość pK. C-końcowej grupy karboksylowej polipeptydu jest większa niż grupy a-karboksylowej odpowiedniego aminokwasu (oznacza to, że grupa karboksylowa peptydu jest słabszym kwasem). Odwrotnie, N-końcowa grupa aminowa polipeptydu jest silniejszym kwasem (ma mniejszą wartość pK) niż grupa aminowa aminokwasu, od którego on pochodzi (tab. 5-1).
Tabela 5-1. Wartości pK glicyny i peptydów glicynowych
9.60 8,17 7,91
Gly
Gly-Gly Gly-Gty-Gly
Filtracja żelowa
W metodzie automatycznego sekwencjono-wania wykorzystuje się niewielką liczbę peptydów o długości 30—100 reszt aminokwaso-wych. Jednakże wiek wielkocząsteczkowych polipeptydów może być nierozpuszczalnych, z powodu odsłonięcia w procesie dcnaturacji uprzednio niedostępnych reszt hydrofobowych. Nierozpuszczalnośc polipeptydów można pokonać przez ich rozpuszczenie w moczniku, alkoholach, kwasach organicznych lub zasadach, ale czynniki te ograniczają stosowanie technik chromatografii jonowymiennej w ich oczyszczaniu. Okazuje się, że filtrację żelową dużych hydrofobowych peptydów można przeprowadzić, używając roztworów kwasu mrówkowego lub octowego w dużych stężeniach (1—4 mol/1).
Wysokociśnieniowa chromatografia cieczowa w fazie odwróconej
Skuteczną techniką tv oczyszczaniu wielkocząsteczkowych, niepolarnych peptydów jest wysokociśnieniowa chromatografia cieczowa (ang. high performance liquid chromatography; skrót — HPLC) na niepolamych nośnikach, z wykorzystaniem polarnych rozpuszczalników. Na rycinie 5-7 przedstawiono 'rozdział bromocyjanowych fragmentów ludzkiej globi-ny płodowej za pomocą tej techniki. Połączenie
PEPTYDY / 53
o
sie lub solami srebra, natomiast polinukleo-lydów — bromkiem etydyny. Popularną odmianą elektroforezy jest PAGE w warunkach denaturujących. Białka przed elektroforeza, poddaje się gotowaniu w odpowiednim buforze, a następnie rozdziela w obecności czynników denaturujących — mocznika lub siarczanu do-decylu sodu (SDS). Ujemny ładunek cząsteczek \ CH1 - (CH,), L - SO3") pokrywa białka w stosunku 1 cząsteczka SDS na 2 wiązania pcptydowe. ["o „obładowanie" białek ładunkami czyni je silnie ujemnymi (anionami). Późniejszy rozdział białek jest oparty na wielkości ich masy cząsteczkowej. Metoda SDS-PAGE jest szeroko stosowana do określania masy cząsteczkowej białek przez porównanie ich ruchliwości elekt-roforetycznej z ruchliwością białek wzorcowych o znanej masie cząsteczkowej.
Ryc. 5-7. Profil elucyjny rozdziału bromocyjano-wych fragmentów ludzkiej globiny pfodowej, uzyskanej metodą HPLC w odwróconej fazie. Przedstawiono oznaczenia dowolnie numerowanych fragmentów ol i 7 łańcuchów (reprodukowano dzięki uprzejmości: J.D. Pearson i wsp.r Deparrment of Biochemistry, Pardue Ur>iversity).
filtracji żelowej i HPLC w odwróconej fazie stosuje się do oczyszczania złożonych mieszanin peptydów, otrzymanych w wyniku częściowego trawienia białek.
Wysokonapięciowa elektroforeza (HVE) na sitach molekularnych
Sączenie molekularne, w połączeniu z rozdziałem związków i wykorzystujące ładunek, ułatwia ich rozdział. Oprócz skrobi i agarozy, najczęściej używanym nośnikiem jest usiedowa-ny polimer akryloamidu (CH2 = CHCONH,,). Podczas elektroforezy w żelu poliakryloamido-wym (ang. polyacrylamide gel electrophoresis; skrót — PAGE) roztwór białka nanosi się na spolimeryzowany akryloamid, w odpowiednim buforze, uformowany w postaci płytki lub w ru-■rkach. Czynnikiem sieciującym złoże poliak-ryloamidu może być 2—10% roztwór metyłe-no-tó-akryloamidu (CH2 = CONH)? - CH2 lub podobne związki sieciujące. Rozdział naniesionych prób, w odpowiednim buforze, dokonuje się pod wpływem prądu elektrycznego. Identyfikację poiipeptydów po rozdziale elektro foretycznym przeprowadza się za pomocą barwienia żeli błękitem brylantowym Coomas-
PIERWSZY ETAP W OKREŚLANIU STRUKTURY PIERWSZORZĘDOWEJ PEPTYDU TO POZNANIE JEGO SKŁADU AMIN0KWASOWEG0
W analizie struktury peptydów najpierw przeprowadza się hydrolizę wiązań peptydo-wych łączących aminokwasy. Ponieważ wiązania te są trwałe w roztworach o obojętnym pH, przeprowadza się hydrolizę kwaśną lub zasadową. Kataliza enzymatyczna jest stosunkowo mało przydatna do przeprowadzenia pełnej hydrolizy wiązań peptydowych. Każdy typ hydrolizy białek przebiega z utratą pewnych reszt aminokwasowych. Metodą z wyboru jest hydroliza w zatopionych, odpowietrzonych probówkach w 6 mol/l roztworze HC1 w temp. HO^C. Podczas takiej hydrolizy wszystkie reszty tryptofanu i cysteiny oraz większość reszt cystynowych ulegają zniszczeniu. W obecności jonów metali dochodzi do częściowej utraty metioniny i tyrozyny. Glutamina i asparagina ulegają deamidacji do kwasów: glutaminowego i asparaginowego. Odzysk seryny i treoniny jest niepełny, a ilość tych aminokwasów zmniejsza się w miarę przedłużania czasu hydrolizy. Niektóre wiązania między resztami aminokwasów obojętnych (Val-Val, Ile-Ile, Val-Ile, Iłe-Val) ulegają rozbiciu tylko w ok. 50% po 20 h hydrolizy. Zwykle przeprowadza się hydrolizę próbek (w powtórzeniach) przez 24, 48, 72 i 96 h, ekstrapolując potem zawartość seryny i treoniny do czasu zerowego. Zawartość waliny i izo-ieucyny oznacza się po 96 h hydrolizy. Amino-
5* / ROZDZIAŁ 5
kwasy dikarboksylowe i ich amidy oznacza się razem i przedstawia łącznie jako „Glx" i „Asx". Cysteina i cystyna przechodzą w trwałą w środowisku kwaśnym pochodną (np. kwas cysternowy) przed hydrolizą. Hydroliza zasadowa, w czasie której ulegają zniszczeniu seryna, treo-nina, arginina i cysteina, a wszystkie aminokwasy ulegają racemizacji, jest stosowana do oznaczania tryptofanu. Po hydrolizie prób ich skład aminokwasowy może być oznaczony podczas automatycznej chromatografii jonowymiennej (p. ryc. 4-11) lub HPLC.
Sekwencjonowanie pierwszego białka przeprowadził Fred Sanger, za pomocą odczynnika mającego od tej pory jego nazwisko
-N-CH -COOH
CH-COOH
Minęło już ponad 30 lat od określenia przez Sangera pełnej sekwencji aminokwasowej hormonu polipeptydowego — insuliny. W metodzie Sangera najpierw rozdzielono insulinę na 2 łańcuchy polipeptydowe A i B, po czym poddano swoistemu trawieniu enzymatycznemu do krótkich peptydów, zawierających odcinki sekwencji zachodzących. Następnie zastosowano związek — i-fluoro-2,4-dinitroben-zen (ryc. 5-8), tworzący pochodne tylko
Ryc. 5-8. Reakcja aminokwasu z 1 -fluoro-2,4--dinitrobenzenem (odczynnikiem Sangera). Odczynniki nazwano od nazwiska laureata nagrody Nobla (1958), biochemika Fryderyka Sangera, który zastnsowal go w celu oznaczenia pierwszo-rzędowej struktury insuliny. Odczynnik ilościowo aryluje wszystkie wolne grupy aminowe, dając intensywnie żółto zabarwione 2,4-dinitrofenyloa-minokwasy. Pochodne te łatwo się oznacza ilościowo za pomocą spektrototometru. Oprócz reszt N-końcowych, fluorod(nitrobenzen reaguje także 'z grupami; e-aminową lizyny, imidazolową his-tydyny, hydroksyl ową tyrozyny i grupą SH cysteiny. Ponieważ grupa dinitrofenylowa nie jesi usuwana podczas kwaśnej hydrolizy, stosuje sieją do analizy N-końcowego aminokwasu polipeptydów.
z N-końcowymi resztami aminokwasowymi peptydów, które po hydrolizie usuwano i identyfikowano. Porównanie sekwencji zachodzących pozwoliło Sartgerowi jednoznacznie wyde-dukować sekwencje obydwu łańcuchów insuliny. Pomimo że metoda Sangera jest wciąż stosowana, 2 inne techniki zrewolucjonizowały oznaczanie struktury pierwszorzędowej polipeptydów (białek). Pierwsza z nich, wprowadzona w 1967 r., to metoda automatycznego usuwania i identyfikowania N-końcowych reszt aminokwa-sowych peptydów, jako ich pochodnych fenylotio-hydantoinowych (degradacja Edmana). Druga — wprowadzona niezależnie przez Sangera oraz Maxama i Gilberta — polega na szybkim sekwencjonowaniu DNA genów kodujących poszukiwane białko. Obecnie optymalną strategią jest stosowanie obydwu równocześnie. Technika automatycznej degradacji Edmana, choć szybsza w analizie sekwencji peptydów od metod Sangera, napotyka trudności i dostarcza wolniej wyników niż metoda sekwencjonowa-nia DNA. Technika sekwencjo no wania DNA nie dostarcza jednak nieomylnych, jednoznacznych wyników dotyczących pierwszorzędowej struktury białek. Najpoważniejsze trudności w sekwencjonowaniu genów eukariotycznych są związane z obecnością w ich obrębie in-tronów — sekwencji nukleotydowych nie ulegających ekspresji w dojrzałym białku. Pomimo to, główna przewaga tej metody polega na stosunkowo łatwej detekcji i sekwencjonowaniu regionów prekursorowych cząsteczek, które mogą „umknąć" wykryciu w technice degradacji Edmana (wskutek dojrzewania przed jego wydzieleniem). Metody sekwencjonowania DNA i degradacji Edmana należy traktować jako uzupełniające się. Zrewolucjonizowały one i daleko posunęły naszą wiedzę o pierwszorzędowej strukturze białek.
Oznaczanie pierwszorzędowej struktury polipeptydów za pomocą techniki automatycznej degradacji Edmana
Ze względu na fakt, że wiele białek składa się z więcej niż jednego łańcucha polipeptydowego, połączonych wiązaniami niekowalencyjnymi lub przez mostki disiarczkowe, pierwszy etap stanowi dysocjacja i rozdzielenie indywidualnych łańcuchów poiipeptydowych. Czynniki denaturujące (mocznik, chlorowodorek guani-dyny) rozbijają wiązania wodorowe i dysoc-jują niekowalencyjnie połączone polipeptydy,
PEPTYDY / 55
a czynniki utleniające i redukujące rozrywają mostki disiarczkowe (ryc. 5-9). Następnie pep-tydy rozdziela się metodami chromatograficznymi.
Wielkocząsteczkowe polipeptydy muszą ulec rozszczepieniu do peptydów o długości nadającej się do automatycznego sekwencjonowania
Aparaty służące do automatycznego sekwencjonowania (sekwentatory) analizują skutecznie polipeptydy o długości 20—60 reszt amino-kwasowych. Ten fakt wpływa na wybór techniki rozcinania polipeptydów oraz oczyszczania otrzymanych fragmentów. Zmierza się więc do uzyskania niewielkiej liczby dłuższych fragmentów (o długości 30—100 reszt aminokwaso-wych). Korzystne jest zatem bardzo swoiste i pełne rozszczepienie łańcucha polipeptydowe-go w określonych miejscach. Takie wymagania spełnia rozszczepienie za pomocą bromocyjanu (CNBr), trypsyny lub o-jodozobenzenu.
=O
O =
CNBr. Reszty cysteiny są najpierw modyfikowane przez kwas jodooctowy. CNBr rozcina
Ryc, 5-9. Rozszczepienie łańcuchów poiipepty-dowych połączonych wiązaniami disiarczkowymi (pole zaciemnione) za pomocą czynników utleniających (kwas nadmrówkowy, strona lewa) bądź redukujących (2-merkaptoetanol; strona prawa) prowadzące do utworzenia 2 peptydów — odpowiednio z resztą kwasu cysteinowego lub resztą cysteiny Iową.
wówczas łańcuch polipeptydowy (w większości ilościowo) tylko w miejscach występowania reszt metioninowych, po ich stronie karbok-sylowej. Ponieważ metionina występuje rzadko w polipeptydach, takie rozszczepienie powoduje powstawanie fragmentów peptyd owych o właściwej długości.
Trypsyna. Trypsyna nacina łańcuchy poh-peptydowe po karboksylowej stronie reszt lizy-ny i argininy. Jeśli reszty lizyny najpierw przeprowadzi się w pochodne za pomocą bezwodnika kwasu cytrakonowego (reakcja odwracalna), w celu zmiany ładunku reszt hzynowych z dodatniego na ujemny, to trypsyna rozszczepia tylko reszty argininowe. Wykorzystanie pochodnych reszt argininowych jest mało użyteczne z powodu stosunkowo dużego „nadmiaru" reszt hzynowych w białkach. Jednakże jest przydatne do kolejnego rozszczepiania CNBr fragmentów.
o- Jodozobenzen. Związek len rozszczepia swoiście i ilościowo miejsca względnie rzadko występujących reszt: Trp-X. Nie wymaga wcześniejszej osłony innych reszt aminokwasowych.
Hydroksyloamina. Hydroksyloamina rozrywa względnie rzadko wiązania: Asn-Gly, chociaż nie w sposób ilościowy.
Proteaza V8. Enzym ze Staphylococcus aureus rozcina peptyd przy resztach Glu-X, z wyraźną wybiórczością w stosunku do reszty X o charakterze hydrofobowym. Wiązanie Glu--Lys nie ulega rozszczepieniu przez tę proteazę. Ta reakcja jest wykorzystywana do kolejnej degradacji CNBr fragmentów.
Łagodna kwaśna hydroliza. W toku takiej hydrolizy ulega rozerwaniu rzadko spotykane wiązanie Asp-Pro. 2 lub 3 procesy trawienia pierwotnego polipeptydu, przy resztach Met, Trp, Arg i Asn-Gly, połączone z odpowiednio nadtrawionymi powstałymi fragmentami, zazwyczaj pozwalają określić pełną sekwencję polipeptydu. Oprócz wyjątkowych trudności w oczyszczaniu fragmentów peptydo-wych, oznaczenie pierwszorzędowej struktury polipeptydu można wykonać, dysponując zaledwie kilkoma jego mikromolami.
Mieszanina peptydów, otrzymana z rozszczepienia polipeptydu, musi być rozdzielona do homogennych peptydów przed ich sek we ncjo no wan i em
Oczyszczanie fragmentów peptydowych przeprowadza się głównie przez ich filtrację
56 / ROZDZIAŁ 5
żelową w kwasie octowym lub mrówkowym, wykorzystując technikę HPLC w odwróconej fazie (ryc. 5-7), albo w toku chromatografii jonowymiennej na fosfocelulozie lub na złożu sulfofenyło-Sephadex w roztworach kwasu ortofosforowego.
Sekwencjonowanie z zastosowaniem odczynnika i reakcji Edmana
Automatyczna analiza sekwencji aminokwasów wykorzystuje fenyloizotiocyjanian (odczynnik Edmana) i reakcje, w wyniku których usuwana jest N-końcowa grupa aminowa pep-tydu, jako pochodna fenylotiohydantoinowa (reakcja Edmana, ryc. 5-10). Metoda tzw. degradacji Edmana polega na kolejnym odłączaniu reszt am i no kwasowych od aminowego końca peptydu. Główną część reaktora stanowi obracające się naczynie szklane, zapewniające przebieg reakcji na ścianie naczynia w postaci cienkiej warstwy. To ułatwia ekstrakcję i kolejne usuwanie rozpuszczalników. W pełni zautomatyzowanym aparacie można zsekwencjono-wać do 30—40 reszt aminokwasowych w 1 ciągu operacyjnym (w wyjątkowych przypadkach do 60 lub nawet 80 reszt). Aparat jest zaprogramowany do wykonywania kolejnych degradacji reszt aminokwasowych od N-końca poli-peptydu. Po usunięciu pierwszego N-końcowe-go aminokwasu, wydzieleniu i identyfikacji powstaje następna w kolejności N-końcowa pochodna Edmana, itd. (ryc. 5-10). Pochodne fenylotiohydantoinowe rozdziela się techniką HPLC i identyfikuje na podstawie ich kolejności i miejsca elucji.
Wnioskowanie o pełnej strukturze pier wszo rzędowej po I i peptydu wymaga sekwencjonowania nakładających się peptydów
W przypadku oznaczenia sekwencji reszt aminokwasowych wszystkich uzyskanych CNBr peptydów wciąż nie znana jest informacja, dotycząca kolejności ułożenia tych peptydów w łańcuchu białkowym. W celu uzyskania jednoznacznych danych o pierwszorzędowej strukturze białka, należy otrzymać i zsek-wencjonować dodatkowe CNBr peptydy, których N-i i C-końcowe reszty nakładają się. Te dodatkowe peptydy otrzymuje się technikami, które rozrywają białko w miejscach innych niż reszta metioninowa (np. przez trawienie chymo-trypsyną). Jednoznaczne określenie struktury pierwszo rzędowej, przez porównanie sekwencji
NH3
R'
R' O
Kwas fenylotlohydanloinowy
H+, nitro-metan
Fsnylolzotlocyjanlan (odczynnik Edrnana) I peptyd
Fenylotiohydantnina I peptyd krótszą o jedną resztę aminokwasową
Ryc. 5-10. Fenyloizotiocyjanian przeprowadza resztę aminokwasową (lub N-końcową resztę poli-peptydu) do fenylotiohydantoiny. Fenyloizotiocy-janian reaguje z grupami aminowymi aminokwasów i peptydów, co prowadzi do powstania kwasów fenylotiohydantoinowych, które po dodaniu kwasu, w rozpuszczalnikach niehydroksytowych, cyklizują do pochodnych fenylotiohydantoinowych. Reakcję tę wykorzystuje się głównie w celu identyfikacji N-końcowych reszt peptydu w metodzie automatycznego sekwencjonowania polipep-tydów.
PEPTYDY / 57
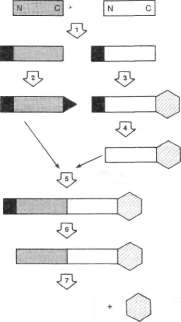
peptydowych, przypomina sposób analogiczny do budowy układanki (ryc. 5-11). W celu umiejscowienia wiązań disiarczkowych peptydy. z kontrolnych oraz zredukowanych lub utlenionych białek, rozdziela się metodą dwuwymiarowej chromatografii lub elektroforezy i chromatografii (tzw. finger-printing). Identyfikacja za pomocą ninhydryny ujawnia w produkcie trawienia białka kontrolnego 2 mniejsze peptydy, zaś w białku, na które działano wymienionymi powyżej czynnikami, 1 nowy peptyd. Mając informację dotyczącą sekwencji tych peptydow, można wyznaczyć w nich położenie wiązań disiarczkowych.
Peptyd Y
Paptyd X
Paptyd Z 1
C —końcowy |
N —końcowy |
fragment |
fragmenl |
peptydu X |
peptyd u Y |
Ryc. 5-11. Nakładające się sekwencje peptydu Z stosuje się w celu stwierdzenia, że peptydy X i Y występują w pierwotnym białku w kolejności X-*Y, a nie Y-»Z.
N |
C |
SYNTEZA PEPTYDOW METODAMI AUTOMATYCZNYMI
Ryc. 5-12. Schematyczne przedstawienie syntezy powstającego dipeptydu za pomocą techniki wprowadznnej przez IWerrifielda. Wyjaśnienie symboli znajdzie Czytelnik w towarzyszącym tekście.
Rycina 5-12 ilustruje procedurę syntezy dipeptydu A -B za pomocą automatycznej, stałofa-zowej techniki Merrifielda, podsumowującej wszystkie reakcje wymagane do zsyntetyzowa-nia peptydu o dowolnej długości. Te etapy syntezy obejmują:
1. Zablokowanie N-końcowego aminokwasu A (otwarty prostokąt) i aminokwasu B (zaciemniony prostokąt) za pomocą grupy /-buty-looksykarbonylowej (t-BOC) (■):
O
II (CH3)—C—O—C—
co prowadzi do utworzenia pochodnych /-BOC-aminokwas A i /-BOC-aminokwas B.
2. Aktywację grupy karboksylowej (-BOC--aminokwas B za pomocą dicykloheksylokar-boimidu(DCC) (A):
N
C = N-C6H(
Reakcje grupy karboksylowej aminokwa
su A (który staje się C-końcową resztą peptydu)
z zaktywowaną, nierozpuszczalny żywicą poli
styrenową ($).Usunięcie grup blokujących z połączeń
f-BOC-aminokwas A, za pomocą kwasu tri-
fluorooctowego w temperaturze pokojowej
(TFA, FjC—COOH).
Uwaga. W praktyce etapy 3 i 4 można pominąć, gdyż żywica z danym ż-BOC-amino-kwasem, połączone wiązaniem estrowym z fe-
58 / ROZDZIAŁ 5
nyloacetamidometylowym (PAM) „łącznikiem" zakotwiczonym w żywicy polistyrenowej, są dostępne w sprzedaży.
Kondensacje zaktywowanej grupy karbo-
ksylowej połączenia t-BOC-aminokwas B z
wolną grupą aminową unieruchomionego ami
nokwasu A.Usunięcie blokującej grupy f-BOC za po
mocą TFA (p. etap 4).Uwolnienie dipeptydu A—B od cząste
czek żywicy przez działanie fluorowodoru (HF)
w dichlorometanie w temp, — 2°C,
Pierwsze osiągnięcia techniki Merrifielda dotyczyły syntezy łańcucha A (21 reszt) i łańcucha B (30 reszt) insuliny w ciągu 11 dni oraz enzymu — rybonukkazy trzustkowej z wydajnością 18%. Kolejne jej udoskonalenia skróciły czas syntezy wiązania peptydowego do ok. 1 h i znacznie zwiększyły wydajność. Technika Merrifielda otworzyła nowe nadzieje nie tylko w badaniach de novo syntezy białek o określonej strukturze pierwszorzędowej, lecz także w immunologii, do produkcji szczepionek i hormonów polipeptydowych i być może także w leczeniu wybranych wrodzonych wad metabolicznych.
PIŚMIENNICTWO
Allen G: Sequencing of Proteins and Peptides. North
Holland, 1981. Bhwon AS: Protein/Peptide Sequence Analysis: Cur-
rent Methodologies. CRC Press, 1988.
Cantor CR, Schimmel PR: Biophyskal Chemistry, Part I: The Conformation of Macromolecules. Freeman, 1980.
Dayhoff M {editor): Atlas of Protein Seąuence and Structure. Vol 5. National Biomedicai Research Foundation, Washington, DC.
Doolittle RF: Ol" uris and orfs: a Primer on How to Analyze Derived Amino Acid Seąuences. Univer-sity Science Books. 1987.
Hewick RM, Hunkapiiler MW. Hood LE, Dryer WJ: A gasliquid solid phase peplide and protein seque-nator. J Biol Chem 1981; 256: 7990.
Hunkapiiler MW, Hood LE: Protein sequence analysis: automated microseąuencing. Science 1983; 219: 650.
LipmanDJ, PearsonWR: Similar amino acid seąuences: chance or common ancestry?/Mo/Bw/19Sl; 16:9.
Mahoney WC, Smith PK, Hermodson MA: Frag-mentation of proteins with o-iodosobenzoic acid: Chemical mechanism and identification of o-iodo-sobenzoic acid as a reactive contaminant that modifies tyrosyl residues. Biochemistry 1981; 20: 443; Methods Enzymol. Vols. 47; 48, 49, 91 (published continuously).
Meriifield RB: Solid phase synthesis. Science 1986; 232: 341.
Pearson JD et al: Reversed-phase supports fó"r thc resolution of iarge denatured protein fragments. / Chromatogr 1981; 207: 325.
Regnier FE. Gooding K.M: High performance liquid chromatography of proteins. Anal Biochem 1980; 103: 1.
Strickler JE, Hunkapiller MW, Wilson KJ: Ulility of the gasphase sequencer for both liąuid- and solid-*phase degradation of proteins and peptides at Iow picomole levels. Anal Biochem 1984; 140: 553.
Białka: struktura i właściwości
6
Victor W. Rodweli, PhD
WPROWADZENIE
Białka to wielkocząsteczkowe polipeptydy. Granica oddzielająca duże polipeptydy od małych białek przebiega zazwyczaj między masami cząsteczkowymi 8000 — 10 000. Białka proste są zbudowane tylko z aminokwasów. Białka złożo-ee^awierąją dodatkowe związki nieamino-kwasowe, takie jak hern, pochodne witamin, lipidy lub węglowodany. Ten rozdział dotyczy białek prostych. W innych rozdziałach zostaną omówione typowe białka złożone, jak hemowe, glikoproteiny i lipoproteiny, a także właściwości białek prostych o szczególnie odrębnej strukturze, takich jak kolagen i białka kurczliwe.
ZNACZENIE BIOMEDYCZNE
Białka odgrywają wiodącą rolę w czynności i budowie komórki. W celach diagnostycznych szeroko stosuje się analizę niektórych białek i enzymów krwi. Dobrym przykładem jest oznaczenie elektroforetyczne stosunku zawartości albumin do globulin osocza, które stanowi integralną część rozpracowania diagnostycznego w chorobach wątroby. Analiza lipoprotein oraz immunoglobulin osocza, za pomocą elektroforezy oraz innych metod, jest powszechnie stosowana w diagnostyce odpowiednio swoistych typów hiperlipoproteinemii i zaburzeń odporności. Prawidłowy ludzki mocz zwykle jest wolny od białek, a więc stwierdzenie znaczącej proteinurii stanowi zazwyczaj ważny sygnał chorób nerek, takich jak różne postacie ich stanów zapalnych.
BIAŁKA SĄ KLASYFIKOWANE W RÓŻNE SPOSOBY W CELU ZILUSTROWANIA RÓŻNORODNYCH WŁAŚCIWOŚCI ICH FUNKCJI STRUKTURALNYCH
Ze względu na brak uniwersalnego, wszechstronnie zadowalającego systemu klasyfikacji białek, powszechnie stosuje się kilka wzajemnie przeciwstawnych sposobów ich podziału. Wszystkie one mają raczej ograniczoną wartość jako pomoc w zrozumieniu wielu kluczowych właściwości białek. Utrzymywanie się w użyciu tych klasyfikacji i terminów — zwłaszcza w laboratoriach klinicznych — zmusza do krótkiej refleksji. Poniżej zostaną przedstawione podstawowe cechy systemów klasyfikacji, opartych na rozpuszczalności białek, ich kształcie, funkcji, właściwościach fizycznych oraz trójwymiarowej strukturze.
Klasyfikacja na podstawie rozpuszczalności
System klasyfikacji białek oparty na ich rozpuszczalności i rozwinięty w latach 1907—1908 jest nadal stosowany w zawężonym stopniu, zwłaszcza w biochemii klinicznej (tab. 6-1). Rozgraniczenia między klasami nie są przekonywające. Wyraźne rozróżnienie, np. miedzy albuminami i globulinami, nie może być przeprowadzone jedynie na podstawie ich rozpuszczalności w wodzie lub roztworach soli.
Klasyfikacja na podstawie kształtu
Dwie obszerne klasy białek mogą być wyodrębnione za pomocą ich stosunku osiowego (długości do szerokości). Białka globularnc, dla których wartość tego stosunku jest mniejsza od 10 i na ogół nie przekracza 3—4, mają łańcuchy polipeptydowe ściśle pofałdowane i zwinięte.
60 / ROZDZIAŁ 6
Albuminy Globuliny
Prolaminy
Histony Skleroprotetny
Tabela 6-1. Klasyfikacja białek oparta na ich rozpuszczalności
Rozpuszczalne w wodzie i roztworach soli. Żadnych szczególnych aminokwasów
Słabo rozpuszczalne w wodzie, ale dobrze w roztworach soli. Żadnych szczególnych aminokwasów
Rozpuszczalne w 70 80% etanolu, ale nierozpuszczalne w wodzie i etanolu absolutnym. Wzbogacone w arginine
Rozpuszczalne w roztworach soli (i kwasów nieorganicznych*)
Nierozpuszczalne w wodzie ani w roztworach soli. Wzbogacone w Gly, Aia, Pro
* Przyp. tłum.
Należą do nich m.in. insulina, albuminy i globuliny osocza oraz wiele enzymów. Białka włó-kienkowe, dla których wartości stosunków osiowych są większe niż 10, zawierają odcinki łańcuchów polipeptydowych zwiniętych śrubowo lub w heliks i połączone krzyżowo wiązaniami kowalencyjnymi lub wodorowymi. Przykładami białek włókienkowych są: kerą.tyna, mio-zyna, kolagen i włóknik.
Tabela 6-2. Główne funkcje biafek
Funkcja |
Białka (przykłady) |
Katalityczna |
Enzymy |
Strukturalna |
Kolagen, elastynar keratyny |
Ochronna |
Włóknik, immunogiobuliny, interferon |
Regulatorowa |
Kalmoduiina |
Regulacja genowa |
Histony, białka represorowe |
Regulacja hormonalna |
Insulina |
Skurcz |
Aktyna, miozyna |
Transport |
Albumina (bilirubina, kwasy tłuszczowe itp.), hemoglobina (tlen)Jipoproieiny (różne lipidy), transferyna (żelazo) |
Klasyfikacja na podstawie czynności
Zgodnie z ich funkcjami biologicznymi, białka mogą być sklasyfikowane np. jako strukturalne, katalityczne lub transportowe (tab. 6-2). Białka katalityczne (enzymy), stanowiące większość tych związków, są ujmowane w klasy według typu reakcji, którą katalizują.
Klasyfikacja na podstawie właściwości
fizycznych
Dla białek budzących zainteresowanie medyczne, wyspecjalizowane systemy klasyfikacji pozwalają rozróżnić ściśle spokrewnione białka. W powszechnym użyciu są np. 2 układy nazewnictwa lipoprotein osocza, a 3. jest rozważany. Pierwszy z nich wyodrębnia ot]-, ot2-, P- i Y-lipo-proteiny na podstawie ich ruchliwości elektro-foretycznej w środowisku o pH 8,6. Lipoprotei-ny są także klasyfikowane pod względem ich zachowania się podczas sedymentacji jako: chy-lomikrony, VLDL, LDL, HDL i VHDL. Ponadto 6 obszernych klas lipoprotein osocza może być rozpoznanych w zależności od występowania w nich apoprotein A, B, C, D, E lub F, które z kolei różnicuje się za pomocą kryteriów immunologicznych.
Klasyfikacja na podstawie struktury trój wy m ia ro wej
Białka mogą być zróżnicowane na podstawie występującej w nich struktury czwartorzędowej lub jej braku (patrz niżej). Ponadto podobieństwa w strukturze, wykryte głównie przez krystalografię promieniami X, dostarczają cennej podstawy do klasyfikacji białek. Na przykład białka łączące się z nukleotydami mają w strukturze trzeciorzędowej wspólną „domenę wiążącą nukleotyd" i mogą być ewolucyjnie spokrewnione. W miarę rozszyfrowywania dalszych struktur krystalicznych możliwe będzie rozpoznanie i ustalenie dodatkowych wspólnych domen.
PIERWSZORZĘDOWA STRUKTURA
BIAŁEK WYWODZI SIĘ
Z KOWALENCYJNEGO POŁĄCZENIA
L-a-AMINOKWASÓW WIĄZANIAMI
PEPTYDOWYMI
Wprawdzie wiodącą rolę wiązań peptydo-wych w białkach wydedukowano dawno temu, na podstawie wielorakich faktów, to jednak najbardziej przekonywającym dowodem nie-
BIAŁKA: STRUKTURA I WŁAŚCIWOŚCI / 61
zbędności jedynie tych wiązań stalą się pełna synteza chemiczna insuliny i rybonukleazy, wyłącznie w wyniku połączenia aminokwasów za pomocą wiązań amidowych.
Wiązanie łączące atomy węgla grupy karbonyiowej i azotu w wiązaniu peptydowym ma częściowo charakter wiązania podwójnego
Chociaż peptydy zapisuje się z pojedynczym wiązaniem łączącym atomy a-karboksylu i a-a-zotu, wiązanie węgiel—azot w rzeczywistości ma częściowo charakter wiązania podwójnego (ryc. 6-!). Nie ma tu żadnego swobodnego
O
O
H
C
H H
Ryc. 6-1. Stabilizacja rezonansowa wiązania pe-ptydowego nadaje mu częściowo charakter wiązania podwójnego i stąd sztywność połączenia C-N.
Ryc. 6-2. Wymiary calkowiciewyciągniętegotań-cucha polipeptydowego. 4 atomy w zacienionych obszarach są wspó! płaszczyzn owe (kop la na me) i tworzą wiązanie peptydowe. Niezacienionymi są: atom węgla a, atom wodoru a i grupa a-R danego aminokwasu. Swobodna rotacja zachodzi wokół wiązań łączących węgiel a z azotem a i węglem cc-karbonylowym (białe strzałki). Rozciągnięty łańcuch poiipeptydowy stanowi więc strukturę półsztywną, w której 2/3 atomów jego szkieletu jest utrzymywanych w stałych wzajemnych powiązaniach płaszczyznowych. Odległość pomiędzy sąsiednimi atomami węgla (/wynosi 0,36 nm. Podano również odległości międzyatomowe i kąty pomiędzy wiązaniami, które nie są równoważne. (Przerysowano i reprodukowano za zgodą z: L Pauiing, L. P. Corey, H. R. Branson: Proc. Nałl. Acad. Sci. USA 1951:37:205).
obrotu wokół wiązania łączącego atomy C i N, a wszystkie 4 atomy' przedstawione na ryc. 6-1, leżą w tej samej płaszczyźnie (tj. są koplanarne). Przeciwnie, rozległa swobodna rotacja zachodzi wokół pozostałych wiązań szkieletu polipep-tydowego. Te koncepcje są przedstawione na ryc. 6-2, gdzie wiązania obdarzone swobodnym obrotem są okółkowane strzałkami, a współ-płaszczyznowe (koplanarne) atomy znajdują się w obszarach zacienionych. Ta półsztywność ma ważne konsekwencje w uporządkowaniu struktury białek powyżej poziomu pierwszego rzędu. W uzupełnieniu do wiązań peptydowych, które tworzą „szkielet" łańcucha polipeptydowego, dodatkowe wiązania kowalencyjne i niekowa-lencyjne przyczyniają się do stabilności polipep-tydów.
Mostki disiarczkowe uformowane przez reszty cysterny tworzą kowalencyjne wiązania w obrębie oraz pomiędzy polipeptydatni niektórych białek
Wiązanie disiarczkowe, utworzone między 2 resztami cysteiny, łączy 2 części łańcucha polipeptydowego za pomocą reszty cystyny. Wiązanie cystynowe jest oporne na warunki powodujące denaturację białka. Kwas nad-mrówkowy (utleniający wiązanie S-S) lub fS-mer-kaptoetanol (redukujący mostki S-S z utworzeniem 2 reszt cysteiny) rozdziela łańcuchy poli-peptydowe połączone wiązaniami disiarczko-wymi bez wpływu na ich strukturę pierwszo-rzędową (ryc. 5-9).
NIEKOWALENCYJNE WIĄZANIA RÓWNIEŻ STABILIZUJĄ CZĄSTECZKĘ BIAŁKA
Trzy główne typy wiązań niekowalencyjnych szczególnie się przyczyniają do utrwalenia struktur białkowych.
Wielokrotne wiązania wodorowe stabilizują ogólną strukturę białek
Wiązania wodorowe. utworzone pomiędzy: resztami w łańcuchach bocznych peptydowo związanych aminokwusówj-iłtomami wodoru i tlenu w samych wiązaniach peptydowych oraz między resztami polarnymi na powierzchni białek i cząsteczkami wody, odgrywają ważną rolę w utrzymaniu struktury białek powyżej pierwszego rzędu (p. niżej: heliks a i struktura P).
62 / ROZDZIAŁ 6
Interakcje hydrofobowe przyczyniają się do stabilizacji wnętrza cząsteczek białkowych
Niepolarne łańcuchy boczne aminokwasów obojętnych w białkach dążą do zasocjowania. Powiązanie nie jest stechiometryczne, wobec tego nie istnieje prawdziwe wiązanie. Tyra niemniej ich duża liczba powoduje, że te oddziaływania odgrywają znaczącą rolę w utrzymaniu struktury białek.
Elektrostatyczne oddziaływania wiążą reszty powierzchniowe
w białkach
Wiązania typu soli, czyli elektrostatyczne, tworzą się albo między przeciwstawnie naładowanymi grupami w łańcuchach bocznych aminokwasów, albo pomiędzy resztami N-i C-końcowymi a innymi ugrupowaniami o przeciwnym ładunku. Na przykład w pH fizjologicznym s-aminowa grupa lizyny dźwiga ładunek netto + 1, zaś nie-a-karboksyl aspara-ginianu i glutamianu — czysty ładunek — 1. Mogą więc one nawzajem oddziaływać elektro-statycznie, stabilizując cząsteczki białka.
Związki denaturujące białka rozrywają w nich wiązania niekowafencyjne, powodując utratę aktywności biologicznej
Denaturacja białka za pomocą takich odczynników, jak: mocznik, siarczan dodecylu sodu (SDS), umiarkowane stężenie jonów H4 lub OH~ rozrywa wiązania wodorowe, hydrofobowe i elektrostatyczne, z wyjątkiem wiązań kowalencyjnych: peptydowych lub disiarczko-wych.
HELIKS a JEST JEDNYM
Z MOTYWÓW STRUKTURALNYCH
UTRZYMUJĄCYCH
UPORZĄDKOWANE KONFORMACJE
W BIAŁKACH
Odkrycie, że łańcuchy polipeptydowe charakteryzują się bardzo uporządkowanymi konformacjami, utrzymywanymi przez wiązania wodorowe, stało się głównym postępem pojęciowym. Wprawdzie istnienie tych bardzo uporządkowanych struktur zostało potwierdzone przez krystalografię rentgenowską, jednak było ono najpierw zaproponowane w wyniku rozważań teoretycznych.
Skok 0,54 nm (3.6 reszt)
Dane rentgenograficzne wykazały obecność powtarzających się jednostek o długości 0,5—0,55 nm wzdłuż długiej osi a-keratyn włosa i wełny. Niestety, żaden wymiar rozciągniętego łańcucha poiipeptydowego nie wynosił 0,5—0,55 nm (ryc. 6-2). Tę oczywistą anomalię rozstrzygnęli Pauling i Corey, którzy zaproponowali ukształtowanie łańcucha polipep-tydowego a-keratyny w formie a-heliksu (ryc. 6-3). W strukturze tej grupy R wystają na zewnątrz od centrum heliksu (ryc. 6-4). Na jedenjJsok śruby (zwój a-heliksu) przyj>adajL&-reszt aminokwasowych, a odległość "przebyta przez jeden skręt (inaczej: odległość między
Ryc. 6-3. Przedstawienie helikalnej konfiguracji głównego łańcucha polipeptydowego wokół osi a-heiiksu.
BiAŁKA: STRUKTURA i WŁAŚCIWOŚCI / 63
0,5 r
Ryc. 6-4. Przekrój poprzeczny heltksu a. Łańcuchy boczne (R) wystają na zewnątrz heiiksu. Promienie van der Waalsa są większe niż wykazano na rycinie, stąd wewnątrz heiiksu prawie nie ma wolnej przestrzeni. (Nieznacznie zmodyfikowano i reprodukowano za zgodą z: Stryer L; Biochemistry, wyd.2, Freeman, 1981, Copyright© 1981 by W. H. Freeman and Co).
zwojami*) wynosi 0,54 nm. Odstęp przypadający na Jedną resztę aminokwasem a wzdłuż osi ct-heliksu wynosi 0,15 nm. Właściwości a-helik-su są następujące:
Strukturę cc-heliksu stabilizują miedzy-
aminokwasowe wiązania wodorowe. Każde
z nich jest utworzone przez atom H połączony
z azotem jednego ugrupowania peptydowego
(NH) i znajdujący się między silnie elektroujem-
nymi atomami (układami*), tj. wymienionym
azotem peptydowym oraz tlenem grupy kar-
bonylowej (ĆO) 4. z kolei reszty aminokwaso-
wej w strukturze pierwszorzędowej.Każde wiązanie peptydowe uczestniczy
w wytworzeniu wiązania wodorowego, co za
pewnia maksymalną stabilność.Wszystkie atomy azotu peptydowego i tle
nu karbonylowego reszt aminokwas owych w
łańcuchu głównym są zaangażowane w wiąza
nia wodorowe, co znacznie zmniejsza hulro-
filowy (zwiększając hydrofobowy) charakter
regionu a-helikalnego.Heliks ot formuje się spontanicznie, ponie
waż — przy najmniejszym zużyciu energii
— stanowi najbardziej stabilną konformację
łańcucha polipeptydowego.
* Przyp. tłum.
Ryc. 6-5. Wiązania wodorowe (kropki} uformowane między atomami H i O stabilizują polipeptyd w konformacji a-heltkalnej. (Przedrukowano za zgodą z; Haggis G. H. i wsp. tntroduetion to Motecular Biology. Witey, 1964).
Tabela 6-3. Wpływ różnycfi reszt aminokwaso-wych na formowanie heiiksu a
Heliks a
stabilizują (utrwalają) |
destabilizują |
kończą (przerywają) |
Ala |
Arg |
Pro |
Asn |
Asp |
Hyp |
Cys |
Glu |
|
Gin |
Gly |
|
His |
Lys |
|
Leu |
Ile |
|
Met |
Ser |
|
Phe |
Thr |
|
Trp |
|
|
Tyr |
|
|
Val |
|
|
64 / ROZDZIAŁ 6
Prawozwojowy heliks występujący w biał
kach jest znacznie bardziej trwały niż lewoskręt-
ny, gdy resztami są L-aminokwasy.Resztami destabilizującymi heliks są: pro-
.liną_(w której atom N jest częścią sztywnego
pierścienia, co uniemożliwia rotację wokół wią
zania N C) oraz aminokwasy z naładowanymi
elektrycznie lub masywnymi grupami R, które
elektrostatycznie lub fizycznie przeszkadzają
w uformowaniu heliksu (tab. 6-3).
RÓWNOLEGŁE
I PRZE CiW RÓWNO LEGŁE
POFAŁDOWANE ŁAŃCUCHY
STANOWIĄ DRUGI TYP
ROZPOZNAWALNIE
UPORZĄDKOWANEJ STRUKTURY
Pauling i Corey zaproponowali również drugie uporządkowanie struktury białka, tj^jtruk-tury p, czyli pofałdowanej kartki (inaczej: pofałdowanego łańcucha, pofałdowanego lub „spli-sowanego" arkusza*) (ang. f3-pleated sheet; p — ponieważ to była druga struktura po opisanym najpierw a-heliksie). W heliksie j. łańcuch polipeptydowy jest zwarty, natomiast w_. strukturze p (inaczej: fałdowej, „harmonijkowej" lub „dywanowej"*) pozostaje on niemal całkowicie rozciągnięty (ryc. 6-6). Struktura P noslnazwę przeciwrównoległej, gdy sąsiadujące łańcuchy polipeptydowe biegną w przeciwnych kierunkach (N-koniec jednego łańcucha do C-końca drugiego) (ryc. 6-6); luedy łańcuchy podążają w tym samym kierunku jest ona nazywana równoległą (nie wykazano).
W wielu białkach występują współbieżne i przeciwbieżne regiony struktury p. Może ją formować 2—5 przylegających łańcuchów poli-peptydowych. Na rycinie 6-7 przedstawiono region rybonukleazy, w której 3 odcinki łańcucha polipeptydowego tworzą strukturę p.
Heliks a jest stabilizowany mostkami wodorowymi między wiązaniami peptydowymi, oddzielonymi od siebie 4 resztami w sensie struktury pierwszorzędowej, podczas gdy strukturę fS utrwalają wiązania wodorowe między segmentami peptydowymi oddalonymi od siebie w sensie struktury pierwszorzędowej (ryc. 6-7).
Ryc. 6-6. W przeciwrównoległej fałdowej strukturze p sąsiednie łańcuchy polipeptydowe biegną w przeciwnych kierunkach. Wiązania wodorowe między grupami NH i CO sąsiednich łańcuchów stabilizują tę strukturę. Łańcuchy boczne (R) aminokwasów znajdują się powyżej i poniżej płaszczyzny kartki. O atomy węgla; © atomy azotu; O atomy wodoru. (Zmodyfikowano i reprodukowano za zgodą z: Siryer L: Biochemistry, wyd. 2, Freeman, 1981, Copyright © 1981 by W. H. Freeman and Co.).
ZAKRĘTY p UMOŻLIWIAJĄ POLIPEPTYDOM PRZYJMOWANIE KSZTAŁTÓW GLOBULARNYCH
Polipeptydy z reguły formują zbite masy globularne, muszą więc istnieć możliwości zmiany kierunku łańcucha polipeptydowego. Jednym z nich jest utworzenie ciasnej pętli zakrętu P (inaczej: skrętu p, zwrotu p1 lub zwrotu typu spinki do włosów*), w której tlen grupy karbonylowej jednej reszty łączy sie wiązaniem wodorowym z wodorem amidowym aminokwasu oddalonego o 3 reszty do przodu.
Często w zakrętach p występują prolina i gli-cyna. Część struktury przestrzennej (geometrii) zakrętu p jest ukształtowana w prolinie, która bardziej niż inne aminokwasy podlega konfor-
Przyp. tłum.
* Przyp. tłum.
BIAŁKA: STRUKTURA I WŁAŚCIWOŚCI / 65
Ryc. 6-7. Cząsteczka rybonukleazy trzustki wołu jest zbudowana z pojedynczego łańcucha 124 reszt połączonego poprzecznie w 4 miejscach za pomocą wiązań disiarczkowych. Region a-heliksu ujęto w kropkowany owal, a region struktury fałdowej zacienione Pozostałe części cząsteczki występują giównie w postaci przypadkowego zwoju. Centrum aktywne enzymu znajduje się w miejscu związania jonu fosforanowego (PO^~). (Zaadoptowano z: Kartha G., Bello J.r Harker D.: Tertiary structure of nbonuclease. Naturę 1967; 213:862) Białko zo-stato w całości z syntetyzowane chemicznie.
macyjnym ograniczeniom. Dla kontrastu mała grupa R. pozwala glicynie działać jako giętki „zawias" między regionami polipeptydu, którego ugrupowania R sprzyjałyby inaczej bardziej rozciągniętej konformacji.
REGIONY BIAŁEK, KTÓRE NIE SĄ UFORMOWANE W HELIKSY, STRUKTURY p LUB ZAKRĘTY % WYSTĘPUJĄ WTZW. KONFORMACJI „PRZYPADKOWEGO" (NIEPLANOWANEGO) ZWOJU
Znaczne części cząsteczki białkowej mogą występować w konformacji przypadkowego (nieplanowanego) zwoju (ang. random-coil; kłębek statystyczny) (ryc. 6-7), Termin „przypadkowy" jest niefortunny, ponieważ może bardziej implikować mniejsze biologiczne znaczenie niż regionów o dużej powtarzalności. Dla funkcji biologicznej regiony przypadkowego zwoju są równie ważne, jak heliks a lub konformacja p.
5 — Biochemia
ASPEKTY STRUKTURY BIAŁEK
ROZWAŻA SIĘ POD KĄTEM
4 POZIOMÓW UPORZĄDKOWANIA
Struktura pierwszorzędowa
Strukturę pierwszego rzędu stanowi, jak w przypadku peptydów, kolejność aminokwasów w łańcuchu lub łańcuchach polipeptydo-wych oraz umiejscowienie wiązań disiarczkowych, jeżeli one w tych łańcuchach występują.
Struktura drugorzędowa
Struktury drugiego rzędu, czyli przestrzenne współzależności między aminokwasami zwartymi w pierwszorzędowym strukturalnym zakresie, mogą być regularne (np. heliks a, struktura f>) lub przejawiać niewiele tej regularności (np. przypadkowy zwój).
Struktura trzeciorzędowa
Ogólne rozmieszczenie i wzajemne powiązanie rożnych regionów lub domen oraz poszczególnych reszt amin o kwas owych w pojedynczym łańcuchu polipeptydowym stanowi trzeciorzędową strukturę białka. Wprawdzie rozgraniczenie pomiędzy strukturą drugo- i trzeciorzędową nie jest wyraźne, struktura trzeciorzędowa wyraża wzajemne przestrzenne usytuowanie reszt aminokwasowych, które — generalnie — są daleko od siebie w- sensie budowy pierwszego rzędu.
Struktura czwartorzędowa
Białka wykazują strukturę czwartego rzędu, jeżeli składają się z 2 lub więcej łańcuchów polipeptydowych połączonych wiązaniami innymi niż kowalencyjne (tj. niepeptydowymi lub disiar-czkowymi*). Siłami stabilizującymi te agregaty są wiązania wodorowe i elektrostatyczne (typu soli) utworzone miedzy resztami aminokwaso-wymi na powierzchniach łańcuchów polipeptydowych. Takie białka noszą nazwę oltgome-rów, a pojedyncze łańcuchy polipeptydowe je
* Cząsteczka białka nie dająca się, przynajmniej potencjalnie, rozdzielić na podjednostki (związane wiązaniami mekowalencyjnymi) nie ma struktury czwartorzędowej. Przykładami białek bez struktury czwartorzędowej są: rybonukleaza (1 łańcuch) i chy-motrypsyna (3 łańcuchy). Przyp. tłum. zaczerpnięty z: Morawiecki A. (red.): Nowe polskie słownictwo biochemiczne. PWN, 1983, s. 113.
66 / ROZDZIAŁ 6
tworzące określa się jako protomery, monomery lub podjednostki.
Wiele białek oligomerycznych zawiera 2 lub 4 protomery, nosząc odpowiednio nazwy di-merów lub tetramerów. Oligomery zbudowane z więcej niż 4 protomerów są również rozpowszechnione, zwłaszcza wśród enzymów regulatorowych (przykładem — karbamoilotransfera-za asparaginianowa). Białka oligomeryczne odgrywają szczególną role w regulacji wewnątrzkomórkowej, ponieważ protomery mogą przyjmować względem siebie różne ułożenia przestrzenne, co powoduje zmiany we właściwościach oligomeru.
Niektóre białka tworzą kompleksy wielkocząsteczkowe
Asocjację różnorodnych białek czynnościowych, z których każde ma 4 poziomy struktury, w wielofunkcjonalne makromolekularne kompleksy spotyka się w transporcie elektronów, biosyntezie kwasów tłuszczowych i metabolizmie pirogronianu.
DRUGORZĘDOWĄ
I TRZECIORZĘDOWĄ STRUKTURĘ
BIAŁEK WYZNACZA SEKWENCJA
AMINOKWASÓW W ŁAŃCUCHU
POLIPEPTYDOWYM
Skoro tylko utworzy się łańcuch polipep-tydowy, grupy R aminokwasów kierują swoistym zwijaniem się poszczególnych jego regionów (struktura drugo rzędowa), jak również zasocjowaniem tych regionów (struktura trzeciorzędowa). Dowodzi tego klasyczne doświadczenie. Zadziałanie na rybonukleazę łagodnym związkiem redukującym (P-merkaptoetanolem) i czynnikiem denaturującym (mocznikiem lub guanidyną; p. niżej) inaktywuje ją tak, że przyjmuje konformację przypadkowego zwoju. Powolne usunięcie czynnika denaturującego i łagodne ponowne utlenienie, w celu rekonstrukcji wiązań S—S, prowadzi do niemal kompletnego odtworzenia aktywności enzymatycznej. Nie jest więc niezbędne postulowanie niezależnej genetycznej kontroli uporządkowania struktury białka powyżej poziomu pierwszego, ponieważ budowa pierwszorzędowa wyznacza drugorzędo-wą, trzeciorzędową oraz (kiedy jest obecna) czwartorzędową strukturę (tj. konformację) cząsteczki białkowej. Rodzimą konformacją białka, takiego jak rybonukleaza, wydaje się być ta,
która jest term o dynamicznie najbardziej stabilna w danym otoczeniu, np. w hydrofilowym w przeciwieństwie do hydrofobowego.
Struktura białka może być modyfikowana podczas po trans! acyjnego dojrzewania, takiego jak konwersja prcproenzyirm w postać katalitycznie aktywną lub usunięcie peptydowego „lidera" (peptydu sygnałowego), który przeprowadza poprzez błonę białka przeznaczone „na eksport'7.
Stosunkowo słabe oddziaływania odpowiedzialne za utrzymanie drugo-, trzecio- i czwartorzędowej struktury białek ulegają łatwo rozerwaniu, powodując utratę ich aktywności biologicznej
Naruszenie rodzimej struktury nosi nazwę denaturacji. Fizycznie denaturacja może być rozpatrywana jako zaburzenia konformacji łańcucha polipeptydowego, nie wpływające aa jego strukturę pierwszorzędowa. Dla protomeru proces ten może być przedstawiony tak, jak na ryc. 6-8. Dla białka oligomerycznego denaturacja może obejmować rozdysocjowanie protomerów, czemu towarzyszą zmiany wich konformacji.
Enzym aktywny (natywny)
Dsnaturacja
Enzym nieaktywny (z denaturowany)
Ryc. 6-8. Zobrazowanie denaturacji protomeru.
Biologiczną aktywność niszczą mocne kwasy lub zasady, wysoka temperatura, detergenty jonowe (amfipatyczne), związki chaotropowe (mocznik, guanidyną), ciężkie metale (Ag, Pb, Hg) lub rozpuszczalniki organiczne. Zdenatu-rowane białka są na ogół gorzej rozpuszczalne i ulegają wytrąceniu. Znajduje to zastosowanie w laboratorium klinicznym. Do krwi lub surowicy, w której oznacza się zawartość małych cząsteczek (np. glukozy, kwasu moczowego, leków), generalnie najpierw dodaje się kwasu trichlorooctowego, fo sforo wolframowego lub fosforomolindenowcgo w celu strącenia białek.
BIAŁKA: STRUKTURA I WŁAŚCIWOŚCI / 67
Po usunięciu ich, przez wirowanie, analizuje się wolny od białek supernatant.
Wrażliwość większości enzymów na ogrzewanie, kwasy oraz proteazy, stanowi podstawę wstępnego badania, stwierdzającego czy daną reakcję katalizuje enzym. Ekstrakt komórek, obdarzony aktywnością enzymatyczną, który traci tę właściwość w wyniku zagotowania, zakwaszenia i ponownego zobojętnienia lub zadziałania proteazą, zapewne zawiera jakiś enzym w charakterze katalizatora. Często na denaturację jakiegoś enzymu wpływa obecność jego substratu. Pojawienie się zmiany konfor-macyjnej podczas związania substratu dowodzi, że nowa konformacja może być trwalsza lub mniej trwała niż poprzednia.
PIERWSZORZĘDOWE STRUKTURY BIAŁEK SĄ OZNACZANE METODAMI STOSOWANYMI DO SEKWENCJONOWANIA PEPTYDOW
Z białek złożonych usuwa się najpierw grupy prostetyczne (np. hem), a wiązanie disiarczkowe utlenia się w celu uzyskania liniowych polipep-tydów (p. ryc. 5-9). Techniki stosowane do
Tabela 6-4. Modyfikacja grup a-COOH i a-NH2 w białkach*
Grupy modyfikowane
a-COOH |
|
||
Amidowa |
N-formylowa |
N-ace tyłowa |
N -metylowa |
|
|
Ala |
Ala |
Asp |
|
Asp |
Asp |
Glu |
|
|
|
Gly |
Gly |
Gly |
Gly |
His |
|
|
|
Met |
Met |
Met |
Met |
Phe |
|
|
|
Pro |
|
|
|
|
|
Ser |
Ser |
|
|
Thr |
Thr |
Tyr |
|
|
|
Val |
|
Val |
|
* Według R. Uy, F. Wold: Posttranslational co-valent modification of proteins, Science 1977; 198:890. Copyright © 1977 American Association for the Advancement of Science, w modyfikacji autora rozdziału za zezwoleniem autorów cytowanej pracy.
Tabela6-5. Zmodyfikowane nie-a-funkcyjne grupy w białkach*
Grupy modyfikowane
—OH |
Mto-et-N |
||
PO3H2 |
N- metylowa |
N dimetylowa |
N-trimetylowa |
Ser Thr Tyr |
Arg His Lys |
Arg Lys |
Lys |
* Według R. Uy, F. Wold: Posttranslational co-valent modification of proteins. Science 1977; 198:890. Copyright © 1977 by the American Association for the Advancement of Science, w modyfikacji autora rozdziału za zezwoleniem autorów tej pracy.
sekwencjonowania tych polipeptydów przedstawiono w rozdz. 5. Wprawdzie większość białek 2awiera tylko aminokwasy wymienione w tab, 4-3, pochodne tych reszt również występują w niektórych białkach (tab. 6-4 i 6-5). Jakkolwiek omówienie metod używanych do zidentyfikowania owych pochodnych amino-kwasowych nie wchodzi w zakres tego rozdziału, jednak ich obecność może komplikować określenie struktury pierwszorzedowej.
Drugorzędowa i trzeciorzędowa struktura białek jest analizowana za pomocą krystalografii rentgenowskiej
Techniki poprzednio stosowane, pozwalające wnioskować o występowaniu w białkach struktur helikalnych (np. optyczna dyspersja rotacyjna, wymiana protonów trytu) zostały wyparte przez rentgenowską analizę krystalograficzną. Promienie X są kierowane na kryształ białka oraz na jedną z jego pochodnych, zawierającą dodatkowo jon ciężkiego metalu. Promienie ulegają rozproszeniu w sposób zależny od gęstości elektronów w różnych częściach cząsteczki białkowej. Obrazy zbioru plamek dyfrakcyjnych, uzyskane na błonie fotograficznej (po jej wywołaniu), są tłumaczone na tzw. mapy rozkładu gęstości elektronowej, które po ułożeniu jednej na drugiej pozwalają krystalografowi skonstruować wiarogodny model danego białka. Krystalografia rentgen o graficzna, chociaż czasochłonna, kosztowna i wymagająca specjalistycznego przeszkolenia, ujawniła szcze-
68 / ROZDZIAŁ 6
gólowe i precyzyjne obrazy z usytuowaniem wszystkich aminokwasów w wielu białkach. Nie można nie docenić jej ogromnego wkładu do naszego dzisiejszego pojmowania struktury białek.
Wyłoniona technika zobrazowania magnetycznego rezonansu (ang. magnetic resonance imaging; skrót — MRI) została ostatnio wykorzystana do zanalizowania trójwymiarowej struktury małego białka. Technika MRI wydaje się mieć szansę zastosowania do większych białek.
Metody fizyczne stosowane do zbadania czwartorzędowej struktury białek
Badanie czwartorzędowej struktury białka oligomerycznego obejmuje określenie liczby i rodzaju obecnych w nich protomerów, ich położenia względem siebie oraz łączących je oddziaływań.
Zakładając, że oJigomery nie ulegają denatu-racji podczas procedury stosowanej do oznaczenia masy cząsteczkowej, wiele metod może dostarczyć o niej danych. Te same techniki mogą być wykorzystane do określenia masy cząsteczkowej protomeru po wcześniejszym zdenaturowaniu oligomeru.
Ultrawirowanie, filtracja żelowa i elektroforeza w żelu pozwalają oznaczyć masę cząsteczkową białek
oi izomerycznych
Rozwinięta przez Svedberga technika ultra-wirowania mierzy szybkość sedymentacji* w polu siły odśrodkowej ok. 105 x g. W ostatnich latach istnieje tendencja, aby zastąpić ją mniej złożonymi metodami.
W pokrewnej technice, tj. ultrawirowaniu w gradiencie gęstości sacharozy, nawarstwia się
białka wzorcowe i badane na gradient stężeń roztworów sacharozy (5—20%), przygotowany w probówce plastikowej. Po ultrawirowaniu przy 105 x g przez kilkanaście godzin (np. w ciągu nocy) przekłuwa się mały otworek w dnie probówki, zbiera jej zawartość w postaci kropel w serii kolejnych małych probówek i oblicza ruchliwości na podstawie względnego położenia białek w gradiencie.
Masę cząsteczkową białka
globu larnego można określić z jego
ruchliwości w sicie molekularnym
Kolumny z żelu Sephadex lub podobne matryce, mające „pory" o wiadomym rozmiarze kalibruje się, stosując białka o znanej masie cząsteczkowej. Poszukiwaną masę cząsteczkową nieznanego białka oblicza się z porównania jego objętości elucyjnej z objętościami elucyj-nymi białek wzorcowych. Można popełnić duże błędy, jeżeli cząsteczka białka jest bardzo asymetryczna albo oddziałuje silnie ze związkami, z których zostały wyprodukowane molekularne sita.
Elektroforeza w żelu poliakrylamidowym (PAGE) to jeszcze jedna metoda oznaczania masy cząsteczkowej
Białka wzorcowe rozdziela się elektroforety-cznie w 5—15% żelach poliakrylamidowych o zmiennej porowatości, wybarwia się je na obecność białek, generalnie błękitem Coomas-sie lub tzw. srebrem, i masę cząsteczkową oznacza względem ruchliwości elektroibretycz-nej wzorców. Najbardziej powszechne zastosowanie tej techniki, PAGE-SDS, pozwala oznaczyć masę cząsteczkową protomeru w wyniku wcześniejszego zdenaturowania oligomeru (np. przez zagotowanie w roztworze detergentu w obecności p-merkaptoetanolu) i rozdzielenie w żelach zawierających detergent jonowy — siarczan dodecylu sodu (SDS).
* Matematycznym wyrazem tej szybkości jest tzw. stała sedymentacji S20, tj. szybkość opadania warstwy roztworu białka pod działaniem jednostki siły odśrodkowej, wyrażona w jednostkach układu cgs, sprowadzona do gęstości i lepkości wody w temp. 20°C. Dla większości białek wartości liczbowe stałej sedymentacji wahają się w granicach l,510~13 — 2010~"; dla cząsteczki białka o kształcie kulistym jednostka stałej sedymentacji (1I0""13) odpowiada masie cząsteczkowej ok. 16 000. (Przyp. tłum. zaczerpnięty z: Skar-żytiski B.: Chemia fizjologiczna, t. I, PWRiL, 3956, s. 85).
KOMPLEKSY BIAŁKOWE MOGĄ BYĆ UWIDOCZNIONE PRZEZ ELEKTRONOWĄ MIKROFOTOGRAFIĘ
Uzyskane w mikroskopie elektronowym powiększenia rozmiarów średnicy nawef 100000 razy pozwalają uwidocznić białka o bardzo dużej masie cząsteczkowej, takie jak cząstki
BIAŁKA: STRUKTURA I WŁAŚCIWOŚCI / 69
Tabela 6-6. Czwartorzędowe struktury wybranych |
enzymów* |
|
|
Enzym (oligomer) |
|
Liczba protomerów |
Masa cząsteczkowa protomeru |
Aminotransferaza asparaginianowa serca kury Syn ta za kwasów tłuszczowych wątroby goiębia Fruktozobisfosfataza wątroby królika |
|
2 2 2" 2** |
50 000 230 000 29 000 37 000 |
Aminotransferaza ornitynowa wątroby szczura Karboksylaza propionylo-CoA serca świni LDH serca, wątroby lub mięśni wołu |
|
4 4 4** |
33 000 175 000 35 000 |
ATPaza mitocriondrrów serca wołu |
|
10 |
26 0O0 |
Syntetaza glutaminowa Escherichia coli Karboksylaza acetylo-CoA wątroby kury |
|
12 2** 10** |
48 500 4 100 000 409 000 |
* Zaadaptowano z: Klotz I. M., Langerman N. R., Darnall D. W.: Cłuaternary structure of enzymes, Annu Rev Biochem 1970; 39:25.
** Niezidentyfikowane podjednostek.
wirusów, kompleksy enzymatyczne i białka oligomeryczne.
W tabeli 6-6 podano przykłady liczby oraz mas cząsteczkowych protomerów stanowiących czwartorzędowe struktury wybranych enzymów.
PIŚMIENNICTWO
Advances in Protein Chemistry. Academic Press, 1944
to datę, [Annual publicalion.] Celis JE. Bravo R: Two-dimensional Gel Electro-
phoresis of Proteins. Methoiłs and Applications.
Academic Press, 1984. Chothia C. Principles that determine the structure of
proteins. Annu Rev Biochem 1984; 53:537. Dunn MJ: Gel Electrophoresis of Proteins. Wright,
1986.
Franks F: Characterization of Proteins. Wright, 1986 One AD, Braun W, Wuthrich K. Studies by
lH NMR and distanoe geometry of the solution
conformation of the a-amylase inhibitor tendami-
stat. / Mol Biol 1986; 189:377. Lennarz WJ (editor): The Biochemistry of Glyco-
proteins and Proteoglycarrs. Plenum Press, 1980. L'Italien JJ: Proteins: Structure and Function. Plenum Press, 1987. Rosę CD, Geselowitz AR, Lesser GJ, Lee RM, Zehfus
MH. Hydrophobiciiy of amino acid residues in
globular proteins. Science 1985; 229:834, Schlessinger DH: Macromolecular Seąuencing and
Analysis: Selected Methods and Applications. AR
Liss, 1988. Schott H: Affinity Chrotnatography. Template Chro-
matography of Nucleic Acids and Proteins. Marcel
Dekker, 1984. Wittmann-Liebold B, Solnikow J, Erdmann VA:
Advancetl Methods in Protein Microseąuence
Analysis. Springer Verlag, 1986.
7
Białka: mioglobina i hemoglobina
Victor W. Rodwell, PhD
WPROWADZENIE
W rozdziale tym omówiono zależność struktury i funkcji na przykładzie mioglobiny i hemoglobiny, białek nie tylko bardzo istotnych fizjologicznie, lecz także obrazujących, w jaki sposób struktura białek decyduje o ich funkcji biologicznej.
ZNACZENIE BIOMEDYCZNE
Białka hemowe biorą udział w transporcie i magazynowaniu tlenu, transporcie elektronów oraz fotosyntezie. Badania mioglobiny i hemoglobiny ujawniły istnienie cech struktury wspólnych dla wielu białek, jak również istnienie zależności między strukturą a funkcją. Ponadto umożliwiły one poznanie molekularnych podstaw chorób genetycznych, takich jak niedo-krwistośc sierpowata (wynik zmienionych właściwości powierzchniowych podjednostki p hemoglobiny) i taJasemie (przewlekłe dziedziczne choroby hemolityczne, charakteryzujące się upośledzeniem syntezy hemoglobiny). Badania wykazały, że cyjanek i tlenek węgla stanowią śmiertelne zagrożenie, ponieważ uniemożliwiają fizjologiczną funkcję odpowiednio oksydazjŁ cytocJLrpmowei-J^hemoglobiny, a stabilizacja struktury czwartorzędowefnieutJenowanej hemoglobiny przez 2,3-bisfosfoglicerynian (BPG) stanowi podstawę mechanizmów choroby górskiej i adaptacji do dużych wysokości.
ZDOLNOŚĆ MIOGLOBINY
I HEMOGLOBINY
DO MAGAZYNOWANIA
I TRANSPORTU TLENU WIĄŻE SIĘ
Z OBECNOŚCIĄ PROSTETYCZNEJ
GRUPY HEMOWEJ
I JONU ŻELAZAWEGO
Grupą proste ty czną mioglobiny i hemoglobiny jest hem, cykliczny tetrapirol nadający tym białkom zabarwienie czerwone. W tetrapirolu 4 pierścienie pirolowe są połączone mostkami tt-metinowytni, tworząc płaski układ cykliczny (ryc. 7-1). Podstawniki w pozycjach P pierścieni
n
Ryc. 7-1. Pirol. Węgle a fączą się za pomocą mostków metinowych tworrac cykliczny tetrapirol. Przy węglach p występują podstawniki charakterystyczne dta swoistych tetrapiroli, np. hemu.
pirolowych określają rodzaj pochodnej tetrapirolu. W hemie w pozycjach p znajdują się grupy metylowe (M), winylowe (V) i pfbpionianpwe (P) ułożone w następującej kolejności: M, V, M, V, M, P, P, M (ryc. 7-2). W centrum tego układu znajduje się atom żelaza w formie jonu żelazawego (Fe2+), Innymi białkami, w których grupą prostetyczną jest tetrapirol zasocjowany z jonem metalu są cytochromy (Fe2+ i Fei+), katalaza, 2,3-dioksygenaza tryptofanowa oraz chlorofil (Mg2+). Funkcja biologiczna cyto-
BIAŁKA: MJOGLOBtNA f HEMOGLOBINA / 71
Ryc. 7-2. Hem. Pierścienie pirolowe, węgle mostków metinowych i atom żelaza Fe'4 + leżą praktycznie w jednej płaszczyźnie. Piąte i szóste wiązanie koordynacyjne Fe2+ są usytuowane prostopadle, znajdując się nad i pod płaszczyzną hemu. Na uwagę zasługuje rodzaj podstawników przy węglach P pierścieni piroiowych, centralnie położony atom zela2a oraz polarne łańcuchy boczne, które są zwrócone na zewnątrz cząsteczki mioglobiny.
chromów wynika z utlenienia i redukcji atomu żelaza. W przeciwieństwie, w przypadku mioglobiny i hemoglobiny utlenienie Fe2+ wiąże się z utrat -a przez te białka ich aktywności biologicz-nej.
Utlenowana mioglobina mięśni stanowi rezerwę tlenu
Fnnk^pt iTiinplrthiTiy jftst ma^Tynnwfinie t1f»-
nu w mięśniach. W warunkach niedoboru tlenu (np. w przypadku intensywnych ćwiczeń fizycznych) tlen utlenowanej miogiobiny jest wykorzystywany w mitochondriach mięśni do syntezy ATP na drodze fosfcrylacji oksydacyjnej.
Poza dwoma wyjątkami reszty polarne znajdują się na zewnątrz, a niepolarne wewnątrz cząsteczki mioglobiny
Mioglobina jest to pojedynczy łańcuch poli-pęptydowy o masie cząsteczkowej 17 000 zbudowany zejj_5J)reszt aminokwasowych rozmieszczonych w "cKaraktery styczny dla białek globularnych sposób. Zewnętrzna cześć cząsteczki białka jest polarna, a wnętrze — niepolarne. Reszty aminokwasowe, zawierające zarówno grupę polarną, jak i niepolarną (np. Thr, Trp i Tyr), są skierowane swoją niepolarną częścią do wnętrza częsteczki. Poza 2 resztami his-tydynowymi, biorącymi udział w wiązaniu tle-
nu, wewnątrz cząsteczki mioglobiny znajdują się wyłącznie aminokwasy niepolarne (np. Leu, Val, Phe i Met).
Mioglobina, pierwsze białko, którego strukturę rozszyfrowano rentgenograficznie, jest bogata w ot-heliks
Mioglobina jest silnie upakowaną, w przybliżeniu kulistą cząsteczką o wymiarach 4,5x3,5 x2,5 nm(ryc. 7-3). Niemniej jednak jej
Ryc. 7-3. Model mioglobiny opracowany na podstawie analizy rentgenograficznej. W modelu zaznaczono wyłącznie węgle s, (Reprodukcja za zgodą z: Dickerson R. E.r w: The Proteins, wyd. 2, t. 2, Neurath H, [red], Academic Press, 1964).
struktura jest nieregularna i wykazuje brak symetrii. Okojo 75% ^"^nyfia wy^t^ry^\yfgarnie R prąwnslfi-^tiiyrh a-heiiksów o długości
7—20 aminokwasów. Licząc od N-końca odcinki helikalne oznacza się kolejnymi literami od A do H, natomiast fragmenty między helikalne literami 2 odcinków helikalnych, które one łączą. Poszczególne aminokwasy określa się za pomocą litery oznaczającej hełiks, w którym dana reszta występuje, oraz liczby wskazującej jej pozycję od końca N w tym heliksie. Na przykład „His F8" odpowiada 8, reszcie w heliksie F zidentyfikowanej jako histydyna. Odległe w rozumieniu struktury pierwszorzędowej reszty aminokwasowe (np. w różnych heliksach) ze względu na ułożenie przestrzenne łańcucha polipeptydowego mogą znajdować się blisko
72 / ROZDZIAŁ 7
His praksymalna (F8)
siebie, jak na przykład histydyna F8 (proksyma-lna) i histydyna E7 (dystalna) {ryc. 7-3).
Drugorzędowa i trzeciorzędowa struktura mioglobiny w roztworze ściśle odpowiada strukturze mioglobiny krystalicznej. Wykazują one identyczne widma absorpcji, obie wiążą tlen. a zawartość cc-heliksów w strukturze mioglobiny w roztworze (oznaczona za pomocą dyspersji skręcalności optycznej i dichroizmu kołowego) jest porównywalna z zawartością ujawniony przez analizę rentgenograficzną.
Pierwszorzędowa struktura a po mioglobiny zapewnia prawidłowe upakowanie białka w obecności hetnu Przekształceniu mioglobiny w pozbawioną hemu apomioglobine, wskutek obniżenia pH do 3,5, towarzyszy zmniejszenie zawartości struktury a-helikalnej, a w obecności mocznika całkowity jej zanik. Usunięcie mocznika poprzez dializę i dodanie hemu przywraca całkowicie strukturę helikalną, a w obecności Fe2+ aktywność biologiczną (wiązanie tlenu). Wskazuje to, że informacja zawarta w strukturze pierwszo-rzędowej apomioglobiny jest odpowiedzialna za swoiste upakowanie białka w obecności hemu. Stwierdzenie, że struktura pierwszorzędowa białka determinuje jego strukturę drugo- i trzeciorzędową okazało się być uniwersalne.
Histydyny F8 i E7 odgrywają unikatową rolę w wiązaniu tlenu przez mioglobinę
W mioglobinie grupa hemowa znajduje się w zagłębieniu między heliksem E i F (ryc. 7-3). Polarne, propionianowe łańcuchy boczne hemu są skierowane na zewnątrz cząsteczki mioglobiny, a jego pozostała część znajduje się we wnętrzu białka, gdzie, z wyjątkiem His_F_8 j.His Ę7, otaczają go reszty niępolarne. Atom żelaza piąj^mwiąza niemkoordvnacvjnvm wiążesie z pierścieniem imidazoTowym His F8 określanej mianem histydyny proksymalnej (ryc. 7-4). Po drugiej stronie płaszczyzny hemu w stosunku do His F8 znajduje się His E7, określaną mianem histydyny dystalnej, która jednak nie zajmuje szóstej pozycji koordynacyjnej żelaza (ryc. 7-4).
Atom żelaza umiejscowiony nieco poza płaszczyzną hemu przesuwa się w jej kierunku po związaniu tlenu
W mioglobinie pozbawionej tlenu atom żelaza jest wysunięty o ok. 0,03 nm (0,3 A) poza płaszczyznę hemu w kierunku His F8. W przy-
His dystalna (B7)
Ryc. 7-4. Wiązanie tlenu z żelazem hemowym
podczas reakcji utlenowania. W reakcji tej istotną
rolę odgrywają pierścienie imidazolowe 2 reszt
histydynowych globiny, łączące się z żelazem he
mowym. (Reprodukcja za zgodą z: Harper H. A.
i wsp., Physiologische Chemie, Springer-Verlarg,
1975).
padku utlenowanej mioglobiny, w którejjjm, zajmuje szósta, pozycje koordynacyjną, żelazo jeśT~ wy sunięte poza płaszczyznę hemu tylko ook. 0,01 nm(0,l A). Utlenowaniu mioglobiny towarzyszy więc ruch atomu żelaza, a w konsekwencji His F8 i reszt kowalencyjnie połączonych z His F8, w stronę płaszczyzny grupy pro-stetycznej. Skutkiem tego zjawiska jest zmiana konformacji części białka.
Apomioglobina zmniejsza powinowactwo żelaza hemowego do tlenku węgla
W utlenowanej mioglobinie wiązanie między atomem tlenu i Fei+ jest prostopadłe~db płaszczyzny nemu. Drugi atom tlenUj w stosunku do płaszczyzny hemu, jest przyłączony natomiast pod kątem 121 stopni z dala od histydyny dystalnej (ryc. 7-5).
Około 25 000 razy silniej niż tlen do wyizolowanego hemu wiąże się tlenek węgla (CO). Chociaż CO znajduje się w atmosferze w ii oś-
BIAŁKA: MLOGLOBINA I HEMOGLOBINA / 73
O III
O I
c
I
Ryc. 7-5. Preferowane kąty wiązania tlenu i tlenku węgla do atomu żelaza w hemie.
ciach śladowych, a podczas prawidłowego kata-bótizmu hemu powstają jedynie niewielkie jego ilości, nasuwa się pytanie, jak to się dzieje, że O2, a nie CO zajmuje szóste miejsce koordynacyjne w żelazie hemu mioglobiny. Przyczyną jest przestrzenna zawada, jaką stanowi otoczenie "Fiemu w mioglobinie. Preferowane ułożenie CO przyłączonego do żelaza hemowego jest dla wszystkich 3 atomów (Fe, C. O) prostopadłe w stosunku do płaszczyzny hemu (ryc. 7-5). Podczas gdy taka orientacja jest możliwa w^przypadku wyizolowanego hemu, w mio-globinie histydy na dystalna stanowi przestrzenną zawadę dla wiązania CO pod tym kątem (ryc. 7-6). Powoduje to, że CO wiąże się w mniej korzystriejTcbnfiguracji, co zmniejsza siłę wiązania hem-CO o ponad 2 rzędy wielkości, do wartości ok. 200 razy przewyższającej wiązanie hem-O2. Pomimo tego w warunkach fizjologicznych niewieika część mioglobiny (ok. 1%) występuje w formie karboksymioglobiny.
KRZYWE DYSOCJACJI TLENOWEJ MIOGLOBINY I HEMOGLOBINY OBRAZUJĄ ICH ODMIENNE FUNKCJE FIZJOLOGICZNE
Mioglobina jest białkiem magazynującym, a nie"transportującym"tlen.llość_przyłączonego do mioglobiny tlenu (wyrażona w „procentach nasycenia") zakżv od jego stężenia (wyrażonego jako pO2, czyli stężenie cząstkowe tlenu) w nąjbliższyjn_gtoczeniu żelaza hemowe^o. Zależność międzypbi i ilością związanego tlenu obrazują graficznie krzywe dysocjacji tlenowej. Dla mioglobiny krzywa dysocjacji tlenowej w. warunkach izotermicznych ma kształt hiperboli (ryc. 7-7). W naczyniach włosowatych płuc,
Ryc. 7-7. Krzywa dysocjacji tlenowej mioglobi
ny. Na uwagę zasługuje zależność stopnia utleno-
wanra i ciśnienia cząstkowego tlenu w płucach
(100 mm Hg), tkankach (20 mm Hg) i pracujących
mięśniach (5 mm Hg).
Ryc. 7-6. Kąty wiązania tlenu i tlenku węgla do atomu żelaza hemu w mioglobinie, Histydyna dystalna E7 stanowi przestrzenną zawadę dla wiązania CO pod preferowanym w stosunku do płaszczyzny hemu kalem (180°),
w których pOj wynosi 100 mm Hg, mioglobina mogłaby skutecznie wiązać tlen. Ponieważ jednak wartość pO2 w żyłach i mięśniach wynosi odpowiednio 40 i 20 mrn_Hgj a mioglobina nie oddaje znacznej części związanego tlenu nawet przy ciśnieniu 20 mm Hg nie może ona służyć jako przenośnik tlenu z płuc do tkanek. Dopiero w warunkach niedoboru tlenu, towarzyszącego dużej aktywności fizycznej, kiedy pO, w mięśniach spada do 5 mm Hg, mioglobina uwalnia związany tlen, który w mitochondriach mięśni jest wykorzystywany do biosyntezy ATP w wyniku fosforylacji oksydacyjnej.
74 / ROZDZIAŁ 7
HEMOGLOBINA TRANSPORTUJE TLEN, DWUTLENEK WĘGLA I PROTONY MIĘDZY PŁUCAMI I TKANKAMI
Hemoglobina, zawarta w erytrocytach kręgowców, spełnia dwie główne funkcje biologiczne; 1) przenosi O2 z płuc do tkanek i 2) przenosi CO2 i protony z tkanek do phic w celu wydalenia. Mimo że biochemia porównawcza hemoglobin kręgowców jest bardzo interesująca, przedmiotem dalszych rozważań są hemoglobiny ludzkie.
KONSEKWENCJĄ
CZWARTORZĘDOWEJ STRUKTURY HEMOGLOBINY SĄ JEJ WŁAŚCIWOŚCI ALLOSTERYCZNE
Właściwości poszczególnych hemoglobin są prawidłowością wynikającą z ich struktury czwartorzędowej (jak również drugo- i trzeciorzędowej). Czwartorzędowa struktura hemoglobiny jest odpowiedzialna za nowe, dodatkowe w stosunku do mioglobiny, właściwości, które warunkują jej unikatową funkcję biologiczną i pozwalają na jej precyzyjną regulację. Właściwości allosteryczne (gr. allos — inna, steros — przestrzeń) hemoglobiny stanowią ponadto model do zrozumienia innych białek allosterycznych.
W przeciwieństwie do mioglobiny, hemogtobina jest tetramerem
Hemoglobina w odróżnieniu od jednołańcu-chowej mioglobiny nie mającej struktury czwartorzędowej, jest tetramerem składającym się z 2 par identycznych łańcuchów polipeptydo-wych, czyli podjednostek (określanych jako a, p, 7,8, S itd.). Podobne pod względem długości polipeptydy a (14l) reszt aminokwas owych) i P (146 jeszt aminokwasowych) hemoglobiny A (HftA) są kodowane przez różne geny i mają odmienną strukturę pierwszorzedową. W przeciwieństwie, łańcuchy p, y i 5 hemoglobin ludzkich charakteryzuje duża konserwatyw-ność ich sekwencji. Najczęściej występujące hemoglobiny mają następujący skład tetrame-ru: HbA j (podstawowa hemoglobina prawidłowa u ludzi dorosłych) = ąj^, ^^(hemoglobina płodowa) = a2Y2, HBS ^hemoglobina sierpowatych krwinek czerwonych) = <x2S2, HbA2 (hemoglobina prawidłowa u ludzi doros-
łych stanowiąca ok. 2,5%* Hb całkowitej) =
Mioglobina i podjednostki
P hemoglobiny mają praktycznie
identyczną strukturę drugorzędową
1 trzeciorzędową
Pomimo różnic w rodzaju i liczbie aminokwasów, mioglobina i łańcuchy polipeptydowe P HbA wykazują praktycznie identyczną strukturę drugorzędową i trzeciorzędową. To ścisłe podobieństwo, dotyczące również umiejscowienia hem u i odcinków helikalnych, jest przynajmniej w części wynikiem substytucji aminokwasów o podobnych właściwościach i równoznacznych pozycjach w strukturze pierwszo-rzędowej mioglobiny i podjednostki P HbA. Nawet występowanie 7 a nie 8 odcinków helikalnych w łańcuchu P nie zmienia zasadniczego podobieństwa strukturalnego tych polipepty-dów. Podobnie jak w przypadku mioglobiny, zarówno podjednostki ot, jak i p HbA charakteryzuje obecność hydrofobowych reszt aminokwasowych wewnątrz cząsteczki (z wyjątkiem
2 reszt His w każdej podjednostce), a hydro-
filowych na zewnątrz cząsteczki.
Uttenowaniu hemoglobiny towarzyszy wsunięcie żelaza w płaszczyznę hemu oraz zmiana konformacji sprzężonej apoproteiny wywołana przemieszczeniem histydyny proksymalnej
Tetramcryczna hemoglobina wiąże 4 cząste-czkftlenu (hem każdej podjednostki wiąże jedną cząsteczkę tlenu), a jej krzywa dysocjacji tlenowej ma kształt sigmoidalny (ryc. 7-8). Przyłączenie O2 przez jeden z hemów ułatwia wiązanie kolejnych O2 przez pozostałe grupy hemowe. To kooperatywne wiązanie tlenu z hemoglobiną jest właściwością pozwalającą na wiązanie maksymalnej ilości O2 w płucach i uwalnianie maksymalnej ilości O3 w tkankach (ryc. 7-8).
Miarą powinowactwa hemoglobiny do tlenu jest wielkość Pso, odpowiadająca ciśnieniu cząstkowemu tlenu, przy którym występuje 50% nasycenia Oa
Wartość P;o jest różna dla różnych organizmów, przy czym jest ona zawsze większa od wartości pO2 w tkankach badanych organiz-
* Przyp. tłum.
BIAŁKA; MIOGLOBINA I HEMOGLOBINA / 75
Utlenowana krew
opuszczająca płuca
Odtlenowana krew powracająca z tkanek
Hemoglobina 1,1
140
20 40 60 80 100 120
Ciśnienie cząstkowe tlenu (mm Hg)
Ryc. 7-8. Krzywedysociacjitlenowej hemoglobiny i mioglobiny. Ciśnienie cząstkowe tlenu w krwi tętniczej wynosi ok. 100 mm Hg; wkrwiżylnej—40 mm Hg; w naczyniach włosowatych — 20 mm Hg; minimum niezbędne dla funkcji cytochromów — 5 mm Hg. Połączenie łańcuchów polipeptydo-wych w strukturę tetrameryczną (hemoglobina) pozwala na zwiększenie, w porównaniu z pojedynczymi łańcuchami, wydajności dostarczania tlenu. (Zmodyfikowana i reprodukowane za zgodą z: StanburyJ, B,,Wyngaarden J. B., Fredrickson D.S. [red.], The Metabolic Bas/s of Inherited Disease, wyd.4, McGraw-Hill, 1978).
Ryc. 7-9. Wiązania poprzeczne międ2y podjed-
nostkarni oraz w obrębie podjednostek nieutleno-
wanej hemoglobiny. Podczas utlenowania te nie-
kowalencyjne, elektrostatyczne wiązania zostają
rozerwane, {Zmodyfikowana i reprodukowana za
zgodą z: Stryer L: Biochemistry. wyd. 2, Freeman,
1981).
go-, trzecio- i czwartorzędowej strukturze białka. Jedna para podjednostek a/P obraca się w stosunku do drugiej pary a/&\ w wyniku czego następuje zbliżenie podjednostek tetramem i zwiększenie powinowactwa hemów do tlenu (ryc, 7-10 i 7-11).
15°
Postać R
mów, czego dobrym przykładem jest ludzka hemoglobina płodowa (HbF). Wartość P50 dla HbA = 26 mm Hg, podczas gdy dla HbF = 2(1 mm Hg. Ta różnica w powinowactwie Hbl i HbA do tlenu umożliwia HbF pobieranie tlenu od HbA w obrębie łożyska. Po porodzie HbF przestaje właściwie spełniać swoją funkcję ponieważ jej duże powinowactwo do O2 utrudnia jego oddysocjowywanie w tkankach.
Najwcześniej w rozwoju osobniczym człowieka są syntetyzowane łańcuchy £ i e. Pod koniec pierwszego trymestru ciąży łańcuch £ zastępuje podjednostka et, a łańcuch e — podjednostka7. Hemoglobina F, pojawiająca się więc w tym okresie życia płodowego, jest zbudowana według wzoru d2 y . Podjednostka % której synteza rozpoczyna się w trzecim trymestrze ciąży, całkowicie zastępuje łańcuch y dopiero kilka tygodni po porodzie.
Utlenowamu hemoglobiny towarzyszą zmiany konformacji białka
PrzyJaszernu O2 towarzyszy rozerwanie wiązań poprzecznych pomiędzy końcami karbo-ksylowymi wszystkich 4 podjednostek hemoglobiny (ryc. 7-9), co powoduje zmiany w dru-
cc, S |
\8 |
1 / |
\ |
i |
V ' |
i |
1 |
|
1 |
. \ |
/ |
|
/ 6 |
|
/ |
|
s ■~~- -■ |
Postać T
Rvc. 7-10. W czasie przejścia postaci T w postać R hemoglobiny jedna para podjednostek (a2/f>2) obraca się o 15° w stosunku do drugiej pary (a1/fl]). Ponieważ oś obrotu jest niewspółśrodkowa, para at2/P2 przesuwa się również nieco w kierunku osi. Na diagramie para a,/^ utrzymująca się w stałej pozycji jest niezaciemniona, a para a2/P: dokonująca obrotu i przesunięcia jest zaciemniona.
Czwartorzędową strukturę częściowo utleno-wanej hemoglobiny określa się jak o stanT(ang. taut — naprężony), a całkowicie utlenowanej hemoglobiny (HbOj) jako stan R (ang. relaxed
76 / ROZDZIAŁ 7
Odtlenowanie (posiać T)
— rozluźniony) (ryc. 7-12). Symbole R i T są również używane w celu określenia czwartorzędowej struktury enzymów allosterycznych, gdzie forma T wykazuje mniejsze powinowactwo do stibstratu.
Utlenowanie hemoglobiny prowadzi
do zmian konformacyjnych
w bezpośrednim otoczeniu grupy
hemowej
Podczas utlenowania hemoglobiny atomy żelaza (Mace ok. 0,06 nm poza płaszczyzną hemów w nieutlenowanej hemoglobinie) wsuwają się w płaszczyznę hemów, pociągając za sobą histydynę proksymalną (F8) i połączone z nią reszty aminokwasowe (ryc. 7-13).
Pod jednostka /)t
Podjednostka a.
TyrC7|«)
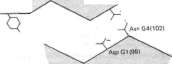
Ryc. 7-11. Zmiany oddziaływań w obrębie ot,/p,
podczas utlenowania. Przesunięcie zazębiających
się obszarów powoduje zmianę miejsc oddziały
wań i „przełączanie" jednych wiązań wodorowych
na inne. Pozostałe wiązania mają charakter niepolar-
ny. (Reprodukcja za zgodą z: Perutz M. F.: Molecu-
lar pathology of human hemoglobin: stereochemi-
cal interpretation of abnormal oxygen affinities,
Naturę 1971; 232:408).
AspG1(94!
Podjednostka f)2
Utlenowanie (postać R)
Postać T
Postać R
Ryc. 7-12. Przejście postaci T w postać R zwiększa prawdopodobieństwo utlenowania każdego
z 4 hemów. Przekształcenie to nie następuje z chwilą przyłączenia określonej liczby cząsteczek tlenu, lecz
dokonuje się wraz z utlenowaniem kolejnych hemów. Stopniowe przyłączanie tlenu powoduje pękanie
(linie cienkie) i osłabianie (linie wężykowate) wiązań poprzecznych łączących podjednostki w postaci T.
Przejście między tymi dwoma postaciami pozostaje pod wpływem różnych czynników, takich jak: protony,
dwutlenek węgla, chlorki, BPG. Im większe stężenie tych czynników, tym więcej tlenu musi zostać
związane, aby zapoczątkować przekształcenie. Schemat nie zawiera całkowicie utłenowanej cząsteczki
w postaci T oraz całkowicie nieutlenowanej cząsteczki w postaci R, ponieważ są one zbyt nietrw'ałe, aby
istnieć w znaczącej liczbie. (Zmodyfikowana i przerysowana za zgodą z: Perutz M. F,: Hemoglobin
structure and respiratory transport, Sci. Am. [Dec], 1978; 239:92),
BIAŁKA: MIOGLOBINA I HEMOGLOBINA / 77
Hlslydyna FB
HeliKs F
Odpychania ' przestrzenne I
Płaszczyzna nam u
Ryc. 7-13. Podczas utlenowania atom żelaza wsuwa się w płaszczyznę hemu. Razem z atomem źeiaza przemieszcza się histydyna F8 i związane z nią reszty aminokwasowe. (Zmodyfikowana i reprodukowana za zgodą z: Stryer L: Biochemistry, wyd. 2. Freeman, 1981).
Po uwolnieniu tlenu w tkankach hemoglobina transportuje dwutlenek węgla i protony do płuc
Oprócz transportu tlenu z płuc do tkanek, hemoglobina bierze udział w przenoszeniu COj
z tkanek do płuc, skąd jest on usuwany podczas oddychania. Bezpośrednio po odłączeniu tlenu, cząsteczka hemoglobinjL.^dąiŁ_CQs_ w ilości stanowiącej~oJć7l5% CO2 transportowanego . Ponadto z chwilą za"SBsorbtyw"ania
pj
COj przez Ićrew dehydrataza węglanowa erytrocytów katalizuje utworzenie kwasu węgiowe-go, który dysocjuje do wodorowęglanu i protonu na skutek przesunięcia równowagi reakcji w kierunku dysocjacji (ryc. 7-14). Funkcję układu buforowego, zabezpieczającego organizm
HCO.-+ H+
co,+
Ryc. 7-14. Tworzenie kwasu węglowego pod wpływem dehydratazy węglanowej i jego dysocja-cja do jonu wodorowęglanowego i protonu.
przed szkodliwym wpływem zwiększenia kwasowości krwi, pełni m.in. hemoglobina._QjL-, łączeniu 4 cząsteczek tlenu z hemoglobiny towarzyszy wiązanie 2 protonów. W płucach natomiast zachodzi zjawisko odwrotne, tj. przyłączenie tlenu do hemoglobiny powoduje uwolnienie protonów. Uwolnione-prolony łączą się z wodorowęglanem, tworząc kwas węglowy, który pod wpływem dehydratazy węglanowej przekształca się w CO2, usuwany w procesie oddychania. Stad wiązanie tlenu zwiększa wydychanie CO2. To odwracalne zjawisko nosf nazwę efektu Bohra (ryc. 7-l5)7EfekTBohTa jest właściwością
Wydychania
2HCO,"
JD.2H
(Buforujące działania
— hemoglobiny)
Płuca
40
Tkanki
Cykl kwasów
trlkarboksylowych
(Krebsa)
Ryc. 7-15. Efekt Bohra. Powstający w tkankach dwutlenek węgla laczy się z wodą, tworząc kwas węglowy, który dysocjuje do protonu i jonu wodorowęglanowego. Nieutlenowana hemoglobina wiąże protony i transportuje je do płuc. W płucach, w wyniku przyłączania tlenu, następuje uwalnianie protonów, które łącząc się z jonem wodorowęg-lanowym tworzą kwas węglowy, przekształcany przez dehydrataze węglanową do dwutlenku węgla usuwanego w procesie oddychania.
charakterystyczną dla tetramerycznej hemoglobiny, 7al1?iB[l ffl jnterakcji hem-hem, czyli kooperatywności wiązania tlenu. Efektu Bohra nie obserwuje się w przypadku mioglobi-ny.
78 / ROZDZIAŁ 7
Rozerwanie wiązań poprzecznych
podczas przyłączania tlenu
do hemoglobiny w postaci T dostarcza
protonów odpowiedzialnych za efekt
Bohra
Protonv_odpowiedzialne za elekt Bohra powstają w wyniku rozerwania \\ i a&łjl „poprzecznych podczas przyłączania tlenu do postaci T, Sa one głównie uwalniane z atomów N pierścieni imidazolowych C-końcowych reszt his-tydyny łańcuchów P HC3 (146). Uwolnione pmtnny pye kształcą ją wodorowęglan w kwaś" węglowy, usuwany pkn m, w naczyniacF
)
węglowy, usuway p
włosowatych pęcherzyków nłucnych fryc. 7-15). Natomiast podczas odłączania tlenu tworzenie wiązań poprzeczaych, pjaslaci T hemoglobiny wymaga przyłączenia protonów do reszt HC3 łańcuchów p. oiwnrrer protonów w tkankach ułatwia więc tworzenie wiązań poprzecznych na skjJteYprotonacji końcowych/reszt Hisw pod-jednostkach p. Odtworzenie wiązań poprzecznych sprzyja wiec uwalnianiu łferm z, ufleno'-wanej (postajLKLJiamo.globiny. Uogólniając, /większenic stężeniajworpntm; powoduje odjacze-nSjttnu.JJOticS^gdy zwiększenie stężenia tlenu powoduje odłączenie protonów. Zależność tęorj-razuje przesunięcie krzywej dysocjacji tlenu na prawo w wyniku zwiększenia zawartości jonów wodorowych (protonów).
W centralnej wnęce cząsteczki nieutlenowanej hemoglobiny 2,3-bisfosfoglicery niań (BPG) tworzy wiązanie poprzeczne, stabilizujące strukturę T
W warunkach niedoboru tlenu w tkankach zwiększa się zawartość 2,3-bisfosfogIicerynianu (BPG) (ryc. 7-16). Związek ten tworzy się
z 1,3-bisfosfoglicerynianu, będącego metabolitem pośrednim glikolizy. BPG wiąże się z hemoglobiną w stosunku 1 cząsteczka BPG na tet-rameryczną cząsteczkę hemoglobiny, a miejscem wiązania jest przestrzeń między 4 podjed-nostkami, znajdująca się w centrum cząsteczki hemoglobiny. Rozmiar tej wnęki jest odpowiedni dla BPG tylko w przypadku postaci T, tj. wtedy, kiedy przestrzeń między heliksami H łańcuchów p jest wystarczająco duża. BPG przyłącza się za pomocą wiązań poprzecznych, twofzą-ćycn się pomiędzy atomami tlenu BPG i dodatnio naładowanymi resztami Val NA1 (grupy aminowe N-koricowe), Lys EF6 oraz His H21 łańcuchów p (ryc. 7-17). BPG stabilizuje
Ryc. 7-17. Sposób wiązania 2,3-bisfosfogltcery-nianu z nieutlenowaną hemoglobiną człowieka. BPG oddziałuje z 3 dodatnio naładowanymi grupami każdego z łańcuchów fi. (Reprodukcja za zgodą z: Arnone A,: X-ray diffraction siudy of binding 2,3-diphosphoglycerate to human deoxyhemoglo-bin. Naturę 1972; 237:146).
Ryc. 7-16.
(BPG).
![]()
o-o-
p
\\
o
Struktura 2,3-bisfosfoglicerynianu
więc postać T lub nieutlenowaną postać hemoglobiny przez tworzenie wiązań poprzecznych
w obrębie łańcuchów p oraz dostarcza dodatkowe wiązania poprzeczne, które muszą ulec zerwaniu przy przechodzeniu postaci T w postać R.
Wiązanie BPG z hemoglobiną płodową jest znacznie słabsze niż z hemoglobiną Judzi dorosłych, ponieważ zamiast His resztą H2T w łańcuchu y hemoglobiny płodowej jest seryna, która nie tworzy wiązań poprzecznych z BPG.
BIAŁKA: MIOGLOBINA I HEMOGLOBINA / 79
wactwa do tlenu (np. hemoglobina Chesapeake). Dostarczanie wskutek tego zbyt małej ilości tlenu do tkanek powoduje hipoksje, która prowadzi do policytemii (zwiększenie liczby erytrocytów).
BPG ma więc mniejszy wpływ na stabilizację formy T w przypadku hemoglobiny płodowej, co
powoduje jej większe w porównaniu z hemoglobiną ludzi dorosłych, powinowactwo do tlenu.
Przejście postaci T w postać R hemoglobiny i odwrotnie jest wyzwalane przez ruch żelaza do wewnątrz i na zewnątrz płaszczyzny układu por-firynowego. Przejście to jest uruchamiane przez czynniki oddziałujące przestrzennie i elektro-statycznie przy nakładzie energii ok. 12 560 J/mol (3000 cal/mol). Niewielka zmiana pozycji Fe2+ w stosunku do płaszczyzny układu po-rfirynowego indukuje więc znaczne zmiany w konformacji hemoglobiny, wpływając decydująco na jej funkcje biologiczną w odpowiedzi na sygnały środowiska.
ZIDENTYFIKOWANO KILKASET MUTANTÓW HEMOGLOBINY WWIĘKSZOŚCI NIE OBJAWIAJĄCYCH ZABURZEŃ FUNKCJI
Mutacje genów kodujących łańcuchy ot lub P mogą wpływać na biologiczną funkcje hemoglobin. Poniżej opisano kilka, spośród kilkuset znanych (w większości łagodnych), mutantów hemoglobin Judzkich o zmienionej funkcji biologicznej, Stan, w którym w wyniku mutacji następuje zaburzenie funkcji biologicznej hemoglobiny określa się mianem hemoglobino-pata.
W hemoglobinach typu M proksymalna lub dystalna reszta histydyny w podjednostkach ot lub ji jest zastąpiona resztą tyrozyny
Zastąpienie proksymalnej lub dystalnej histydyny resztą tyrozyny przyczynia się do stabilizacji żelaza hemowego w formie Fe3+, na skutek tworzenia przez niego trwałego połączenia z anionem fenolanowym Tyr. Obecność hemu w formie żelazowej nie wiążącej 02 powoduje met hemoglobinemię. W przypadku wariantów hemoglobin M dotyczących łańcuchów a obserwuje się przesunięcie równowagi przejścia R-T w kierunku formy T, zmniejszenie powniowaćTwa do tlemi i zniesienie efektu Boh-raTNatomiast w przypadku wariantów dotyczących łańcuchów P efekt Bohra jest zachowany, ponieważ nie ulega zakłóceniu przejście R-T.
Mutacje, które sprzyjają tworzeniu się postaci R charakteryzują się zwiększeniem powino-
W hemoglobinie Sj^ęsztŁ l
gl j^ęs
glutaminowego w pozycji 6 łańcucha
{> jest zastąpiona resztą waliny
Hemoglobina S powstaje w wyniku zastąpie-uiśLCjju A2 (ó)p\ tj, reszty 6 w łańcuchu (3 przez ^al. Reszta A2 (Glu lub Val) znajduje się na powierzchni cząsteczki, odpowiadając za jej kontakty z wodą. Zastąpienie polarnego kwasu glutaminowego niepolarną waliną powoduje powstanie na powierzchni łańcucha P „lepkich miejsc". „Lepkie miejsca" występują zarówno w utlenowanej, jak i nieutlenowanej postaci hemoglobiny S; nie ma ich n^ bT
glojbinic: AJJ koTei na powierzchni hemoglobiny nieutlenowanej występują miejsca komplementarne do „lepkich miejsc". Są one niedostępne w hemoglobinie utlenowanej (ryc. 7-18).
IMieutlenowana hemoglobina S
może tworzyć włókna,
które zniekształcają erytrocyty
„Lepkie miejsca" nieutlenowanej hemoglobinySmogą łączyć się z miejscami komplementarnymi innej cząsteczki nieutlenowanej hemoglobiny. Oddziaływania te powodują polimeryzację nieutlenowanej hemoglobiny S, w wyniku czego powstają długie, wytrącające się agregaty zniekształcające erytrocyty (erytrocyty przybierają kształt sierpowaty), odpowiedzialne za ich liżę i inne objawy kliniczne. Utrzymanie hemoglobiny S w postaci utlenowanej lub zmniejszenie do minimum jej stężenia w postaci nieutlenowanej zapobiegałoby tworzeniu się polimerów, a tym samym sierpowatości krwinek. Jest bowiem wiadomo, że polimeryzacji ulega postać T hemoglobiny S. Interesujący, chociaż nie do zastosowania terapeutycznego, jest fakt, że forma żelazowa hemoglobiny S (methemoglobi-na S) nie tworzy polimerów. Podobnie bowiem jak w przypadku methemoglobiny A, jon żelazowy pozostaje w płaszczyźnie układu porfiryno-wego, stabilizując strukturę R hemoglobiny.
Chociaż nieutlenowana hemoglobina A zawiera miejsca komplementarne do „lepkich miejsc" występujących w utlenowanej i nieutlenowanej hemoglobinie S, to jej przyłączenie do hemoglobiny S uniemożliwia dalsze wy-dhiżanie polimerów na skutek braku na jej
80 / ROZDZIAŁ 7
Utleń owa na A
Nieutlenowana A
U tlen owa na S
Nieutlenowana S
lllllllllll
|
a. |
a |
e |
Nieutlenowana A Nieutlenowana S
Ryc. 7-18. Schemat przedstawiający „lepkie miejsca" (A) w hemoglobinie S i ich „receptory" wnieutlenowanej hemoglobinie A i S. Komplementarność oddziałujących powierzchni powoduje faczenie się n te utleń owa n ej hemoglobiny S w polimery. Wydłużanie polimerów przerywa nieuttenowana hemoglobina A nie zawierająca „iepkich miejsc". (Zmodyfikowana i reprodukowana za zgodą z: Stryer L: Biochemistry, wyd. 2, Freeman, 1981).
6,2 nm
powierzchni „iepkich miejsc" sprzyjających dalszemu wiązaniu (ryc. 7-18). Przyłączenie nieu-tlenowanej hemoglobiny A do hemoglobiny S zarówno w formie R, jak i T stanowi barierę do dalszej polimeryzacji.
W,_wyniku agregacji nieutlenowanej hemoglobiny S tworzą się włókna o helikalnej strukturze, w których każda cząsteczka hemoglobiny oddziałuje z 4 przylegającymi cząsteczkami (ryc. 7-19). Włókna te deformują erytrocyty tak, że przybierają ońlTEsżtałt sieTpowaty (ryc. 7-20). Krwinki te wykazują zwiększoną podatność do hemolizy w czasie przechodzenia przez zatoki śledzionowe.
Talasernie są niedokrwistościamj charakteryzującymi się upośledzeniem syntezy łańcuchów cc lub p hemoglobiny
W talasemiach zmniejsza się synteza łańcuchów a (a-talasemie) lub łańcuchów p (fi-ta-lasemie) hemoglobiny. Wywołuje to często groźną w skutkach niedokrwistość. Wyniki badań ostatnich lat wniosły duży wkład do naszej wiedzy o molekularnych mechanizmach odpowiedzialnych za talasernie.
RyC-7-19, Proponowana helikaIna struktura włókna utworzonego w wyniku agregacji nieutlenowa-nej hemoglobiny S, (Reprodukcja za zgodą z: Maugh T. II: A new understanding of sickle celi emerges. Science 1981; 211:265, Copyright © 1981 by the American Associatiort for the Advancement of Science).
BIAŁKA: MI0GL0B1NA I HEMOGLOBINA / 81
Ryc. 7-20. Obraz erytrocytu prawidłowego (A) oraz sierpowatego (B)w skaningowym mikroskopie elektronowym. Obserwowane różnice strukturalne są wynikiem zmiany w obrębie łańcuchów (i będącej skutkiem mutacji punktowej w DNA. Zamiana T na A powoduje, że w łańcuchu {J zamiast kwasu glutaminowego znajduje się walina.
PIŚMIENNICTWO
Bunn HF, Forget BG: Hemoglobin: Molecular, Gene-tic, and Ciinical Aspects, Saunders, 1986.
Dickerson RE, Geis I: Hemoglobin. Benjamin/Cum-mjngs, 1983.
Embury SH: The ciinical pathology of sickle-ceil disease. Anmt Rcv Med 1986;37:36[.
Embury SH, Scharf SJ, Saiki RK, Gholson MA, Golbus M, Arnheim N, Erlich HA: Rapid prenatal diagnosis of sickle celi anemia by a new method of DNA analysis. New Engl J Med 1987;316:ó56.
Fermi G, Perutz MF, Shaanan B, Fourme R: The crystal structure of hunian deoxyhemoglobin at 1.74 angstrom resolution. J Mol Biol I984;175: 159.
Friedman JM: Structure, dynamics, and reactivity in hemoglobin. Science 1985;228:1273.
Saiki RK, Scharf S, Faloona F, MuBis KB, Horn GT, Erlich HA, Arnheim N: Enzymatic amplification of beta-globin genomic: seąuences and restriction site analysis for diagnosis of sickle-cell anemia. Science, 1985;230:1350.
Schacter L, Wartti JA, Gordon EM, Prasad A, Klein BL: Altered araount and activity of superoxide dismutase in sickle celi anemia. FASEB J 198S;2:237.
Weatherall DJ, Clegg JB, Higgs DR, Wood WG: The hemoglobinopathies. In The Metabolk Basis of InheritedDisease, 6th Ed. Serwer CR, Beaudet AL, Sly WS, Valle D (editors). McGraw-Hill, p. 2281, 1989.
6 — Biochemia
Wyszukiwarka