Hemostaza (wg Colmana)
Jest to zespół mechanizmów fizjologicznych, które zapewniają:
sprawne hamowanie krwawienia po przerwaniu ciągłości ściany naczyń krwionośnych,
szczelność łożyska naczyniowego,
płynność krążącej krwi.

Hemostaza pierwotna
Jest to wstępna faza aktywacji układu hemostazy:
płytki krwi „rozpoznając” uszkodzenie naczynia tworzą czop płytkowy uwalniając substancje wspomagające układ krzepnięcia,
dodatkowo dochodzi do obkurczenia uszkodzonego naczynia krwionośnego.
Tworzy się skrzep „biały”- pierwotny.
Hemostaza wtórna
Uszkodzona ściana naczynia jest źródłem czynnika tkankowego - TF, który uaktywnia układ krzepnięcia składający się z enzymów (proteaz serynowych) tworzących sprzężony układ generujący trombinę.
Końcowym efektem działania układu jest przekształcenie przez trombinę rozpuszczalnego fibrynogenu w nierozpuszczalną fibrynę. Następuje wzmocnienie czopu płytkowego „białego” i tworzenie czopu hemostatycznego.
Głównymi elementami zapewniającymi hemostazę są:
ściana naczyń krwionośnych,
płytki krwi (inne elementy morfotyczne),
osoczowy układ krzepnięcia,
osoczowy układ fibrynolizy,
układ fagocytarny (siateczkowo-śródbłonkowy).


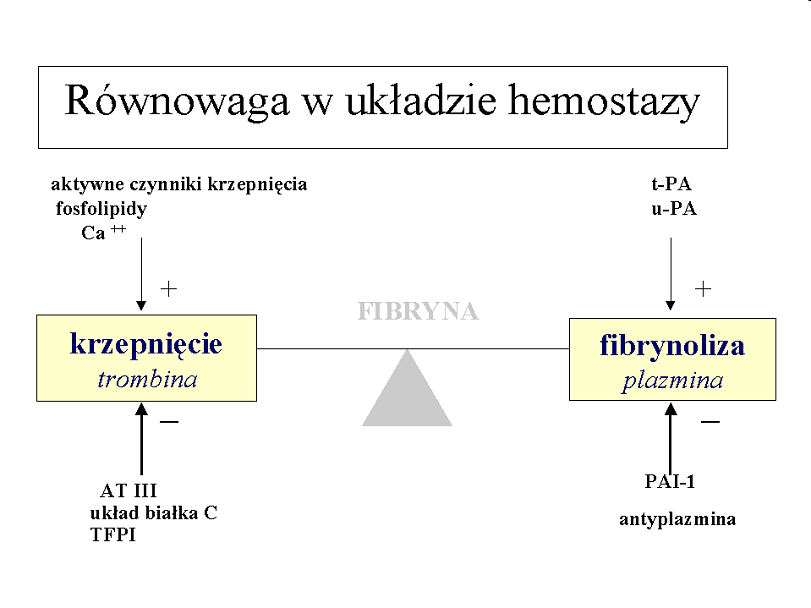
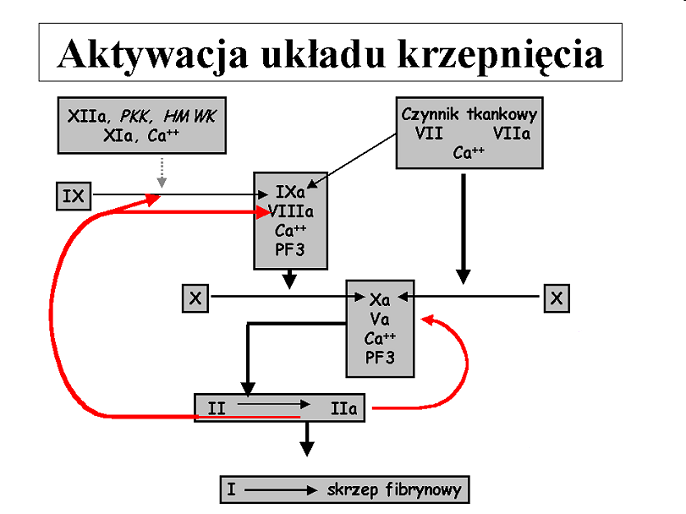
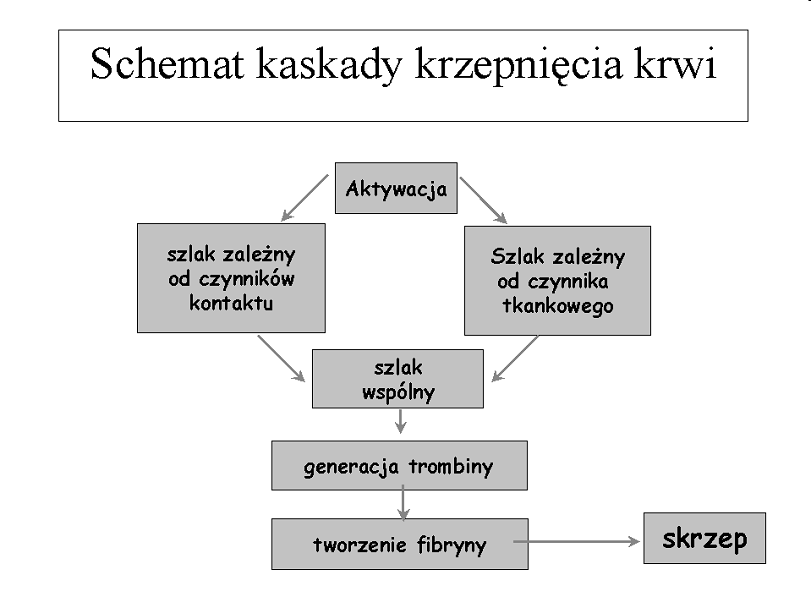

Najważniejsze składniki układu antykoagulacyjnego
1. Ściana naczyń krwionośnych
2. Inhibitory osoczowe układu krzepnięcia
3. Układ fibrynolityczny

Inhibitory osoczowe układu krzepnięcia
Układ białka C
TFPI
ATIII - najlepiej poznany inhibitor krzepnięcia, uaktywniany przez wydzielaną przez komórki tuczne heparynę, inaktywuje wszystkie proteazy krzepnięcia, najsilniej czynnik X i trombinę.
HC II (kofaktor II heparyny) - inhibitor "rezerwowy", inaktywuje głównie trombinę.
- Neksyna 1, hamująca trombiny, plazminy i urokinazy.
- Aneksyna V - białko o wysokim powinowactwie do fosfolipidów, współzawodniczy z czynnikami krzepnięcia o dostęp do fosfolipidów błon komórkowych.

Składowe układu białka C:
- Trombomodulina - białko związane z błoną komórek śródbłonka
- Białko C - występuje w osoczu jako zymogen proteazy serynowej (zależnej od wit. K)
- Białko S - glikoproteina zależna od wit. K, kofaktor białka C
Układ białka C
Działanie układu inicjowane jest przez trombinę przyłączoną do związanej ze śródbłonkiem trombomoduliny. Trombina po związaniu z trombomoduliną nabywa zdolności aktywowania białka C, a to z kolei, w obecności kofaktora, którym jest białko S, degraduje i inaktywuje aktywne czynniki VIII i V. Dodatkowo, aktywne białko C ma własności profibrynolityczne, ponieważ wiąże inhibitor fibrynolizy - PAI-1.
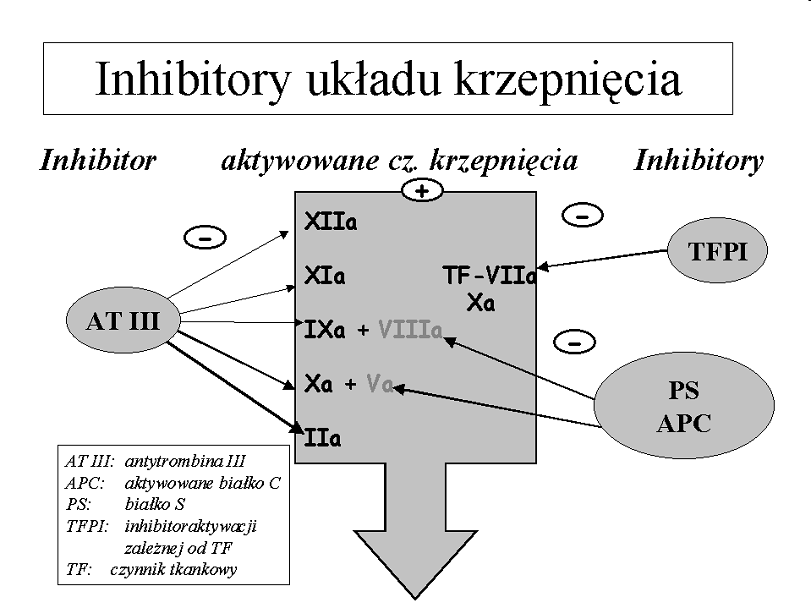
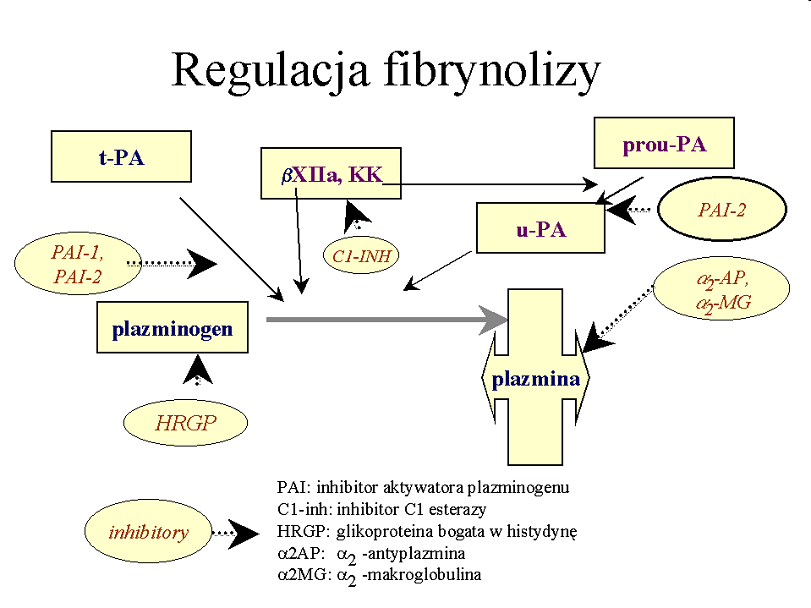
Inhibitory fibrynolizy
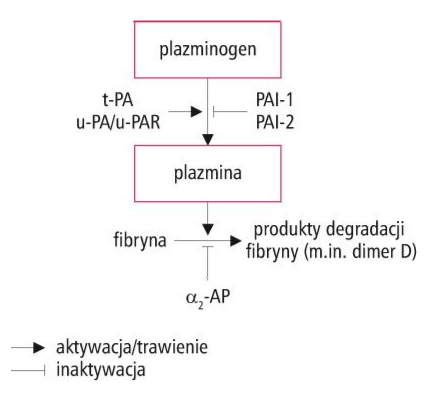
- PAI-1- główny inhibitor aktywatora pazminogenu t-PA. W jego obecności t-PA także jest błyskawiczne kompleksowany. Powstają kompleksy t-PA/PAI-1,
- PAI-2- ma mniejsze znaczenie inaktywuje wybiórczo u-PA.
- a2-antyplazmina- główny osoczowy inhibitor plazminy. W jego obecności wolna plazmina jest natychmiast inaktywowana i powstają kompleksy PAP, znane jako markery aktywacji układu fibrynolotycznego,
- a2-makroglobulina- inaktywuje pozostałą ilość wolnej plazminy (inhibitor rezerwowy),
- C1-inh: inhibitor C1 esterazy blokuje profibrynolityczną aktywność kompleksu czynników kontaktu,
- HRGP: glikoproteina bogata w histydynę, która wiąże plazminogen.
Układ kininogenezy w hemostazie
- Na powierzchni błony komórkowej komórek śródbłonka występuje wieloproteinowy receptor kininogenu (cytokreatyna, receptor urokinazowy+gC1qR0).
- Prekalikreina (PK) i czynnik XI krążą w osoczu związane z wysocząsteczkowym kinininogenem (HK). Kompleks PK - HK wiąże się specyficznie do komórek śródbłonka za pośrednictwem tego receptora w obecności Zn2+. Do kompleksu przyłącza się czynnik XII, przyśpieszając aktywację PK do kalikreiny.
- W obrębie tego kompleksu aktywowany jest także czynnik XI, czyli składnik układu krzepnięcia. W warunkach fizjologicznych ma to małe znaczenie. Aktywacja zależna od czynników kontaktu nabiera znaczenia w czasie kontaktu ze sztucznymi powierzchniami.
W opisanym kompleksie kalikreina uwalnia bradykininę, a ta stymuluje uwalnianie t-PA i syntezę prostacykliny.
Skazy krwotoczne definicja
Są to zaburzenia, w których występuje niewydolność jednej lub kilku składowych ustrojowego układu hemostazy.
Inaczej mówiąc, skaza krwotoczna to skłonność do krwawień:
długotrwałych,
zbyt obfitych,
pojawiających się bez istotnej przyczyny.
Skazy krwotoczne mogą być uwarunkowane genetycznie (wrodzone) lub mają charakter nabyty.

Objawy kliniczne skaz krwotocznych
Naczyniowe skazy krwotoczne
etiologia i patogeneza
Związane są z zaburzeniami budowy i czynności drobnych naczyń krwionośnych - tętniczek, naczyń włosowatych i małych naczyń żylnych.
U podłoża zaburzeń leżą:
wrodzone zaburzenia budowy naczyń,
nabyte uszkodzenie ściany naczyniowej na tle immunologicznym, w wyniku działania toksyn, leków, w przebiegu zakażeń,
zmiany naczyń związane z procesem starzenia, działania urazów mechanicznych, czy też nie znanych dotąd czynników.
Naczyniowe skazy krwotoczne
objawy
Skaza objawia się najczęściej:
wybroczynami, plamicą lub krwotocznymi wykwitami na skórze i błonie śluzowej,
krwawieniami z dziąseł i łatwym siniaczeniem,
rzadziej występują krwawienia z nosa, dróg rodnych, dróg moczowych czy przewodu pokarmowego.
Naczyniowe skazy krwotoczne
badania dodatkowe
W badaniach laboratoryjnych nie stwierdza się odchyleń w zakresie:
badań płytek krwi,
osoczowych czynników krzepnięcia i fibrynolizy.
Występuje natomiast dodatni objaw opaskowy (niska swoistość).
Naczyniowe skazy krwotoczne
podział
Wrodzone
CHOROBA RENDU-OSLERA
SKAZY KRWOTOCZNE W PRZEBIEGU WRODZONYCH CHORÓB TKANKI ŁĄCZNEJ
Nabyte
PLAMICA ALERGOTOKSYCZNA
PLAMICA NACZYNIOWA
NACZYNIOWE SKAZY KRWOTOCZNE WRODZONE
CHOROBA RENDU-OSLERA
(autosomalnie dominująco dziedziczona anomalia naczyń krwionośnych, anomalia genu TGFၢ)
PATOGENEZA : osłabienie ściany naczyń włosowatych i drobnych żył
OBJAWY:
rodzinne występowanie
poszerzenie naczyń w postaci guzków w skórze i błonach śluzowych
krwawienia z nosa, krwioplucie, smoliste stolce
krwotoki płucne
niedokrwistość
zgon.
SKAZY KRWOTOCZNE W PRZEBIEGU WRODZONYCH CHORÓB TKANKI ŁĄCZNEJ
zespół MARFANA
zespól EHLERSA-DANLOSA
NACZYNIOWE SKAZY KRWOTOCZNE NABYTE
PLAMICA ALERGOTOKSYCZNA (zespół SCHÖNLEINA - HENOCHA)
Aseptyczne zapalenie ściany naczyń na podłożu immunologicznym:
- występuje przeważnie u dzieci
- poprzedzone zazwyczaj ostrym zakażeniem dróg oddechowych (30% Streptococcus)
PATOGENEZA:
kompleksy immunologiczne w surowicy zawierające IgA
biopsja skóry - aseptyczne zapalenie naczyń z ogniskami martwicy fibrynoidowej z naciekami granulocytarnymi, złogi IgA.
PATOMECHANIZM: odkładanie kompleksów immunologicznych zawierających IgA z aktywacja komplementu.
OBJAWY: krwiomocz (uszkodzenie nerek), wysypka krwotoczna (wysiękowo-grudkowa), bóle brzucha, krwiste stolce, bóle obrzęki stawów
PLAMICA NACZYNIOWA
PRZYCZYNY:
1. niedobór witaminy C
2. LEKI: SULFONAMIDY, FUROSEMID, ALLOPURINOL, SOLE ZŁOTA, CHLORPROPAMID, ZWIĄZKI JODU.
OBJAWY: wybroczyny i wylewy w skórze z wtórnymi zmianami zanikowymi i barwnikowymi.
PŁYTKOWE SKAZY KRWOTOCZNE
podział
związane z nieprawidłową liczbą płytek (małopłytkowości, nadpłytkowości)
Wrodzone
Nabyte
związane z zaburzeniami czynności płytek krwi (trombocytopatie)
Wrodzone
Nabyte
MAŁOPŁYTKOWOŚĆ (TROMBOCYTOPENIA)
Etiologia
Zmniejszenie wytwarzania płytek
Nadmierne niszczenie płytek
Sekwestracja płytek (np. hipersplenizm)
TROMBOCYTOPENIA
Patomechanizm
zmniejszone wytwarzanie płytek
Wrodzone:
- Wrodzona hipoplazja szpiku (zespół Fanconiego),
- Brak trombopoetyny,
- Dziedziczne trombocytopenie: związane z płcią i upośledzeniem odporności- zespół Wiscota-Aldricha; ze współistniejącą głuchotą- zespół Alporta,
- W przebiegu wrodzonej białaczki, siatkowicy, różyczki noworodków,
- Związana z leczeniem matki tiazydami.
Nabyte:
- Aplazaja megakariocytowa,
- Niedokrwistość aplastyczna,
- Uszkodzenie szpiku przez promienie jonizujące, leki,
- Białaczki, plazmocytoma, histiocytoza,
- Mielofibroza,
- Niedobór wit B12 i kwasu foliowego,
- Zakażenia wirusowe,
- Niewydolność nerek.
TROMBOCYTOPENIA
Patomechanizm
nadmierne niszczenie płytek
Wrodzone:
- immunologiczne- alloimunologiczna małopłytkowość noworodków; związana z samoistną plamicą małopłytkową u matki,
- nieimmunologiczne- choroba hemolityczna noworodków, wcześniactwo, zatrucie ciążowe u matki, naczyniaki olbrzymie, wrodzone defekty skracające czas życia płytek krwi.
Nabyte:
- immunologiczne- samoistna plamica małopłytkowa, małopłytkowość polekowa (HIT), potransfuzyjna, w przebiegu tocznia układowego, niedokrwistości hemolitycznych, chorób rozrostowych.
TROMBOCYTOPENIA
Patomechanizm
złożony lub nie w pełni jasny
- Zakażenia, DIC, obfite krwotoki, krążenie pozaustrojowe, masywne przetoczenia krwi, oparzenia.
- Plamica małopłytkowa zakrzepowa.
- Zespół hemolityczno-mocznicowy.
- Nocna napadowa hemoglobinuria.
- Cykliczna małopłytkowość.
PŁYTKOWE SKAZY KRWOTOCZNE
objawy
Charakterystycznymi cechami skazy małopłytkowej są:
krwawienia skórno-śluzówkowe - pojawienie się wybroczyn na skórze kończyn, tułowia, rzadziej twarzy, oraz błonie śluzowej jamy ustnej,
krwawienia z nosa, dziąseł i dróg rodnych u kobiet,
nadmierne krwawienia po zabiegach chirurgicznych,
wybroczyny w miejscach niewielkich urazów,
do ciężkich powikłań należą krwawienia z przewodu pokarmowego lub krwawienia śródczaszkowe.
TROMBOCYTOPENIE NABYTE
SAMOISTNA PLAMICA MAŁOPŁYTKOWA (ITP)
ETIOLOGIA:
występuje zwykle u dzieci po zakażeniach wirusowych.
PATOGENEZA:
- u dzieci antygen wirusowy wzbudza syntezę przeciwciał, które reagują z antygenem zaadsorbowanym na powierzchni płytek,
- u dorosłych - powstają przeciwciała przeciwpłytkowe.
1) ZESPÓŁ HEMOLITYCZNO- MOCZNICOWY
OBJAWY:
nagle występująca małoplytkowość,
hemoliza,
ostra niewydolność nerek z bezmoczem.
WYSTĘPOWANIE:
niemowlęta, małe dzieci, u kobiet w ciąży, po porodzie.
PATOGENEZA :
- przypuszczalnie zakażenie bakteriami Gram (-),
- podobny zespól może wystąpić po cytostatykach: cyklosporynie i mitomycynie.
2) U chorych HIV dodatnich- objawy jak w ITP + dodatni test w kierunku zakażenie wirusem HIV.
ZABURZENIA CZYNNOŚCI PŁYTEK KRWI
Wrodzone:
CHOROBA von WILLEBRANDA.
TROMBASTENIA GLANZMANNA.
Zespół Bernarda-Souliera.
Zespół Scotta.
Nabyte:
Towarzyszące zespołom chorobowym,
Polekowe.
ZABURZENIA CZYNNOŚCI PŁYTEK wrodzone
CHOROBA von WILLEBRANDA
skaza krwotoczna dziedziczona autosomalnie dominująco spowodowana ilościowymi (TYP I) lub jakościowymi (TYP II) nieprawidłowościami czynnika von WILLEBRANDA
Multimery czynnika vW są niezbędne do:
- adhezji płytek do struktur podśródbłonkowych
- tworzenia kompleksow z czynnikiem VIII (niedobór - hemofilia A).
Najczęściej występująca wrodzona skaza krwotoczna. Występuje ona w postaci heterozygotycznej z częstością 1 na 8000 przypadków, czyli dwukrotnie częściej niż hemofilia A. Jako zespół heterogenny o często łagodnym przebiegu jest rzadko wykrywany.
anomalie błony płytkowej:
zespół Bernarda-Souliera:
niedobór GPIb- (przedłużony czas krwawienia, nieznaczna małopłytkowość, olbrzymie i morfologicznie nieprawidłowe płytki), zaburzenia adhezji płytek
trombastenia Glanzmanna:
niedobór GPIIIa- (przedłużony czas krwawienia, brak agregacji płytek pod wpływem ADP, kolagenu), zaburzenia procesów agregacyjnych płytek,
zespół Scotta
zaburzenie prokoagulacyjnej aktywności płytek- zmiany konformacyjne fosfolipidów płytkowych, uniemożliwiające bądź utrudniające kontakt z Ca2+ i osoczowymi czynnikami krzepnięcia.
zaburzenia sekrecji ziarnistości płytkowych:
- choroba puli magazynowej, zaburzenia mechanizmu sekrecji.
TROMBASTENIA GLANZMANNA
najczęściej występująca wrodzona anomalia płytek,
dziedziczona autosamalnie recesywnie,
wynika z anomalii receptora dla fibrynogenu w błonie komórkowej płytek (białko GpIIb-IIIa)
OBJAWY:
krwawienia z przewodu pokarmowego i naczyń mózgowych.
ZABURZENIA CZYNNOŚCI PŁYTEK nabyte
Towarzyszą:
- zespołom mielodysplastycznym
- zespołom mieloproliferacyjnym
- mocznicy
- makroglobulinemii
- szpiczakowi mnogiemu
- marskości wątroby
Polekowe: CEFALOSPORYNA, PENICYLINA, inne.
SKAZY OSOCZOWE
PODZIAŁ
Wrodzone:
- CHOROBA von WILLEBRANDA
- HEMOFILIA A
- HEMOFILIA B
- HEMOFILIA C
Nabyte
- niedobór wit. K
- choroby wątroby
- DIC
SKAZY OSOCZOWE wrodzone
Hemofilie
1. HEMOFILIA A - niedobór czynnika VIII (80% hemofilii)
2. HEMOFILIA B - niedobór czynnika IX
3. HEMOFILIA C - niedobór czynnika XI
OBJAWY ciężkiej hemofilii:
krwawienia podskórne
krwawienia z pępowiny
krwawienia po zabiegach
krwotoki wewnętrzne (do mięśni, do stawów).
DIC-najczęstsza skaza osoczowa (złożona) nabyta
Jest to wewnątrznaczyniowa aktywacja układu krzepnięcia przebiegająca z tworzeniem rozsianych mikrozakrzepów w końcowych odcinkach łożyska naczyniowego.
DIC rozpoczyna się po przedostaniu się do krwi substancji o aktywności tromboplastyny tkankowej inicjującej proces krzepnięcia.
DIC jest następstwem:
Powikłań położniczych
Zakażeń Gram-ujemnych
Nowotworów ( trzustki, stercza)
W czasie rozpoznawania i monitorowania DIC, oprócz D-dimerów, które w zespole ostrym wielokrotnie przekraczają zakresy referencyjne, obserwujemy spadek miana płytek i aktywności antytrombiny III.
TROMBOFILIA
definicja
Trombofilia to wrodzone lub nabyte zaburzenia mechanizmu hemostazy, które usposabiają do powstawania zakrzepów.
Trzy przyczyny warunkujące rozwój zakrzepicy (triada Virchowa):
zmiany w przepływie krwi (zmiany reologiczne),
zmiany dotyczące ściany naczyniowej,
zmiany składu krwi.
APCR (mutacja Leiden)
- Powodem oporności na białko C jest punktowa mutacja genu dla osoczowego czynnika V.
- Mutacja ta polega na zamianie pojedynczego aminokwasu argininy na glutaminę w pozycji 506. Podstawienie to istotnie utrudnia hydrolizę czynnika V przez aktywne białko C.
- Aktywne białko C inaktywuje czynnik VIII, lecz nie może sprawnie i wydajnie unieczynniać czynnika V. W wielu przypadkach jest to powodem pojawiania się incydentów zakrzepowych już w młodym wieku.
APCR
- Zaburzenia zakrzepowe ujawniają się najczęściej po zabiegach chirurgicznych, w czasie ciąży, połogu, w czasie długotrwałego unieruchomienia czy stosowania środków antykoncepcyjnych.
- Jednak w ponad 30% przypadków nawet osobom homozygotycznym pod względem tej mutacji udaje się ustrzec incydentów zakrzepowych.
Hiperhomocysteinemia
Homocysteina jest aminokwasem powstającym podczas metabolizmu metioniny. W ostatnich latach postrzegana jako czynnik cytotoksyczny dla komórek śródbłonka, przyczyniający się do powstawania blaszki miażdżycowej. Sugeruje się udział homocysteiny w przyspieszonym złuszczaniu komórek śródbłonka i utleniania LDL.
Sądzi się, że hiperhomocysteinemia może powodować:
obniżoną aktywność trombomoduliny,
nasiloną ekspresję czynnika tkankowego i czynnika V,
obniżoną aktywność fibrynolityczną.
Zespół antyfosfolipidowy
Dokładny mechanizm działania przeciwciał antyfosfolipidowych nie został do końca poznany. Uważa się, że charakteryzują się one silnym powinowactwem do fosfolipidów niezbędnych w kluczowych reakcjach układu antykoagulacyjnego, prawdopodobnie blokują działanie układu białka C.
Przeciwciała LA mogą występować u około 10% pacjentów z ogólnoustrojowym toczniem rumieniowatym, jak również może towarzyszyć wszelkim innym chorobom autoimmunologicznym, takim jak np. zakażenie wirusem HIV (około 50% pacjentów) czy choroba Castleman'a. U pacjentów z toczniem rumieniowatym i antykoagulantem tocznia ryzyko wystąpienia choroby zakrzepowej szacuje się na około 40%, podczas gdy u osób charakteryzujących się jedynie podwyższonym mianem LA ryzyko to jest o około 10% niższe. Zjawisko spontanicznych krwawień na skutek podwyższonego miana LA odnotowuje się rzadziej niż u 1% pacjentów. U około 40% pacjentów stwierdza się małopłytkowość.
Przeciwciała antyfosfolipidowe antykoagulanta tocznia (Lupus anticoagulant antibodies, LA) obejmują różne klasy immunoglobulin osocza krwi. W przeciwieństwie do innych inhibitorów układu krzepnięcia, LA nie hamują aktywności większości specyficznych czynników osoczowych krzepnięcia. Autoprzeciwciała skierowane są przeciwko istotnym w regulacji hemostazy ujemnie naładowanym fosfolipidom (fosfatydyloseryna-a2-GPI).

Skazy osoczowe
Skazy płytkowe i naczyniowe
Najczęstsze objawy
wylewy krwi do mięśni i stawów, późne krwawienia pourazowe
plamica, skłonność do siniaczenia, krwawienia z błon śluzowych
Krwawienia pooperacyjne
często późne krwawienia, bardzo niebezpieczne
krwawienia podczas zabiegu, mało niebezpieczne
Wybroczyny i sińce
wybroczyny występują rzadko, sińce pojedyncze
często liczne wybroczyny i sińce
Wynik ucisku miejsca krwawienia
po zwolnieniu ucisku nawrót krwawień
krwawienie zatrzymuje się często na stałe
Wyszukiwarka