Ćwiczenie 11: Zaburzenia biało- i czerwonokrwinkowe.
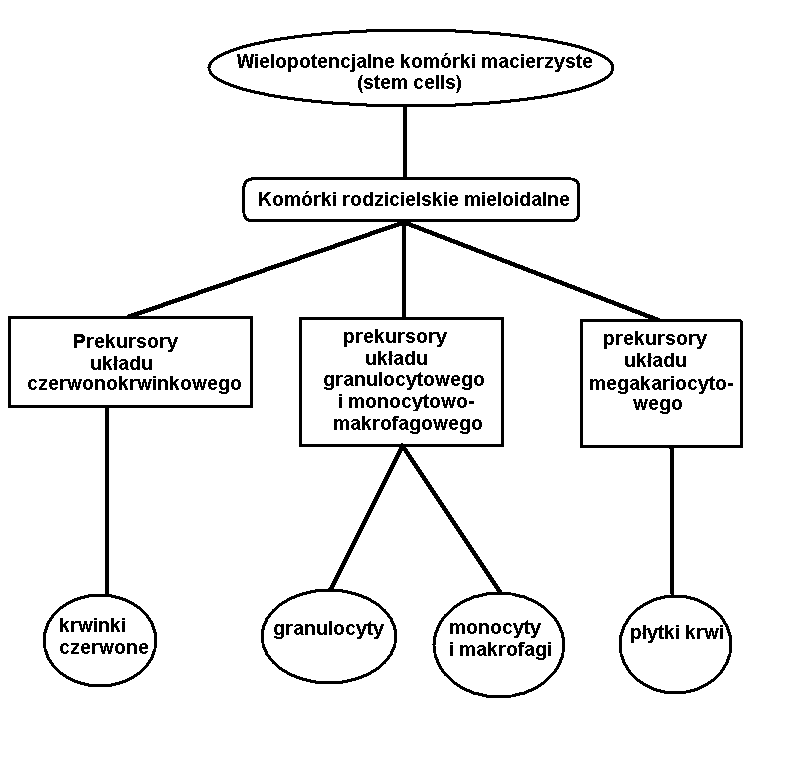
Hematopoeza
Krwiotworzenie - ciągły proces w wyniku którego z wielopotencjalnej komórki macierzystej hematopoezy znajdującej się w szpiku kostnym powstają wyspecjalizowane komórki krążące we krwi obwodowej.
Narządy krwiotwórcze:
- Pęcherzyk żółtkowy
- Wątroba
- Śledziona
- Szpik kostny - podejmuje funkcję krwiotwórczą w ostatnim trymestrze ciąży
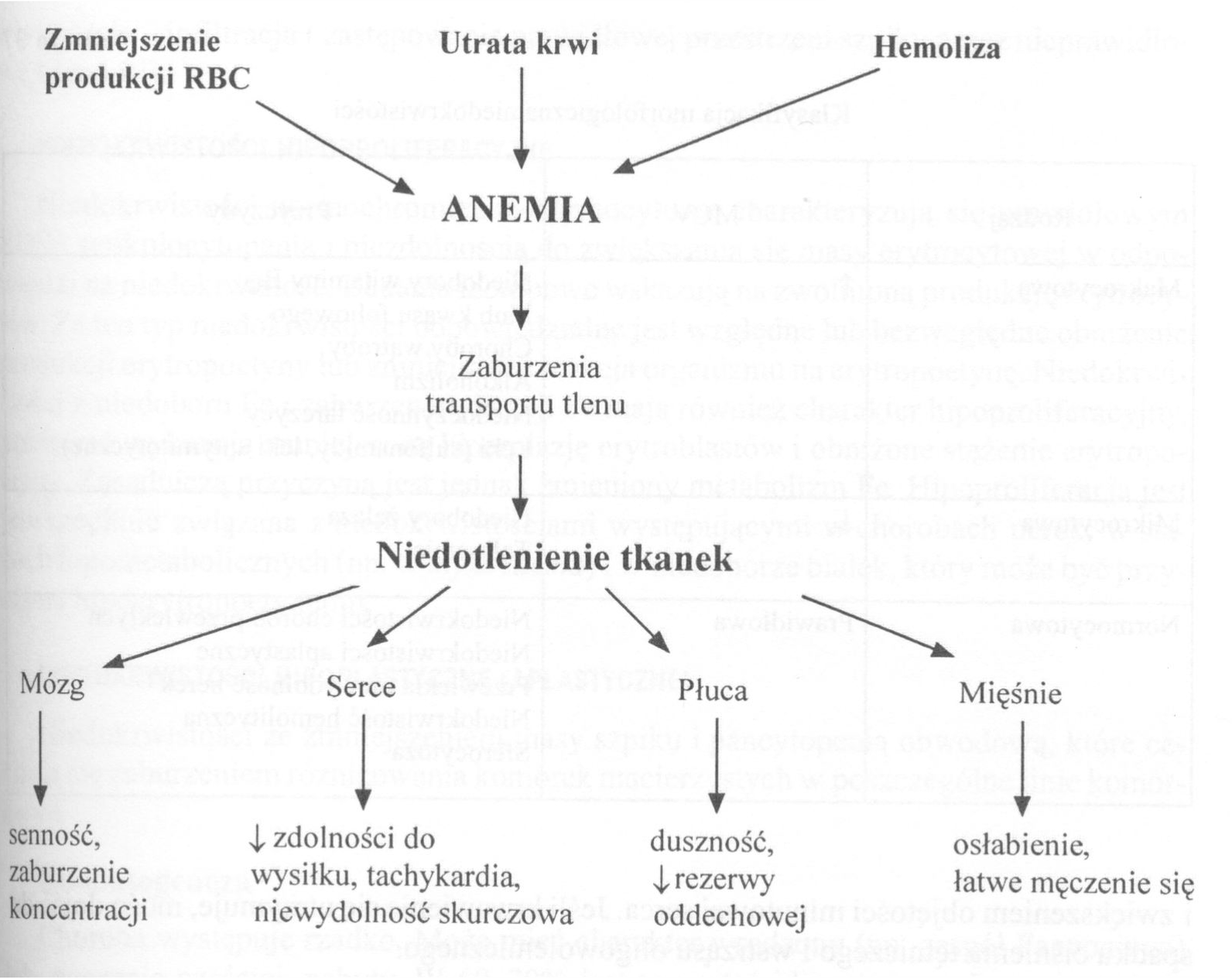
Niedokrwistość - zmniejszenie stężenia Hb, Ht lub liczby erytrocytów poniżej dolnej granicy przedziału wartości referencyjnych dla wieku i płci badanego.
U zdrowych osób występuje silna korelacja pomiędzy:
- Stężeniem Hemoglobiny
- Liczbą erytrocytów
- Wartością hematokrytu
Niedokrwistość jest manifestacją choroby a nie jej ostatecznym rozpoznaniem!
Stężenie Hb (g/dl) a wiek
- Krew pępowinowa 13,5 - 20,5
- 1. dzień życia 15,0 - 23,5
- Dzieci 6 m.ż. - 6 r.ż. 11,0 - 14,5
- Dzieci 6 r.ż. - 12 r.ż. 12,0 - 15,5
- Dorośli mężczyźni 13,0 - 17,0
- Dorosłe kobiety 12,0 - 15,5
- Kobiety w ciąży 11,0 - 14,0
Objawy niedokrwistości
1. Podmiotowe:
- Osłabienie, łatwe męczenie się
- Duszność po wysiłku, kołatanie serca
- Dusznica bolesna, chromanie przestankowe
- Bóle i zawroty głowy
- Zaburzenia widzenia, senność
- Nudności
- Zaburzenia miesiączkowania, utrata libido
2. Przedmiotowe:
- Bladość skóry
- Tachykardia
- Duża amplituda tętna z tętnieniem naczyń kapilarnych
- Szmery serca
- Objawy prawokomorowej niewydolności krążenia
Przyczyny niedokrwistości
Utrata krwi (niedokrwistość pokrwotoczna)
Skrócenie czasu przeżycia krwinek czerwonych (niedokrwistość hemolityczna)
Defekt wrodzony (wrodzona sferocytoza, niedokrwistość sierpowatokrwinkowa)
Defekt nabyty (polekowa, malaria)
Zaburzenie wytwarzania krwinek czerwonych (nieefektywna erytropoeza)
Gromadzenie i niszczenie krwinek czerwonych w powiększonej śledzionie
Zwiększenie objętości osocza (ciąża)
Podział niedokrwistości w zależności od wielkości erytrocytów (MCV)
Mikrocytowe - MCV < 80 μm3
Normocytowe - MCV 80 - 94 μm3
Makrocytowe - MCV > 94 μm3
Podział patogenetyczny niedokrwistości
Niedokrwistości aregeneracyjne - z niewystarczającej podaży erytrocytów
Niedoborowe: Fe, B12, kwasu foliowego
Aplazja szpiku
Dyserytropoeza (zaburzenie tworzenia)
Niedokrwistości z nadmiernej utraty erytrocytów
Hemolityczne
Pokrwotoczne
Niedokrwistości z innych, często wtórnych przyczyn
Żelazo (Fe)
- Ilość w organizmie dorosłego człowieka 3 - 5 g
- Stężenie we krwi 50 - 175 μg/dl
- Kofaktor enzymów transportujących elektrony w metabolizmie tlenowym oraz białek transportujących tlen w ustroju
Pula magazynowa Fe
Ferrytyna - m.cz. 440 kDa
- Rozpuszczalna w wodzie apoferrytyna wiążąca Fe
- Fe stanowi 23% cząsteczki
- Obecna w: enterocyty, hepatocyty, komórki śledziony, szpik kostny, osocze krwi
Hemosyderyna - nierozpuszczalny agregat cząstek ferrytyny
- Fe w ilości 30%, uwalniane zacznie wolniej
- Obecna w: hepatocyty, komórki trzustki, mięsień sercowy, nadnercza
Transferyna
- Glikoproteina z frakcji β1 - globulin
- m. cz. 80 kD
- Syntetyzowana w wątrobie
- Zaangażowana w transport Fe we krwi
- Jedna cząsteczka transferyny wiąże 2 x Fe3+
- Receptory na erytroblastach rozpoznają transferynę - następuje endocytoza i zużycie żelaza na potrzeby syntezy Hb.
Podział etiologiczny stanów przeładowania żelazem organizmu
Hemochromatoza pierwotna
Hemochromatoza wtórna
Hemosyderoza miejscowa
Nadmiar Fe początkowo odkłada się w fizjologicznych magazynach (wątroba, jelito cienkie, śledziona, szpik) a w dalszej kolejności w wielu innych życiowo ważnych organach
- Hemosyderoza - zwiększona ilość Fe w organizmie
- Hemochromatoza - gdy na skutek nadmiaru Fe dochodzi do uszkodzenia tkanki
Stężenie Fe w surowicy zależy od:
- Wchłaniania w przewodzie pokarmowym
- Magazynowania w jelicie, śledzionie i szpiku kostnym
- Syntezy i rozpadu Hb
- Utraty Fe z ustroju
Hemochromatoza pierwotna
- Autosomalna recesywna; gen HFE na krótkim ramieniu chromosomu 6.
- Częstość występowania 3 - 10 osób / 100
- Nosicielstwo dotyczy 10% populacji
- Objawy kliniczne ujawniają się zazwyczaj po 40 r.ż.; częściej dotyczą mężczyzn
- U kobiet naturalne mechanizmy utraty krwi eliminują znaczne ilości patologicznie wchłoniętego żelaza, co opóźnia wystąpienie i stopień ciężkości choroby
- Najczęściej i najszybciej uszkodzeniu ulega wątroba (30 - 94% przypadków prowadzi do marskości wątroby; konsekwencją może być rak wątrobowokomórkowy)
- Odkładanie się Fe w innych narządach manifestuje się: cukrzycą, kardiomiopatią, zapaleniem stawów, niedoczynnością przysadki, niewydolnością kory nadnerczy (ciemne zabarwienie skóry)
- Leczenie - upusty krwi; farmakologicznie - chelatory Fe
Wtórny nadmiar Fe
- Wielokrotne transfuzje krwi
- Nadmierna podaż dożylnych i domięśniowych preparatów żelaza
- Zwiększone wchłanianie żelaza w jelicie cienkim (megadawki wit.C > 1g/ dobę)
Przyczyny wzrostu stężenia Fe
- Nadmierna podaż dożylnych i domięśniowych preparatów Fe
- Częste transfuzje krwi
- Hemochromatoza pierwotna
- Niedokrwistości hemolityczne i aplastyczna
- Niedokrwistość złośliwa
- MDS
- wzw, ostre uszkodzenia wątroby
- Zapalenia nerek
- Doustne środki antykoncepcyjne
Stopnie niedoboru żelaza
Io
- Zmniejszenie puli magazynowej żelaza (obniżenie stężenia ferrytyny w surowicy) bez zaburzeń czynności ustroju
- Świadczy tylko o podatności organizmu na niedobory żelaza
- Ryzyko rozwoju niedokrwistości jest minimalne - organizm może zwiększyć wchłanianie żelaza w miarę zmniejszania się jego zapasów
IIo
- Ilość Fe w ustroju jest niewystarczająca do syntezy Hb i innych substancji zawierających żelazo
- Stopień wysycenia transferyny żelazem obniża się i wzrasta stężenie protoporfiryny w erytrocytach
- Stężenie Hb w normie - niedobór Fe bez niedokrwistości
IIIo
- Jawna niedokrwistość
Przyczyny niedokrwistości z niedoboru żelaza
1. Utrata żelaza z krwią
- Obfite krwawienia miesiączkowe
- Inne niż miesiączkowe krwawienia z dróg rodnych
- Krwawienia do światła przewodu pokarmowego
- Inne krwawienia
2. Dietetyczne
- Niedostateczna podaż u noworodków i niemowląt
- Dieta uboga w żelazo
- Nadmiar substancji utrudniających wchłanianie żelaza (chelatory, garbniki)
3. Zespoły złego wchłaniania
4. Zakażenia pasożytnicze przewodu pokarmowego
5. Stany zapalne i infekcje
6. Nowotwory
Objawy szczegółowe:
- Łamliwość włosów i paznokci
- Zmiany w kącikach ust
- Zmiany na błonie śluzowej języka, gardła i przełyku - ból i pieczenie przy przełykaniu
Niedokrwistości megaloblastyczne
- Niedobór witaminy B12 - zdecydowana większość przypadków)
- Niedobór kwasu foliowego (rzadziej)
- Megaloblastoza wynika z nieprawidłowej syntezy DNA w komórkach prekursorowych linii czerwonokrwinkowej, białokrwinkowej i płytkowej
Witamina B12
- Źródłem są pokarmy pochodzenia zwierzęcego (wątroba cielęca)
- Zawartość w organizmie 2 - 5 mg
- Dzienne zapotrzebowanie 1 - 3 μg
- Magazyn witaminy B12 w wątrobie zapewnia jej dostarczanie przez 3 lata
- Wchłanianie w jelicie krętym w postaci kompleksów z czynnikiem wewnętrznym produkowanym przez komórki okładzinowe żołądka
- Po wchłonięciu ze światła jelita jest transportowana do wątroby w połączeniu z transkobalaminami I, II i III
Przyczyny niedoboru witaminy B12
Zaburzenia wchłaniania
choroba Addisona - Biermera
Resekcja części lub całości żołądka
Zespół złego wchłaniania
Tasiemczyce (bruzdogłowiec szeroki)
Niedostateczna podaż w diecie - ścisły wegetarianizm
Rzadko - niedobór wrodzony
Zespół Imerslund Grasbeck - defekt receptora dla czynnika wewnętrznego w jelicie cienkim
Choroba Addisona - Biermera
* Podłoże immunologiczne
* Trzy rodzaje autoprzeciwciał:
- Typ 1 - przeciwko czynnikowi wewnętrznemu
- Typ 2 - przeciwko kompleksowi IF-B12
- Typ 3 - przeciwko komórkom okładzinowym żołądka
Objawy kliniczne niedokrwistości z niedoboru B12
- Neurologiczne: parestezje, osłabienie odruchów ścięgnistych, zaburzenia widzenia, objawy psychotyczne, zaburzenia pamięci, objawy zwyrodnienia sznurów bocznych rdzenia (niepewny chód, osłabienie napięcia mięśniowego, objawy niedowładu, mrowienie dłoni i stóp)
- Ze strony przewodu pokarmowego: zanikowe zapalenie błony śluzowej żołądka w niedokrwistości złośliwej, zapalenie błony śluzowej języka (glossitis Hunteri) - język piekący i żywoczerwony
Kwas foliowy
- Zawartość w organizmie 50 mg
- Minimalne dobowe zapotrzebowanie 50 μg, optymalne 400 μg
- Źródło - pokarmy pochodzenia roślinnego
- Magazyn wątrobowy wystarcza na 2 - 4 m-ce
Przyczyny niedoboru kwasu foliowego
- Niedobory dietetyczne (dieta uboga w produkty roślinne, obróbka termiczna pokarmów)
- Zwiększone zapotrzebowanie: ciąża, laktacja)
- Zaburzenia wchłaniania
- Przewlekły alkoholizm
- Zwiększone zużycie: nadczynność tarczycy, choroby mieloproliferacyjne
- Niektóre leki interferują z przemianami kwasu foliowego (metotreksat, trimetoprim, fenobarbital, doustne środki antykoncepcyjne)
Obraz kliniczny
- Podobny jak w niedoborze witaminy B12
- Brak objawów neurologicznych
Następstwa niedoboru kwasu foliowego u ciężarnych
- Defekty cewy newowej płodu prowadzące n.p. do rozszczepu kręgosłupa
- Przedwczesne odklejenie łożyska
- Krwawienia wewnątrzmaciczne
Niedokrwistości hemolityczne
- Istotą jest skrócenie czasu przeżycia krwinek na skutek czynników wewnątrzkrwinkowych lub pozakrwinkowych
- Charakter wrodzony lub nabyty
Przyczyny niedokrwistości hemolitycznych
1. Wewnątrzkrwinkowe
- Defekt błon erytrocytów (np. sferocytoza wrodzona)
- Defekty enzymów wewnątrzkrwinkowych (np. niedobór G-6-PD)
- Zaburzenia tworzenia hemoglobiny (hemoglobinopatie, talasemie)
- Nocna napadowa hemoglobinuria
2. Zewnątrzkrwinkowe
- Działanie czynników biologicznych: zakażenie zarodźcem malarii, hemolizyny paciorkowcowe i gronkowcowe, hemolizyny w jadach, zakażenia wirusowe
- Działanie czynników chemicznych: ołowica, leki, rozpuszczalniki organiczne
3. Immunizacja
- Obecność przeciwciał, głównie IgM i IgG, rzadko IgA jako zjawisko pierwotne lub wtórne w przebiegu schorzeń z autoimmunizacji, np. uogólnione choroby tkanki łącznej
- Napadowa hemoglobinuria z oziębienia - IgG
- Odczyny poprzetoczeniowe, choroba hemolityczna noworodków
4. Czynniki mechaniczne
- Hemoliza marszowa
- Sztuczne zastawki serca
5. Połaczenie działania czynników wewnątrz- i zewnątrzkrwinkowych
- Fawizm (niedobór G-6-PD i spożycie roślin strączkowych)
- Hemolizy polekowe lub w warunkach niedotlenienia
Objawy kliniczne
- Niedokrwistość o charakterze najczęściej normocytowym normobarwliwym
- Skóra blada z żółtawym odcieniem
- Żółtaczka
W przełomach hemolitycznych:
- Gorączka
- Bóle głowy, mięśni i stawów
- Powiększenie śledziony
Sferocytoza wrodzona
- Dziedziczona autosomalnie dominująco
- Defekt genów białek błonowych: spektryny i ankryny (najczęściej)
- Błony erytrocytarne o zmniejszonych właściwościach elastycznych koniecznych do przechodzenia przez mikrokrążenie śledziony - erytrocyty są zatrzymywane w śledzionie i tam niszczone
- Leczenie z wyboru - splenektomia
Niedokrwistość z niedoboru G-6-PD
- G-6-PD - dehydrogenaza glukozo - 6 - fosforanowa
- Dziedziczona w sprzężeniu z chromosomem X
- Niedobór G-6-PD prowadzi do spadku wytwarzania zredukowanego, glutationu, który chroni erytrocyty przed uszkadzającym dzialaniem utleniaczy
Hemoglobinopatie
- Hemoglobina - metaloproteina złożona z części niebiałkowej - hemu i białkowej - globiny
- Tetramer złożony z dwóch par jednakowych łańcuchów peptydowych
Podstawowe warianty hemoglobiny
- HbA1 - hemoglobina dorosłych - łańcuchy α2β2,
- HbA2 - α2δ2 do 2% Hb dorosłych
- HbF - hemoglobina płodowa - łańcuchy α2γ2
Hemoglobiny patologiczne
- HbM - methemoglobinemia (Hb utleniona)
- HbS - zmniejszenie powinowactwa do tlenu, protekcja heterozygot na zakażenie Plasmodium falciparum
- Hb Chesapeake - wzrost powinowactwa Hb do tlenu
Gdy zmiana AA w łańcuchu Hb nie powoduje zmian w transportowaniu tlenu to taką Hb określa się jako hemoglobinę naturalną.
Niedokrwistość sierpowatokrwinkowa
- Dziedziczona autosomalnie dominująco
- Patologiczna HbS: kwas glutaminowy w pozycji 6 w obu łańcuchach β jest zastąpiony przez walinę
- W stanie niedotlenienia, gorączki, obniżenia pH dochodzi do agregecji cząsteczek Hb i powstawania erytrocytów sierpowatych
- Erytrocyty sierpowate źle przeciskają się przez naczynia włosowate co prowadzi do zaburzeń w mikrokrążeniu
Talasemia β (choroba śródziemnomorska)
-Zaburzenie wytwarzania łańcuchów β globiny z kompensacyjnym wzrostem wytwarzania łańcuchów γ lub δ
-Dziedziczenie autosomalne dominujące
-Dwa typy: homozygotyczna (ciężka) i heterozygotyczna (łagodniejsza)
-Nieprawidłowa budowa łańcucha β powoduje wytrącanie Hb
-Erytrocyty o kształcie tarcz strzeleckich o upośledzonej zdolności transportu tlenu są eliminowane w śledzionie i wątrobie
Nocna napadowa hemoglobinuria
-Erytrocyty o zwiększonej wrażliwości na lityczne działanie dopełniacza
-Hemoliza zachodzi zwykle w czasie snu, kiedy pH krwi ulega obniżeniu
-Osłabienie, hemoglobinuria (ciemny mocz), bóle brzucha, głowy, pleców
-Zwiększona skłonność do zakrzepicy wynikająca z aktywności tromboplastycznej fragmentów erytrocytów
Niedokrwistości autoimmunohemolityczne
Etiologia:
Pierwotne (samoistne) - ok.70% przypadków
Wtórne (objawowe) - towarzyszą innym chorobom
Zakażenia: mononukleoza zakaźna, HIV
Choroby limfoproliferacyjne: chłoniaki, ziarnica złośliwa, szpiczak mnogi
Choroby autoimmunologiczne: toczeń, RZS, twardzina
Dwa rodzaje autoprzeciwciał
- Typu ciepłego - głównie IgG, rzadziej IgA lub IgM
Główny mechanizm hemolizy: wiązanie przeciwciał opłaszczających erytrocyty z receptorami Fc makrofagów, fagocytoza erytrocytów i liza cytotoksyczna w śledzionie
- Typu zimnego - IgM
Główny mechanizm hemolizy - aktywacja układu dopełniacza
Choroba hemolityczna noworodków (erythroblastosis foetalis)
- Zwykle rezultat niezgodności grup krwi matki i płodu (konflikt serologiczny)
- Antygenem najczęściej odpowiedzialnym za konflikt serologiczny jest antygen D z układu antygenów grupowych Rh
- Hemoliza erytrocytów następuje wtedy, gdy przeciwciała matki przejdą przez łożysko i zwiążą się z antygenami krwinek czerwonych płodu
W zależności od nasilenia hemolizy, ChHN przebiega jako:
- Niedokrwistość hemolityczna
- Żółtaczka hemolityczna noworodków
- Ciężki uogólniony obrzęk płodu
Porody martwe dotyczą 15% przypadków; śmierć wewnątrzmaciczna zwykle ok. 20 tygodnia ciąży
Profilaktyka
- Wykrywanie potencjalnego ustawienia konfliktowego między matką a ojcem
- Oznaczanie miana przeciwciał anty-D (12 i 28 tydzień ciąży)
- W razie potrzeby - podawanie matce surowicy anty-D
- Leczenie - transfuzja wymienna i objawowe zwalczanie niewydolności krążeniowo - oddechowej i kwasicy
Reakcje poprzetoczeniowe
- Próba zgodności serologicznej /próba krzyżowa/ wykonywana przed przetoczeniem krwi to uzyskanie pewności, że w osoczu biorcy krwi nie ma przeciwciał, które mogłyby reagować z jakimkolwiek z antygenów krwinek czerwonych dawcy.
- Metoda wykrywania przeciwciał oparta na zjawisku aglutynacji erytrocytów pod wpływem przeciwciał pozostaje praktycznie niezmieniona od prawie 150 lat. Po raz pierwszy aglutynację zaobserwował A. Creite w Getyndze w 1869 r.
- Obecnie w diagnostyce powszechnie stosuje się test antyglobulinowy Coombsa bezpośredni i pośredni.
- W przypadku przetoczenia krwi niezgodnej grupowo dochodzi do masywnej hemolizy wewnątrznaczyniowej ze wszystkimi konsekwencjami rozpadu erytrocytów i podaży wolnej hemoglobiny co nieuchronnie prowadzi do wstrząsu. Po przeżyciu pierwszej reakcji rozwija się ostra niezapalna niewydolność nerek.
Niedokrwistość hemolityczna mikroangiopatyczna
- Występuje w przebiegu chorób, w których dochodzi do uogólnionego uszkodzenia śródbłonka naczyń (cukrzyca, nadciśnienie tętnicze, naczyniaki, protezy zastawek) lub odkładania na wewnętrznej powierzchni naczyń skupień komórkowych lub kompleksów immunologicznych i agregatów płytkowych (nowotwory, kolagenozy)
- Erytrocyty przeciskając się przez zmienione drobne naczynia ulegają rozpadowi
- Szczególna postać - zespół hemolityczno - mocznicowy
Niedokrwistość aplastyczna
- Upośledzenie hemopoezy z postępującym zanikiem komórek jądrzastych w szpiku. Utkanie hematopoetyczne szpiku zastępowane jest tkanką tłuszczową
Podział niedokrwistości aplastycznych
Wrodzone
Zespół Fanconiego
Zespół Blackfana - Diamonda
Nabyte
Infekcyjne (EBV, HBV, HCV)
Toksyczne (benzen, chloramfenikol, NLPZ, sole złota)
Autoimmunizacja
Nowotwory
Niedokrwistość aplastyczna
- Objawy wynikające z niedokrwistości
- Skaza krwotoczna małopłytkowa (wybroczyny, sińce, krwawienia z nosa, dziąseł, dróg rodnych)
- Infekcje związane z neutropenią
Niedokrwistość Fanconiego
- Autosomalna recesywna, ujawnia się najczęściej w 5 - 10 r.ż.
- Obraz kliniczny niejednolity (postaci choroby A - G)
- Postępująca niewydolność szpiku
- Wady rozwojowe dotyczą ok. 70% chorych
- Małogłowie, mikrostomia, zniekształcenia kości kończyn
- Niska masa urodzeniowa, opóźniony wzrost
- Zaburzenia pigmentacji skóry, hipoplazja jąder, zez
Niedokrwistość Blackfana - Diamondsa
- Autosomalna recesywna lub dominująca
- Wybiórcza aplazja układu czerwonokrwinkowego w szpiku
- Wady rozwojowe u ok. 30% chorych
Nadkrwistości
Nadkrwistość (polycythaemia) - stan manifestujący się przede wszystkim zwiększeniem liczby erytrocytów i hemoglobiny we krwi obwodowej oraz podwyższeniem wartości hematokrytu.
Podział nadkrwistości
Bezwzględne (prawdziwe)
Pierwotna (samoistna)
Czerwienica prawdziwa
Wtórna (objawowa)
Wskutek niedotlenienia (obniżenie pO2 w atmosferze, przewlekłe choroby płuc, wady wrodzone serca)
Wskutek zwiększenia wytwarzania Epo (nienowotworowe choroby nerek, np. torbiele, nowotwory nerek i inne
Względne (rzekome) - z powodu odwodnienia, zmian w rozmieszczeniu płynów ustrojowych, zespół Gaisboecka
Ściana naczyń krwionośnych
Płytki krwi
Układ krzepnięcia i fibrynolizy
Hemostaza pierwotna
Skurcz naczynia krwionośnego
Aktywacja płytek krwi i wytworzenie czopu płytkowego
Hemostaza wtórna
Stabilizacja wiotkiego czopu płytkowego siatką włóknika = wytworzenie czopu hemostatycznego
Fibrynoliza
ogranicza narastanie czopu płytkowego zapewniając drożność naczynia
Warstwa zewnętrzna
Warstwa środkowa - mięśniowa
Warstwa wewnętrzna
Śródbłonek pokryty glikokaliksem
Warstwa podśródbłonkowa
Udział w hemostazie pierwotnej - tworzenie czopu płytkowego
Uczestnictwo w reakcjach kaskady krzepnięcia - aktywowane płytki udostępniają cz.III krzepnięcia czyli fosfolipidy płytkowe (PL)
Adhezję płytek
Aktywację płytek i zmianę kształtu
Degranulację ziarnistości
Hemofilia A - niedobór cz.VIII (80% wszystkich przypadków hemofilii); 1 : 16.000
Hemofilia B - niedobór cz.IX; 1 : 110.000
Hemofilia C - niedobór cz.XI, przebiega jako łagodna lub umiarkowana skaza krwotoczna
Wydłużony czas krwawienia (w typie 1 może być prawidłowy)
Przedłużony APTT (w typie 1 i 2 może być prawidłowy)
Ilościowe = trombocytopenia, małopłytkowość
Zmniejszona produkcja płytek (niedokrwistość aplastyczna, niedobory B12 i kwasu foliowego, promieniowanie jonizujące, dziedziczne małopłytkowości)
Zmniejszone przeżycie płytek
Immunologiczne (idiopatyczne, toczeń rumieniowaty, polekowe, plamica małopłytkowa noworodków)
Hipersplenizm
Plamica małopłytkowa
Zespół hemolityczno - mocznicowy
Defekty jakościowe płytek = trombopatie
dziedziczne
Zespół Bernarda - Souliera (defekt adhezji)
Trombastenia Glanzmanna (defekt agregacji)
Nabyte: zespoły mielodysplastyczne i mieloproliferacyjne, mocznica, marskość wątroby, polekowe
Polekowe (kwas acetylosalicylowy, propranolol, heparyna, penicylina, betablokery, blokery kanału wapniowego, cytostatyki)
Towarzyszące chorobom krwi
Zakażenia bakteryjne (posocznica); infekcje wirusowe, grzybicze, pierwotniakowe
Powikłanie położnicze, np. z powodu przedwczesnego odklejenia łożyska czy zapalenia błon płodowych
Nowotwory
Rozległe uszkodzenia tkanek (urazy, rozległe zabiegi operacyjne)
Hemoliza wewnątrznaczyniowa
Antytrombina III (ATIII) - inaktywacja trombiny i cz.Xa;
APC - proteoliza cz.Va i VIIIa;
TFPI - inaktywacja kompleksu TF/VIIa.
Wynikające z uszkodzenia narządów wskutek niedokrwienia (wątroba, nerki, OUN, płuca)
Wynikające z ciężkiej skazy krwotocznej - charakterystyczne są krwawienia ze wszystkich naturalnych otworów ciała, krwawienia podskórne i podśluzówkowe
Czerwienica prawdziwa
Proliferacyjna choroba układu krwiotwórczego ze szczególnie intensywnym rozrostem układu czerwonokrwinkowego przy jednocześnie wzmożonej proliferacji układu białokrwinkowego i płytkowego. Zaliczana do przewlekłych zespołów mieloproliferacyjnych.
Epidemiologia
- Częstość 0,6 - 1,8/ 100 tys. mieszkańców rocznie
- Ujawnia się najczęściej między 55 a 60 r.ż.
- Nieco częstsza u mężczyzn
Patogeneza czerwienicy prawdziwej
- Pierwotnym zjawiskiem jest mutacja wielopotencjalnej komórki macierzystej, która nie podlegając fizjologicznym mechanizmom regulacji hemostazy daje początek nowotworowemu klonowi komórek (rozrost klonalny)
- Poziom Epo jest w normie!!!
- Patologiczny klon komórek może wykazywać abberacje chromosomowe (najczęściej trisomia 8 lub 9, delecja 20q, translokacja chromosomu 1)
Obraz kliniczny
- Objawy wynikają głównie ze zwiększenia masy erytrocytarnej, wzrostu lepkości i objętości krwi i przeciążenia układu krążenia
- Częste udary mózgu, ChNS, zawały serca, zakrzepica żylna
Objawy
- Bóle i zawroty głowy
- Szum w uszach
- Zaburzenia widzenia
- Osłabienie, łatwe męczenie się
- Duszność
- Świąd skóry
- Czerwonosine zabarwienie skóry
- Przekrwienie spojówek i błon śluzowych
- Powiększenie śledziony
- Niekiedy podwyższone ciśnienie tętnicze
Powikłania czerwienicy prawdziwej
- Zakrzepy, zatory
- Udary mózgu, zawał serca
- Krwawienia
- Nadciśnienie tętnicze
W rozpoznaniu ważne jest wykluczenie czerwienicy wtórnej (związanej z hipoksemią lub nieprawidłową produkcją Epo)
Leczenie:
- Podstawowe ma na celu obniżenie wartości hematokrytu poniżej 46% - upusty krwi
- Utrzymanie wartości Htk w granicach 42-46% - chemioterapia (hydroksymocznik), radioterapia
Nadkrwistości rzekome
- Odwodnienie
- Zmiany w rozmieszczeniu płynów ustrojowych (enteropatie, zastoinowa niewydolność krążenia)
- Zespół Gaisbőcka - pseudonadkrwistośc występująca zazwyczaj w stresie o charakterze przejściowym i krótkotrwałym
Leukopenia
- Obniżenie liczby krążących leukocyów do < 4000/μl
- Najczęściej spowodowana jest obniżeniem liczby granulocytów obojętnochłonnych
Neutrofile:
- 76% w puli rezerwowej w szpiku
- 20% w puli prekursorów w szpiku
- 2% we krwi
- 3% w puli brzeżnej we krwi
Neutropenia (<1500/ ml)
- Zmniejszone wytwarzanie, nieefektywna granulocytopoeza
- Skrócenie czasu przeżycia neutrofilów
- Zmieniona dystrybucja - zwiększenie puli brzeżnej (pseudogranulocytopenia)
Zmniejszone wytwarzanie neutrofilów
Wrodzone
Zespół Kostmana (zahamowanie granulopoezy na szczeblu promielocyta)
Zespół Schwachmana-Diamonda
Cykliczna neutropenia
Zespół Chediaka-Higashiego
Nabyte
Niedokrwistość aplastyczna i megaloblastyczna
Rozrost nowotworowy w szpiku
Polekowe (leki przeciwnowotworowe, antybiotyki, sole złota)
Promieniowanie jonizujące
Zespoły mielodysplastyczne
Skrócenie czasu przeżycia neutrofilów
- Hipersplenizm
- Polekowe, o podłożu immunologicznym
- Skutki chorób autoimmunologicznych (toczeń rumieniowaty układowy, RZS, zespół Felty'ego)
- infekcje wirusowe (CMV, EBV, hepatitis virus, HIV-1) i bakteryjne (supresja szpiku przez toksyny, migracja do ogniska zapalnego)
- posocznica
Neutropenia
- Łagodna 1000-1500/ mm3
- Umiarkowana 500 - 1000/ mm3
- Ciężka = agranulocytoza <500/ mm3
Agranulocytoza
- Liczba neutrofilów < 500/ mm3
- Stan zagrożenia życia
- Niedobór odporności nieswoistej
- Zwiększona podatność na zakażenia bakteryjne i grzybicze
Białaczki
- Choroby nowotworowe układu białokrwinkowego charakteryzujące sięnieprawidłową proliferacją, dojrzewaniem i uwalnianiem krwinek białych ze szpiku kostnego i obecnością niedojrzałych postaci tych komórek we krwi obwodowej oraz nacieków narządowych i tkankowych.
Czynniki wywołujące białaczki
- Promieniowanie jonizujące, niektóre środki chemiczne (benzen, cytostatyki)
- Wirusy (EBV, HTLV-1)
- Predyspozycje genetyczne (zespół Downa, niedokrwistość Fanconiego)
- Teoria aktywacji onkogenów (genów kodujących powstawanie receptorów dla czynników sterujących podziałami komórkowymi)
Właściwości komórek białaczkowych
- Klon białaczkowy jest genetycznie niestabilny
- kle podziałowe komórek białaczkowych są najczęściej wolniejsze, a przyrost mniejszy w porównaniu z prawidłowymi komórkami szpiku
- przewaga klonu jest możliwa dzięki nagromadzeniu się komórek białaczkowych, które nie różnicują się i nie dojrzewają tak jak komórki prawidłowe
Właściwości komórek białaczkowych
- Komórki białaczkoe mogą pozostawać w fazie G0 cyklu komórkowego
- Zaburzenia syntezy adhezyn powodują, że komórki białaczkowe z zaburzeniami podziałów przechodzą do krwi obwodowej
Hemostaza - zespół procesów fizjologicznych, które zapewniają sprawne hamowanie krwawienia po przerwaniu ciągłości ściany naczynia oraz utrzymują płynność krążącej krwi.
Główne elementy hemostazy
Trzy fazy procesu hemostazy
Mechanizmy zapobiegające tworzeniu zakrzepów w krążeniu
- Integralność śródbłonka i produkcja substancji o charakterze przeciwzakrzepowym
- Endogenne inhibitory krzepnięcia (ATIII, HC II, białko C i białko S, TFPI)
- Fibrynoliza
Ściana naczyń krwionośnych
Śródbłonek naczyniowy - syntetyzuje, magazynuje, uwalnia lub unieczynnia szereg substancji: kontrolujących napięcie naczyń, adhezję i agregację płytek, substancji pro- i przeciwzakrzepowych.
Substancje o działaniu przeciwzakrzepowym
- Glikokaliks
- Prostacyklina (PGI2) i NO - działają rozkurczająco na naczynia krwionośne, hamują adhezję, agregację i aktywację płytek
- t-PA - główny aktywator plazminogenu
- TFPI - inhibitor szlaku czynnika tkankowego
- ADP-aza - degraduje ADP
- Trombomodulina
- Antytrombina III (ATIII)
Substancje o działaniu prozakrzepowym
- Endotelina 1 (ET1) - obkurcza naczynia
- Czynnik von Willebranda - udział w adhezji i agregacji płytek krwi
- Inhibitor tkankowego aktywatora plazminogenu (PAI-1)
- Trombospondyna - przyspiesza adhezję i agregację płytek
- Czynnik tkankowy (TF) - inicjator krzepnięcia
- Czynnik aktywujący płytki krwi (PAF) - zwiększa przepuszczalność naczyń, wpływa na adhezję i agregację płytek
- Interleukiny: IL-1, IL-5, IL-6, IL-8 - działanie prozapalne i prozakrzepowe
Rola płytek krwi
Interakcja płytek z uszkodzoną ścianą naczynia uruchamia szereg procesów:
Adhezja płytek do kolagenu:
1.Bezpośrednio
2. Za pośrednictwem VWF - główny mechanizm wiązania VWF wiąże się z jednej strony z receptorem płytkowym gp Ib/V/IX a z drugiej ze ścianą naczynia
Aktywatory płytek krwi
- Kolagen
- Trombina
- Epinefryna
- ADP
- TXA2
- PAF
Aktywacja płytek prowadzi do:
- Zmiany kształtu
- Degranulacji ziarnistości płytkowych
- Generacji TXA2
- Udostępnienia fosfolipidów powierzchni płytek (PL niezbędne w procesie krzepnięcia)
- Tworzenia agregatów płytkowych
Gp IIb/ IIIa - eksponowany na płytkach po ich aktywacji, wiąże się z fibrynogenem jak i z VWF. TWORZY SIĘ CZOP PŁYTKOWY
Rola płytek krwi w hemostazie
- Poprzez uwalnianie adrenaliny, noradrenaliny, serotoniny i tromboksanu powodują obkurczanie drobnych naczyń krwionośnych po urazie i zmniejszenie krwawienia
- Poprzez agregację i tworzenie czopu płytkowego zatrzymują krwawienie
- Aktywowane płytki stanowią główne źródło fosfolipidów, niezbędnych w kaskadzie krzepnięcia
- Dostarczają miejsc wiążących dla protrombiny i cz.IX
- Uwalniają fibrynogen, cz.V i VIII
- Synteza α2-antyplazminy i PAI-1 (osłabienie fibrynolizy)
- Uwalniają TXA2, PAF i VWF - silne aktywatory agregacji płytek
Krzepnięcie krwi
- Kaskada reakcji, których istotą jest przekształcenie rozpuszczalnego białka osocza - fibrynogenu - w nierozpuszczalną fibrynę
- Zachodzi przy udziale kilkunastu osoczowych czynników krzepnięcia, jednego integralnego białka błon komórkowych (TF), fosfolipidów (głównie płytkowych), jonów wapnia i cynku oraz pośrednio witaminy K
Fibrynogen
- Sytetyzowany głównie w wątrobie
- Zaliczany do dodatnich białek ostrej fazy
- Kofaktor agregacji płytek krwi; uczestniczy w tworzeniu czopu płytkowego
- Prekursor fibryny
- Homodimer; pojedyńcza podjednostka złożona z trzech łańcuchów peptydowych: Aα, Bβ, γ.
- Trobmina uwalnia fibrynopeptydy A i B z cząsteczki fibrynogenu powodując utworzenie monomerów fibryny
- Agregacja monomerów fibryny - tworzenie siatki włóknika
Inhibitory krzepnięcia
- Antytrombina III (ATIII) - wiąże trombinę w nieaktywne kompleksy. Inaktywuje również cz.XIIa, XIa, Xa, IXa.
- Trombomodulina - trombina w kompleksach z trombomoduliną traci swe właścwości prozakrzepowe. Takie kompleksy aktywują białko C, które przy udziale kofaktora - białka S degraduje cz. Va i VIIIa.
- TFPI (Tissue Factor Pathway Inhibitor) - hamuje tworzenie kompleksów TF - cz.VIIa
Wrodzona trombofilia
Zaburzenia dotyczące fizjologicznych endogennych inhibitorów krzepnięcia (ATIII, białka C i białka S)
- Wrodzone niedobory ATIII, białka C i S
- Oporność na aktywowane białko C (czynnik 5 Leiden)
Defekt budowy cz.V - w miejscu Arg 506 - Glu (mutacja punktowa genu cz.V). Mutacja powoduje oporność na aPC.
- Mutacja G20210A genu protrombiny - zwiększenie stężenia protrombiny we krwi
Nabyte przyczyny niedoborów ATIII, białka C i S
- Choroby wątroby
- DIC
- Zespół nerczycowy
- Ciąża (zmniejszenie białka S)
- Leczenie heparyną (zmniejszenie ATIII)
Proces fibrynolizy
- Kluczowym enzymem jest plazmina
- Plazmina powstaje z zymogenu - plazminogenu w reakcji ograniczonej proteolizy
- Główny aktywator plazminogenu to t-PA
- Glu-Plazminogen (Glu-Pg) - natywna cząsteczka plazminogenu. Plazmina przekształca Glu-Pg w Lys-Pg, który jest 20-krotnie lepszym substratem dla t-PA.
- Powstające w osoczu niewielkie ilości plazminy są niemal natychmiast inaktywowane przez fizjologiczne inhibitory. Fibryna poprzez blokowanie miejsc wiążących α2-antyplazminę chroni plazminę przed inaktywacją.
Plazmina wbudowana w strukturę skrzepu jest chroniona przed działaniem inhibitorów!!!
Skazy krwotoczne
- Skłonność do krwawień długotrwałych i nadmiernie obfitych w stosunku do wywołującego je urazu lub pojawiających się niespodziewanie.
Dzielimy na skazy:
1. Płytkowe
2. Osoczowe
3. Naczyniowe
4. Mieszane
Skazy naczyniowe
1. Wrodzone
- choroba Rendu - Oslera - Webera
2. Nabyte
- Choroba Schonleina - Henocha
Choroba Rendu - Oslera - Webera
- Dziedziczna naczyniakowatość krwotoczna
- Autosomalna dominująca
- Defekt budowy ściany małych naczyń tętniczych i żylnych: ścieńczenie ściany naczyń, występowanie nieregularnych rozszerzeń, w tętniczkach zmniejszenie grubości warstwy mięśniowej i sprężystej, naczynia łatwo pękają
- Nawracające krwawienia z nosa, krwotoki płucne, krwotoki z przewodu pokarmowego, do OUN
Zespół Schonleina - Henocha
- Uogólnione zapalenie naczyń o podłożu immunologicznym (paciorkowiec β-hemolizujący)
- Głównie u dzieci 3 - 7 r.ż.; chłopcy
Objawy (występują najczęściej razem)
- Skórne: wyczuwalne palpacyjnie wybroczyny na kończynach, pośladkach, nigdy na tułowiu;
- Stawowe: duże stawy (skokowe, kolanowe, barkowe, łokciowe)
- Brzuszne: kolka jelitowa, smoliste stolce, fusowate wymioty
- Mogą wystąpić powikłania nerkowe
Awitaminoza C (gnilec, szkorbut)
- Upośledzenie tworzenia w naczyniach substancji międzykomórkowej tkanki łącznej
Skazy osoczowe
Hemofilie
Spowodowane dziedzicznymi niedoborami osoczowych czynników krzepnięcia:
Dziedziczenie recesywne w sprzężeniu z płcią
Obraz kliniczny
Zależny od stopnia ciężkości choroby, określonego na podstawie stężenia czynników krzepnięcia w osoczu:
- Ciężka < 2% cz.VIII lub IX
- Umiarkowana 2 - 5 % normy cz.VIII lub IX
- Łagodna 6 - 30 % normy cz.VIII lub IX
>30% - taki stan określa się jako nosicielstwo
U kobiet nosicielek genu hemofilii stężenie cz. VIII czy IX jest zmniejszone o nawet 50%, co jednak zapewnia prawidłową hemostazę
Krwawienia do stawów
- Kolanowy 44%
- Łokciowy 25%
- Skokowy 15%
- Barkowy 8%
Powikłanie - artropatia hemofilowa
Krwawienia domięśniowe
- Udo, pośladki - niebezpieczne ze względu na swoją obfitość (wstrząs pokrwotoczny)
- Mięśnie dna jamy ustnej, języka - grozi uduszeniem
- Ucisk krwiaka na naczynie lub nerw - niedokrwienie, porażenie
Leczenie
Leczenie substytucyjne - uzupełnianie brakującego czynnika
- Przetaczanie krwi (1840 r. - Lane)
- Przetaczanie osocza (1923 r. - Feislly)
- Krioprecypitat (1959 - 1966; Pool, Łopaciuk) - do leczenia hemofilii A
- Koncentraty cz.VIII z osocza
- Koncentraty rekombinowanego cz.VIII - 1992 r
- Terapia genowa
Rozpoznanie hemofilii
- Wywiad rodzinny
- Wydłużenie czasu APTT przy prawidłowym czasie krwawienia i PT
- Oznaczanie stężenia cz.VIII i IX krzepnięcia
Choroba von Willebrandta
- Najczęstsza skaza krwotoczna wrodzona na świecie
- Dziedziczona autosomalnie dominująco lub recesywnie
- Częstość występowania 1 : 8000
- Mutacje w obrębie genu dla cz.VW, defekt ilościowy lub jakościowy VWF
Rola VWF
- Multimery o bardzo wysokiej masie cząsteczkowej uczestniczą w adhezji płytek krwi do kolagenu poprzez receptor płytkowy gp Ib
- Rola w krzepnięciu - multimery o zróżnicowanej masie cząsteczkowej wiążą się we krwi z cz.VIII chroniąc go przed proteolizą (zapewnienie prawidłowego poziomu cz.VIII we krwi)
Typy choroby von Willebrandta
Typ 1 - najczęstszy, niewielki ilościowy niedobór VWF
Typ 2 - defekt jakościowy VWF
Typ 3 - recesywny, znaczny niedobór ilościowy VWF
Typ 2 - osłabienie aktywności VWF
- 2A - defekt uwalniania VWF z komórek śródbłonka lub nadmierna proteoliza multimerów VWF w osoczu
- 2B - zwiększenie powinowactwa VWF do gp Ib płytek krwi. Występuje spontaniczne wiązanie VWF do płytek i usuwanie powstających kompleksów płytkowych, co prowadzi do trombocytopenii i utraty dużych aktywnych multimerów VWF
- 2M - zmniejszenie powinowactwa VWF do gp Ib płytek krwi
- 2N - osłabienie lub brak zdolności wiązania cz.VIII przez VWF. Obniżenie aktywności cz.VIII w osoczu z objawami hemofilii
Obraz kliniczny
- Typ 1 - skaza krwotoczna o przebiegu łagodnym, czasem utajona. Krwawienia śluzówkowe (z nosa, przewodu pokarmowego, dziąseł), nadmierne krwawienia miesiączkowe, wylewy podskórne, siniaczenie
- Typ 2 - objawy podobne, bardziej nasilone
- Typ 1 i 2 - dramatyczne pogorszenie po zażyciu aspiryny
- Typ 3 - defekt VWF i niedobór cz.VIII. Występują krwawienia skórno - śluzówkowe, siniaczenie ale również krwawienia do mięśni, stawów (jak w hemofilii).
Diagnostyka
Testy przesiewowe:
Skazy krwotoczne płytkowe
Trombocytopatie dziedziczne
Zespół Bernarda - Souliera
- Dziedziczony autosomalnie recesywnie
- Anomalia gp Ib/V/IX
- Małopłytkowość, przedłużony czas krwawienia, obecność olbrzymich płytek krwi
- Upośledzenie adhezji płytek
Trombastenia Glanzmanna
- Dziedziczona autosomalnie recesywnie i dominująco
- Anomalia gp IIb/IIIa
- Upośledzenie agregacji płytek
- Upośledzenie retrakcji skrzepu
- Wydłużenie czasu krwawienia
- Prawidłowa liczba i wielkość płytek
Trombocytopatie nabyte
Małopłytkowości (płytki < 150 tys/ml)
- Charakter wrodzony lub nabyty
- Amegakariocytowa - upośledzenie produkcji płytek krwi w szpiku
- Megakariocytowa - zwiększone niszczenie płytek krwi lub ich zużycie
- Niewłaściwe rozmieszczenie lub gromadzenie płytek krwi na obwodzie (splenomegalia, naczyniaki)
Objawy kliniczne małopłytkowości
- Mnogie wybroczyny krwotoczne na skórze kończyn i twarzy
- Drobne wybroczyny rozsiane w miejscach niewielkich urazów
- Krwawienia z błon śluzowych (nosa, przewodu pokarmowego, dróg moczowych, narządów płciowych, nadmierne krwawienia po zabiegach chirurgicznych)
Samoistna plamica małopłytkowa na tle immunologicznym (ITP)
- U dzieci po zakażeniach wirusowych
- U dorosłych - przewlekły przebieg, bez wyraźnego czynnika wywołującego
- Wybroczyny skórne, plamica, niewielkie krwawienia z błon śluzowych
- Trombocytopenia we krwi, obecność dużych płytek
- W szpiku wzrost liczby megakariocytów
Małopłytkowości przypominające ITP.
- Immunotrombocytopenie w przebiegu kolagenoz (np. SLE) lub chorób limfoproliferacyjnych
- Płytkowe reakcje poprzetoczeniowe (antygen PLA-1)
Małopłytkowość w przebiegu leczenia heparyną
- Rozwija się wskutek wiązania kompleksów heparyna - przeciwciało przez receptory błonowe płytek
- Heparyna wiąże się z PF4 w kompleksy posiadające właściwości antygenowe; powstają IgG przeciwko tym kompleksom
- Powstają agregaty płytkowe (zużycie płytek)
DIC (Disseminated Intravascular Coagulation)
zespół charakteryzujący się uogólnioną wewnątrznaczyniową aktywacją krzepnięcia prowadzącą do odkładania złogów fibryny w krążeniu. Depozycja fibryny przyczynia się do niedokrwienia i niedotlenienia tkanek. Masywna i trwająca w czasie aktywacja krzepnięcia skutkuje zużyciem płytek krwi i czynników krzepnięcia i rozwojem koagulopatii ze zużycia. Skaza krwotoczna nasilana jest dodatkowo przez wtórną aktywację fibrynolizy i pojawienie się we krwi inhibitorów krzepnięcia (produkty degradacji fibryny)
DIC - ciężkie powikłanie szeregu stanów patologicznych:
Patofizjologia DIC
- Uogólniona aktywacja krzepnięcia
- Upośledzenie funkcji fizjologicznych inhibitorów krzepnięcia
- Nasilenie fibrynolizy
- Cytokiny
Aktywacja krzepnięcia - szlak TF
Czynnik tkankowy (TF) produkowany przez:
- Komórki śródbłonka naczyniowego
- Monocyty
Także:
- Płuca, nerki, nabłonki
Generacja trombiny w DIC zachodzi głównie w szlaku zależnym od TF.
Regulacja generacji trombiny
W DIC każdy z tych poziomów regulacji jest upośledzony
Cytokiny w rozwoju DIC
- IL-6 i IL-1 pośredniczą w aktywacji krzepnięcia przez TF/VIIa
- TNF-α:
- Dysregulacja fizjologicznych mechanizmów antykoagulacyjnych i fibrynolizy
- Moduluje aktywność IL-6
Objawy kliniczne DIC
Diagnostyka
- Wydłużenie czasu krzepnięcia
- Wydłużenie czasów: PT, APTT, trombinowego
- Zmniejszenie stężenia fibrynogenu
- Zmniejszenie stężenia ATIII
- Wzrost stężenia D-dimerów i FDP
- Zmniejszenie liczby płytek krwi
Specyficzne markery krzepnięcia i fibrynolizy
- Wzrost PF4
- Wzrost fragmentu protrombinowego F 1+2
- Wzrost fibrynopeptudu A
- Wzrost kompleksów TAT
- Zmniejszenie stężenia plazminogenu
- Zwiększenie ilości kompleksów plazmina - antyplazmina gdy aktywacja fibrynolizy
Wyszukiwarka