K W A S Y K A R B O K S Y L O W E
Aleksander Kołodziejczyk 2007.12
Charakterystyczną cechą kwasów karboksylowych jest obecność w cząsteczce grupy karboksylowej: -COOH, dzięki której związki te pozyskują właściwości kwasowe.
Budowa grupy karboksylowej
Atom węgla grupy karboksylowej ma hybrydyzację sp2. Tworzy on wiązanie podwójne z atomem tlenu (tak jak w grupie karbonylowej) i dwa pojedyncze wiązania, jedno z funkcją OH i drugie z resztą alkilową (kwasy alifatyczne) lub arylową (kwasy aromatyczne). Grupa karbonylowa i hydroksylowa razem z atomem węgla, do którego są przyłączone nazywana jest funkcją (grupą) karboksylową.

W najprostszym kwasie karboksylowym - w kwasie mrówkowym (metanowym) grupa karboksylowa połączona jest z atomem wodoru.
Parametry grupy karboksylowej

W kwasie mrówkowym kąty wiązań wychodzących od karboksylowego atomu węgla przyjmują wartości zbliżone do 120o, chociaż widoczne są wyraźne odchylenia. Ta deformacja wynika z obecności małego atomu wodoru związanego z grupą karboksylową;
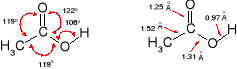
W kwasie octowym takich różnic już nie ma.
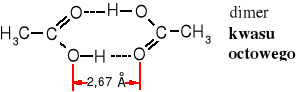
Grupa karboksylowa, podobnie jak karbonylowa jest płaska i silnie spolaryzowana. Obecność atomu wodoru związanego z atomem tlenu grupy hydroksylowej sprzyja powstawaniu wiązań wodorowych i tworzeniu dimerów, co wpływa na podwyższenie temperatury wrzenia tych związków. Ich cząsteczki nawet w stanie pary są dimerami.
Występowanie
Kwasy karboksylowe są bardzo rozpowszechnione w naturze. Występują w stanie wolnym (np. kwas mrówkowy czy octowy), ale w największej ilości w postaci estrów (tłuszcze, woski, substancje zapachowe) oraz innych pochodnych, np. amidów. Amidami są peptydy i białka.
Kwasy tłuszczowe będące składnikami tłuszczów i innych lipidów zawierają parzystą liczbę atomów węgla, a ich łańcuch alifatyczny, zwykle nierozgałęziony, bywa nasycony lub zawiera 1 albo więcej podwójnych wiązań. Najmniejszym kwasem zaliczanym do kwasów tłuszczowych jest kwas masłowy (C3H7COOH), następnie zaś kapronowy (C5H11COOH).

Najpopularniejszym naturalnym kwasem tłuszczowym jest kwas oleinowy (C17H33COOH), a pośród nasyconych - palmitynowy (C15H31COOH).
Do popularnych kwasów zaliczają się też kwas stearynowy (C17H35COOH) i linolowy (C17H31COOH).
Nienasycone kwasy tłuszczowe tłuszczów jadalnych powinny mieć wiązania podwójne niesprzężone, typu cis-, tak jak w kwasie oleinowym, linolowym czy linolenowym (C17H29COOH). Znane są także naturalne kwasy karboksylowe zawierające sprzężone podwójne wiązania typu trans-, np. kwas oleostearynowy, stereoizomer kwasu linolenowego, występujący w oleju tungowym (chińskim). Jest on szkodliwy dla zdrowia, ale znalazł praktyczne zastosowanie - służy do produkcji artystycznych farb olejnych.
Podczas modyfikacji tłuszczów naturalnych, np. w trakcie ich uwodorniania (w procesie produkcji margaryn twardych), a także w czasie długotrwałego smażenia i poddaniu ich na działanie tlenu dochodzi do izomeryzacji wiązań podwójnych: cis- → trans-, co pogarsza się ich wartość odżywczą. Nienasycone kwasy tłuszczowe trans- są szkodliwe dla zdrowia.
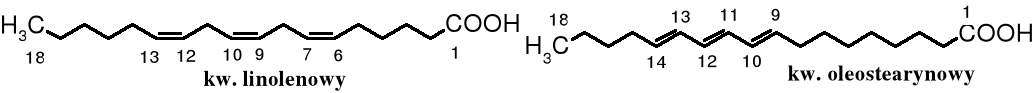
Pytanie: dlaczego izomery cis- łatwiej przechodzą w trans-, a nie odwrotnie?
Kwasy wchodzące w skład wosków są zwykle nasycone i mają ponad 20 atomów węgla.
Od nienasyconego dwudziestowęglowego kwasu arachidonowego wywodzą się ważne dla prawidłowego funkcjonowania organizmu prostaglandyny.
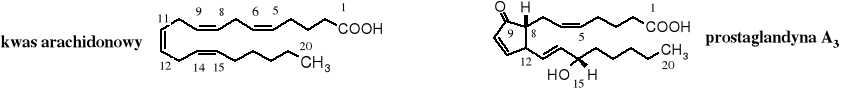
W skład żółci wchodzą kwasy żółciowe, należą one do steroidów. Najpopularniejszym jest kwas cholowy.
Znane są też aromatyczne kwasy karboksylowe. Kwas benzoesowy pełni rolę środka konserwującego; znajduje się w niektórych owocach, np. w żurawinach. Kwas salicylowy występuje w stanie wolnym, w postaci estrów, a także glikozydów. Ma działanie przeciwgorączkowe.
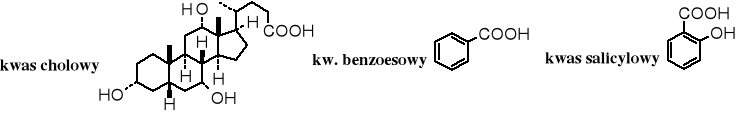
Znany jest kwas migdałowy i cynamonowy; ich nazwy wskazują pochodzenie.

Nomenklatura
1.Nazwy zwyczajowe
Dla wiele kwasów karboksylowych powszechnie używa się nazw zwyczajowych, ponieważ kwasy należą do wcześnie odkrytych związków organicznych, a wówczas nie było jeszcze ustalonych reguł nomenklaturowych. Nazwy często nawiązywały do źródła, z którego związek został po raz pierwszy wyizolowany. Z tego powodu do dzisiaj popularne są takie nazwy jak kwas: mrówkowy, octowy, masłowy, walerianowy, laurowy, oleinowy, arachidowy, migdałowy czy cynamonowy. Stosowane są one również w literaturze naukowej.
2. Nomenklatura systematyczna
Według zasad IUPAC nazwy alifatycznych kwasów karboksylowych tworzy się dodając do słowa kwas przymiotnikową nazwę węglowodoru macierzystego (węglowodoru zawierającego taki sam szkielet węglowy, co kwas).
Przykłady (w nawiasach nazwy zwyczajowe):
HCOOH - kwas metanowy (mrówkowy) CH3COOH - kwas etanowy (octowy)
CH3CH2COOH - kwas propanowy (propionowy) CH3CH2CH2COOH - kwas butanowy (masłowy)
CH3CH2CH2CH2COOH - kwas pentanowy (walerianowy) CH3(CH2)4COOH - kwas heksanowy (kapronowy)
CH3(CH2)6COOH - kwas oktanowy (kaprylowy) CH3(CH2)4COOH - kwas dekanowy (kaprynowy)
CH3(CH2)4COOH - kwas heksadekanowy (palmitynowy) CH3(CH2)16COOH - kwas oktadekanowy (stearynowy)
Kwasy nienasycowne
CH2=CHCOOH - kwas prop-2-enowy (akrylowy)
CH3CH=CHCOOH - kwas but-2-enowy (krotonowy - trans-)
CH3(CH2)7C=C(CH2)7COOH - kwas oktadeka-9-enowy (oleinowy - cis-)
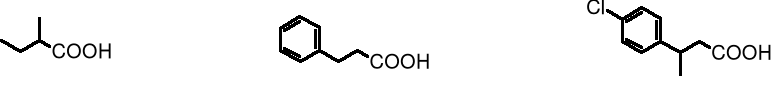
kwas 2-metylobutanowy kwas 3-fenylopropanowy kwas 3-(p-chlorofenylo)butanowy
W tym sposobie nazewnictwa atomowi węgla grupy karboksylowej jest przypisany lokant 1.
Kwasy dikarboksylowe otrzymują końcówkę -diowy, która jest połączona ze rdzeniem łącznikiem „o”.
HOOCCOOH - kwas etanodiowy (szczawiowy)
HOOCCH2COOH - kwas propanodiowy (malonowy)
HOOC(CH2)2COOH - kwas butanodiowy (bursztynowy)
HOOC(CH2)3COOH - kwas pentanodiowy (glutarowy)
HOOC(CH2)4COOH - kwas heksanodiowy (adypinowy)
HOOC(CH2)5COOH - kwas heptanodiowy (pimelinowy)
3. Drugi systematyczny sposób nazywania kwasów karboksylowych, także zatwierdzony przez IUPAC, polega na tym, że do zestawu słów kwas .......karboksylowy dodaje się nazwę węglowodoru, do którego została dołączona grupa karboksylowa. Według tej zasady kwas octowy nazywa się kwasem metanokarboksylowym, a kwas bursztynowy - etanodikarboksylowym.
CH3-COOH - kwas metanokarboksylowy (octowy)
HOOC-(CH2)2-COOH - kwas etanodikarboksylowy (bursztynowy)
Sposób wykorzystujący zestaw słów „kwas alkanokarboksylowy” pozwala na łatwe tworzenie nazw kwasów cyklicznych - kwasy cykloalkanokarboksylowe i kwasów acyklicznych, o bardziej rozbudowanych łańcuchach.
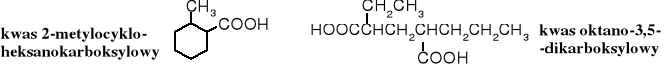
4. Nazewnictwo podstawnikowe
Grupę karboksylową można traktować jako podstawnik i tworzyć nazwy związków dodając do rdzenia przedrostek karboksy- wraz z odpowiednim lokantem. W przypadku skomplikowanych związków można łączyć dwa systemy.
Właściwości fizyczne i fizjologiczne
Niższe alifatyczne kwasy monokarboksylowe (do nonanowego) są cieczami. Ich temperatury wrzenia są wysokie, wyższe nawet niż temperatury wrzenia alkoholi, znacznie wyższe niż aldehydów, eterów i węglowodorów o podobnej masie cząsteczkowej. Przyczyną tego jest tendencja cząsteczek kwasów do tworzenia dimerów stabilizowanych stosunkowo silnymi wiązaniami wodorowymi. Kwas mrówkowy wrze w temp. 100,5oC, a octowy, propionowy i masłowy odpowiednio: 118, 141 i 163 oC.
Kwas dekanowy topnieje w temperaturze 31oC. Rozgałęzienia łańcuchów i wiązania nienasycone znacząco obniżają tt. kwasów. Kwas oleinowy topnieje w temp. w 16oC, kwas linolowy, w -12oC, a dla porównania stearynowy - w 70oC. Pierwsze cztery kwasy z szeregu homologicznego do masłowego włącznie mieszają się z wodą w każdym stosunku. Od kwasu walerianowego rozpuszczalność w wodzie szybko spada. Począwszy od kwasu dodekanowego są praktycznie nierozpuszczalne w wodzie. Kwasy tłuszczowe rozpuszczają się dobrze w wodnych roztworach alkalicznych (tworzą sole) i w rozpuszczalnikach organicznych. Sole wyższych kwasów tłuszczowych nazywane są mydłami; mają właściwości detergentów, dlaczego?
Zadanie: dlaczego mydła są związkami powierzchniowo czynnymi?
Mieszanina stałych kwasów tłuszczowych nosi nazwę stearyny. Jest ona używana do wyrobu świec (stearynowych, są jeszcze znane świece parafinowe i woskowe). Kropla roztopionej stearyny (lub stopionego stałego kwasu) wlana na powierzchnię wody, tak żeby utworzyła się cienka warstwa zastyga w płatek, którego powierzchnie różnią się wyglądem i właściwościami. Ta część, która stykała się z wodą jest hydrofilowa (zwilża się wodą), a ta druga jest hydrofobowa - kropelki wody spływają po niej. Zjawisko to polega na tym, że w cienkiej warstwie ciekłego jeszcze kwasu dochodzi do orientacji jego cząsteczek. Grupy karboksylowe (hydrofilowe) nawiązały kontakt z wodą, a łańcuchy węglowodorowe zostały skierowane nad wodę.
Zapach kwasów mrówkowego, octowego i propionowego jest ostry, duszący, kwaśny. Kwas masłowy i jego średniocząsteczkowe homologi mają bardzo nieprzyjemny, odrażający zapach (odór). Zjełczałe masło wydziela zapach kwasu masłowego; im bardziej rozłożone do wolnych kwasów, tym bardziej staje się cuchnące. Wyższe kwasy tłuszczowe (od kwasu dodekanowego) i kwasy aromatyczne są bezzapachowe.
Otrzymywanie
1. Źródła naturalne1
Wiele kwasów tłuszczowych jest pochodzenia naturalnego; pozyskuje się je głównie z tłuszczów, które są triacyloglicerolami, czyli estrami z glicerolem. Można je uwalniać z tłuszczów za pomocą hydrolizy kwaśnej lub zasadowej. Częściej stosowana jest hydroliza zasadowa, zwana zmydlaniem tłuszczów, ponieważ tworzące się sole są mydłami.
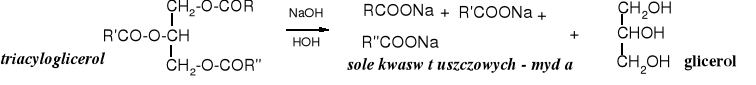
Naturalne triacyloglicerole zawierają zwykle różne reszty acylowe (R ≠ R' ≠ R''), może to być mieszanina reszt kilku, a nawet kilkunastu kwasów. Czyste kwasy karboksylowe pozyskuje się z tłuszczów, w których jeden z kwasów występuje w znacznej przewadze, np. z oleju oliwkowego wyodrębnia się kwas oleinowy, a z łoju wołowego kwas stearynowy. Często tak otrzymane kwasy muszą być dodatkowo doczyszczane. Robi się to różnymi sposobami. Małe ilości najlepiej za pomocą chromatografii. Na skalę techniczną przeprowadza się destylację kwasów lub lepiej ich estrów metylowych w warunkach mocno obniżonego ciśnienia, za pomocą tzw. destylacji molekularnej. Kwasy stałe można krystalizować.
2. Otrzymywanie kwasu octowego
Kwas octowy jako produkt naturalny w postaci skwaśniałego wina czy soków był znany od dawna. Jako tzw. ocet winny (rozcieńczony wodny roztwór AcOH) stosowano go w kuchni od niepamiętnych czasów. Powstawał on w wyniku bakteryjnego utlenienia etanolu (wina). Do dzisiaj w procesie otrzymywania spożywczego kwasu octowego wykorzystuje się bakterie. Utlenia się 15% wodny roztwór etanolu przepuszczając go przez złoże (wióry) zawierające odpowiednie bakterie. Powstały w ten sposób rozcieńczony (~10%) roztwór kwasu octowego zwany octem spirytusowym, w odróżnieniu od octu winnego jest bezbarwny i pozbawiony specyficznego aromatu tego drugiego.
Etanol można też katalitycznie utlenić tlenem z powietrza do kwasu octowego.
Kwas octowy ma ogromne zastosowanie w przemyśle, głównie do wytwarzania różnych estrów. Jego światowa produkcja przekracza 4 mln ton rocznie. Współcześnie za najlepszą metodę otrzymywania kwasu octowego uznaje się karbometylowanie metanolu.
CH3OH + CO _→ CH3COOH
Popularną technologią jest też utlenianie powietrzem takich węglowodorów, jak butan, buteny, benzyna, a nawet cięższe alifatycznej frakcje ropy naftowej. Obecnie, ze względu na wysoką cenę surowca, nie stosuje się utlenienia etanalu.
3. Otrzymywanie kwasu mrówkowego
Kwas mrówkowy wytwarzany jest w skali o rząd mniejszej niż kwas octowy. Otrzymuje się go obecnie poprzez hydrolizę mrówczanu metylu, powstającego w reakcji karbonylowania metanolu.
HOH
CH3OH + CO _→ HCOOCH3 _→ HCOOH + CH3OH
HCOOH
Ta technologia wyparła uprzednio stosowane karbonylownie wodorotlenku sodu.
>150oC H2SO4
CO + NaOH __→ HCOONa __→ HCOOH
4. Utlenianie aldehydów prowadzi do kwasów karboksylowych. W praktyce nie jest to jednak często wykorzystywany sposób otrzymywania kwasów, ponieważ aldehydy są trudno dostępne i to raczej pochodne kwasów stanowią substraty w ich syntezie. Powszechne, uprzednio wykorzystywane przemysłowe utlenianie etanalu do kwasu octowego zostało zastąpione innymi dogodniejszymi metodami. Acetaldehyd utleniało się tlenem z powietrza w roztworze kwasu octowego, wobec katalizatora zapewniającego wysoką wydajność.
powietrze
CH3CHO + 0,5 O2 ___→ CH3COOH 95%
<60oC, MnAc3
5. Utlenianie alkanów
Utlenianie węglowodorów (najczęściej odpowiednich frakcji ropy naftowej, najlepiej parafiny zawierającej do 30 atomów C)) powietrzem wobec katalizatorów prowadzi do mieszaniny kwasów karboksylowych. Zawiera ona kwasy od mrówkowego do ejkozanowego i większe. Poszczególne kwasy wyodrębniania się metodą destylacji frakcjonowanej.
powietrze
RCH2-CH2R' ___→ RCOOH + R'COOH
Mn3+
Ten sposób otrzymywania kwasów był próbą zastąpienia kwasów naturalnych syntetycznymi. Można ją uznać za próbę raczej nieudaną, ponieważ podaż tłuszczów naturalnych jest wystarczająca, a kwasy syntetyczne wytwarzane tą technologią zawierają cząsteczki zarówno z parzystą jak i nieparzystą liczbą atomów węgla. Takie kwasy nie nadają się, np. do produkcji detergentów, ponieważ te drugie nie ulegają biodegradacji.
6. Utlenianie alkiloarenów
Utlenianie związków aromatycznych zawierających alkilowe łańcuchy boczne jest powszechnym sposobem otrzymywania kwasów aromatycznych z grupą karboksylową (grupami) związaną bezpośrednio z pierścieniem aromatycznym. Wykorzystuje się je zarówno w przemyśle, jak i w laboratoriach. W przemyśle do utleniania stosuje się najczęściej tlen z powietrza, a w laboratorium KMnO4. Każdy łańcuch alifatyczny, niezależnie od jego długości, zostaje w warunkach tej reakcji utleniony do grupy karboksylowej związanej z pierścieniem.
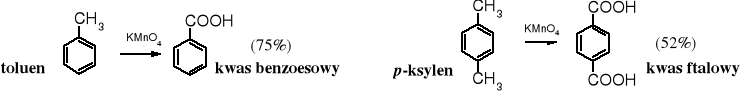
Wydajności powyższych reakcji nie są wysokie, ponieważ substraty pełnią zarazem rolę rozpuszczalnika i nie mogą być utlenione do końca. Zastosowanie katalizatorów międzyfazowych znacznie przyspiesza reakcję i umożliwia otrzymywanie kwasów z wydajnością prawie ilościową.
7. Karboksylowanie związków Grignarda
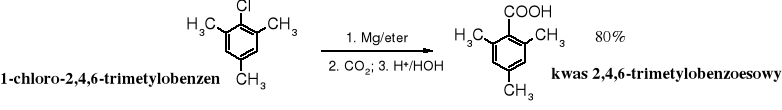
Addycja ditlenku węgla do związków Grignarda stanowi uniwersalną metodę otrzymywania kwasów karboksylowych. Surowcami wykorzystywanymi w tej syntezie są stosunkowo łatwo dostępne halogenki alkilowe lub arylowe. Z uwagi na koszty i stosowanie niebezpiecznego eteru etylowego jest to raczej laboratoryjny sposób wytwarzania kwasów.

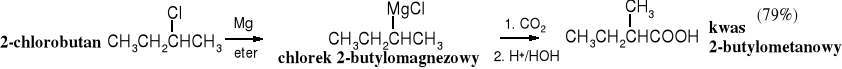
Karboksylowanie związków Grignarda jest dogodnym sposobem otrzymywania ,-dipodstawionych kwasów karboksylowych (zatłoczonych), np. kwasu piwalowego (trimetylooctowego) czy 2,4,6-trimetylobenzoesowego, trudnych do syntezy na innej drodze.
![]()
Ditlenek węgla można przyłączać również do sodo- lub litopochodnych organicznych.
8. Hydroliza pochodnych kwasów karboksylowych
Pośród pochodnych kwasowych najczęściej hydrolizowanymi są nitryle, ponieważ są łatwe w syntezie. W tej metodzie najczęściej pierwotnymi substratami są halogenki alkilowe.
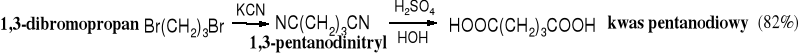
Do otrzymywania kwasów wykorzystywane są też inne pochodne kwasowe, najczęściej te, które są pochodzenia naturalnego, a więc estry. Pośród nich największe znaczenie jako surowce mają tłuszcze i woski.
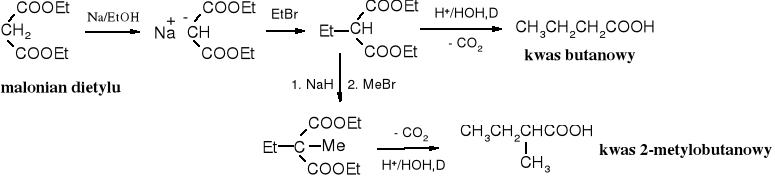
9. Synteza malonowa
Alkilowanie pochodnych kwasowych zawierających ruchliwe atomy wodoru jest często stosowanym sposobem otrzymywania kwasów z rozgałęzionymi łańcuchami, kwasów dikarboksylowych, a także zawierających w położeniu dodatkową grupę funkcyjną, np. aminową. Częstym substratem w tej reakcji jest ester malonowy. Synteza polega na alkilowaniu sodopochodnej estru malonowego, hydrolizie i dekarboksylacji (lub nie) produktu.
10. Metody mikrobiologiczne i enzymatyczne
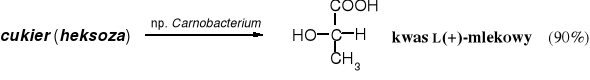
W przemyśle, do otrzymywania kwasów karboksylowych wykorzystuje się często mikroorganizmy (zwykle bakterie) i enzymy. Poza kwasem octowym metody te są stosowane raczej do produkcji kwasów wielofunkcyjnych, głównie aminokwasów (lizyny, fenyloalaniny i innych) oraz hydroksykwasów (kwasu cytrynowego, mlekowego czy glukonowego). Kwas winowy też powstaje w procesie fermentacji, ale jest to raczej produkt uboczny. Metody mikrobiologiczne i enzymatyczne zapewniają wysoką chiralną czystość produktów.
W fermentacyjnych procesach kwaszenia mleka, kapusty, ogórków i innych produktów bakterie utleniają cukry do kwasu mlekowego. W zależności od szczepu bakterii może powstać kwas mlekowy L-, D- lub racemiczny. Rocznie produkuje się ponad 50 000 ton kwasu mlekowego, z czego 70% metodą fermentacyjną, a resztę na drodze syntezy chemicznej.
Właściwości chemiczne
Kwasy karboksylowe są kwasami słabymi; w wodzie dysocjują tworząc anion karboksylanowy.
![]()
Stała kwasowości dla tej równowagi Ka i pKa wyrażają się wzorami:
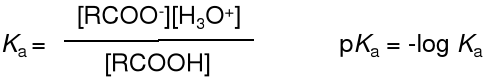
Stała kwasowości kwasów karboksylowych przyjmuje wartość około 10-5, co oznacza, że jedynie ~ 0,1% ich cząsteczek jest zdysocjowana. Dla kwasu octowego wartość Ka wynosi 1,76.10-5, wobec czego pKa = 4,75. Są to więc kwasy zdecydowanie słabsze, od np. kwasu solnego (pKa = -7), ale silniejsze niż kwas węglowy i znacznie silniejsze od alkoholi (pKa etanolu = 16).
Kwasy karboksylowe reagują z węglanami, przy czym wydziela się CO2, a jeżeli wolny kwas jest trudno rozpuszczalny w wodzie, to zwykle przechodzi on w rozpuszczalną w sól. To proste doświadczenie jest dowodem, że kwasy karboksylowe są silniejszymi kwasami od kwasu węglowego.
![]()
Kwasy karboksylowe z zasadami tworzą sole.
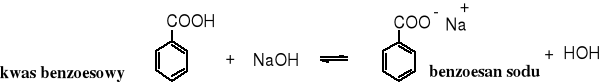
trudno rozpuszczalny w wodzie sól rozpuszczalna w wodzie
Wartości Ka i pKa wybranych kwasów Tabela 13.1
Nazwa kwasu |
wzór |
Ka |
pKa |
względna moc |
solny |
HCl |
107 |
-7 |
b. mocny
b. słaby |
trifluorooctowy |
CF3COOH |
0,59 |
0,2 |
|
trichlorooctowy |
CCl3COOH |
0,23 |
0,6 |
|
dichlorooctowy |
CHCl2COOH |
3,3.10-2 |
1,5 |
|
fluorooctowy |
CH2FCOOH |
2,6.10-3 |
2,6 |
|
chlorooctowy |
CH2ClCOOH |
1,4.10-3 |
2,9 |
|
bromooctowy |
CH2BrCOOH |
2,1.10-3 |
2,7 |
|
jodooctowy |
CH2ICOOH |
7,5.10-4 |
3,1 |
|
mrówkowy |
HCOOH |
1,8.10-4 |
3,8 |
|
glikolowy |
HOCH2COOH |
1,5.10-4 |
3,8 |
|
benzoesowy |
PhCOOH |
6,5.10-5 |
4,2 |
|
akrylowy |
H2C=CHCOOH |
5,6.10-5 |
4,3 |
|
fenylooctowy |
PhCH2COOH |
5,2.10-5 |
4,3 |
|
octowy |
CH3COOH |
1,8.10-5 |
4,8 |
|
propionowy |
CH3CH2COOH |
1,3.10-5 |
4,9 |
|
etanol |
CH3CH2COH |
1.10-16 |
16 |
|
Z wartości podanych w tabeli 12.1 wynika, że na moc kwasów karboksylowych znaczący efekt wywierają podstawniki ulokowane blisko grupy karboksylowej. Podstawniki elektroakceptorowe zwiększają kwasowość - im silniej wyciągają elektrony, tym mocniej zwiększają kwasowość. Kwas fluorooctowy jest około 100 razy mocniejszy od kwasu octowego, a trifluorooctowy aż 32 000 razy. Natomiast Podstawniki elektrodonorowe obniżają kwasowść. Kwas propionowy jest nieznacznie słabszym kwasem niż kwas octowym, ale też efekt elektrodonorowy grupy metylowej jest bardzo słaby.
Moc kwasów karboksylowych jest związana z efektem mezomerycznym stabilizującym jon karboksylanowy. Podstawniki elektroakceptorowe zwiększają rozmycie ładunku ujemnego, a tym samym stabilizują jon karboksylanowy. Podstawniki elektrodonorowe mają odwrotne działanie.
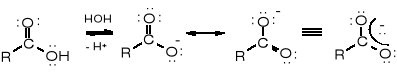
dysocjacja mezomeria
Rozmycie ujemnego ładunku jonu karboksylanowego spowodowane mezomerią stabilizuje ten jon, a tym samym ułatwia dysocjację grupy karboksylowej do anionu karboksylanowego. W jonie alkoholanowym nie ma możliwości mezomerii; jon ten nie jest stabilizowany, a przez to alkohole nie są kwasami.
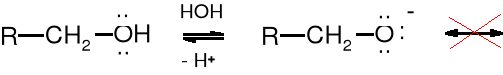
Podstawniki wyciągające elektrony (EWG - electron withdrowing group) przez rozmycie ładunku zwiększają stabilizację anionu karboksylanowego, zaś podstawniki elektrodonorowe (EDG - electron donoring group) zwiększają ładunek ujemny anionu i destabilizują go.

Moc kwasu zależy od odległości podstawnika od grupy karboksylowej. Efekt ten można prześledzić na przykładzie kwasów -, -, i γ- chloromasłowych. Łatwo zauważyć, że im bliżej znajduje się chlor tym większy jest jego wpływ kwasowość izomerów kwasu chloromasłowego.
Kwasowość izomerów kwasu chloromasłowego Tab. 13.2
Nazwa kwasu |
wzór |
Ka |
pKa |
-chloromasłowy |
CH3CH2CH(Cl)COOH |
1,4.10-3 |
2,9 |
-chloromasłowy |
CH3CH(Cl)CH2COOH |
9,0.10-5 |
4,1 |
γ-chloromasłowy |
ClCH2CH2CH2COOH |
3,0.10-5 |
4,5 |
masłowy |
CH3CH2CH2COOH |
1.5.10-5 |
4,8 |
Większa liczba podstawników elektroujemnych w większym stopniu zwiększa moc kwasu, ponieważ efekty sumują się wektorowo.
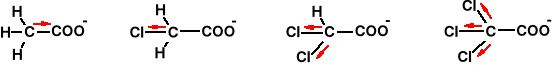
pKa: 4,7 2,9 1,3 0,6
Wpływ podstawników w pierścieniu na moc kwasu benzoesowego
Uprzednie stwierdzenie, że wielkość wpływu podstawników na moc kwasów karboksylowych zależy od ich odległości od grupy karboksylowej nie sprawdza się w przypadku kwasów aromatycznych, ponieważ to stwierdzenie jest prawdziwe jedynie dla efektu indukcyjnego, którego oddziaływanie szybko się zmniejsza wraz z odległością. Natomiast efekty mezomeryczne, które dominują w układach aromatycznych, są mniej wrażliwe na odległości, jako że przenoszą się wzdłuż wiązań. Mezomeryczne efekty tych samych podstawników w pozycji para- i orto- będą zbliżone. Jedynie inne efekty (indukcyjny i orto-) będą modyfikowały ich oddziaływanie. W pozycji meta- efekt mezomeryczny nie występuje.
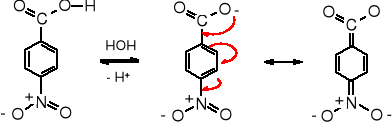
Wartość pKa kwasu benzoesowego wynosi 4,2, podczas gdy p-nitrobenzoesowego 3,4, co oznacza, że ten drugi jest 8 razy mocniejszy. Natomiast kwas p-metoksybenzoesowy, zawierający podstawnik elektrodonorowy jest kwasem słabszym niż benzoesowy, jego pKa wynosi 4,5.
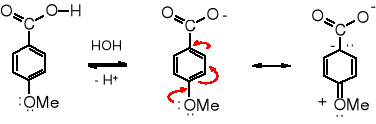
Pomiar kwasowości jest łatwy do przeprowadzenia, dzięki czemu na podstawie zmiany stałej kwasowości Ka można ilościowo oznaczyć wpływ podstawników na kwasowość grupy karboksylowej kwasu benzoesowego. Zmiana kwasowości analogu kwasu benzoesowego w porównaniu z kwasowością kwasu benzoesowego wywołana obecnością podstawnika w położeniu para- odzwierciedla efekt mezomeryczny tego podstawnika. Należy brać pod uwagę wpływ grupy obecnej w położeniu para-, ponieważ w meta- nie ma podobnego oddziaływania, a w orto- daje o sobie znać efekt indukcyjny i tzw. efekt orto-. Można założyć, że oznaczony na podstawie zmiany kwasowości kwasu benzoesowego efekt mezomeryczny danego podstawnika będzie podobnej wielkości w reakcjach SE cząsteczki aromatycznej zawierającej ten sam podstawnik.
Takie oznaczenia zostały przeprowadzone i na ich podstawie został zrobiony podział na podstawniki aktywujące reakcje SE, a zarazem obniżające kwasowość kwasu benzoesowego i podstawniki dezaktywujące reakcje SE. Równocześnie ilościowe zmiany kwasowości pozwalają na ułożenie tych podstawników w kolejności zależnej od wielkości wywoływanego efektu. Taka lista przedstawiona jest w tabeli 12.3.
Wpływ podstawników w położeniu para- na kwasowość na kwasowość pochodnych benzenu Tabela 12.3
Moc kwasu |
Y |
Ka |
p Ka |
|
|
-OH |
3,3.10-5 |
4,5 |
grupy aktywujące |
|
-OCH3 |
3,5.10-5 |
4,5 |
|
|
-CH3 |
4,3.10-5 |
4,3 |
|
|
-H |
6,5.10-5 |
4,2 |
|
|
-Cl |
1,0.10-4 |
4,0 |
grupy dezaktywujące |
|
-Br |
1,1.10-4 |
4,0 |
|
|
-CN |
2,8.10-4 |
3,6 |
|
|
-NO2 |
2,9.10-4 |
3,4 |
|
Jakie praktyczne korzyści daje znajomość wartości mocy analogów podstawionego kwasu benzoesowego? Otóż na podstawie tych wartości można oceniać właściwości podstawników, np. ich wpływ na reakcje SE.

Redukcja grupy karboksylowej
Grupa karboksylowa jest grupą polarną i do jej redukcji wymagane są polarne reduktory, takie jak tetrahydroglinian litu albo borowodór lub specjalne katalizatory umożliwiające wykorzystanie wodoru gazowego. Grupa karboksylowa nie ulega redukcji pod wpływem tetrahydroboranu sodu ani wodoru w obecności Pt lub Pd. Dzięki tym właściwościom redukcję grupy karboksylowej można przeprowadzić selektywnie w obecności wiązań podwójnych. Tetrahydroglinian litu redukuje zarówno -COOH, jak i -NO2.
![]()
Ten sam kwas redukowany katalitycznie wodorem wobec Pd lub Pt zostaje przekształcony z kwasu nienasyconego (w tym wypadku kwasu oleinowego) w kwas nasycony (kwas stearynowy).
Zadanie: napisz równanie powyższej reakcji.
Selektywną redukcję grupy karboksylowej w obecności nitrowej przeprowadza się za pomocą borowodoru.
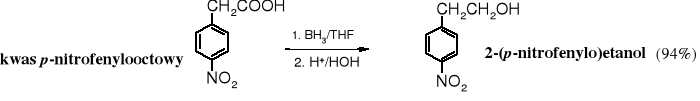
W przemyśle, na ogromną skalę przeprowadza się selektywną redukcję nienasyconych kwasów tłuszczowych za pomocą wodoru. Redukuje się je wodorem w obecności katalizatora niklowego do mniej nienasyconych lub nasyconych kwasów tłuszczowych (np. z kwasu linolenowego można otrzymywać etapami kwas linolowy, oleinowy, a wreszcie stearynowy) lub wodorem wobec katalizatorów miedziowo-chromowych do odpowiednich alkoholi (alkoholi tłuszczowych).
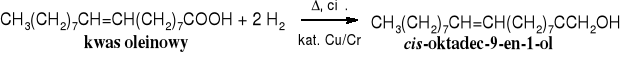
Zadanie: napisz ciąg reakcji prowadzących od kwasu linolenowego do stearynowego.
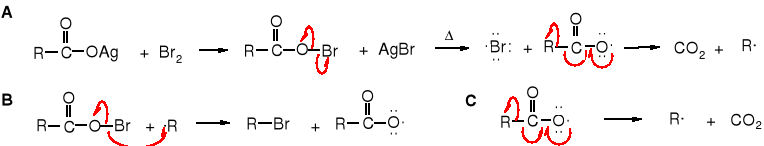
Reakcja Hunsdieckera - dekarboksylacja
Sole kwasów karboksylowych z rtęcią lub srebrem ogrzewane w obecności halogenów ulegają dekarboksylacji tworząc halogenek alkilu zawierający o jeden atom C mniej niż wyjściowy kwas karboksylowy.
![]()
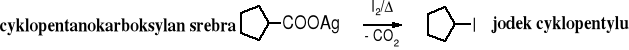
Mechanizm reakcji
Reakcja biegnie rodnikowo, mechanizmem łańcuchowym. W pierwszym etapie - A -powstaje O-bromokwas, który pod wpływem podwyższonej temperatury rozkłada się na rodnik bromkowy i rodnik karboksylanowy, ten z kolei na rodnik alkilowy i CO2. Rodnik alkilowy w drugim etapie - B - reaguje z O-bromokwasem tworząc halogenek alkilu i rodnik karboksylanowy, a ten rozkłada się tak samo na rodnik alkilowy jak w etapie A.
Reakcja Hunsdieckera wykorzystywana jest do otrzymywania halogenków alkilowych wówczas, kiedy odpowiednie kwasy karboksylowe są łatwo dostępne, a inne metody syntezy tych halogenków niedogodne.
Zadanie: z kwasu heksanowego otrzymać bromek heksylu i bromek pentylu.
Reakcja Hella-Volharda-Zielińskiego
W kwasach karboksylowych atomy wodorów H ulegają substytucji bromem lub chlorem, w wyniku czego powstają -halogenokwasy. Reakcja zachodzi pod wpływem bromu lub chloru w obecności czerwonego fosforu lub PBr3 (PCl3).
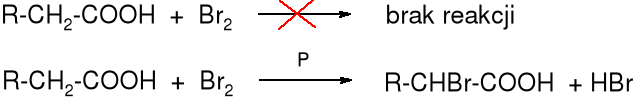

Mechanizm reakcji
Reakcja zaczyna się od utworzenia tribromku fosforu: 3 Br2 + 2 P _→ 2 PBr3
Fosfor dodawany jest w ilości katalitycznej.
Powstały PBr3 przekształca część kwasu karboksylowego w bromek kwasowy:
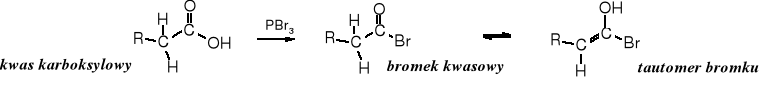
W formie tautomerycznej bromku kwasowego znajduje się podwójne wiązanie, do którego przyłącza się cząsteczka bromu, po czym następuje eliminacja HBr i tworzy się bromek -bromokwasu.
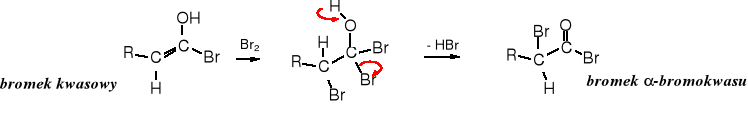
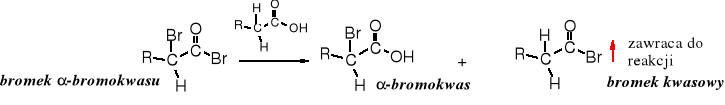
Bromek -bromokwasu w reakcji z kwasem karboksylowym przekształca się w -bromokwas i bromek kwasowy, który zawraca do reakcji przyłączając cząsteczkę bromu. Dzięki tej wymianie wystarcza katalityczna ilość P lub PBr3.
-Halogenokwasy są wykorzystywane w syntezie organicznej zarówno w reakcjach na grupie karboksylowej, jak i poprzez wymianę halogenu na różne funkcje, np. aminową czy hydroksylową.
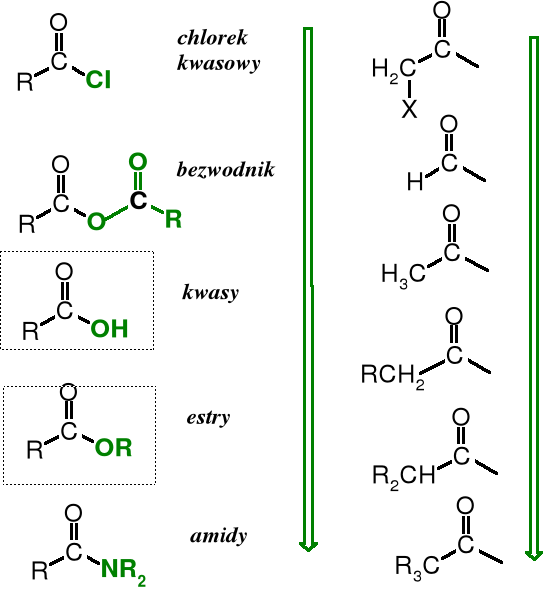
Pochodne kwasów karboksylowych
Funkcję -OH w grupie karboksylowej można wymienić, np., na -X, -OR, -NH2, -NHNH2 czy inne ugrupowania. Otrzymuje się wówczas pochodne zwane odpowiednio: halogenkami kwasowymi, estrami, amidami i hydrazydami. Jeżeli w grupie karboksylowej oba atomy tlenu zostaną zastąpione przez atom azotu powstaje nitryl.
Poniżej przedstawione są wzory strukturalne tych pochodnych.
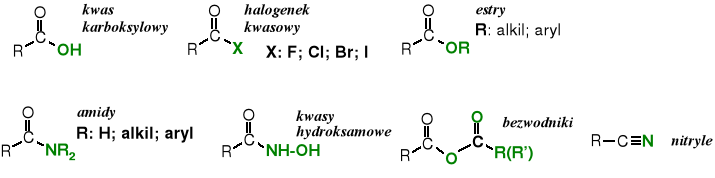
Reszta cząsteczki kwasu karboksylowego pozbawiona grupy -OH nosi nazwę acylu.

Takie same reszty powstają z estrów po usunięciu funkcji -OR, z amidów bez -NR2, aldehydów bez -H grupy aldehydowej i ketonów bez jednego -R. Formalnie reszty acylowe wywodzi się od kwasów karboksylowych i od kwasów pochodzą ich nazwy (z nazw angielskich).

formyl acetyl propionyl butyryl oksalil malonyl sukcynyl glutaryl
Aroil to ogólna nazwa reszt kwasów aromatycznych.
Atak nukleofilowy na acylowy atom węgla
Zarówno kwasy karboksylowe, jak i pochodne kwasowe zawierają w grupie karboksylowej dodatnio naładowany atom węgla, zwany acylowym atomem węgla. Jest on podatny na atak nukleofilowy. Addukt powstający po przyłączeniu do -COY odczynnika nukleofilowego :Nu- ulega stabilizacji, najczęściej poprzez odszczepienie łatwo odchodzącej grupy :Y-.
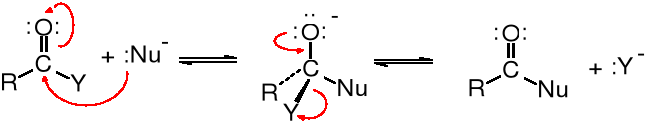
Grupy odchodzące Y: F; Cl; Br; I; lub HY: HOR; HSR; HOCOR; HOH; HNR2
Reaktywność pochodnych kwasowych z odczynnikami nukleofilowymi zależy od wielkości ładunku dodatniego na acylowym atomie węgla - im bardziej elektroujemny jest atom lub grupa Y, tym większy ładunek dodatni znajduje się na atomie C i tym bardziej reaktywna staje pochodna kwasowa.
bardziej reaktywne w reakcjach z :Nu-
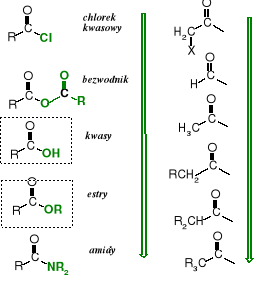
Względna reaktywność pochodnych kwasowych
w reakcji z odczynnikami nukleofilowymi
(aktywność w reakcjach acylowania)
mniej reaktywne w reakcjach z :Nu-
Halogen (w halogenkach kwasowych) i reszty acylowe (w bezwodnikach) jako EWG powiększają lukę elektronową na acylowym atomie węgla, czyniąc go tym samym bardziej podatnym na atak odczynników nukleofilowych (zwiększają aktywność acylującą pochodnej kwasowej). Reszty alkilowe R wywierając w estrach efekt indukcyjny +I, a w amidach +M lub efekt +M i +I (w grupie -NR2) obniżają aktywność acylującą.
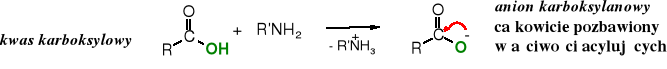
W powyższym diagramie nie ma kwasów karboksylowych, które powinny sytuować się pomiędzy bezwodnikami i estrami, ponieważ oddziaływanie atomu wodoru grupie -OH na karboksylowy atom węgla można uznać za obojętne w porównaniu do elektroakceptorowej reszty acylowej w bezwodnikach i elektrodonorowej grupy alkilowej w estrach. Tak faktycznie jest, kiedy kwas reaguje z alkoholem, natomiast w przypadku reakcji kwasu z amoniakiem lub aminą tworzy się sól karboksylanowa, której anion jest całkowicie pozbawiony właściwości acylujących, jako że posiada ładunek ujemny.
Na aktywność acylującą estrów duży wpływ ma charakter grupy -R. Reszty alkilowe w wyniku efektu indukcyjnego +I obniżają właściwości acylujące estrów, natomiast grupy arylowe zwiększają ich reaktywność z odczynnikami nukleofilowymi. Wprowadzenie do pierścienia aromatycznego dodatkowych ugrupowań elektroakceptorowych, np. -NO2, halogenów czy reszt acylowych zwiększa aktywność acylującą takich estrów. Estrom o podwyższonej aktywności acylującej nadano nazwę estrów aktywnych. Do tej grupy odczynników acylujących zalicza się też związki o podobnej budowie - RCOONR, w których atom tlenu grupy karboksylowej połączony jest z resztą organiczną R poprzez atom azotu zamiast węgla. Nie należą one do estrów, ponieważ nie są pochodnymi kwasów z alkoholami. Pomimo tego związki te (nieprawidłowo) też zwane są estrami aktywnymi. Zawierają one, takie ugrupowania, jak np. N-hydroksysukcynoimidyl czy N-hydroksybenzotriazyl.
Estry aktywne
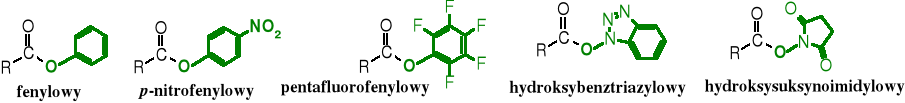
Estry aktywne służą do otrzymywania amidów, szczególnie często są wykorzystywane w syntezie peptydów. Można też za ich pomocą otrzymywać inne estry.
15
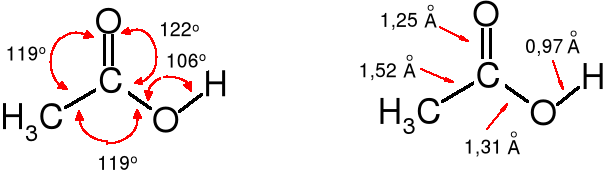
Wyszukiwarka