LEK - to substancja lub jej metabolit( to produkt metabolizmu, przemian chemicznych zachodzących w organizmach- czasem metabolit działa silniej niż związek macierzysty, a czasem dopiero metabolit wykazuje działanie farmakologiczne - forma PRO-DRUG-podany lek jest prekursorem właściwie działającego związku) wywołująca w organizmie określony efekt terapeutyczny, profilaktyczny lub diagnostyczny.
Leki dostępne w Polsce ujęte są w Urzędowym Wykazie Produktów Leczniczych Dopuszczonych do Obrotu na Terytorium Rzeczypospolitej Polskiej.
Zgodnie z ustawą z dnia 6 września 2001 r. Prawo farmaceutyczne ;
•lekiem aptecznym - jest produkt leczniczy sporządzony w aptece zgodnie z przepisem przygotowania zawartym w Farmakopei Polskiej lub farmakopeach uznawanych w państwach członkowskich Unii Europejskiej przeznaczony do wydawania bezpośrednio w tej aptece,
• lekiem gotowym - jest produkt leczniczy wprowadzony do obrotu pod określoną nazwą i w określonym opakowaniu,
• lekiem recepturowym - jest produkt leczniczy sporządzony w aptece na podstawie recepty lekarskiej,
Surowiec farmaceutyczny - substancja lub mieszanina substancji pochodzenia chemicznego, roślinnego, zwierzęcego lub mineralnego przeznaczona do sporządzenia - w praktyce receptury aptecznej bądź wytworzenia na skalę przemysłową w laboratorium galenowym lub zakładach farmaceutycznych produktów leczniczych stosowanych w lecznictwie u ludzi i zwierząt. W pewnych sytuacjach nieprzetworzony surowiec farmaceutyczny może stanowić produkt leczniczy (np. jodoform, dermatol, kseroform - do zasypywania ran; wazelina i in.). Informacje o lekach;
Baza bloz
Do podstawowych zadań naszej bazy należy:
- uporządkowanie informacji o lekach i środkach ochrony zdrowia dostępnych na polskim rynku farmaceutycznym,
- zapewnienie automatycznej wymiany informacji (zamówienia, faktury, raporty, itp.) pomiędzy aptekami, hurtowniami, importerami, producentami leków oraz NFZ i lekarzami,
- zautomatyzowanie wymiany danych o parametrach leków, zmienianych okresowo przez Ministerstwo Zdrowia (Wykazy dotyczące: grup odpłatności, cen urzędowych, limitów, itd.),
- ujednolicenie sprawozdawczości o dystrybucji leków w Polsce.
International Nonproprietary Name, w skrócie INN (Międzynarodowa Nazwa Niezastrzeżona) - oficjalna niezastrzeżona, generyczna nazwa środka farmaceutycznego zalecana w nazewnictwie przez Światową Organizację Zdrowia. Jest ona stosowana dla ułatwienia ze względu na bardzo dużą ilość nazw handlowych leków i środków farmaceutycznych. Na przykład dla leku o nazwie handlowej Relanium (Międzynarodowa Nazwa Niezastrzeżona) to diazepam. Lek generyczny, lek odtwórczy, generyk - określenie leku będącego zamiennikiem leku oryginalnego, zawierającym tę samą substancje czynną. Leki generyczne, aby mogły być wprowadzone do obrotu, muszą mieć udowodniony : tę samą postać,.tę samą dostępność biologiczna (biodostępność)
takie samo działanie farmakologiczne
W Polsce na prośbę pacjenta farmaceuta ma obowiązek wydać lek generyczny w tej samej dawce i odpowiedniej ilości, niezależnie od tego, czy na recepcie widnieje nazwa międzynarodowa czy handlowa leku.
Losy leku w organizmie.
Po zażyciu leków następuje wiele procesów fizykochemicznych i enzymatycznych w trakcie których leki ulegają wchłonięciu, rozmieszczeniu w organizmie, wywierają działanie lecznicze, następnie ulegają metabolizmowi i wydaleniu. O tym jak szybko i w jakiej ilości lek pojawia się w miejscu działania oraz jak długo tam przebywa decyduje nie tylko podana dawka leku, ale także szereg procesów związanych z jego przemieszczaniem się (kinetyką)w organizmie obejmujących wchłanianie leku z miejsca podania do krążenia ogólnego, dystrybucję w płynach i tkankach organizmu oraz eliminację na drodze biotrasformacji i wydalania
Przedmiotem badań farmakokinetycznych są wymienione tu procesy, szybkość ich przebiegu, analiza wpływu na stężenie leku(także metabolitu) we krwi innych leków jednocześnie zażywanych oraz stanów chorobowych, oddziałujących na metabolizm i eliminacje leków z organizmu. Przebieg tych procesów w farmakokinetyce jest wyrażany w oparciu o matematyczne i analityczne metody. Wyznaczone na podstawie obliczeń matematycznych zależności pomiędzy podana dawką a stężeniem leku we krwi pozwalają scharakteryzować właściwości farmakokinetczne danego leku za pomocą takich parametrów jak; dostępność biologiczna, objętość dystrybucji, biologiczny okres półtrwania.
Farmakokinetyka (farmakon- lek, kinesis- ruch).
zajmuje się matematyczną oceną ilościową procesów kinetycznych jakim lek podlega w organizmie
obejmuje wchłanianie, dostępność biologiczną, rozmieszczenie w tkankach, biotransformację i wydalanie.
Farmakokinetyka ma duże znaczenie w farmacji i farmakologii (ocena biofarmaceutyczna leków, tj. określenie ich dostępności biologicznej) oraz w klinice (racjonalizacja i optymalizacja farmakoterapii).
Farmakodynamika
bada wpływ leku na organizm i poszczególne narządy
bada mechanizm działania leku
- w zależności od metody wyróżniamy: farmakodynamikę biochemiczną, molekularną, genetyczną.
Różnice w działaniu leków u różnych ludzi wynikają głównie (choć nie wyłącznie) z tzw. defektów enzymatycznych.Wiele leków (np. izoniazyd, sulfonamidy, prokainamid) ulega biotransformacji przez przyłączenie do ich cząsteczki kwasu octowego. Proces ten nazywa się acetylowaniem i prowadzi do dezaktywacji leków. Wśród populacji ludzi odróżniamy 2 grupy pod względem sprawności acetylowania - szybko acetylujących i wolno acetylujących. U szybko acetylujących podanie przeciętnej dawki INH (izoniazyd - lek przeciwgruźliczy) - nie wywiera działania leczniczego u wolno acetylujących - może powodować kumulację leku i zatrucie (np. zapalenie wielonerwowe po stosowaniu izoniazydu występuje prawie 4-krotnie częściej u wolnych acetylatorów). U przedstawicieli rasy białej jest po ok. 50% szybko- i wolno-acetylujących. U Japończyków i Eskimosów przeważają wolno-acetylujący.
Genetycznie uwarunkowana jest również nadwrażliwość na sukcynylocholinę - lek zwiotczający mięśnie prążkowane stosowany w chirurgii. Przyczyną tego jest dziedziczna enzymopatia dotycząca osoczowej acetylocholinoesterazy. U ludzi z tą enzymopatią prawidłowa dawka sukcynylocholiny może spowodować długotrwaly bezdech, prowadzący niekiedy nawet do zejścia śmiertelnego. Świadomość tego pozwala dobrać właściwą dawkę sukcynylocholiny.
LOSY LEKÓW W ORGANIZMIE
Od momentu podania leku do organizmu dochodzi do licznych następujących po sobie i związanych z sobą procesów, które obejmują dzieje leku w organizmie. Całość toczących się procesów określamy skrótem . LADME od pierwszych liter angielskich nazw;
L- liberation(uwolnienie);
absorbtion(wchłanianie);
D- distribution (rozmieszczenie);
M- metabolism (metabolizm);
E- excretion (wydalanie);
L -uwolnienie warunkiem działania leku jest jego uwolnienie z danej postaci. Proces ten polega na rozpadzie postaci leku (tabletki, drażetki etc.) rozpuszczeniu substancji czynnej i dyfuzji tej substancji z miejsca podania do miejsca wchłaniania. Szybkość uwalniania leków zależy od ich postaci i od szybkości transportu leku z miejsca podania do miejsca wchłaniania. Etap uwalniania się substancji leczniczej do otaczających lek płynów ustrojowych jest kluczowy dla skuteczności terapii lekiem.
Ilość uwolnionego z preparatu i przechodzącego do roztworu leku określa się jako oddawalność co determinuje jego dostępność biologiczną.
Leki mogą działać miejscowo lub ogólnie. Leki o działaniu miejscowym nie wchłaniają do krwioobiegu, działając tylko na powierzchnie na które zostały zastosowane. W odróżnieniu od leków działających ogólnie, mają określone stężenie lecznicze wyrażone w procentach. Działanie miejscowe wykazują środki antyseptyczne, miejscowo znieczulające, niektóre z nich mogą ulec wchłonięciu do krwioobiegu, np. w wypadku przekrwienia skóry po kąpieli, łatwiej wchłaniają się też leki przez skórę niemowląt, ze względu na jej wyjątkową chłonność i wrażliwość.
Leki które przenikają przez skórę, błony śluzowe lub wchłaniają się z tkanki mięśniowej, podskórnej po dostaniu się do krwioobiegu wywołują działanie ogólne.
A-wchłanianie obejmuje procesy związane z przejściem leku z miejsca podania do krążenia ogólnego. O wchłanianiu decydują ;
-droga podania- sposób wprowadzenia leku do ustroju
-postać leku- Postać leku może wpływać na szybkość wchłaniania, lek podany w postaci roztworu ulega szybszemu wchłonięciu niż po podaniu w postaci stałej.
- właściwości fizykochemiczne leku; masa cząsteczkowa, lipofilność, rozpuszczalność, stopień zjonizowania(Substancje zjonizowane silniej wchłaniają się wolniej i w mniejszym stopniu. Ładunek albowiem komplikuje przenikanie poprzez membrany biologiczne) , pH środowiska(Skala pH to ilościowa skala kwasowości i roztworów zasadowości wodnych związków chemicznych)
-rodzaj i właściwości barier miejsca wchłaniania (budowa morfologiczna i skład chemiczny)
- indywidualne cechy pacjenta
- Najczęściej leki podaje się droga doustną, ale działanie tak podanych leków jest słabsze i następuje wolniej. Leki podane droga doustną ulegają wchłonięciu w górnym odcinku przewodu pokarmowego, głównie na zasadzie dyfuzji biernej. Stąd leki o charakterze kwaśnym (kwas acetylosalicylowy, fenobarbital)przede wszystkim tam ulegają wchłonięciu, natomiast większość będących słabymi zasadami ulega wchłonięciu w jelicie cienkim. Zmiany pH w przewodzie pokarmowym pod wpływem czynników fizjologicznych (pożywienie)oraz patologicznych (nadkwasota ) mogą w istotny sposób zmieniać wchłanianie poszczególnych leków. Inne czynniki wpływające na szybkość tego procesu to zmiana motoryki przewodu pokarmowego(opróżnianie żołądka , perystaltyka jelit) ora choroby przewodu pokarmowego(celiakia).
Wchłanianie leku po domięśniowym podaniu przebiega również na przenikania drodze substancji czynnej poprzez błony biologiczne do krwiobiegu. Tu również ma szczebel znaczenie zjonizowania wchłanianej substancji.
Niektóre leki podane doustnie działają wyłącznie w świetle przewodu pokarmowego (np. sulfoguanidyna, neomycyna) niewielka rozpuszczalność tych leków powoduje że nie przechodzą one do krążenia ogólnego. Inne (gentamycyna,streptomycyna, strofantyna) nie mogą być stosowane, ponieważ wchłaniają się z przewodu pokarmowego w różnym stopniu. Jeszcze inne (benzylopenicylina)ulegają szybkiemu rozkładowi w kwaśnym środowisku żołądka lub nawet strawieniu w przewodzie pokarmowym(insulina).
Leki podane droga doustną pokonując ścianę żołądka i jelit przechodzą do krążenia wrotnego i z nią są transportowane do wątroby. Ich wychwyt przez wątrobę i metabolizm jakiemu ulegają podczas pierwszego pasażu przejścia są określane jako efekt pierwszego przejścia.
Efekt pierwszego przejścia - zmniejszenie się ilości leku po jego wchłonięciu z przewodu pokarmowego, a przed dotarciem do krążenia ogólnego.
Do leków ulegających intensywnemu metabolizmowi w efekcie pierwszego przejścia należą in. kwas acetylosalicylowy, imipramina, propranolol, nifedypina, werapamil.
Niektóre leki np. nitrogliceryna ulega w wątrobie unieczynnieniu aż w 95%(dlatego stosuje się ja podjęzykowo).
Jedyną drogą podania leku, przy której trudno mówić o jego wchłanianiu jest dożylne podanie wprowadzające lek wprost do krążenia ogólnego, gdzie wprost mieszają się z krwią. W tym przypadku nie zachodzą też procesy uwalniania. Dożylnie wstrzykuje się leki w roztworach jałowych, zwykle izotonicznych czasem hipertonicznych. Nie można natomiast dożylnie stosować roztworów hipotonicznych, ani olejowych (zatory).
PARAMETREM FARMAKOKINETYCZNYM CHARAKTERYZUJĄCYM PROCES WCHŁANIANIA JEST DOSTĘPNOŚC BIOLOGICZNA KTÓRA OKREŚLA UŁAMEK-PROCENT DAWKI, JAKI DOCIERA DO KRĄZENIA OGÓLNEGO ORAZ SZYBKOŚĆ Z JAKĄ TEN PROCES ZACHODZI , PO POZANACZYNOWYM PODANIU LEKU. Przyjmuje się, że po dożylnym podaniu leku wartość tego ułamka wynosi jeden (100%), natomiast po pozanaczyniowym podaniu jest mniejsza od jedności.
Jest to spowodowane m.in. niecałkowitym uwalnianiem leku z danej postaci, niecałkowitym wchłanianiem z miejsca podania lub efektem pierwszego przejścia przez wątrobę.
Parametry określające biodostępność: AUC, Cmax, tmax.
AUC (area under the curve) - POLE POD KRZYWĄ STĘŻEŃ - miara ilości leku jaka dociera do krążenia ogólnego w postaci nie zmienionej.
Czas stężenia maksymalnego (tmaks) - czas, mierzony od chwili pozanaczyniowego podania leku, po którym jego stężenie we krwi osiąga wartość maksymalną.
Stężenie maksymalne (Cmaks) - największe stężenie leku obserwowane we krwi natychmiast po dożylnym jego podaniu lub po pewnym czasie, po podaniu pozanaczyniowym.
Stężenie minimalne (Cmin) - wartość stężenia leku obserwowana w trakcie jego wielokrotnego podawania tuż przed podaniem kolejnej dawki.
D - dystrybucja leków w organizmie w chwili gdy lek przedostanie się do krwi
następuje jego dystrybucja, czyli rozmieszczenie w płynach i tkankach organizmu. Głównymi barierami, jakie pokonuje lek są:bariera krew-łożysko, bariera krew-mózg, bariera krew-tkanka,
bariera krew-ciecz wodnista oka.
O procesie dystrybucji decyduje:
- szybkość transportu poprzez błony biologiczne
-wiązanie leku z białkami krwi i tkanek
- lipofilność leku
-szybkość krwi przepływu poprzez poszczególne tkanki i organy
-szybkość przepływu krwi przez poszczególne tkanki i narządy
Z uwagi na to, że przepływu szybkość krwi przez poszczególne tkanki i organy jest zróżnicowana, umiejscowienie leków w danych tkankach i narządach jest rózne. Czynnikiem determinującym to rozmieszczenie wewnątrz organizmu i przenikanie do tkanek, w których dochodzi do interakcji lek-receptor jest ukrwienie danej tkanki.
Stężenie leku w dobrze ukrwionych tkankach ustala się szybciutko, chwilkę po wchłonięciu, na niezmiennym poziomie, stężenia w słabo kompartmencie ukrwionym osiągane są później. Do nienagannie ukrwionych narządów należą: serce, mózg, nerki, płuca jak i gruczoły wydzielania wewnętrznego; do słabo ukrwionych: tkanka kostna, tłuszczowa.
Transport wielu leków odbywa się szybko i stosunek stężeń w tych tkankach do stężeń we krwi osiąga wartość stałą. Tkanki te z kinetycznego punku widzenia tworzą kompartment centralny. Tkanki słabo ukrwione tworzą tzw. Kompartment tkankowy.
Kompartment - objętość, przestrzeń organizmu, w której lek ulega równomiernemu rozmieszczeniu, hipotetyczna przestrzeń organizmu obejmująca zespół tkanek i narządów (na ogół nie określonych pod względem anatomicznym), w których lek z kinetycznego punktu widzenia zachowuje się podobnie. np. szybkość zmian stężenia leku w czasie jest taka sama.
-szybkość transportu przez błony biologiczne.
Transport poprzez leków błony biologiczne odbywa się jednakowo jak transport innych substancji, na zasadzie:
dyfuzja bierna - podstawowy mechanizm przenikania większości leków (Dyfuzja to samorzutna procedura rozprzestrzeniania się cząsteczek lub energii w danym ośrodku, np. w gazie, cieczy lub ciele niezmiennym) konsekwencją będący przypadkowych zderzeń dyfundującej cząsteczek substancji między sobą lub z cząsteczkami otaczającego ją ośrodka. Tkanka to ekipa kom
przebiega zgodnie z różnicą stężeń leków tzn. od stężenia większego do mniejszego
przenikająca przez błonę ilość leku wskutek dyfuzji biernej jest proporcjonalna do powierzchni błony, a odwrotnie proporcjonalna do jej grubości
szybkość dyfuzji w układzie zamkniętym maleje w miarę zmniejszania się różnicy stężeń, aż ustaje całkowicie po wyrównaniu stężeń po obu stronach
mając do czynienia z układami (kompartamentami) otwartymi, w których z jednej strony następuje stała dostawa leku, z drugiej-równoważna jego eliminacja to wówczas dyfuzja będzie przebiegać stale ze stałą szybkością
dyfuzja przez pory - przenikanie leku rozpuszczonego w wodzie przez wypełnione wodą kanaliki tzw.pory w ścianie błony komórkowej (dyfuzja bierna leków)
kierunek i szybkość tej formy transportu zależy od różnicy ciśnień hydrostatycznych, liczby porów w danej błonie oraz ich średnicy i grubości
głównym czynnikiem limitującym i determinującym dyfuzję przez pory jest wielkość cząsteczek (łatwo dyfundujące są związki o masie cząsteczkowej do 150,natomiast związki o masie cząsteczkowej do 400 dyfundują trudniej i to tylko przez niektóre pory)
dyfuzja ułatwiona - jego mechanizm jest związany z obecnością w błonie specjalnego systemu przenośnikowego- białek błonowych
przenośnik - składnik komórki mający określone powinowactwo do wiązania chemicznego z transportowaną substancją
kompleks przenośnik - lek dyfunduje z jednej strony błony na drugą, gdzie ulega ponownej dysocjacji uwalniając cząsteczkę leku do fazy wodnej
przenośnik powraca do swego pierwotnego położenia, gdzie ponownie może przyłączyć następną cząsteczkę
wyróżniamy dwa rodzaje transportu:
transport czynny
- transport substancji wbrew różnicy stężeń, przy udziale nośników i nakładzie energetycznym
-lek przenikający przez błonę może na skutek transportu czynnego być przerzucony z roztworu o mniejszym stężeniu do roztworu o większym stężeniu wbrew różnicy stężeń
- transport ten wymaga dostarczenia energii, a jest hamowany przez trucizny metaboliczne, niedobór tlenu i obniżenie temperatury
-jeśli dwie substancje są transportowane przez ten sam system przenośników to wówczas jedna z nich hamuje, współzawodnicząc o przenośnik, transport drugiej
pinocytoza - swoista forma transportu przypominająca fagocytozę drobnoustrojów przez makrofagi
małe kuleczki cieczy lub małe cząsteczki stałe wnikają w postaci wodniczki do błony, wędrują na jej drugą stronę i tam zostają usunięte do fazy wodnej
transport głównie kwasów tłuszczowych, fragmentów białek, kwasów nukleinowych
w przypadku leków nie ma istotnego znaczenia
- Wiązanie leku z białkami krwi i tkanek
leki po wniknięciu do krwi rozpuszczają się w całej objętości osocza (czas trwania ~1min.)
we krwi lek zostaje w mniejszym lub większym stopniu związany w sposób
odwracalny z białkami krwi, głównie albuminami (w mniejszym stopniu z niektórymi globulinami) .Albumina to białko rozpuszczalne w wodzie, w osoczu krwi, kreowane poprzez wątrobę, zarówno hepatocyty.
lek związany z białkiem jest w zasadzie nieczynny
- nie bierze udziału w dystrybucji
- nie jest metabolizowany ani wydalany
- nie przenika do tkanek
- nie wywiera działania farmakologicznego, tylko wolna frakcja może wywierać efekt farmakologiczny
- stanowi formę magazynu, z którego jest wolno uwalniany (w miarę zmniejszania się frakcji wolnej leku w płynach ustrojowych)
Wiązanie leków z białkami jest procesem odwracalnym, jest reakcją odwracalną, zależy od Ph środowiska, temperatury oraz właściwości fizykochemicznych leku.
Kompleks lek białko nie może przechodzić przez błony biologiczne, stanowi magazyn leku. Część leku związana z białkiem staje się biologicznie nieczynna. Tylko wolna frakcja może wywierać efekt farmakologiczny. W przypadku silnego wiązania leku z białkiem np. w 98%, tylko 2% z wprowadzonej dawki leku pozostaje jako wolna frakcja i tylko ta część może przenikać do tkanek. Szczebel wiązania leku z osocza białkami jest jednym z czynników zasadniczych warunkujących czas i siłę jego działania. W przypadku silnego wiązania leku z białkami, np. 98%, jedynie blisko 2% dawki wchłoniętej leku jest we krwi jako frakcja wolna i jedynie ta część może przenikać do tkanek, a więc wywierać rezultat farmakologiczny. Obserwuje najrozmaitsze się powinowactwo danych leków do wiązania z białkami, zjawisko to jest jeden z zachodzenia powodów między lekami interakcji.
Leki tworzące w sporym procencie kompleks z białkami osocza działają długo, albowiem krok po kroku uwalniają się z takiego kompleksu. Leki , które wiążą się z białkami w niewielkim stopniu, wydalane są szybciutko i wywierają działanie krótkotrwałe.
Wiązanie leków z białkami krwi i tkanek jest ważnym czynnikiem warunkującym ich rozmieszczenie w organizmie i wpływa na objętość dystrybucji Vd która określa zależność między ilością leku w organizmie(A) a stężeniem leku we krwi.
Vd = A/C
Vd jest parametrem charakteryzującym proces dystrybucji leków.
BIOLOGICZNY OKRES PÓŁTRWANIA LEKU (t0.5) - czas, po upływie którego stężenie leku obserwowane we krwi zmaleje o połowę w wyniku eliminacji po zakończeniu procesu wchłaniania i dystrybucji.
M- Metabolizm leków, biotransformacja
Większość leków ulega w organizmie chemicznej zmianie do związków, które są lepiej rozpuszczalne w wodzie i mogą być dzięki temu znacznie łatwiej wydalane z organizmu przez nerki. Jeżeli tak by się nie działo, wiele leków mogłoby nieograniczenie długo przebywać w organizmie i nadal wywierałoby swoje działanie, gdyż związki rozpuszczalne w tłuszczach, po przesączeniu przez kłębki nerkowe, wchłaniają się zwrotnie w kanalikach nerkowych i nie mogą być wydalone. Te chemiczne zmiany określa się metabolizmem lub biotransformacją.
Proces ten zachodzi przede wszystkim w mikrosomach wątroby, które zawierają układy enzymatyczne. Biotransformacji nie podlegają jedynie związki trudno rozpuszczalne w płynach ustrojowych i nie ulegające wchłanianiu z przewodu pokarmowego (węgiel leczniczy, parafina). Metabolizm leków przebiega także przy pomocy enzymów zlokalizowanych w osoczu krwi, płucach, łożysku, jądrach , jajnikach, nerkach, skórze, siatkówce oka i w jelitach, ale przede wszystkim przeprowadzany jest przez enzymy, które znajdują się w membranach retikulum endoplazmatycznego komórek wątroby - hepatocytach. Metabolizm leków jest procesem enzymatycznym (lek musi połączyć się z enzymem co jest warunkowane określonym powinowactwem),warunkiem zapoczątkowania reakcji enzymatycznej jest wniknięcie leku do wnętrza mikrosomów
Aktywność takich enzymów zależy od bardzo wielu czynników, konkretnie takich jak: temperatura ciała, płeć, wiek, a także stany patologiczne ustroju, m.in. choroby miąższu wątroby.
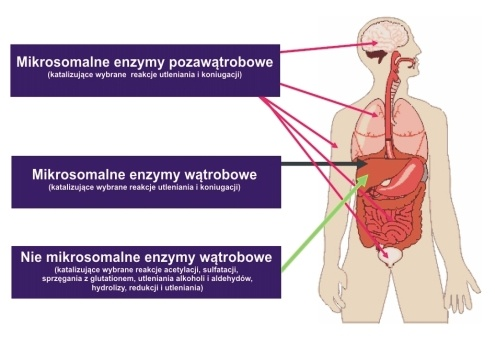
Reakcje biochemiczne związane z metabolizmem leków zachodzą w dwóch fazach(wg, Williamsa);
-Reakcje I fazy to to procesy rozkładu, nierzadko prowadzące do chemicznej zwiększenia reaktywności cząsteczki leku. Zalicza się do nich: utlenianie, redukcję, hydrolizę itp. Procesy rozkładu: mogą spowodować dezaktywację leku, mogą stanowić przygotowanie do II fazy biotransformacji lub zmienić styl leku to kierunek i siłę działania.
W konsekwencji zachodzących przemian I powstające fazy metabolity powinny być farmakologicznie nieaktywne i jako takowe zostają z organizmu wydalone lub powinny tworzyć związki o działaniu silniejszym, słabszym lub innym, w innym kierunku, niż pierwotnie wskazany lek. Przykładem czynnych metabolitów jest morfina, metabolit kodeiny.
Większość reakcji utleniania (głównej reakcji fazy pierwszej) jest przeprowadzana przez enzymy grupy cytochromu P450. Największe stężenie tych enzymów stwierdza się w wątrobie i w jelicie cienkim. Cytochrom P450 nie jest pojedynczą jednostką, ale w rzeczywistości jest to bardzo duża rodzina pokrewnych izoenzymów. Około 30 z nich zostało odkrytych w tkance wątroby człowieka. U człowiek wyróżnia się co najmniej 12 rodzin tych enzymów Za najbardziej istotne izoenzymy metabolizujące 90% leków uważa się: CYP1A2, CYP2C9, CYP2C19, CYP2D6, , CYP2E1 i CYP3A4, z czego dwa najważniejsze to CYP3A4 i CYP2D6. Poszczególni ludzie różnią się typem posiadanych enzymów ctochromu P450, w wyniku czego poszczególne leki są u różnych pacjentów metabolizowane z różną szybkością co wprowadza element dodatkowej niepewności, jeśli chodzi o określenie terapeutycznej dawki leku.
Należy zauważyć, że leki mogą być metabolizowane tylko przez jeden izoenzym
np. omeprazol/CYP2C19 lub jeden lek może być metabolizowany przez wiele różnych izoenzymów np. imipramina/CYP2D6, CYP1A2, CYP3A4, CYP2C19
Reakcje II fazy to procesy syntezy, do których zalicza się: acetylację, sprzęganie z związkami endogennymi aminokwasami, kwasem glukuronowym czy siarkowym, i prowadzi do powstania zazwyczaj związków nieaktywnych, przygotowanych do wydalenia z organizmu z moczem lub żółcią. Głównym celem obu faz jest zwiększenie rozpuszczalności leków w wodzie, a co za tym idzie ułatwienie ich wydalania.
Mniej wiadomo jest na temat enzymów odpowiedzialnych za reakcje sprzęgania drugiej fazy. UDP-glukuronylotransferaza (UGT), metylotransferaza i N-acetylotransferaza (NAT) są przykładami enzymów biorących udział w procesach drugiej fazy metabolizmu.
Rysunek przedstawia uproszczony schemat przemian jakim może ulegać lek w procesach biotransformacji.
.
Czynniki wpływające na metabolizm leków
- czynniki genetyczne
-płeć to decydujące znaczenie ma aktywność większa enzymów mikrosomalnych wątroby u mężczyzn niż u kobiet, szybszy dlatego metabolizm leków u mężczyzn
-wiek to mniejszą stwierdzono aktywność enzymów mikrosomalnych u dzieci, co powoduje, że wiele leków jest dla nich toksyczna. Aktywność enzymów takich redukuje się również w starszym wieku.
-stany patologiczne to prowadzą do upośledzenia biotransformacji leków i przedłużenia czasu ich działania
-droga podania to po podaniu doustnym wielu leków stwierdza się nasilenie metabolizmu wątrobowego, rezultat pierwszego przejścia,
-interakcje to leki wpływające na enzymów aktywność mikrosomalnych powinny metabolizm zmieniać innych podawanych zarazem leków,
Interkacje na etapie metabilizmu
Indukcja enzymatyczna
W czasach kiedy barbiturany były szeroko stosowane jako leki nasenne odkryto, że należy w czasie leczenia zwiększać dawki leku tak, aby osiągnąć ten sam efekt nasenny. Powodem takiego stanu rzeczy jest zwiększenie aktywności enzymów mikrosomalnych przez barbiturany, w wyniku czego stopień metabolizmu i wydalania tych leków jest zwiększony. Ten fenomen „stymulacji” lub indukcji enzymów, nie tylko wpływa na potrzebę zwiększenia dawki, ale jeśli inny lek, który jest metabolizowany przez tę samą grupę enzymów jest również obecny, to także jego metabolizm enzymatyczny jest zwiększony i potrzebne są wtedy większe dawki, aby uzyskać ten sam efekt terapeutyczny.
Najczęściej indukowanym szlakiem metabolicznym jest I faza utleniania, w której pośredniczą izoenzymy cytochromu P450. Główne grupy leków odpowiedzialne za indukcje najważniejszych klinicznie izoenzymów cytochromu P450 ; tytoń, Etanol Fenobarbital,(Luminal ), Ryfampicyna, Izoniazyd, Karbamazepina, (Amizepin, Tegretol),Fenytoina Epanutin Parenteral), Wyciągi z dziurawca
Procesy drugiej fazy metabolizmu mogą być również indukowane. Przykładem może być indukcja sprzęgania z kwasem glukuronowym zydowudyny (Retrovir) wywołana przez ryfampicynę (Rifampicyna).
Zazwyczaj potrzeba paru tygodni aby móc zaobserwować w pełni rozwinięty efekt działania induktora enzymatycznego. Efekt ten może utrzymywać się przez podobny czas nawet wtedy, gdy zaprzestano podawania środka indukującego.
Jeżeli jeden lek zmniejsza działanie innego przez indukcje enzymatyczną , możliwe byłoby dostosowanie interakcji przez odpowiednie dawkowania leku, którego działanie ulega zmianie. Przykładem może być ryfampicyna, która aktywuje enzymy mikrosomalne wątroby zmieniając metabolizm wielu leków. Utrzymanie odpowiedniego stężenia terapeutycznego leków, po dołączeniu ryfampicyny, może wymagać odpowiedniej zmiany ich dawkowania, np. ryfampicyna przyspiesza metabolizm m.in. leków hormonalnych, pacjentki stosujące doustne środki antykoncepcyjne podczas leczenia ryfampicyną powinny stosować alternatywne, niehormonalne metody antykoncepcji.
Indukcja enzymatyczna jest powszechnym mechanizmem interakcji i nie ogranicza się tylko do leków. Czynnikami, które powodują indukcje enzymów mogą być insektycydy polichlorowe takie jak lindan i DDT czy też palenie papierosów.
Inhibicja enzymatyczna
Bardziej rozpowszechniona od indukcji enzymów jest ich inhibicja. Wynikiem inhibicji enzymatycznej jest zmniejszenie metabolizmu danego leku. Lek taki może zacząć kumulować się w organizmie, a efekt jest zazwyczaj tak samo istotny jak w przypadku gdybyśmy
zwiększyli dawkowanie. Inhibicja enzymatyczna występuje bardzo szybko. Zaraz po podaniu pierwszej dawki inhibitora blokuje on metabolizm równocześnie podanego leku. Najczęściej hamowanym szlakiem metabolizmu jest reakcja utleniania I fazy metabolizmu, która przebiega przy udziale izoenzymów cytochromu P450.
Leki, które powodują istotne klinicznie interakcje; Cymetydyna(Cimetidine, Altramed), Fluwoksamina(Fevarin), Tyklopidyna (Aclotin, Ticlo) Gemfibrozyl (Lipozil), Montelukast (Singulair), Fluoksetyna (Andepin, Bioxetin),Ketoconazol (Ketokonazol), Lansoprazol (Lanzul), Omeprazol(Gasec, Omar), Amiodaron (Cordarone, Opacorden), Flukonazol(Flumycon), Klomipramina (Anafranil, Hydiphen) Haloperydol(Haloperidol)
Metadon (Methadone Hydrochloride) ,Paroksetyna (Seroxat),Chinidyna (Chinidinum sulfuricum) Klarytromycyna(Klacid, Fromilid), Erytromycyna(Erythromycinum)
Diltiazem(Dilzem, Oxycardil),Fluwoksamina(Fevarin), Itrakonazol (Orungal
Sok z grejpfruta
Dla przykładu zastosowanie cymetydyny w dawce 800mg będącej nieswoistym inhibitorem cytochromu CYP3A4 jednocześnie z sildenafilem (Viagra) w dawce 50mg powoduje wzrost stężenia sildenafilu w surowicy o 56|%. Podobnie sok grejpfrutowy będący słabym inhibitorem CYP3A4 w ścianie jelit, może powodować nieznaczne zwiększenie stężenia sildenafilu w surowicy.
Klinicznie istotne interakcje inhibicji enzymatycznej zależą od zakresu, do którego zostanie podniesione stężenie leku w osoczu. Jeżeli poziom stężenia leku w osoczu pozostaje w zakresie terapeutycznym, interakcja taka może nie być istotna klinicznie.
Inhibicja lub indukcja efektu pierwszego przejścia.
W komórkach nabłonka jelit (enterocytach) znajdują się enzymy metabolizujące. Są to głównie izoenzymy cytochromu P450. Istnieją dowody, że niektóre leki mogą wywierać znaczny wpływ na stopień metabolizmu pierwszego przejścia poprzez inhibicję lub indukcję izoenzymów cytochromu P450 w ścianie jelit lub w wątrobie. Przykładem jest efekt działania soku grejpfrutowego. Zawarte w nim składniki (szczególnie należąca do fumarokumaryn - 6'7'dihydroksybergamotyna) hamują aktywność izoenzymu 3A4 cytochromu P450, głównie w jelicie cienkim. Wynikiem tego działania jest zmniejszenie metabolizmu statyn (np. symwastatyny), doustnych blokerów kanałów wapniowych (np. felodypiny, nifendypiny, nitrendypiny.), cyklosporyny, estradiolu, diazepamu, triazolamu, midazolamu.
W przypadku spożycia soku grejpfrutowego w ilości 240 ml efekt jest minimalny (13-to procentowy wzrost aktywności hamującej reduktazę HMG-CoA) i nie ma znaczenia klinicznego. Jednakże spożywanie bardzo dużych ilości soku (ponad 1 litr na dobę) znacznie zwiększa osoczową aktywność hamującą reduktazę HMG-CoA. Innym przykładem może być felodypina, w przypadku której picie soku grejpfrutowego może powodować nawet dwukrotne zwiększenie stężenia maksymalnego (Cmax) felodypiny.
Z powyższych powodów najsłuszniejszym postępowaniem przy zażywaniu leków jest ich popijanie czystą niegazowaną wodą.
Pomimo zmiany w ilości leku „wchłoniętego”, interakcje te są zazwyczaj rozpatrywane jako interakcje metabolizmu leków.
E - wydalanie leków
Leki mogą być wydalane z ustroju z moczem, żółcią, potem, mlekiem matki oraz z wydychanym powietrzem Najważniejszą drogą eliminacji leków z organizmu są nerki. Uszkodzenie nerek, związane ze zmniejszeniem przepływu nerkowego może znacznie zwalniać wydalanie leku, stąd konieczna jest modyfikacja jego dawkowania.
Wątroba jest drugim po nerkach narządem odgrywającym zasadniczą rolę w eliminacji leku. Biotransformacja w wątrobie nie ma na celu jak się to często uważa -,,odtruwania” organizmu , podstawowym jej celem jest przekształcenie związku w substancje bardziej hydrofilną łatwiej wydalana przez nerki. Niektóre leki są wydalane w znacznych ilościach z kałem, są to związki niekompletnie wchłaniane z przewodu pokarmowego. Wydalanie leków przez płuca i za pośrednictwem gruczołów wydzielania wewnętrznego odgrywa mniejsza rolę w procesie eliminacji leku z organizmu.
KLIRENS LEKU (CL) - objętość krwi oczyszczana z leku w jednostce czasu. Jest miarą wydajności eliminacji leku.
WYKAZ LEKÓW, KTÓRYCH WYDALANIE JEST UZALEŻNIONE GŁÓWNIE OD CZYNNOŚCI NEREK; Acyklovir ,Amikacyna ,Cefalorydyna .Cefazolina ,Colistin ,Doksycyklina
Gentamycyna, Streptomycyna .Wankomycyna ,Węglan litu
WYKAZ LEKÓW, KTÓRYCH WYDALANIE PRZEBIEGA GŁÓWNIE DROGĄ ŻÓŁCIOWĄ LUB INNYMI DROGAMI NIENERKOWYMI
5-fluorouracyl ,Acetaminofen, Azatiopryna ,Diazepam, Klindamycyna,Chinidyna ,Digitoksyna
Leki stosowane u matek karmiących mogą być wydalane z mlekiem, stąd mogą w mniejszy lub większy sposób wpływać na ustrój noworodka.
Do mleka może przedostać się z krwi matki wiele leków przy czym nabłonek pęcherzyków gruczołu sutkowego zachowuje się jak błona lipidowa rozdzielająca krew.Do mleka łatwo przenikają:
- dobrze rozpuszczalne w tłuszczach leki znieczulające ogólnie
- niektóre barbiturany
- nikotyna
- chloramfenikol
- leki p/zakrzepowe
- p/padaczkowe -
- p/cukrzycowe
- niektóre środki przeczyszczające
- benzodiazepiny i moczopędne tiazydy
- kofeina, salicylany - w ilościach nieznacznych
FARMAKOKINETYKA LINIOWA(I°) - opisuje procesy farmakokinetyczne, których szybkości są liniową funkcją stężenia, a wielkości parametrów farmakokinetycznych nie zależą od podanej dawki leku.
FARMAKOKlNETYKA NIELINIOWA (0°) - opisuje procesy farmakokinetyczne, których szybkości nie są liniową funkcją stężenia leku, lecz zmieniają się w sposób opisany przez równanie Michaelisa-Menten. Konsekwencją nieliniowości tych procesów jest zmienność parametrów farmakokinetycznych w zależności od podanej dawki leku.
Działania leków.
Każdy lek, każda substancja lecznicza wywiera określone działanie lecznicze tj. znosi przyczynę choroby, są to więc leki przyczynowe- etioterapeutyczne lub znosi objawy choroby i są to leki objawowe. Do leków działających przyczynowo zaliczamy antybiotyki i chemioterapeutyki, ponieważ niszcząc lub hamując drobnoustroje chorobotwórcze likwidują przyczynę choroby.
Leki przeciwkaszlowe, przeciwgorączkowe nie usuwają przyczyny choroby ale jej objawy, działają więc objawowo.
Leki mogą wykazywać działanie ośrodkowe dotyczy to OUN- ośrodkowego układu nerwowego lub obwodowe, na inne tkanki lub narządy poza OUN.
Miejscowe działanie leku wynika z ich miejscowego zastosowania na powierzchnię skóry lub błon śluzowych w postaci maści, zasypek, leki nie ulegają wchłonięciu do krwioobiegu.
Wybiórcze działanie leku polega na wyłącznym oddziaływaniu na czynność jednego narządu, tkanki czy receptor, np. metoprolol wykazuje wybiórcze działanie hamujące na receptory β1- adrenergiczne w mięśniu sercowym a nie działa na receptory β 2-adrenergiczne znajdujące się w oskrzelach.
Oprócz pożądanego działania terapeutycznego, które stanowi podstawę zakwalifikowania leku do danej grupy, może on wpływać także szkodliwie na czynność i strukturę narządów, mówimy wtedy o działaniu niepożądanym lub toksycznym.
Działania niepożądane zgodnie z definicją Światowej Organizacji Zdrowia (WHO) to szkodliwy, niezamierzony skutek działania leku obserwowany w dawkach, które służą zapobieganiu , diagnozie, leczeniu, lub modyfikacji czynności fizjologicznych. W tej definicji nie rozważa się następstw przedawkowania leków lub umyślnego czy też przypadkowego zażycia, chodzi o to że działania niepożądane leku są charakterystyczne dla konkretnego leku i mogą wystąpić lecz nie muszą, wówczas gdy jest on stosowany w dawce leczniczej, zaleconej przez lekarza, zgodnie ze wskazaniami.
Działania niepożądane leków są bardzo różne i zależą od wielu czynników. W zasadzie nie istnieją leki całkowicie pozbawione tego typu działań. O przydatności leku w terapii konkretnego schorzenia decyduje bilans korzyści i strat wywoływanych przez ten lek.
Klasyfikacja kliniczna działań niepożądanych(wikipedia)
Typ A. Działania uboczne, które można przewidzieć - wynikają z mechanizmu działania farmakologicznego. Ich nasilenie zależy od zastosowanej dawki. Ustępują po zmniejszeniu dawki lub odstawieniu leku. Przykłady: Kaszel po lekach obniżających ciśnienie krwi z grupy I-ACE, krwawienia po przedawkowaniu leków przeciwzakrzepowych
Typ B. Działania uboczne nieprzewidywalne, niezależne od podanej dawki leku. Działania te nie wynikają z mechanizmu działania farmakologicznego. Często są wynikiem reakcji immunologicznej (uczulenia) na lek. Przykłady: uszkodzenia szpiku kostnego po zastosowaniu chloramfenikolu,
zespół Stevesna-Johnsona po sulfonamidach, agranulocytoza po metamizolu
wstrząs anafilaktyczny po penicylinie,pokrzywka po ampicylinie, Działania tego typu stanowią ok. 20% wszystkich obserwowanych działań niepożądanych leków i często są przyczyną wycofania leku z obrotu, lub ograniczeń w jego stosowaniu.
Typ C. Działania uboczne występujące po długotrwałym stosowaniu leku (najczęściej w terapii chorób przewlekłych). Mechanizm ich powstawania najczęściej jest znany i wynika z właściwości farmakologicznych leku. Przykłady; uszkodzenie wątroby po przewlekłym stosowaniu paracetamolu, choroba wrzodowa po przewlekłym stosowaniu NLPZ-tów,
Choroba zakrzepowa występująca u kobiet zażywających doustne środki antykoncepcyjne. Badania epidemiologiczne wykazały, że stosowanie pigułek antykoncepcyjnych zwiększa ryzyko wystąpienia choroby zakrzepowej, ale w przypadku indywidualnej pacjentki nie można udowodnić, że środek antykoncepcyjny spowodował chorobę.
Typ D. Działania uboczne pojawiające się po długim czasie od zastosowania leku, niezależnie od tego jak długo stosowany był sam lek, a nawet po jego zakończeniu. Przykłady: nowotwory narządów rodnych u kobiet, których matki w czasie ciąży leczone były stilbestrolem,działanie teratogenne np. talidomidu, leflunomidu, rzekomobłoniaste zapalenie jelit po antybiotykoterapii
Typ E. Działanie niepożądane występujące po nagłym odstawieniu leku. Zwykle jest to po prostu zaostrzenie choroby, następujące po zaniechaniu terapii. Leki wykazujące działania niepożądane typu E powodują zwiększenie (zaostrzenie) objawów choroby po odstawieniu leku ponad poziom na którym te objawy byłyby, gdyby leku w ogóle nie zastosowano. Przykłady: zwiększenie poziomu gastryny prowadzące do wyrzutu kwasu solnego w żołądku po odstawieniu antagonistów receptorów H2 w żołądku (leków hamujących wydzielanie kwasu solnego)
Tego typu działaniom niepożądanym leków łatwo zapobiec - wystarczy odstawiać leki powoli, stopniowo zmniejszając dawkę.
Ośrodek Monitorowania Działań Ubocznych Leków powstał w 1971 r., powołany przez Ministerstwo Zdrowia i Opieki Społecznej. Obecnie jednostka pracuje w ramach Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych jako Wydział Monitorowania Niepożądanych Działań Produktów Leczniczych.
Zgodnie z obowiązującymi przepisami w zgłaszaniu ndl powinni brać udział lekarze, farmaceuci i pielęgniarki i położne.
Główną rolę powinni pełnić lekarze rodzinni, jako osoby mogące „całościowo” przyjrzeć się swoim pacjentom, zebrawszy informacje o wszelkich zalecanych przez specjalistów terapiach oraz o produktach, jakie chorzy zdecydowali się przyjmować w ramach samoleczenia.
Zobowiązują do tego następujące akty prawne: Ustawa z dnia 5 grudnia 1996 o zawodzie lekarza, Ustawa z dnia 6 września 2001 Prawo Farmaceutyczne, Rozporządzenie Ministra Zdrowia z dnia 17 lutego 2003 w sprawie monitorowania bezpieczeństwa produktów leczniczych, Dyrektywa Unii Europejskiej 2001/83/EC.
Działanie niepożądane powinno być zgłoszone podmiotowi odpowiedzialnemu za wprowadzenie na rynek leku lub substancji, a w przypadku niemożności jego ustalenia do Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych. Zgłoszenia krajowe są następnie przekazywane do międzynarodowej bazy danych, która mieści się w Uppsali w Szwecji.
Podstawowym sposobem jest obserwacja i zgłaszanie ndl. W tym celu można posłużyć się formularzem -„żółtą kartą” opracowaną przez Wydział lub międzynarodową kartą CIOMS (formularz do zgłaszania pojedynczych przypadków niepożądanych działań produktów leczniczych, opracowany przez Radę Międzynarodowych Towarzystw Medycznych CIOMS - Council for International Organizations of Medical Sciences) - w wersji polskiej lub angielskiej.
Formularze o niepożądanych działaniach leków prosimy przesyłać na adres:
Wydział Monitorowania Niepożądanych Działań Produktów Leczniczych
Urząd Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych.
ul. Ząbkowska 41
03-736 Warszawa
tel. (022) 49 21 300 oraz (022) 49 21 301; fax (022) 49 21 309
Nie są honorowane raporty od pacjentów, nie potwierdzone prze lekarza sprawującego opiekę nad chorym.
Należy zgłaszać ndl po lekach zarejestrowanych w Polsce:
-wszystkie ciężkie reakcje, jakie wystąpiły na terenie Polski w ciągu 15 dni kalendarzowych od daty powzięcia informacji o ich wystąpieniu
-wszystkie ciężkie i jednocześnie niespodziewane reakcje, które wystąpiły poza terenem naszego kraju w ciągu 15 dni kalendarzowych od daty powzięcia informacji o ich wystąpieniu (dotyczy wytwórcy leku)
-wszystkie pozostałe działania niepożądane włączone do okresowego raportu o bezpieczeństwie (dotyczy wytwórcy leku).
Jakie reakcje uznaje się za ciężkie ?
Jest to każde niezamierzone działanie produktu leczniczego, które po podaniu człowiekowi jakiejkolwiek dawki powoduje: zgon pacjenta, zagrożenie życia, konieczność leczenia szpitalnego lub jego przedłużenie, trwałe lub znaczne inwalidztwo, wady rozwojowe płodu.
Pod uwagę należy brać także inne działania niepożądane leków takie jak;
- reakcje uczuleniowe, które są wyrazem nadwrażliwości organizmu na niektóre leki np. antybiotyki, surowice. Reakcje uczuleniowe mogą mieć charakter miejscowych zmian( pokrzywka, wysypka0 lub ogólnych(opóźnone lub natychmiastowe).Objawy mogą dotyczyć układu nerwowego, układu krążenia, oddechowego, pokarmowego, nie mające związku z zasadniczym kierunkiem działania leku.
- działanie na płód, przed zastosowaniem leku należy rozważyć czy lek może przenikać do płodu i czy może działać teratogennie , najbardziej niebezpieczny dla płodu jest pierwszy trymestr ciąży.
Z danych statystycznych wynika ze większość kobiet w czasie ciąży stosuje leki, w tym 5 % stanowią leki o udowodnionym działaniu uszkadzającym płód. Przystosowanie organizmu matki do przebiegu ciąży i porodu związane jest ze zmianami czynności wielu układów i narządów. W ustroju ciężarnych tworzy się dodatkowy kompartment(płód) o właściwościach konmpartmentu wolnowywiennego. Ciąża to faza życia kobiety wymagająca szczególnej ostrożności podczas stosowania leków. Istniejąca triada matka-płód- łożysko stanowi biologiczną , farmakokinetyczna i farmakodynamiczna całość.
W działaniu farmakologicznym leku u płodu musimy uwzględniać kinetykę jego przemian w organizmie matki jak i w obrębie łożyska. Rozmieszczenie leku w organizmie wraz z jego metabolizmem w łonie matki warunkują dostępność leku dla płodu. Lek z organizmu matki przechodzi do płodu z krwi tętniczej przez przestrzenie międzykosmkowe do naczyń włosowatych płodu znajdujących się w kosmkach i dalej przechodzi do płodu przez żyłę pępowinową.
Wpływ leków na farmakokinetykę; zwiększenia pojemności wyrzutowej serca i przepływu przez nerki, zwiększenie eliminacji przez nerki, zwiększenie metabolizmu wątrobowego, spadek stężenia białek krwi-III trymestr , osłabienie wiązania z białkami, zwiększenia wolnej frakcji leków, następuje hormonalna indukcja aktywności enzymów zależnych od cytochromu P-450.
Lożysko jest tkanką aktywna metabolicznie. Transport leków przez łożysko odbywa się na zasadzie dyfuzji prostej, dyfuzji ułatwionej i pinocytozy.
Szkodliwy wpływ na płód zależy m.in. od ;genetycznej wrażliwości zarodka, okresu ciąży, dawki i czasu podawnia leków, chorób przewlekłych, niedoborów kwasu foliowego, żelaza, czynników matczynch;wiek <17 rż i >35 rż.
Uszkodzenie płodu może nastąpić w okresie ;
- blastogenezy(pomiędzy zapłodnieniem a 18 dniem ciąży), obumarcie zarodka , wady rozwojowe ( bliźnieta syjamskie)
- embriogenezy( (z greckiego embrion - zarodek) - niepożądane działanie występujące po podaniu leku u kobiet w ciąży w okresie embrionalnym (do ok. 60 dnia ciąży-12 tydzień), największe narażenie teratogenne. Każdy z narządów się tworzących osobno ma własny czas różnicowania i rozwoju, jednak czasami okresy te ulegają nałożeniu z czego wynika powstawanie mnogich wad rozwojowych typu ciężkich uszkodzeń( rozszczep kręgosłupa, wady serca i naczyń)
--fetogenezy- (od 12 tygodnia do porodu-2 ostatnie trymestry ciąży) dosłownie potworotwórczość od teratos - potwór - właściwość teratogenów powodująca wady (potworności) w rozwoju płodu (mają ją np. promieniowanie, wirusy, ksenobiotyki, niektóre lekarstwa). W okresie płodowym kształtuje się głównie układ płciowy i zachodzi końcowe różnicowanie i dojrzewanie OUN. Wówczas toksyczne działanie leków stosowanych w tym okresie może spowodować upośledzenie umysłowe, zaburzenia rozwoju i czynności płciowych.
Do leków o działaniu teratogennym należą; cytostatyki, leki przeciwpadaczkowe(fenytoina, karbamazepina, kwas walproinowy)sole litu, retinoidy, alkohol!!!
Klasyfikacja FDA leków stosowanych w ciąży - klasyfikacja mająca na celu określenie ryzyka uszkodzenia płodu w wyniku działania leku, który jest przyjmowany przez matkę w ciąży zgodnie z zaleceniem. Klasyfikacja nie zawiera żadnych informacji o ryzyku związanym z lekiem i jego metabolitami zawartymi w mleku matki.
Klasyfikacja FDA leków stosowanych w ciąży
Kategoria Definicja
A Badania z grupą kontrolną nie wykazały istnienia ryzyka dla płodu w I trymestrze, możliwość uszkodzenia płodu wydaje się mało prawdopodobna
B Badania na zwierzętach nie wskazują na istnienie ryzyka dla płodu, ale nie przeprowadzono badań z grupą kontrolną u ludzi, lub badania na zwierzętach wykazały działanie niepożądane na płód, ale badania w grupie kobiet ciężarnych nie potwierdziły istnienia ryzyka dla płodu
C Badania na zwierzętach wykazały działanie teratogenne lub zabójcze dla płodu, ale nie przeprowadzono badań z grupą kontrolną kobiet, lub nie przeprowadzono odpowiednich badań ani na zwierzętach, ani u ludzi
D Istnieją dowody na niekorzystne działanie leku na płód, ale w pewnych sytuacjach klinicznych potencjalne korzyści z jego zastosowania przewyższają ryzyko (np. w stanach zagrażających życiu lub chorobach, w których inne, bezpieczne leki nie mogą być zastosowane lub są nieskuteczne)
X Badania przeprowadzone na zwierzętach lub u ludzi wykazały nieprawidłowości płodu w wyniku stosowania danego leku bądź istnieją dowody na niekorzystne działanie leku na płód ludzki i ryzyko zdecydowanie przewyższa potencjalne korzyści z jego zastosowania
- działania rakotwórcze , właściwości rakotwórcze- karcynogenne mogą wykazywać leki przeciwnowotworowe, immunosupresyjne( azatopiryna z prednizolonem, jest związana z ryzykiem chłoniaków), niektóre sulfonamidy, związki metali -arsenu, chromu, niklu, ołowiu, kobaltu. Nowotwory wywołane przez leki występują prawdopodobne bardzo rzadko, ponieważ przemysł farmaceutyczny stara się nie dopuścić do obrotu środków rakotwórczych. Mechanizmy zaangażowane w chemiczna karcynogenezę są zazwyczaj nieznane.
-Działanie toksyczne występują w wyniku przedawkowania leku, czyli przekroczenia maksymalnej dawki leczniczej. Terapia monitorowana.!!
W Wielkiej Brytanii najbardziej pospolitymi lekami powodującymi zgony w wyniku samozatrucia są trójpierścienowe leki przeciwdepresyjne, paracetamol. Nierzadko występują samozatrucia z wykorzystaniem 2 lub więcej leków i alkoholu(50% przypadków). Przypadkowe samozatrucie występuje głównie u małych dzieci, (poniżej 5r.ż)i zazwyczaj jest spowodowane przez leki lub środki chemiczne używane w gospodarstwie domowym(np. wybielacze) pozostawione w zasięgu ręki. Chorzy z objawami zatrucia musza być poddania wstępnej ocenie ,ważne jest wykluczenie innych przyczyn śpiączki lub nienormalnego zachowania (padaczka, urazy głowy, cukrzyca).Większość chorych wymaga leczenia w warunkach szpitalnych.
Mechanizmy działania leku.
Pod pojęciem mechanizm działania leku rozumiemy jego wpływ na żywy organizm, w wyniku czego dochodzi do zmian czynności komórek i tkanek organizmu i w efekcie do powstania określonego działania farmakologicznego.
Leki mogą wywierać działania o charakterze fizykochemicznym, zmieniając np. przepuszczalność błon komórkowych, tak działają środki neutralizujące kwas solny w żołądku, osmotyczne środki przeczyszczające, niektóre leki znieczulające ogólnie, które powodują fizykalne zmiany błon komórkowych, niektóre leki np. kurara powodują zmiany ładunku elektrycznego komórki, niektóre leki np. kurara powodują zmiany ładunku elektrycznego komórki, porażając mięśnie prążkowane.
Mechanizm biochemiczny polega na reakcji chemicznej zachodzącej pomiędzy lekiem a receptorem, którym może być enzym- struktura białkowa, która działa w organizmie żywym jak katalizator lub substrat- związek który stanowi materiał wyjściowy w reakcji katalizowanej enzymatycznie. Wynikiem tej reakcji jest zmiana czynności komórki, narządu na skutek hamowania lubnasilenia aktywności enzymów np. fizostygmina hamuje aktywność acetylocholinoesterazy co powoduje zwiększenie poziomu nierozłożonej przez ten enzym acetylocholiny, która to działając na odpowiednie receptory zmienia czynność narządów.
Siła działania większości leków jest wynikiem ich wiązania z receptorami błonowymi a w przypadku hormonów z receptorami wewnątrzkomórkowymi i jest to mechanizm receptorowy. Leki łącząc się z receptorami prowadzą do zmiany procesów metabolicznych komórek i tym samym wpływają na funkcje poszczególnych tkanek i narządów. Zgodnie z teorią receptorową warunkiem działania leku jest jego reakcja z białkiem komórki -receptorem , w wyniku czego dochodzi do zmian jej czynności.
Receptorem nazywamy swoiste miejsce wiązania leku z komórką zazwyczaj na jej powierzchni które pośredniczą w działaniu leku. Większość receptorów to białka transbłonowe, stanowiące miejsca wiązania naturalnych endogennych przekaźników; serotoniny, adrenaliny. Wiązanie przekaźnika powoduje zmianę kształtu receptora i jest to początek procesu w wyniku którego do komórki dociera odpowiedni sygnał, dzięki temu można wpłynąć na zmianę funkcjonowania organizmu. W ustroju człowieka występuje wielka liczba rozmaitych receptorów, odzdiałujących z różnymi chemicznymi przekaźnika np. receptory adrenergiczne aktywowane przez adrenalinę lub dopaminergiczne aktywowane przez dopaminę. Każdy typ receptora reaguje selektywnie na jeden rodzaj przekaźnika, ponieważ ich miejsca wiążące różnią się kształtem, struktura i składem aminokwasów. Leki ,,wykorzystują” te same receptory, naśladując działanie endogennych neuroprzekaźników lub hamując ich działanie. Lek aby zadziałać musi osiągnąć odpowiednie stężenie i zdolność wiązania się z receptorem czyli powinowactwo do receptora. Leki o dużym powinowactwie łatwo łączą się z receptorem i mogą wypierać z połączeń z receptorem leki o mniejszym powinowactwie. Aby lek mógł wpływać na czynność komórek oprócz powinowactwa musi posiadać aktywność wewnętrzną.
Związki łączące się z receptorem, czyli mające powinowactwo nazywamy ligandami. W wyniku związania się liganda z receptorem może dojść do pobudzenia lub zablokowania receptora.
Pobudzenie następuje wtedy gdy w wyniku reakcji ligand- receptor dochodzi do zmiany kształtu receptora i uruchomienia procesów fizykochemicznych dających w końcowym efekcie zmianę potencjału błony komórkowej, procesów metabolicznych komórki lub ekspresji genów.
Blokada następuje wówczas gdy ligand przyłączy się do receptora ale nie zmienia jego konfirmacji i uniemożliwia endogennemu przekaźnikowi łączenie się z receptorem.
Ligandy pobudzające receptory to agoniści a blokujące receptory to antagoniści.
Antagonizm farmakologiczny występuje wtedy gdy antagonista uniemożliwia agoniście połączenie się z receptorem w celu wywołania efektu farmakologicznego.
Rozróżniamy;
Antagonizm konkurencyjny-kompetencyjny, gdy antagonista konkurencyjnie współzawodniczy w sposób odwracalny z agonistą o ten sam receptor np. noradrenalina z propranololem o receptor adrenergiczny.
Antagonizm niekonkurencyjny -niekompetencyjny, gdy antagonista i agonista mają inne punkty uchwytu działania. Nie ogranicza się on do receptorów w obrębie jednego narządu, ale ma szerszy zakres działania. np. zmniejszone oddziaływanie acetylocholiny w obecności leku przeciwhistaminowego- mepipraminy.
Antagonizm czynnościowy gdy zastosujemy dwie substancje o różnych punktach uchwytu i wywołujące przeciwstawne efekty. np. noradrenalina zwęża naczynia a acetylocholina je rozszerza, czyli działanie to zachodzi przez różne receptory.
Biorąc pod uwagę budowę i biochemiczny mechanizm działania receptory dzielimy na;
• receptory metabotropowe, są to receptory β- α -adrenergiczne oraz cholinergiczny receptor muskarynowy.
• receptory jonotropowe występujące w OUN i zwojach układu autonomicznego.
Niektóre receptory narażone na powtarzające się działanie agonisty czyli leków pobudzających, mogą stać się niewrażliwe lub ich liczba może się zmniejszyć, mówimy wtedy o ujemnej regulacji(down regulation).
Natomiast przy przewlekłym podawaniu antagonisty czyli leku blokujacego receptory może dojść do zwiększenia wrażliwości receptorów lub ich liczby i mówimy wtedy o dodatniej regulacji (up regulation).
Interakcje leków.
Interakcja (interferencja) polega na wzajemnym oddziaływaniu leków w żywym organizmie, oznacza wpływ jednego leku na końcowy wynik działania drugiego równocześnie stosowanego leku .Skutków działania wielu leków, zwłaszcza wówczas gdy są podawane jednocześnie , nie można przewidzięć znając jedynie działanie każdego z nich z osobna. Wzajemne oddziaływanie leków ma istotne znaczenia w terapii, może dojść z jednej strony do korzystnego zwiększenia siły działania farmakodynamicznego podanych równocześnie preparatów leczniczych i w końcowym efekcie poprawić efekt terapeutyczny z drugiej strony może dojść do osłabienia lub zniesienia działania jednego leku przez drugi lek, co może być przyczyna wystąpienia silnych objawów niepożądanych. Częstość występowania interakcji leków zależy bezpośrednio od liczby stosowanych jednocześnie leków. Przy kilku lekach wynosi ona 3-5 % w przypadku stosowania dziesięciu lub więcej leków, może wzrosnąć do 20%.
Polipragmazja - termin medyczny określający sytuację, w której chory przyjmuje więcej niż kilka leków jednocześnie. Jest to jeden z częstszych błędów w leczeniu, prowadzący m.in. do znacznego zwiększenia występowania niezamierzonych interakcji lek-lek lub lek-pożywienie.
Rozróżnia się trzy typy interakcji;
- farmaceutyczną
- farmakodynamiczną
- farmakokinetyczną.
Interakcje farmaceutyczne występują podczas przygotowywania leku, przed podaniem choremu. Na te interakcje składają się niezgodności recepturowe fizyczne i chemiczne.
Interakcje farmakokinetyczne dotyczą wpływu jednego leku na procesy farmakokinetyczne drugiego leku, czyli na wchłanianie, wiązanie z białkami, transport przez błony biologiczne, biotransformację, wydalanie.
(Np. -interakcje na etapie dystrybucji) temperatura(wzrost temperatury ciała obniża zdolność wiązania leków z białkami).
Po podaniu choremu kilku leków dochodzi di ich wzajemnego wypierania się z połączeń z białkami. Leki które uzyskują we krwi większe stężenie lub maja większą zdolność wiązania się z białkami, wypierają leki o mniejszym powinowactwie do białek i leki mające mniejsze stężenie we krwi. Lek wyparty z wiązania z białkami ma zwiększone stężenie frakcji wolnej i dlatego zwiększa się jego aktywność farmakologiczna, może to doprowadzić do wystąpienia działań niepożądanych. Wyparcie leku z wiązań z białkami powoduje jego szybsza eliminacje z krwioobiegu. Kwaśny odczyn leku sprawia że zwiększa się jego powinowactwo do białek. Leki te zwane są ,,wypieraczami” np. salicylany, wypierają leki o mniejszym powinowactwie. Kwas acetylosalicylowy wypiera z białek sulfonamidy zwiększając ich aktywność;
Interakcje farmakodynamiczne (koergizm).
W wyniku zastosowania leków równocześnie może dojść do wystąpienia synergizmu lub anatgonizmu w ich działaniu. W przypadku synergizmu leków używa się także pojęcia- potęgowanie działania leków. Jeśli synergizm wystąpi w wyniku działania leków na ten sam receptor, to jest to synergizm swoisty, jeśli działanie obejmuje różne receptory to jest to synergizm czynnościowy.
Rozróżniamy pojęcia;
-synergizm addycyjny, polega na tym że efekt działania dwóch lub więcej leków podanych razem jest równy sumie działania poszczególnych składników np. mieszaniny leków nasennych i przeciwbólowych;
- synergizm hiperaddycyjny, występuje wtedy gdy efekt działania leków podanych razem jest większy niż suma działania poszczególnych składników np. jony wapnia i glikozydy nasercowe.
W zakresie antagonizmu wyróżnia się pojęcia;
- antagonizm konkurencyjny( kompetencyjny) zachodzi wówczas, gdy da leki agonista i antagonista współzawodniczą o wiązanie z tym samym receptorem, np. nalokson i morfina o recepto opioidowy.
- -antagonizm czynnościowy(funkcjonalny)polega na tym że dwa leki o różnym punkcie uchwytu wywołują przeciwny efekt np. naradrenalina
Literatura; ,,Farmakologia'' pod red. G. Rajtar -Cynke wyd, Czelej
,,Chemia leków'' G. Patrick wyd. PWN
,, Farmakologia w zarysie; tłum. W Janiec wyd. PZWL
,, Farmakologia kliniczna” pod red. A Chodery ,Z.S Hermana
Int. Wikipedia
Czasopismo aptekarskie Nr 10(20009) A. Zimmermann,, Zgłaszanie działan niepożądanych produktów leczniczych -obowiązek farmaceuty”