P1100173

16.7. SPEKTRALNA ANALIZA ILOŚCIOWA podstawą spektralnej analizy ilościowej jest i?w. równanie Łonutkina~SchęłbeQ0 ofcre>Mjaet? zależność między procentowym siotcniem (f) pierwiastka o mlrnsnr. noścłą (O jego Unii
1 “ **f* (M.tZ)
gdzie u, b — stałe dla danej linii spektralnej i danych warunków doiwiadczalnyd Srała b zalety od procesu wzbudzania w źródle i jest określona szcęcgćfilfe reabsorpcją własnego promieniowania przez nie wzbudzone atomy pierwiastka. Może ociągać maloysaafną wartość I dla linii powstałej przy przeskoku z poziomu W.-cowcgo, stosunkowo wy so kocnergctyczncgo E>la linii intcn<ywniej*zycb w kcn*o fcu-encji własnej absorpcji wartość stałej b speda poniżej jedności. Stała o jest zw% rana z przejściem pierwiastka z próbki w stanie stałym lub ciekłym do plazmy.
Przy za łożeniu stałych warunków wzbudzenia energia emitowana przez ara li* /owany pierwiastek jest proporcjonalna do liczby jego atomów wzbudzonych, a więc także do jego stężenia w próbce.
Postaci próbki do celów ilościowych należy poświęcić wiele uwagi, ponieważ we wzbudzeniu bierze udział tylko niewielka część substancji i analiza ma raczej charakter analizy lokalnej.
Metale są najczęściej analizowane w postaci walcowatych prętów, przy czym jest ważny kształt i sposób obróbki końców elektrod, który powinien być bliski tzw. kształtowi normalnemu (rys. I6.7a).
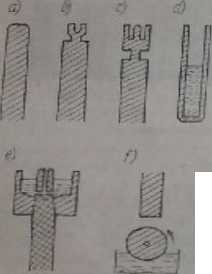
Rys. H5.7. Niektóre kształty elektrod stosowanych w emisyjnej analizie spektralnej: a) elektroda metalowa o normalnym karuk eie. b, c) elektrody do analizy materiałów proszkowych, <0 elektroda > eldmanna r porowatym dnem do analizy roztworów, c) elektroda kapilarna według Zinka, f) układ elektrod w metodzie wirującego dysku
W analizie metali i stopów, szczególnie jeżeli do wzbudzenia używa się wyła-dowanjii iskrowego, jest potrzebny określony czas, aby ustaliło się równowaga mię* tlzy $ tężeniem oznaczanych pierwiastków w próbce i ich stężeniem w plazmie. Ten czas, kiedy widma nic są jeszcze zdejmowane, nazywa się czasem przedisk rżenia, a sam proces - przed i skrzeniem. Czas przcdi skrzenia rzadko przekracza 1 min i określa się go z tzw. krzywych przediskrzenia, tj. graficznej zależności stosunku
intensywności dwóch analitycznych linii (humaloguwnej pary linii) od czasu (cras liczy się cd momentu włączenia iródła wzbudzeniu). Kształt krzywych przediskrze* nia w okresie przodiskrzenia znacznie zmienia się w czasie, a ich ustalony przebieg w dalszym przedziale czasu określa, kiedy można zacząć właściwą ekspozycję. W okresie przediskrżeniu jednocześnie powinien być osiągnięty stan, kiedy można założyć, że próbka jest już przeprowadzona w tzw. średnią próbkę.
Materiały nicprzewc d/ące, najczęściej w postaci proszków, odparowuje się Z zagłębień elektrod węglowych (rys. 16.7b, c). Proszki przed analizą rozdrabnia się do potrzebnej ziarnistości i miesza się z substancjami pomocniczymi, które mają na celu wyeliminowanie wahań temperatury źródła, podwyższenie przewodności, ewentualnie mają jeszcze inne działanie. Do próbek proszkowych mogą również być stosowane sposoby ciągłęgo wprowadzania proszku do łuku poziomego lub pionowego.
Roztwory są analizowane po odparowaniu na powierzchniach ciekited węglowych lub metalowych. Lepszo wyniki otrzymuje się przez zastosowanie różnych typów elektrod kapilarnych, w których roztwór ul bo przesiąka przez porowate dno (rys, I6,7d), albo dostaje się na powierzchnię elektrody wskutek kapilarnego przenoszenia kanalikami elektrody. Najlepsza dla roztworów jest jednak metoda wirującego dysku (rys. 16.70* Analizowany roztwór można wprowadzać do łuku również W postaci aerozolu otrzymanego za pomocą różnych typów rozpylaczy.
16.7.1. Metody pńlilościowe
Do wyznaczania przybliżonej zawartości danego pierwiastka w próbce często stosuje się mniej doki: dne metody — tzw. pćłilcściowe. Ich średni błąd waha się w granicach ± 10 20% lub dostarczają one tylko informacji o granicach stężenia, co w wielu przypadkach całkowicie wystarcza (np. zawartość Cr w stali stopowej w granicach 0.2-0,35 %; w granicach 1,5-2% itp.).
Najprostszy sposób półilościowcj oceny zawartości pierwiastka w próbce polega ni porównywaniu widm. Razem z widmem badanej próbki fotografuje się kilka wzorców o różnych stężeniach i wizualnie za pomocą projektora określa się rząd wielkości poszukiwanego stężenia, przyrównując do stężenia tego wzorca, którego intensywność określonej linii oznaczanego pierwiastka jest najbliższa intensywności tej samej linii w próbce.
W tak zwanej metodzie linii ostatnich wykorzystuje się znnne już stwierdzenie, że linie ostatnie nio występują w widmie, jeżeli stężenie pierwiastka spada poniżej określonej granicy, ewentualnie dlsi określonych stężeń występuje tylko określona, ograniczona liczbo linii. Z tablic linii ostatnich z odpowiadającymi im zakresumi stężeń pierwiastków można ocenić rząd wielkości stężenia, tj. rozróżnić stężenia 0,1; 0,01; 0,001 %. Dla większych stężeń metoda tn nie jest dogodna, ponieważ odczyt i identyfikacja dużej liczby linii są trudne.
Dokładniejsza niż poprzednie dwie metody jest meteda homologicznych par Unii. Homologiczną parą Unii nazywa się dwie linie spektralne, które nnleżą do d" óch różnych pierwiastków. Z reguły mają one podobny mechanizm wzbudzenia, a wartość ich intensywności zależy tylko od stężenia pierwiastków, w małym stopniu od
217
Wyszukiwarka